

ABSTRACT
Long noncoding RNAs (lncRNAs) have emerged as critical regulators for gene expression in multiple levels and thus are involved in various physiological and pathological processes. Sirtuin 1 (SIRT1) has been established to exert key roles in the diverse biological process through deacetylation of substrates, including DNA damage repair. Nevertheless, the regulatory relationship between SIRT1 and lncRNAs, and the effect of lncRNA on SIRT1-mediated functions were still far to be elucidated. We herein uncovered that lncRNA miR17HG was notably down-regulated in SIRT1-deficient cells, and significantly up-regulated after ectopic expression of SIRT1. Subsequently, the results of dual luciferase reporter (DLR) showed that SIRT1 dramatically enhanced the promoter activity of the miR-17-92 cluster. Furthermore, we specifically knocked down the previous demonstrated transcription factor for the miR-17-92 cluster, C-Myc, which was the validated substrate of SIRT1. As expected, miR17HG and miR-17-92 miRNAs were evidently down-regulated after silencing of C-Myc; and silencing of C-Myc significantly reversed the effect of SIRT1 on miR17HG expression, suggesting that SIRT1 endowed cells with elevated miR17HG expression through stabilization of C-Myc. What is more, silencing of miR17HG significantly inhibited the repair of DNA DSBs, while enforced expression of miR17HG promoted DSBs repair. Fascinatingly, overexpression of miR17HG evidently enhanced the deacetylation activity of SIRT1, while silencing of miR17HG conferred diminished deacetylation activity. In addition, the results of RIP unraveled the physical interaction between miR17HG and SIRT1. Taken together, we presented evidences that miR17HG and SIRT1 probably formed a positive feedback loop, which exerted a crucial effect on DSBs repair.
KEYWORDS: SIRT1, lncRNA, miR17HG, DNA damage, double stranded breaks
Introduction
Long non- coding RNA (lncRNAs) is currently defined as RNA molecules that are longer than 200 nt and lack the protein-coding capacity, which is divided into at least five categories, including sense, antisense, intergenic, intronic and bidirectional lncRNAs [1]. Although mammalian genomes encode more than 10,000 lncRNAs, lncRNAs are traditionally viewed as the by-products generated from the background noise of transcription [2]. Nevertheless, growing evidences in recent years have unveiled the powerful effects of lncRNAs on the regulation of gene expression at multiple levels, including epigenetic chromatin modification, transcription, post- transcription and translation level, though few have been fully elucidated [3–6]. A plenty of studies have demonstrated that lncRNAs exerted vital biological functions such as DNA damage repair [7], tumorigenesis [8], and the deregulation of lncRNAs has been connected to diverse human diseases [9].
Sirtuins are the members of NAD+-dependent, highly conserved histone deacetylase family [10]. Silent information regulator 1 (SIRT1) is the most widely studied member of mammalian sirtuins, which participates in a broad set of biological processes including DNA damage repair [11], apoptosis [12], aging [13] and autophagy [14], and exerts a central role in protection against various human diseases, including neurodegenerative diseases, cardiovascular diseases, metabolic syndromes and tumorigenesis [15,16]. In addition to direct deacetylate transcription factors, repair enzymes or other cellular proteins such as p53, RAD51 and Ku70 [17,18], and thereby impact on their activity, SIRT1 has also been established to repress gene expression by silencing the chromatin structure through deacetylation of histone [19]. Thus, we speculated that SIRT1 probably modulated the lncRNA profile through deacetylation of transcription factors or histone, which might exert an important effect on SIRT1-mediated functions.
DNA damage arises from numerous endogenous cellular events and exogenous environmental agents, which severely compromises genomic integrity [20]. Double-stranded breaks (DSBs) are the most destructive DNA damage which closely associated with the development of cellular senescence, metabolism disorder and tumorigenesis [21]. To counteract the deleterious effect of DNA damage and maintain the genomic integrity, diverse conserved DNA damage response (DDR) and repair pathways have been evolved. In addition, SIRT1 has been demonstrated to exert a pivotal role in the repair of various types of DNA damage including DSBs, through deacetylation of the substrates involved in homologous recombination (HR) and non-homologous end joining (NHEJ) [22,23]. Moreover, a handful of lncRNAs has been shown to participate in the repair of DSBs [24]. Hence, it is reasonable to speculate that SIRT1-regulated lncRNAs might probably exert a crucial role in modulating the DSBs repair. In this study, we sought to identify SIRT1-related lncRNAs, and further deepen into the effect and underlying mechanism of SIRT1-related lncRNAs on DSBs repair.
Materials and methods
Cell culture
HEK293 was an embryonic kidney cell line and was cultured in Dulbecco’s Modified Eagle Medium (DMEM) medium (GIBCO) supplemented with 10% fetal bovine serum (FBS) (GIBCO) staying at 5% CO2 and 37°C.
Chemicals
Neocarzinostatin (NCS, 0.5mg/ml) was purchased from Sigma (N9162). The cells were treated with 0.5μg/ml NCS (at a dilution of 1:1000) for 8 h to induce DNA damage or 48 h to induce apoptosis.
Cell transfections
The small interference RNAs (siRNA) targeting SIRT1 mRNA (Genbank accession no. NM_012238.4) was designated as siSIRT1-1 (Sense Strand: 5ʹ- GCGGG AAUCCAAAGGAUAATT-3ʹ), siSIRT1-2 (Sense Strand: 5ʹ- CCCUGUAAAGCUUU CAGAATT-3ʹ), and siSIRT1-3 (Sense Strand: 5ʹ- GGAGAUGAUCAAGAGGCAATT −3ʹ). siRNA targeting C-Myc mRNA (Genbank accession no. NM_002467.5) was designated as siC-Myc-1 (Sense Strand: 5ʹ- GUGCAGCCGUAUUUCUACUTT-3ʹ), siC- Myc -2 (Sense Strand: 5ʹ- GAACACACAACGUCUUGGATT-3ʹ), and siC-Myc-3 (Sense Strand: 5ʹ- GGAAACGACGAGAACAGUUTT-3ʹ). The control RNA duplex, designated as siNC (Sense Strand: 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ) was nonhomologous to any human genome sequences. The above RNA oligoribonucleotides were purchased from Genepharma (China). In addition, siRNA targeting lncRNA miR17HG (Ensembl accession no. ENST00000400282.2) was designated as simiR17HG-1 (Sense Strand: 5ʹ- GCAACTTCCTGGAGAACAA -3ʹ), simiR17HG-2 (Sense Strand: 5ʹ-TGTAGTTTGAAGACACACT-3ʹ), and simiR17HG-3 (Sense Strand: 5ʹ- CCACTTGAGACTTCAGATT -3ʹ), which were purchased from Ribo (China).
For siRNA transfection, the RNA oligoribonucleotide (s) was transfected using Lipofectamine RNAiMAX (Invitrogen, USA), following the manufacturer’s protocol. Fifty nM RNA duplex was utilized in each transfection unless otherwise indicated. In the experiment of expressing exogenous SIRT1, cells were transfected with 400 ng plasmids in a 24-well plate, using FuGene HD Transfection Reagent (Promega, USA) according to the manufacturer’s protocol. The SIRT1-overexpression plasmid and the control vector (pReceiver-M02) were purchased from Genecopoeia (USA).
RNA isolated and real- time quantitative PCR
Total RNA was extracted from cells utilizing Trizol reagent (Life, USA) following the manufacturer’s protocol, and the quantity and quality were measured by DS-11 spectrophotometer (DeNovix, USA).
For mRNA or lncRNA detection, RNA was transcribed to be cDNA with PrimerScript ™RT reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, Japan) according to the manufacturer’s protocol. Real-time quantitative PCR was carried out in LightCycler 96 (Roche, Switzerland) with SYBR Select Master Mix (Life, USA). The follow primer sequences for SIRT1, C-Myc, miR17HG, GAPDH were listed in Table 1. GAPDH gene was used as an internal control.
Table 1.
Sequences of primers for real-time qPCR.
| Name | Sense Primer (5ʹ-3ʹ) | Antisense Primer (5ʹ-3ʹ) |
|---|---|---|
| SIRT1 | TAGCCTTGTCAGATAAGGAAGGA | ACAGCTTCACAGTCAACTTTGT |
| C-Myc | GTCAAGAGGCGAACACACAAC | TTGGACGGACAGGATGTATGC |
| miR17HG-001 | GGAATGGTATTTGCTAAGTGGA | AAAACAGTCCTCAGTCACACAGA |
| miR17HG-002 | TCTGATTGTCAGTGATTTATAAGCT | GACTTAGAGTGGGGTGGC |
| miR17HG-003 | TGAAAAGGAAGTTTTCTGGAATGGT | GAGAGGGTTTACCTAACTGTGTATC |
For miRNA detection, cDNA was synthesized from the purified RNA with All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia, USA). Following the manufacturer’s instructions, real-time qPCR was performed on LightCycler 96 (Roche, Switzerland) with All-in-One miRNA qRT-PCR Detection Kit. All miRNA primers, which included miR-17, miR-18a, miR-19a, miR-19b, miR-20, miR-92a, U48, were purchased from GeneCopoeia. U48 was used as an internal control.
Western blotting
All the proteins were separated on 10% or 12% SDS polyacrylamide and electrophoretically transferred to polyvinylidene difluoride membranes (PVDF) (0.22 μm pore size) (Millipore, USA). After blocked with 3% Bovine serum albumin (BSA) for 1 h at room temperature or 4°C overnight, the membrane(s) was incubated with the primary antibodies overnight at 4°C, followed by the respective secondary antibodies conjugated with horseradish peroxidase (HR) and subjected to a commercial-enhanced chemiluminescence (ECL) kit (Pierce, USA). The following primary antibodies were included in this study: anti-SIRT1 antibody (13161-1-AP, Protein Tech, China), anti-C-Myc antibody (10828-1-AP, Protein Tech, China), anti-Acetyl-H4K16 antibody (13534S, Cell Signaling Technology, USA), anti-PARP1 antibody (13371-1-AP, Protein Tech, China), and anti-γH2A.X antibody (2577S, Cell Signaling Technology, USA). Protein loading was estimated using mouse anti-Tubulin monoclonal antibody (T5168, Sigma-Aldrich, USA) or mouse anti-GAPDH monoclonal antibody (60004-1-Ig, Protein Tech, China).
Dual luciferase assay
Dual luciferase reporter assay (pEZX-GA01, GeneCopoeia, USA) was comprised of two kinds of luciferase expression, one was Gluc containing the promoter region of the miR17HG and miR-17-92 host gene, and another was the constitutive expressed SeAP. Firstly, the HEK293 cells were plated in a 48- well plate, then cotransfected with 50 nM siNC or siSIRT1, or 100 ng of pReceiver-M02 or SIRT1-overexpression plasmid (designated as SIRT1), and 100 ng of pEZX-GA01-promoter1 (designated as Promoter1) or pEZX-GA01-promoter2 (designated as Promoter2) using Lipofectamine 2000 (Life, USA) according to the manufacturer’s protocol. After transfection for 48 h, cell culture medium was collected and detected using the Secrete-Pair Dual Luminescence Assay Kit (GeneCopoeia, USA). Luciferase activity was detected by FB12 Luminometer (Berthold). Gluc luciferase activity of each sample was normalized by SEAP luciferase activity.
Apoptosis assays
Apoptosis was evaluated by determination of the activity of Caspase-3/7, PARP-1 cleavage, and TUNEL assay. The activity of Caspase-3/7 was detected in 96-well format using the Caspase Glo3/7 Assay (Promega). One hundred microliter of the Caspase- Glo3/7 reagent were added to each well and then incubated at room temperature for 1 h. The luminescence was measured using Synergy 2 microplate fluorescence reader (BioTek, USA). The PARP-1 cleavage was detected by western blot using the primary anti- PARP 1 antibody (13371-1-AP, Protein Tech, China). Terminal dUTP nick end labeling (TUNEL) was carried out using the one-step TUNEL apoptosis assay kit according to the manufacturer’s instructions (C1090, Beyotime Institute of Biotechnology). Briefly, cells were fixed with 4% paraformaldehyde. Then, the fixed cells were treated with 0.2% Triton X-100 to permeabilize cells, followed incubation with the Cy3-conjugated TUNEL reaction mixture. Finally, cells were stained with DAPI (Sigma-Aldrich, USA) as a counterstain, and read under a fluorescence microscope (IX73, Olympus).
RNA-binding protein immunoprecipitation (RIP)
The RIP was performed utilizing the Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore). Briefly, cells were collected by trypsinization and centrifugal separation. Then, the collected cells were lysed using the RIP Lysis Buffer. The washed magnetic beads were incubated with the antibody (5 µg per immunoprecipitation) for 30 min at room temperature, and then the magnetic beads were incubated with the above-prepared cell lysate overnight at 4°C. After immunoprecipitation, the RNA-protein complex was washed and dissociated with magnetic beads. Finally, the immunoprecipitated RNA of the complex was extracted after the removal of the protein by Proteinase K.
Immunofluorescence staining
Immunofluorescence (IF) staining for γH2A.X was performed as follows. Initially, cells were washed and fixed by 4% paraformaldehyde at room temperature or 100% pre-cold methyl at 4°C. Then, the fixed cells were incubated with the primary γH2A.X antibody (2577S, Cell Signaling Technology, USA) overnight at 4°C, followed by Alexa Fluor 594-conjugated Goat Anti-Mouse IgG (H + L) (SA00006-3, Protein Tech, China). Cells were stained with 4ʹ-6ʹ-diamidino-2-phenylindole (DAPI; Sigma- Aldrich) as a counterstain and read under a confocal microscope (Leica SP8, Germany).
Statistical analysis
All data are presented as mean±SEM from at least three separate experiments. Unless otherwise noted, the differences between two groups were analyzed using Student’s two- tailed t test, while the differences between more than two groups were assessed by one-way analysis of variance (ANOVA). P <0.05 was considered statistically significant.
Accession number
Genbank Accession numbers for SIRT1 mRNA sequence (NM_012238.4), C-Myc mRNA sequence (NM_002467.5) are found at the National Center for Biotechnology Information (http://www.ncbi.nlm. nih.gov/). The sequence of lncRNA miR17HG (ENST00000400282.2) is obtained in the ENSEMBL database (http://www.ensembl.org/). The sequences of miR-17 (MIMAT0000070), miR-18 (MIMAT0000072), miR-19a (MIMAT0000073), miR-19b (MIMAT0000074), miR-20 (MIMAT0000075) and miR-92a (MIMAT0000092) described in this paper have been deposited in miRBase (http://www.mirbase.org/).
Results
1. The expression of miR17HG and miR-17-92 miRNAs was positively correlated with SIRT1
Initially, in order to identify SIRT1-related non-coding RNAs, we specifically knocked down the SIRT1 expression in HEK293 cells using designed siSIRT1 (siSIRT1-1/2/3). Cells were transfected with siNC or siSIRT1-1/2/3, and 48 h later analyzed for endogenous SIRT1 mRNA and protein expressions by qPCR and western blot. All three RNAi fragments significantly diminished the SIRT1 expression, but only siSIRT1-1 transfected cells exhibited elevated H4K16 acetylation level (Figure 1(a,b)). Thus, siSIRT1-1 was utilized in the following experiments and designated as siSIRT1. Subsequently, lncRNA and miRNAs microarray were performed to profile lncRNA and miRNA expression levels in siNC and siSIRT1 transfected cells. The data of microarray uncovered that miR17HG (ENST00000400282.2) and the six miR-17-92 miRNAs (miR-17, miR-18, miR-19a, miR-19b, miR-20 and miR-92a) were all significantly down-regulated in SIRT1-deficient cells, which were all transcribed from the miR17–92 cluster host gene (Table 2).
Figure 1.
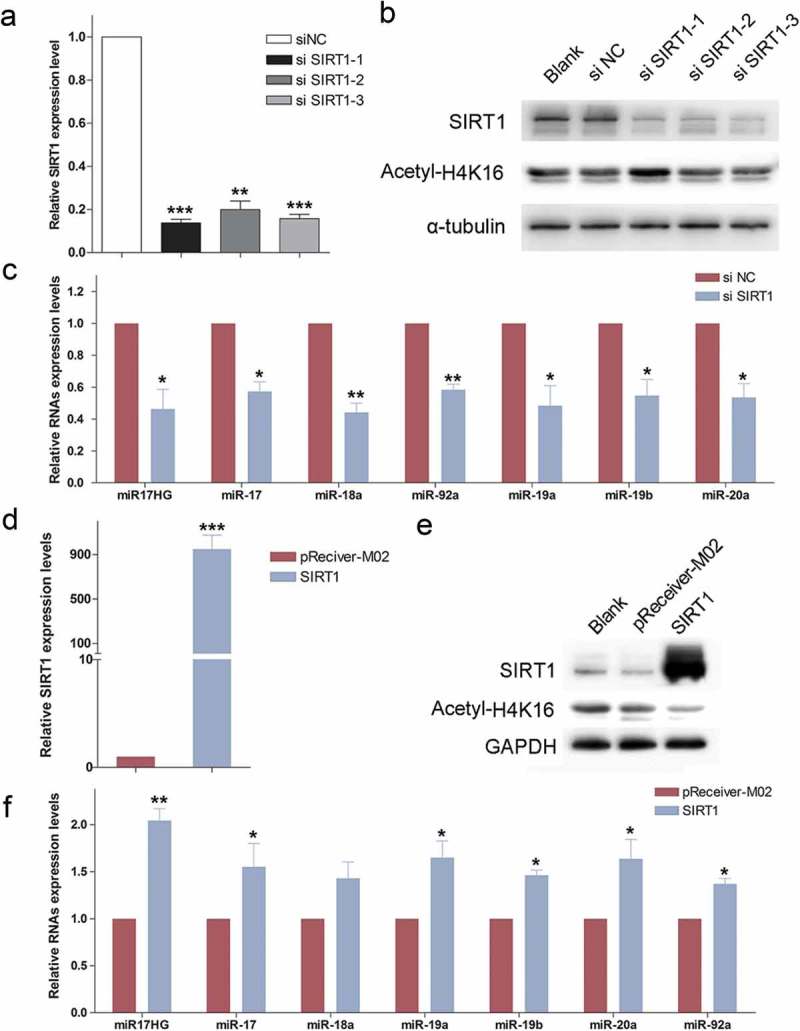
The expression of miR17HG was positively correlated with SIRT1. (a) Real-time qPCR was used to detect the expression levels of SIRT1 mRNA in 293 cells transfected with siNC or siSIRT1-1/2/3. Columns, mean of at least three independent experiments; bars, SEM. **, P< 0.01; ***, P < 0.001, comparison between two groups as indicated. (b) Western blot was performed to monitor the expression levels of SIRT1 and Acetyl-H4K16 in cells transfected with siNC or siSIRT1-1/2/3. (c) Expression of miR17HG and miR-17–92 miRNAs in siNC or siSIRT1 transfected cells. Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05; **, P< 0.01, comparison between two groups as indicated. (d) Expression of SIRT1 in pReceiver-M02 or SIRT1-overexpression plasmid (SIRT1) transfected cells. Columns, mean of at least three independent experiments; bars, SEM. ***, P < 0.001, comparison between two groups as indicated. (e) Western blot was performed to monitor the expression levels of SIRT1 and Acetyl-H4K16 in cells transfected with pReceiver-M02 or SIRT1-overexpression plasmid (SIRT1). (f) Expression of miR17HG and miR-17–92 miRNAs in pReceiver-M02 or SIRT1 transfected cells. Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05; **, P< 0.01, comparison between two groups as indicated.
Table 2.
The expression of miR17-92 cluster after silencing of SIRT1 in HEK293 cells.
| Name | Probe Signal (raw) |
Fold change | |
|---|---|---|---|
| siNC | siSIRT1 | ||
| miR17HG (ENST00000400282) | 255.0722 | 127.5364 | −1.962295942* |
| hsa-miR-17-5p | 713.8784 | 347.8665 | −2.133189372 |
| hsa-miR-18a-5p | 200.4057 | 82.6517 | −2.520438218 |
| hsa-miR-19a-3p | 640.029 | 339.885 | −1.957426354 |
| hsa-miR-20a-5p | 1238.002 | 722.454 | −1.781266596 |
| hsa-miR-19b-3p | 1206.524 | 732.559 | −1.712029068 |
| hsa-miR-92a-3p | 229.4998 | 158.3216 | −1.506814943 |
* -,means down-regulation.
To confirm the validity of our screening technique, real-time qPCR was carried out to evaluate the expression of miR17HG and miR-17–92 miRNAs in siNC or siSIRT1 transfected cells, and validated the down-regulation of miR17HG and these miRNAs in SIRT1-deficient cells (Figure 1(c)). Meanwhile, the constructed vector for SIRT1 overexpression was transfected into HEK293 cells. The ectopic expression of SIRT1 was confirmed, which endowed cells with decreased H4K16 acetylation level (Figure 1(d,e)). The results of real-time qPCR showed that enforced expression of SIRT1 dramatically enhanced the expression of miR17HG and miR-17-92 miRNAs (Figure 1(f)).
Furthermore, the cells were treated with SIRT1 activator SRT1720 (Selleck, USA) and inhibitor EX527 (Selleck, USA), which effectively altered the deacetylase activity of SIRT1 (SFigure 1A). Consistently, SRT1720 treatment significantly enhanced the miR17HG expression level, while EX527 decreased the expression of miR17HG (SFigure 1B), which further suggested the positive correlation between SIRT1 and miR17-92 cluster encoded miR17HG and miRNAs.
2. SIRT1 promoted the expression of miR17-92 cluster through C-Myc
To gain the insights into the detailed mechanism of how SIRT1 regulated the expression of miR-17-92 cluster, we further investigated the effect of SIRT1 on the promoter activity of the miR-17-92 cluster. As described previously, the transcription of miR-17-92 cluster not only initiates from a host gene promoter region upstream of exon 1 [25] but also originates from an intronic A/T-rich region upstream of the miRNA coding sequences [26] (Figure 2(a)). Hence, two dual luciferase reporter system were prepared utilizing cloning the two promoter regions upstream gaussia luciferase (GLuc) reporter, respectively. Then, the HEK293 cells were co-transfected with the constructed dual luciferase reporters and siSIRT1 or SIRT1-overexpressing plasmid. As shown by luciferase reporter assay, silencing of SIRT1 evidently diminished the relative fluorescence intensity of the luciferase reporters containing both promoter regions (Figure 2(b)), while ectopic expression of SIRT1 notably enhanced the relative fluorescence intensity of both reporters (Figure 2(c)), suggesting that SIRT1 significantly promoted the promoter activity of the miR-17-92 cluster gene.
Figure 2.
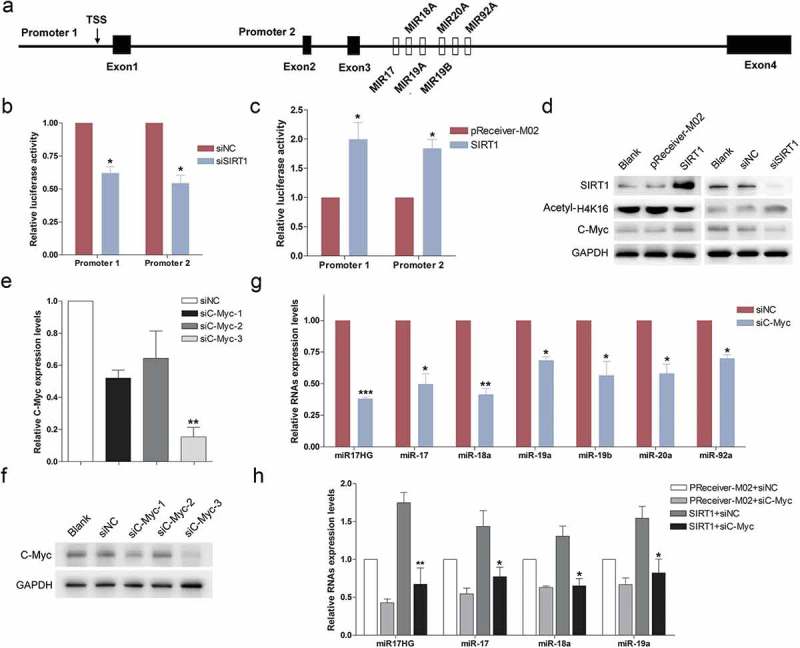
SIRT1 enhanced the expression of miR17–92 cluster through stabilization of C-Myc. (a) Schematic diagram of the two promoter regions of miR17–92 cluster. (b) Determination of luciferase activity. Cells were co-transfected with Promoter1 or Promoter2 with siNC or siSIRT1. Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05, comparison between two groups as indicated. (c) Determination of luciferase activity. Cells were co-transfected with Promoter1 or Promoter2 with pReceiver-M02 or SIRT1-overexpression plasmid (SIRT1). Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05, comparison between two groups as indicated. (d) Western blot was carried out to monitor the expression levels of SIRT1, C-Myc and Acetyl-H4K16 in cells transfected with siNC/siSIRT1, or pReceiver-M02/SIRT1-overexpression plasmid (SIRT1). (e) Real-time qPCR was used to detect the expression levels of C-Myc mRNA in 293 cells transfected with siNC or siC-Myc-1/2/3. Columns, mean of at least three independent experiments; bars, SEM. **, P < 0.01, comparison between two groups as indicated. (f) Western blot was performed to evaluate the expression levels of C-Myc in cells transfected with siNC or siC-Myc-1/2/3. (g) Expression of miR17HG and miR-17-92 miRNAs in siNC or siC-Myc transfected cells. Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001, comparison between two groups as indicated. (h) Expression of miR17HG and miR-17-92 miRNAs in cells cotransfected with siNC/siSIRT1, or pReceiver-M02/SIRT1-overexpression plasmid (SIRT1). Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05; **, P < 0.01, comparison between two groups as indicated.
Previous studies have demonstrated that C-Myc was a substrate of SIRT1, and the half-life of c-Myc was elevated through SIRT1-mediated deacetylation [27]. The effect of SIRT1 on C-Myc abundance was further confirmed (Figure 2(d)). Additionally, C-Myc has been established to be the pivotal transcription factor for the miR-17-92 cluster, which triggered the expression of miR-17-92 miRNAs [28]. Thus, we speculated that SIRT1 promoted the expression of miR17HG and miR-17-92 miRNAs through stabilization of C-Myc. Subsequently, we knocked down C-Myc expression using designed siC-Myc-1/2/3. Among the three RNAi fragments, only siC-Myc-3 effectively knocked down the expression of endogenous C-Myc in HEK293 cells (Figure 2(e,f)). As expected, silencing of C-Myc significantly decreased the expression of miR17HG and miR-17-92 miRNAs (Figure 2(g)). Furthermore, to probe into whether SIRT1 impacted on the promoter activity of the miR-17-92 cluster through regulation of C-Myc, cells were cotransfected with siC-Myc and SIRT1-overexpression vector. The results showed that silencing of C-Myc significantly reversed the effect of SIRT1 on up-regulation of miR17HG and miR-17-92 miRNAs (Figure 2(h)). To sum up, we herein presented the evidences that SIRT1 probably promoted the expression of miR-17-92 cluster encoded miR17HG and miRNAs through stabilization of C-Myc.
3. miR17HG facilitated the repair of NCS-induced DNA DSBs
Previous literatures have established the critical role of SIRT1 in DSBs (Double-stranded breaks) repair. Thus, we further digged into the effect of the SIRT1-regulated miR17HG on DSBs repair, in order to shed light on SIRT1-related lncRNAs on SIRT1-mediated functions. To specifically knock down miR17HG expression, cells were transfected with three designed siRNAs targeting miR17HG (simiR17HG-1/2/3), and 48 h later analyzed for endogenous miR17HG expressions by qPCR. All three RNAi fragments significantly suppressed the miR17HG expression (>90%) but exhibited a minor effect on the expression of two other miR17HG isoforms (Figure 3(a)). NCS (neocarzinostatin) is a radiomimetic anti- tumor drug, which generates DSBs in cells [29]. We then monitored the γH2A.X levels in cells transfected with simiR17HG-1/2 or siNC with or without NCS treatment (0.5μg/ml). The results of western blot and immunofluorescence revealed that silencing of miR17HG dramatically elevated the γH2A.X level (Figure 3(b,c)). Moreover, ectopic expression of miR17HG conferred cells with decreased γH2A.X levels with or without NCS treatment (Figure 3(d–f)). In addition, we noted that silencing of miR17HG evidently enhanced the PARP1 cleavage and over-expression of miR17HG suppressed PARP1 cleavage under NCS treatment, indicating the protection effect of miR17HG against apoptosis (Figure 3(c,e)). Then, the caspase 3/7 activity was further determined to evaluate the apoptosis in cells transfected with miR17HG-overexpressing plasmid or simiR17HG under NCS treatment. The results uncovered that silencing of miR17HG dramatically elevated the caspase 3/7 activity, while ectopic expression of miR17HG decreased the caspase 3/7 activity (Figure 3(g,h)). Taken together, our data suggested that miR17HG significantly facilitated the repair of NCS-induced DSBs, and exerted protection effect against apoptosis.
Figure 3.
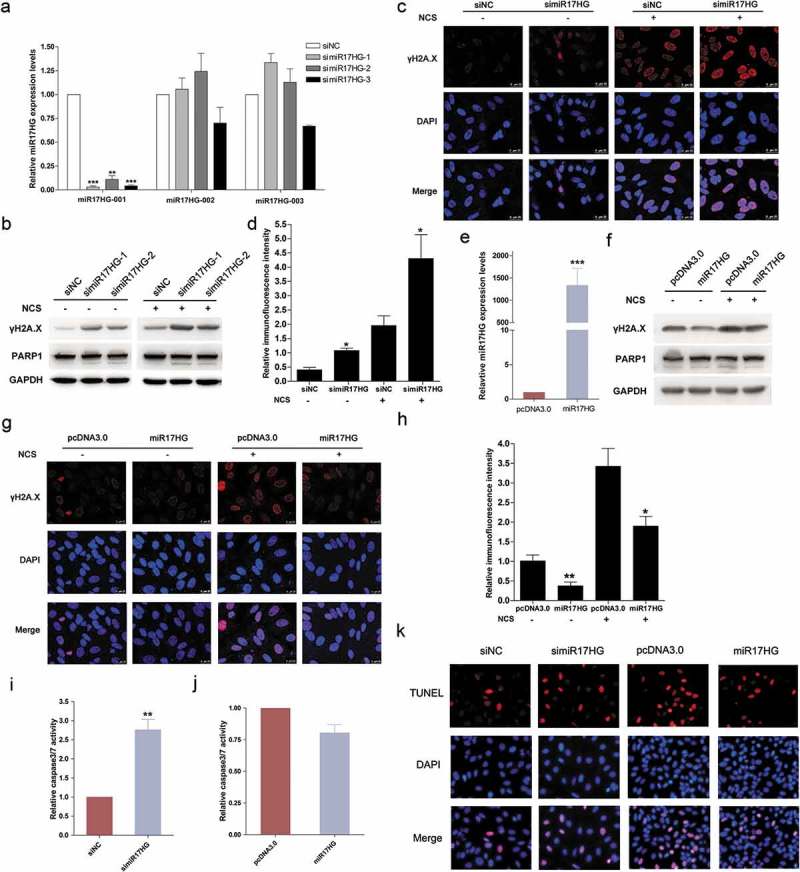
miR17HG facilitated the repair of NCS-induced DNA DSBs. (a) Real-time qPCR was used to detect the expression levels of three miR17HG isoforms (ENST00000400282, designated as miR17HG-001; ENST00000582141, designated as miR17HG-002; ENST00000581816, designated as miR17HG-003) in 293 cells transfected with siNC or simiR17HG-1/2/3. Columns, mean of at least three independent experiments; bars, SEM. **, P < 0.01; ***, P < 0.001, comparison between two groups as indicated. (b) The γH2A.X expression levels and PARP-1 cleavage in cells transfected with siNC or simiR17HG-1/2 with or without NCS treatment. (c) Representative images of cells stained with DAPI (blue fluorescence) and γH2A.X (red fluorescence) in siNC or simiR17HG transfected cells with or without NCS treatment. (d) The relative fluorescence intensity of cells stained with DAPI (blue fluorescence) and γH2A.X (red fluorescence) in siNC or simiR17HG transfected cells with or without NCS treatment. Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05, comparison between two groups as indicated. (e) The miR17HG expression levels in cells transfected with pcDNA3.1 or miR17HG-overexpression plasmid (designated as miR17HG). Columns, mean of at least three independent experiments; bars, SEM. ***, P < 0.001, comparison between two groups as indicated. (f) The γH2A.X expression levels and PARP-1 cleavage in cells transfected with pcDNA3.1 or miR17HG-overexpression plasmid (miR17HG). (g) Representative images of cells stained with DAPI (blue fluorescence) and γH2A.X (red fluorescence) in pcDNA3.0 or miR17HG-overexpression plasmid (miR17HG) transfected cells with or without NCS treatment. (h) The relative fluorescence intensity of cells stained with DAPI (blue fluorescence) and γH2A.X (red fluorescence) in pcDNA3.0 or miR17HG-overexpression plasmid (miR17HG) transfected cells with or without NCS treatment. Columns, mean of at least three independent experiments; bars, SEM. *, P < 0.05; **, P < 0.01, comparison between two groups as indicated. (i) and (j) The caspase3/7 activity of cells transfected with siNC or simiR17HG (i), or pcDNA3.0 or miR17HG-overexpression plasmid (j). Columns, mean of at least three independent experiments; bars, SEM. **, P < 0.01, comparison between two groups as indicated. (k) The TUNEL assay of cells transfected with siNC or simiR17HG, or pcDNA3.0 or miR17HG-overexpression plasmid under NCS treatment.
4. SIRT1 exerted a critical role in miR17HG-mediated effect on DSBs repair
We further probed into the potential molecular mechanism underneath the effect of miR17HG on DSBs repair. Initially, since miR17HG (Gene ID: 407975) is the host gene for six miR-17-92 cluster miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20 and miR-92a), we speculated that the lncRNA miR17HG might exert its function through altering the expression levels of these miRNAs. Thus, we constructed an artificial miRNA sponge vector for six miR-17-92 cluster miRNAs. The inhibition of the six miRNAs by miRNA sponge was confirmed (SFigure 2A). As expected, the inhibition of the miR-17-92 cluster miRNAs dramatically elevated the γH2A.X levels with or without NCS treatment, which was consistent with the effect of silencing of miR17HG on DSBs repair (SFigure 2B). Nonetheless, the results of qPCR showed that specifically silencing of miR17HG only conferred modest effect on the expression of miR-17-92 miRNAs (SFigure 2C), indicating that the effect of miR17HG on DSBs repair was not mainly due to miR-17-92 miRNAs.
Additionally, considering the demonstrated function of SIRT1 in DSBs repair and the protection effect against apoptosis, we speculated that SIRT1 might play an important role in these miR17HG-mediated functions. Initially, cells were transfected with siNC or siSIRT1 or SIRT1-overexpressing plasmid (designated as SIRT1) or control plasmid (pReceiver-M02). Our data verified that silencing of SIRT1 significantly enhanced the γH2A.X levels and apoptotic rates (Figure 4(a)), while ectopic expression of SIRT1 notably suppressed the γH2A.X levels and apoptotic rates (Figure 4(b)). Furthermore, cells were co-transfected with siNC or simiR17HG, and the SIRT1-overexpressing plasmid (designated as SIRT1) or control plasmid (pReceiver-M02). The results showed that enforced expression of exogenous SIRT1 significantly reversed the effect of silencing of miR17HG on the γH2A.X levels (Figure 4(c,d)) and apoptotic rates (Figure 4(e)), suggesting that miR17HG probably facilitated DSBs repair and protected the cell against apoptosis through SIRT1.
Figure 4.
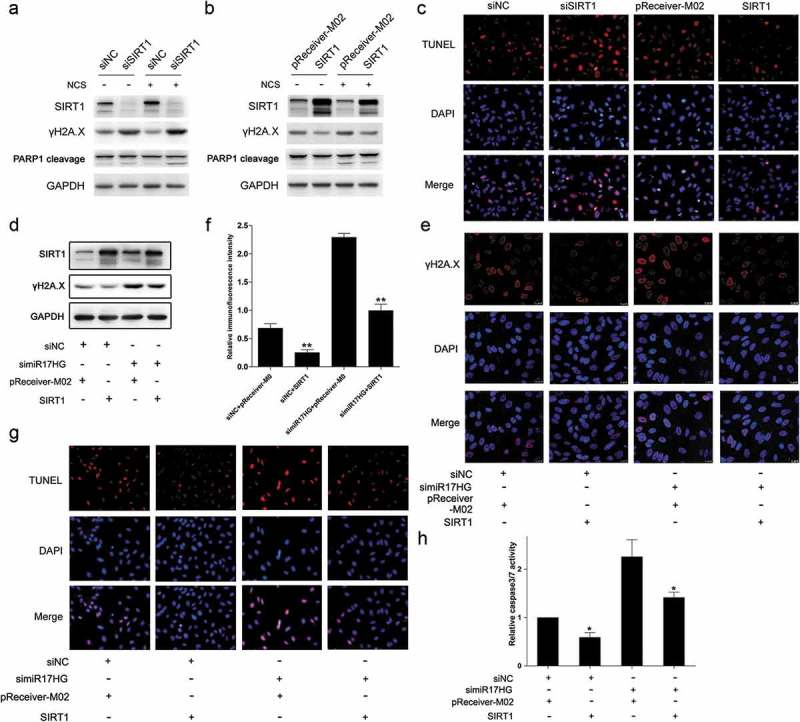
SIRT1 exerted a critical role in miR17HG-mediated effect on DSBs repair. (a) Western blot was performed to detect the expression levels of SIRT1, γH2A.X expression levels and PARP-1 cleavage in cells transfected with siNC or siSIRT1 with or without NCS treatment. (b) Western blot was performed to detect the expression levels of SIRT1, γH2A.X expression levels and PARP-1 cleavage in cells transfected with pReceiver-M02 or SIRT1 transfected cells with or without NCS treatment. (c) The TUNEL assay of cells transfected with siNC or siSIRT1, or pReceiver-M02 or SIRT1 under NCS treatment. (d) The SIRT1 and γH2A.X expression levels in cells cotransfected with siNC/simiR17HG, or pReceiver-M02/SIRT1 under NCS treatment. (e) Representative images of cells stained with DAPI (blue fluorescence) and γH2A.X (red fluorescence) in cells cotransfected with siNC/simiR17HG, or pReceiver-M02 or SIRT1 under NCS treatment. (f) The relative fluorescence intensity of cells stained with DAPI (blue fluorescence) and γH2A.X (red fluorescence) in cells cotransfected with siNC/simiR17HG, or pReceiver-M02/SIRT1 under NCS treatment. Columns, mean of at least three independent experiments; bars, SEM. **, P < 0.01, comparison between two groups as indicated. (g) The TUNEL assay of cells cotransfected with siNC/simiR17HG, or pReceiver-M02/SIRT1 under NCS treatment. (h) The caspase3/7 activity of cells cotransfected with siNC/simiR17HG, or pReceiver-M02 or SIRT1 under NCS treatment. Columns, mean of at least three independent experiments; bars, SEM. *, P< 0.05, comparison between two groups as indicated.
5. miR17HG and SIRT1 formed a positive feedback loop
Fascinatingly, we noted that silencing of miR17HG dramatically enhanced the H4K16 acetylation level, while ectopic expression of miR17HG significantly decreased the H4K16 acetylation level, though the endogenous SIRT1 expression levels exhibited no significant difference (Figure 5(a)). To further deepen into the association between miR17HG and SIRT1, we constructed a GFP-SIRT1 expressing plasmid by cloning the coding sequence of the fusion protein GFP-SIRT1 into pcDNA3.1 (designated as pcDNA3.1-GFP-SIRT1). The expression of this constructed vector for GFP-SIRT1 was confirmed (Figure 5(b)). Cells were transfected with pcDNA3.1-GFP-SIRT1 or control plasmid (pcDNA3.1-GFP), then RIP (RNA-binding protein immunoprecipitation) assay was performed 48 h later. The results of RIP assay uncovered the direct interaction between miR17HG and SIRT1 (Figure 5(b)), indicating that miR17HG might impact on the stability or activity of SIRT1 through physical interaction. These results revealed that the physical interaction between miR17HG and SIRT1 effectively enhanced the deacetylase activity of SIRT1, thus promoted the DSBs repair. In aggregate, the above results suggested that miR17HG and SIRT1 formed a positive feedback loop, which exerted a pivotal role in DSBs repair and the protection effect against apoptosis (Figure 5(c)).
Figure 5.
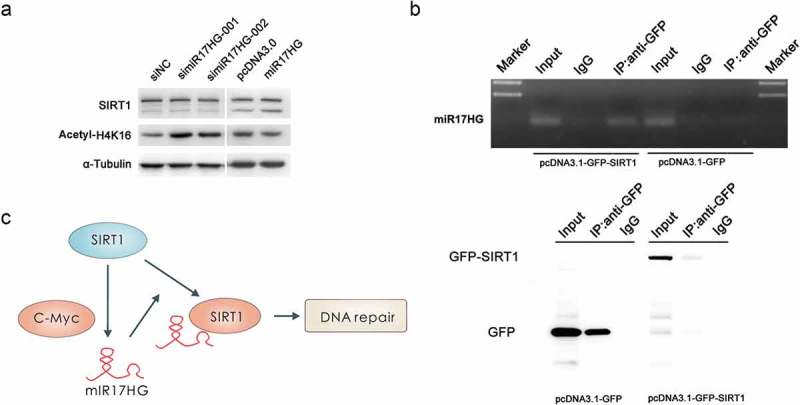
The positive feedback loop between miR17HG and SIRT1. (a) Western blot was performed to monitor the expression levels of SIRT1 and Acetyl-H4K16 in cells transfected with siNC, simiR17HG1-1/2, pcDNA3.1, or miR17HG. (b) RIP-coupled PCR assay of the interaction between SIRT1 and miR17HG in 293 cells. (c) Schematic diagram of the positive feedback loop of SIRT1 and miR17HG, and the effect of this loop in DSBs repair.
Discussion
SIRT1 exerted a critical role in various cellular processes through deacetylation of substrates. Additionally, we herein uncovered that SIRT1 promoted DSBs repair via induction of lncRNA – miR17HG, which formed a positive feedback loop with SIRT1. And the positive feedback loop between SIRT1 and miR17HG, effectively promoted the repair of NCS-induced DSBs, and protected cells against apoptosis.
SIRT1 has been established to be a transcriptional silencer of various gene expressions through deacetylation of N-terminus tails of multiple acetylated histones, H3K9ac, H4K16ac, H1K26ac and H3K56ac [30,31]. In addition, a broad range of non-histone proteins has been identified as substrates of SIRT1, including p53 [17], PGC-1α [32], C-Myc [27] and FOXOs [33]. Thus, SIRT1 exerts a critical role in the regulation of the expression of targets genes and direct impacts on the binding ability, stability, activity or function of substrates via deacetylation. We herein presented the evidences that SIRT1 was involved in the modulation of lncRNA expression (miR17HG), further enlarged the knowledge of SIRT1 on the regulation of gene expression.
A growing body of evidences has unraveled that lncRNAs have emerged as novel gene regulators at multiple levels, and thus exerted a pivotal role in a variety of cellular processes, including DNA repair. LncRNA- PCAT-1 impaired the homologous recombination (HR) for DSBs repair through the repression of BRCA2 [34]. LncRNA-TODRA was located at the RAD51 Locus but was transcribed in the opposite direction, which effectively enhanced the RAD51-dependent DSBs Repair [35]. Moreover, lncRNA-DDSR1 was induced by NCS treatment in an ATM-NF-κB pathway-dependent manner, which promoted HR through direct interaction with BRCA1 and hnRNPUL1 [36]. In addition, LINP1 promoted DSBs repair after IR through interacting with NHEJ proteins (Ku80 and DNA-PKcs) [37]. Our demonstration that miR17HG facilitated the repair of NCS-induced DNA damage through physical interaction with SIRT1, further verified the close relationship between lncRNA and DNA repair, and uncovered the complexity of the underlying mechanisms for the maintenance of genome integrity.
miR17HG was derived from the mir-17-92a-1 cluster host gene (also known as c130rf25), which was located at the classical 13q31 chromosomal level. In addition to the lncRNA miR17HG, the c13orf25 transcript generated six functional miRNAs as well, including miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1 [38]. This region has been reported to be involved in the regulation of cell survival, proliferation, differentiation, and angiogenesis, and was closely associated with the development of various types of human cancers [39]. One group has reported that miR-17/20a promoted DNA repair through the direct target the death-associated protein kinase3 (DAPK3), a p53 activating kinase [40]. In our study, the constructed miRNA sponge that significantly inhibited the expression of these six miRNAs evidently suppressed the repair of NCS-induced DNA damage as well (SFigure 2B). Nonetheless, the effect of the lncRNA miR17HG on DNA damage repair and its underlying mechanism were still far to be elucidated. We herein presented the evidence that miR17HG exerted a critical role in NCS-induced DSBs repair, which further verified the function of the mir-17-92a-1 cluster in DNA repair. In addition, our results (data not shown) unveiled that silencing of miR17HG significantly accelerated the process of cellular senescence, which further demonstrated the protective effect of miR17HG.
Long noncoding RNAs have emerged as key regulators of gene expression at multiple levels [41]. Additionally, lncRNAs modulate the localization, stability, and activity of the protein through direct binding to specific protein partners [42]. For instance, the long non-coding RNA LSINCT5 significantly increased the stability of HMGA2 via physical interaction, and thus promoted malignancy of non-small cell lung cancer [43]. LncRNA FEZF1-AS1 could bind and increase the stability of the pyruvate kinase 2 (PKM2), and thus promote the colorectal cancer proliferation and metastasis [44]. Our results uncovered that miR17HG significantly enhanced the deacetylase of SIRT1 through direct interaction and promoted the repair of NCS-induced DNA damage, which further confirmed this functional mechanism of lncRNA.
In aggregate, we firstly presented evidences that SIRT1 significantly enhanced the expression levels of lncRNA miR17HG through deacetylation of C-Myc, which exerted a critical role in NCS-induced DSBs repair, and thus effectively protected cells against apoptosis. Hence, we herein identified a positive feedback loop of SIRT1 and miR17HG, which not only confirm the pivotal role of SIRT1 in the maintenance of genome integrity but also deepened the understanding of the molecular mechanisms of SIRT1 in various biological processes.
Funding Statement
This work was supported by the grants from the National Natural Science Foundation of China [31600976, 81671399, 81170327], the Natural Science Foundation of Guangdong Province [2016A030313684, 9252402301000002], the Innovation Team Construction Projects in Ordinary University of Guangdong Province [2015KCXTD022], the Unique Innovative Projects in Ordinary University of Guangdong Province [2015KTSCX049], the International Scientific and Technological Cooperation of Dongguan City [2016508102001], the Science & Technology Innovation Fund of Guangdong Medical University [STIF201102], the “Climbing” Program of Guangdong Province [pdjh2019b0214, pdjhb0223].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Rinn JL, Chang HY.. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. PubMed PMID: 22663078; PubMed Central PMCID: PMC3858397. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013. July 03;154(1):26–46. S0092-8674(13)00759-9 [pii]. PubMed PMID: 23827673; PubMed Central PMCID: PMC3924787. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Carpenter S. Long noncoding RNA: novel links between gene expression and innate immunity. Virus Res. 2016;212:137–145. [DOI] [PubMed] [Google Scholar]
- [4].Olive V, Li Q, He L. mir-17-92: a polycistronic oncomir with pleiotropic functions. Immunol Rev. 2013. May;253(1):158–166. PubMed PMID: 23550645; PubMed Central PMCID: PMC3972423. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xue M, Chen W, Li X. Urothelial cancer associated 1: a long noncoding RNA with a crucial role in cancer. J Cancer Res Clin Oncol. 2016. July;142(7):1407–1419. 10.1007/s00432-015-2042-y [pii]. PubMed PMID: 26341664; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. 2014. January;9(1):21–26. 27472 [pii]. PubMed PMID: 24335342; PubMed Central PMCID: PMC3928181. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wan G, Hu X, Liu Y, et al. A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. Embo J. 2013. October 30;32(21):2833–2847. emboj2013221 [pii]. PubMed PMID: 24097061; PubMed Central PMCID: PMC3817469. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017. August 01;77(15):3965–3981. 0008-5472.CAN-16-2634 [pii]. PubMed PMID: 28701486; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013. March 14;152(6):1298–1307. S0092-8674(13)00201-8 [pii]. PubMed PMID: 23498938; PubMed Central PMCID: PMC3651923. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mimura T, Kaji Y, Noma H, et al. The role of SIRT1 in ocular aging. Exp Eye Res. 2013. November;116:17–26. S0014-4835(13)00215-7 [pii]. PubMed PMID: 23892278; eng. [DOI] [PubMed] [Google Scholar]
- [11].Ming M, Shea CR, Guo X, et al. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci U S A. 2010. December 28;107(52):22623–22628. 1010377108 [pii]. PubMed PMID: 21149730; PubMed Central PMCID: PMC3012476. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ling L, Gu S, Cheng Y. Resveratrol inhibits adventitial fibroblast proliferation and induces cell apoptosis through the SIRT1 pathway. Mol Med Rep. 2017. February;15(2):567–572. PubMed PMID: 28101569; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007. May 25;100(10):1512–1521. 01.RES.0000267723.65696.4a [pii]. 10.1161/01.RES.0000267723.65696.4a. PubMed PMID: 17446436; eng. [DOI] [PubMed] [Google Scholar]
- [14].Liu J, Bi X, Chen T, et al. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015;6(7):e1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012. March 07;13(4):225–238. PubMed PMID: 22395773; PubMed Central PMCID: PMC4872805. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Giblin W, Skinner ME, Lombard DB. Sirtuins: guardians of mammalian healthspan. Trends Genet. 2014. July;30(7):271–286. S0168-9525(14)00069-9 [pii]. PubMed PMID: 24877878; PubMed Central PMCID: PMC4077918. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee JT, Gu W. SIRT1: regulator of p53 deacetylation. Genes Cancer. 2013. March;4(3–4):112–117. 10.1177_1947601913484496 [pii]. PubMed PMID: 24020002; PubMed Central PMCID: PMC3764473. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jeong J, Juhn K, Lee H, et al. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007. February 28;39(1):8–13. 200702282 [pii]. 10.1038/emm.2007.2. PubMed PMID: 17334224; eng. [DOI] [PubMed] [Google Scholar]
- [19].Oberdoerffer P, Michan S, McVay M, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008. November 28;135(5):907–918. S0092-8674(08)01317-2 [pii]. PubMed PMID: 19041753; PubMed Central PMCID: PMC2853975. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Papamichos-Chronakis M, Peterson CL. Chromatin and the genome integrity network. Nat Rev Genet. 2013. January;14(1):62–75. nrg3345 [pii]. PubMed PMID: 23247436; PubMed Central PMCID: PMC3731064. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Marnef A, Legube G. Organizing DNA repair in the nucleus: DSBs hit the road. Curr Opin Cell Biol. 2017. June;46:1–8. S0955-0674(16)30178-8 [pii]. 10.1016/j.ceb.2016.12.003. PubMed PMID: 28068556; eng. [DOI] [PubMed] [Google Scholar]
- [22].Li K, Casta A, Wang R, et al. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J Biol Chem. 2008. March 21;283(12):7590–7598. M709707200 [pii]. PubMed PMID: 18203716; eng. [DOI] [PubMed] [Google Scholar]
- [23].Dobbin MM, Madabhushi R, Pan L, et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013. August;16(8):1008–1015. nn.3460 [pii]. PubMed PMID: 23852118; PubMed Central PMCID: PMC4758134. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dianatpour A, Ghafouri-Fard S. The role of long non coding RNAs in the repair of DNA double strand breaks. Int J Mol Cell Med. 2017. Winter;6(1):1–12. PubMed PMID: 28868264; PubMed Central PMCID: PMC5568187. eng. [PMC free article] [PubMed] [Google Scholar]
- [25].Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007. January 26;282(4):2130–2134. doi: C600252200 [pii]. 10.1074/jbc.C600252200. PubMed PMID: 17135268; eng. [DOI] [PubMed] [Google Scholar]
- [26].Thomas M, Lange-Grunweller K, Hartmann D, et al. Analysis of transcriptional regulation of the human miR-17-92 cluster; evidence for involvement of Pim-1. Int J Mol Sci. 2013. June 07;14(6):12273–12296. ijms140612273 [pii]. PubMed PMID: 23749113; PubMed Central PMCID: PMC3709785. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Menssen A, Hydbring P, Kapelle K, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A. 2012. January 24;109(4):E187–96. 1105304109 [pii]. PubMed PMID: 22190494; PubMed Central PMCID: PMC3268300. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].O’Donnell KA, Wentzel EA, Zeller KI, et al. C-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005. June 9;435(7043):839–843. nature03677 [pii]. 10.1038/nature03677. PubMed PMID: 15944709; eng. [DOI] [PubMed] [Google Scholar]
- [29].Chin DH, Li HH, Kuo HM, et al. Neocarzinostatin as a probe for DNA protection activity–molecular interaction with caffeine. Mol Carcinog. 2012. April;51(4):327–338. PubMed PMID: 21538576; eng. [DOI] [PubMed] [Google Scholar]
- [30].Vaquero A, Scher M, Erdjument-Bromage H, et al. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007. November 15;450(7168):440–444. nature06268 [pii]. 10.1038/nature06268. PubMed PMID: 18004385; eng. [DOI] [PubMed] [Google Scholar]
- [31].Bosch-Presegue L, Vaquero A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. Febs J. 2015. May;282(9):1745–1767. PubMed PMID: 25223884; eng. [DOI] [PubMed] [Google Scholar]
- [32].Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem. 2005. April 22;280(16):16456–16460. M501485200 [pii]. 10.1074/jbc.M501485200. PubMed PMID: 15716268; eng. [DOI] [PubMed] [Google Scholar]
- [33].Lee D, Goldberg AL. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem. 2013. October 18;288(42):30515–30526. M113.489716 [pii]. PubMed PMID: 24003218; PubMed Central PMCID: PMC3798522. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Prensner JR, Chen W, Iyer MK, et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014. March 15;74(6):1651–1660. 0008-5472.CAN-13-3159 [pii]. PubMed PMID: 24473064; PubMed Central PMCID: PMC4009928. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gazy I, Zeevi DA, Renbaum P, et al. TODRA, a lncRNA at the RAD51 locus, is oppositely regulated to RAD51, and enhances RAD51-dependent DSB (double strand break) repair. PLoS One. 2015;10(7):e0134120 PONE-D-14-31719 [pii]. PubMed PMID: 26230935; PubMed Central PMCID: PMC4521930. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sharma V, Khurana S, Kubben N, et al. A BRCA1-interacting lncRNA regulates homologous recombination. EMBO Rep. 2015. November;16(11):1520–1534. embr.201540437 [pii]. PubMed PMID: 26412854; PubMed Central PMCID: PMC4641504. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang X, Liu H, Shi L, et al. LINP1 facilitates DNA damage repair through non-homologous end joining (NHEJ) pathway and subsequently decreases the sensitivity of cervical cancer cells to ionizing radiation. Cell Cycle. 2018;17(4):439–447. PubMed PMID: 29527968; PubMed Central PMCID: PMC5927633. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Molinari C, Salvi S, Foca F, et al. miR-17-92a-1 cluster host gene (MIR17HG) evaluation and response to neoadjuvant chemoradiotherapy in rectal cancer. Onco Targets Ther. 2016;9:2735–2742. ott-9-2735 [pii]. PubMed PMID: 27226732; PubMed Central PMCID: PMC4866748. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhu H, Han C, Wu T. MiR-17-92 cluster promotes hepatocarcinogenesis. Carcinogenesis. 2015. October;36(10):1213–1222. bgv112 [pii]. PubMed PMID: 26233958; PubMed Central PMCID: PMC4794620. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cai Z, Cao R, Zhang K, et al. Oncogenic miR-17/20a forms a positive feed-forward loop with the p53 kinase DAPK3 to promote tumorigenesis. J Biol Chem. 2015. August 7;290(32):19967–19975. M115.661504 [pii]. PubMed PMID: 26117336; PubMed Central PMCID: PMC4528155. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012. August;40(14):6391–6400. gks296 [pii]. PubMed PMID: 22492512; PubMed Central PMCID: PMC3413108. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018. January 25;172(3):393–407. S0092-8674(18)30048-5 [pii]. 10.1016/j.cell.2018.01.011. PubMed PMID: 29373828; PubMed Central PMCID: PMC5978744. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tian Y, Zhang N, Chen S, et al. The long non-coding RNA LSINCT5 promotes malignancy in non-small cell lung cancer by stabilizing HMGA2. Cell Cycle. 2018;17(10):1188–1198. PubMed PMID: 29883241; PubMed Central PMCID: PMC6110604. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bian Z, Zhang J, Li M, et al. LncRNA-FEZF1-AS1 promotes tumor proliferation and metastasis in colorectal cancer by regulating PKM2 signaling. Clin Cancer Res. 2018. October 1;24(19):4808–4819. 1078-0432.CCR-17-2967 [pii]. PubMed PMID: 29914894; eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.