

Abstract
Solid-phase resins functionalized with poly-deoxythymidine (dT) oligos facilitate purification of poly-adenylated molecules from solution through high affinity, high selectivity base-pairing interactions. These resins are commonly used to purify messenger RNA (mRNA) from complex biological mixtures as well as mRNA-protein fusion molecules for mRNA Display selections. Historically, dT-conjugated cellulose was the primary resin for poly-dA purification, but its scarcity has prompted the development of alternative resins, most notably dT-functionalized magnetic beads. In order to develop a cost-effective alternative to commercially available poly-dT resins for large-scale purifications of mRNA-protein fusions, we investigated the purification properties of dT25-conjugated Oligo Affinity Support resin (dT25-OAS) alongside poly-dT14 magnetic beads and dT25-cellulose. dT25-OAS was found to have the highest dA21 oligo binding capacity at 4 pmol/μg, followed by dT14-magnetic beads (1.1 pmol/μg) and dT25-cellulose (0.7 pmol/μg). To determine the resin specificity in the context of a complex biological mixture, we translated mRNA-protein fusions consisting of a radiolabeled Her2 affibody fused to its encoding mRNA. Commercial dT25-cellulose showed the highest mRNA-affibody purification specificity, followed by dT25-OAS and dT14-magnetic beads. Overall, dT25-OAS showed exceptionally high binding capacity and low background binding, making it an attractive alternative for large-scale mRNA purification and mRNA Display library enrichment.
Keywords: dT purification, OAS resin, mRNA Display, Affibody
Graphical Abstract
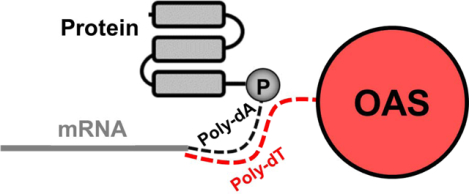
Purification of poly-adenylated molecules is a key step in several laboratory workflows. Typically, a solid-phase resin functionalized with poly-deoxythymidine (dT) binds tightly to polyadenylated molecules via complementary base-pairing. After washing, poly-A-containing molecules are selectively eluted in water or mild base. dT-cellulose was originally developed for the purpose of purifying nucleotides from cell lysates1. Since then, its use has expanded to the purification of mRNA for the preparation of cDNA libraries2, detection of microbial contamination in water samples3 and for purification of mRNA-protein fusion molecules in mRNA Display selections4–6.
In mRNA Display, dT-based purifications are essential for the isolation of mRNA-protein fusions from complex in vitro translation mixtures. DNA libraries are transcribed into mRNA libraries which are then ligated to a poly-dA linker terminated at the 3’ end with puromycin. When RNA-DNA-puromycin templates are translated in vitro, fusion of the nascent peptide with its encoding mRNA is facilitated by entry of the pendant puromycin in the A site of the ribosome and subsequent amide bond formation with the C-terminus of the protein. mRNA-protein fusions are then isolated by binding of the poly-dA linker to a poly-dT resin followed by elution in water or mild base4. In order to preserve library complexity, this poly-dT purification step needs to be nearly quantitative and elution must be gentle enough to preserve the integrity of the labile mRNA component.
In recent years, low demand for dT-cellulose has resulted in diminished commercial availability. A low-cost synthesis of dT-cellulose was recently reported7 although this requires access to oligonucleotide synthesizers which may be unavailable for this purpose. Alternatively, dT-conjugated magnetite beads have become commercially available and eliminate the need for centrifugation8. However, this option can be prohibitively expensive for large-scale mRNA purifications and isolation of high-diversity mRNA Display libraries.
We sought to determine the utility of poly-dT-conjugated Oligo Affinity Support resin (dT25-OAS) as a cost-effective alternative for purification of poly-A-containing molecules. OAS resins are commercially available polystyrene supports in which the 3’ terminal nucleotide is covalently tethered to the resin via the nitrogenous base9. The oligo can then be synthesized through standard phosphoramidite chemistry to the desired length10. During the terminal deprotection step, the oligonucleotide-resin linkage remains intact allowing solid-phase display of the newly-synthesized oligonucleotide. OAS resins have been previously used for the display of complex DNA structures and subsequent affinity purification of DNA-binding ligands11.
dT25-OAS was synthesized by phosphoramidite chemistry and deprotected with 10% (v/v) diethylamine in acetonitrile to remove the cyanoethyl protecting groups on the phosphate backbone. The binding capacity of the resin-bound oligo was measured and compared with dT25-cellulose and dT14-magnetic beads. In order to measure resin selectivity, all three resins were incubated with [35S]-labeled mRNA-protein fusions in rabbit reticulocyte lysate and the selective binding and elution of mRNA-protein fusions measured by scintillation counting. Of the three resins tested, dT25-OAS showed the highest binding capacity and minimal non-specific binding indicating its suitability for large-scale purification of poly-adenylated oligonucleotides from complex biological mixtures.
A poly-deoxyadenosine binding assay was used to determine the binding capacity of dT25-OAS, dT25-cellulose, and dT14-magnetic beads. 1–50 μg resin was incubated with 83 pmol of dA21 ssDNA in binding buffer with agitation for 1 hour at room temperature. The resin samples were pelleted and the absorbance of the supernatant measured at 260 nm. The amount of dA21 bound to the dT-resin was calculated, graphed against input resin mass, and the slope of the linear regression used to calculate binding capacity (Figure 1A). dT25-OAS was found to have a binding capacity of 4.0 pmol/μg which was significantly higher than dT14-magnetic beads (1.1 pmol/μg) and dT25-cellulose (0.7 pmol/μg). A time course then was used to determine the rate of dA21 oligo binding to each resin (Figure 1B). The dT14-magnetic beads reached 90% binding at 0.7 min while dT25-OAS and dT25-cellulose reached capacity at 13.3 min and 13.5 min, respectively. Elution efficiency was evaluated by treating resin-bound dA21 oligo with 1 mM NaOH. dT14-magnetic beads had the highest elution efficiency (44.8%), followed by dT25-cellulose (39.8%) and dT25-OAS (34.3%, Figure 1C). Although we observed modest differences in the dA21 elution efficiency of each resin, these differences did not rise to the level of statistical significance as assessed by one-way ANOVA.
Figure 1. Poly-dA binding properties of dT-resins.

A: The binding capacity of each resin was measured using a poly-deoxyadenosine binding assay. 83 pmol dA21 oligo was incubated with a 1–50 μg of dT-resin for 1 h and the A260 of the supernatant used to calculate pmol of bound dA21. The slope of the curve at non-saturating conditions describes the binding capacity in pmol dA21/μg resin. The dotted line represents the A21 oligo input. B: Time course of poly-deoxyadenosine binding assay with 20.8 μg dT25-OAS, 76.8 μg dT14-magnetic beads and 125.8 μg dT25-cellulose. The time-series data was normalized to the mass of poly-dA bound at 30 min and fit using a one-site specific binding model in GraphPad Prism (solid lines). C: The elution efficiency was measured by treating 50 μg dA21-loaded resin with 1 mM NaOH and calculating the % elution based on the mass of input dA21.
In addition to binding capacity, binding specificity is critical for many poly-A purifications, particularly mRNA Display selections. mRNA Display exploits the fusion of in vitro translated proteins and peptides with their encoding mRNA to covalently link genotype with phenotype. These mRNA-protein fusions contain a poly-dA sequence (pF30P, dA21-Spacer 93-ACC-Puromycin) between the mRNA and protein to facilitate purification on poly-dT resins4. Although fusion formation is efficient (3.5–12% yield)12, a significant amount of non-fused protein is produced during in vitro translation. Non-specific binding of un-fused protein to the poly-dT resin could result in carryover into the binding selection step and unwanted target binding competition with the mRNA-protein fusion library.
In order to measure the non-specific binding of each poly-dT resin in the context of mRNA-protein fusions, we synthesized an mRNA-protein fusion of the Her2-binding affibody (ZHER2:477). This affibody is 58 residues in size and binds to the Her2 ectodomain with mid-low picomolar affinity13. We reasoned that the size of the affibody protein and mRNA-affibody fusion would provide a robust test of dT-resin selectivity. Her2 affibody DNA was transcribed to RNA, ligated to pF30P ssDNA and then translated using rabbit reticulocyte lysate (Figure 2A). The resulting fusions were purified by dT25-OAS, treated with RNAse A or micrococcal nuclease, and analyzed by SDS-PAGE followed by autoradiography. As seen in Figure 2B, a radiolabeled band corresponding to the mRNA-pF30P-affibody fusion was observed close to the expected size of 86 kDa. Treatment with RNase A yielded a band corresponding to the pF30P-affibody fragment, expected at 15 kDa. Additional treatment with micrococcal nuclease, which degrades both DNA and RNA, yielded a radiolabeled band corresponding to the affibody protein, expected at 7.3 kDa. These experiments confirm proper formation and purification of the mRNA-affibody fusion.
Figure 2. Generation of mRNA-affibody fusions.

A: mRNA encoding the Her2 affibody was enzymatically ligated to a poly-dA oligo bearing puromycin at the 3’ end (pF30P) and translated in rabbit reticulocyte lysate to generate a mixture of [35S]-affibody and [35S]-mRNA-affibody fusion. Purification on poly-dT resin yields the pure [35S]-mRNA-affibody fusion for selection. B: [35S]-mRNA-affibody fusions purified by dT25-OAS resin and incubated alone, with RNase A, or with RNase A plus micrococcal nuclease and separated by SDS-PAGE. Autoradiography shows the radiolabeled products resulting from RNase A (degrades RNA), and micrococcal nuclease (degrades DNA) treatment, confirming the structural characteristics of the mRNA-affibody fusions.
mRNA-pF30P templates were translated in rabbit reticulocyte lysate the presence of [35S]-methionine as described above to generate mRNA-affibody fusions. A control translation was performed with un-ligated mRNA (without the poly dA puromycin linker) to generate [35S]-affibody protein lacking poly dA for purification. Both translation reactions were incubated with an excess (150 μg) of dT25-cellulose, dT14-magnetic beads, or dT25-OAS for 1 hour at 4 °C, washed and eluted with 1 mM NaOH. 35S activity in the eluted fractions and post-elution resins was measured by liquid scintillation counting. Figure 3A shows that all resins effectively bind and release mRNA-affibody fusions upon treatment with mild base. However, in the absence of poly-dA target, as was the case in the mRNA only control, dT14-magnetic beads still capture and release a substantial amount of radiolabeled material. This may include radiolabeled affibody, free [35S]-methionine, or [35S]-labeled aminoacyl tRNA. Non-specific capture (background binding) and release of this material leads to significantly lower elution specificity for the dT14-magnetic beads compared to the dT25-OAS and dT25-cellulose resins (Figure 3B).
Figure 3. Binding specificity of dT-resins.

dT-resins were incubated with in vitro translated affibody or mRNA-affibody protein fusions. Following washing, bound materials were eluted with 1 mM NaOH. Radioactivity of eluate and resin were measured on a liquid scintillation counter. A: Raw counts per minute (CPM) of eluate and resin for each in vitro translation. B: Elution specificity of each resin ([CPM mRNA-affibody eluate – CPM affibody eluate]/CPM mRNA-affibody eluate) × 100.
A summary of dT-resin characteristics can be found in Table 1. dT25-OAS showed the highest binding capacity of the three resins tested which is likely due to substantially larger loading capacity of this resin as reported by the manufacturer. Indeed, dT25-OAS has over an order of magnitude higher loading capacity than dT14-magnetic beads and dT25-cellulose. dT25-OAS had substantially lower non-specific binding than dT14-magnetic beads and was second only to dT25-cellulose in elution specificity. The difference in elution efficiency between resins (Figure 1C) is modest leading us to conclude that this property plays a minor role in the elution specificity of the poly-dT resins. The dT14-magnetic beads showed more rapid on-rate kinetics than either dT25-OAS or dT25-cellulose which, when combined with magnetic separation, may facilitate high throughput poly-A purifications. However, dT25-OAS resin was found to sediment quickly without centrifugation and the large particle size enabled easy pipetting and manipulation of the supernatant. In contrast, dT25-cellulose required centrifugation at 10,000 × g and careful pipetting to avoid disturbing the soft, disperse pellet.
Table 1.
Characteristics of the dT-resins described in this study.
| Resin | Estimated Loading Capacity (pmol poly-dT/μg) | Binding Capacity (pmol dA21/μg) | Time to 90% Bound (min) | A21 Elution Efficiency (%) | Elution Specificity (%) | Ease of Use |
|---|---|---|---|---|---|---|
| dT25-OAS | 25 | 4.0 | 13.3 | 34.3 | 99.2 | ++ |
| dT14-magnetic beads | 0.3 | 1.1 | 0.7 | 44.8 | 75.8 | +++ |
| dT25-cellulose | 0.6 | 0.7 | 13.5 | 39.8 | 99.8 | + |
These results demonstrate that dT25-OAS is well-suited for purification of high diversity mRNA-protein libraries prior to selection experiments where high non-specific binding and/or low purification yields in the early rounds may result in the loss of rare functional sequences. High capacity and low non-specific binding are also critical when mRNA-protein fusions are subjected to successive purifications following post-translational chemical modifications such as cyclization14,15 and small molecule conjugation16,17. In addition, the polystyrene-based OAS resin is significantly more stable to organic solvents and harsh reaction conditions than cellulose, facilitating its use in mixed-phase reactions where library modifications are performed on-resin. These characteristics make dT25-OAS a suitable substitute for dT-cellulose in applications where chemical stability, high binding capacity, and low non-specific binding are required.
Materials
Oligo A25 was purchased from Integrated DNA Technologies. OAS resin (Oligo-Affinity Support PS, catalog number 26-4101-41) can be purchased from Glen Research (Sterling, VA). dT14-SeraMag™ magnetic beads were purchased from Sigma Aldrich. dT25-OAS and pF30P ssDNA (A21-Spacer 93-ACC-Puromycin) were purchased from Keck Biotechnology Resource Lab at Yale University. Retic Lysate IVT™ Kit was purchased from Thermo Fisher Scientific. Easy Tag Express Protein Labeling Mix [35S]-Methionine was purchased from Perkin Elmer. The dT25-cellulose was purchased previously from Amersham Biosciences. RNAse A was purchased from Millipore and micrococcal nuclease was purchased from New England Biolabs Inc.
Polyadenosine binding assay
Resins were washed 5x with dT binding buffer (20 mM Tris, pH 8.0; 1M NaCl; 1 mM EDTA; 0.2% [v/v] Triton-X 100) and resuspended at 10 mg/mL concentration. These stock solutions were serially diluted 2-fold with dT binding buffer from 50 μg to 0.78 μg in 10 μL total volume. To these were added 83 pmol of A21 ssDNA oligo to a total volume of 20 μL in dT binding buffer. After shaking for 1 hour at room temperature, resins were pelleted and the A260 measured on a NanoDrop1000. The pmol of bound oligo was calculated using the extinction coefficient of 255,400 L/(mole × cm) at 260 nm and graphed against input resin. The slope of the linear regression provided the binding capacity in pmol A21/μg resin. The binding time course assay was performed as above, but with 20.8 μg dT25-OAS, 76.8 μg dT14-magnetic beads and 125.8 μg dT25-cellulose input resin. 1 μL of supernatant was sampled after 1, 3, 5, 10, 15 and 30 min of incubation and 260 nm absorbance measured. Non-linear regression for One Site-Specific binding was calculated and used to determine the time at which 90% of binding had occurred. Linear and non-linear regressions performed with GraphPad Prism 8. The elution assay was performed by incubating 50 μg of each resin with A21 oligo, calculating the total bound oligo, washing the resin once with dT binding buffer and then eluting twice with 200 μL 1 mM NaOH. The eluate was ethanol precipitated in the presence of 50 μg of linear acrylamide at −80 °C for 1 hour followed by 16,000 × g for 30 min. The supernatant was discarded and the pellet dried by speedvac. DNA pellet was reconstituted in 20 uL dT binding buffer and the amount of dA21 oligo measured by absorbance at 260 nm. The elution efficiency was calculated by dividing the mass of eluted oligo by the mass of input oligo and multiplying by 100.
Resin binding specificity assay
Her2 affibody DNA was purchased as a gBlock® from Integrated DNA Technologies and PCR amplified with primers containing a T7 promoter (forward primer) and a DNA ligation spacer sequence (reverse primer). Following T7 RNA polymerase transcription, the mRNA was splint ligated to pF30P ssDNA and purified by Urea-PAGE. Fusion templates were translated in rabbit reticulocyte lysate using 20x high salt mixture without methionine according to manufacturer’s instructions. For each translation reaction, 15 pmoles of mRNA or mRNA-pF30P template were translated in the presence of 15 μCi of [35S]-methionine. Translation mixtures were divided into three aliquots, and each aliquot incubated with 150 μg of washed resin (dT25-OAS, dT14-SeraMag™ magnetic beads, or dT25-cellulose) in 500 μL total volume of dT binding buffer for 1 hour at 4°C on an end-over-end rotator. Following binding, the resins were washed three times with 600 μL and once with 300 μL of dT binding buffer. Resins then were washed twice in 300 μL dT wash buffer (20 mM Tris, pH 8.0; 0.3 M NaCl). Resin elution was performed with two cycles of 3-minute room temperature incubations with 250 μL 1 mM NaOH. Post-elution resins and eluates were diluted in equal volumes of scintillation fluid and analyzed on a liquid scintillation counter.
Autoradiography of Fusions
Affibody fusions (20 picomoles input) purified via dT25-OAS were ethanol precipitated, resuspended in 40 μL of water, split into thirds and incubated either alone, with RNAse A (10 μg), or RNAse plus micrococcal nuclease (12,500 gel units) with supplied buffer overnight at room temperature. Reaction solutions were separated by reducing tris-glycine SDS-PAGE, gel washed in water, dried onto filter paper, exposed on autoradiography film and imaged on a FLA 5100 Scanner (FujiFilm).
Acknowledgements
We would like to thank the Keck Oligo Synthesis Facility at Yale for their helpful suggestions.
Funding
This work was supported by MD Anderson Startup Funds (SWM), a GE In-kind Multi-investigator Imaging (MI2) Research Award (SWM), the MDACC Ovarian Cancer SPORE (P50 CA2167685), The National Institutes of Health (2R44CA206771 SWM, 1T32CA196561 BJG, F32EB024379 BJE), and the MD Anderson Cancer Center Support Grant (5P30CA016672).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests Statement
The authors declare no competing interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References and Notes
- 1.Gilham PT, Robinson WE. The Use of Polynucleotide-Celluloses in Sequence Studies of Nucleic Acids. J Am Chem Soc. 1964;86(22):4985–4989. doi: 10.1021/ja01076a050 [DOI] [Google Scholar]
- 2.Fellmann F, Pretet J-L, Fellmann D. Simplified Protocol of Solid-Phase cDNA Libraries for Multiple PCR Amplification. Biotechniques. 1996;21(5):766–770. doi: 10.2144/96215bm02 [DOI] [PubMed] [Google Scholar]
- 3.Stinear T, Matusan A, Hines K, Sandery M. Detection of a single viable Cryptosporidium parvum oocyst in environmental water concentrates by reverse transcription-PCR. Appl Environ Microbiol. 1996;62(9):3385–3390. http://www.ncbi.nlm.nih.gov/pubmed/8795230. Accessed September 9, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94(23):12297–12302. doi: 10.1073/pnas.94.23.12297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiacco SV, Kelderhouse LE, Hardy A, et al. Directed Evolution of Scanning Unnatural-Protease-Resistant (SUPR) Peptides for in Vivo Applications. Chembiochem. 2016;17(17):1643–1651. doi: 10.1002/cbic.201600253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson CA, Liao H-I, Sun R, Roberts RW. mRNA display selection of a high-affinity, modification-specific phospho-IkappaBalpha-binding fibronectin. ACS Chem Biol. 2008;3(8):480–485. doi: 10.1021/cb800069c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sau SP, Larsen AC, Chaput JC. Automated solid-phase synthesis of high capacity oligo-dT cellulose for affinity purification of poly-A tagged biomolecules. Bioorg Med Chem Lett. 2014;24(24):5692–5694. doi: 10.1016/j.bmcl.2014.10.065 [DOI] [PubMed] [Google Scholar]
- 8.Leon AJ, Banner D, Xu L, et al. Sequencing, Annotation, and Characterization of the Influenza Ferret Infectome. J Virol. 2013;87(4):1957–1966. doi: 10.1128/jvi.02476-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Champdoré M, De Napoli L, Di Fabio G, Messere A, Montesarchio D, Piccialli G. New nucleoside based solid supports. Synthesis of 5′,3′-derivatized thymidine analogues. Chem Commun. 2001;1(24):2598–2599. doi: 10.1039/b107200p [DOI] [Google Scholar]
- 10.Caruthers MH, Barone AD, Beaucage SL, et al. Chemical synthesis of deoxyoligonucleotides by the phosphoramidite method. Methods Enzymol. 1987;154(9):287–313. doi: 10.1016/0076-6879(87)54081-2 [DOI] [PubMed] [Google Scholar]
- 11.Musumeci D, Amato J, Randazzo A, et al. G-quadruplex on oligo affinity support (G4-OAS): An easy affinity chromatography-based assay for the screening of G-quadruplex ligands. Anal Chem. 2014;86(9):4126–4130. doi: 10.1021/ac500444m [DOI] [PubMed] [Google Scholar]
- 12.Liu R, Barrick JE, Szostak JW, Roberts RW. Optimized synthesis of RNA-protein fusions for in vitro protein selection. Methods Enzymol. 2000;318:268–293. http://www.ncbi.nlm.nih.gov/pubmed/10889994. [DOI] [PubMed] [Google Scholar]
- 13.Orlova A, Magnusson M, Eriksson TLJ, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521 [DOI] [PubMed] [Google Scholar]
- 14.Millward SW, Takahashi TT, Roberts RW. A general route for post-translational cyclization of mRNA display libraries. J Am Chem Soc. 2005;127(41):14142–14143. doi: 10.1021/ja054373h [DOI] [PubMed] [Google Scholar]
- 15.Menegatti S, Hussain M, Naik AD, Carbonell RG, Rao BM. mRNA display selection and solid-phase synthesis of Fc-binding cyclic peptide affinity ligands. Biotechnol Bioeng. 2013;110(3):857–870. doi: 10.1002/bit.24760 [DOI] [PubMed] [Google Scholar]
- 16.Li S, Roberts RW. A Novel Strategy for In Vitro Selection of Peptide-Drug Conjugates. Chem Biol. 2003;10(3):233–239. doi: 10.1016/S1074-5521(03)00047-4 [DOI] [PubMed] [Google Scholar]
- 17.Horiya S, Bailey JK, Krauss IJ. Directed Evolution of Glycopeptides Using mRNA Display In: Methods in Enzymology. Vol 597 Academic Press Inc.; 2017:83–141. doi: 10.1016/bs.mie.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]