

Abstract
WD repeat domain 5 (WDR5) is a member of the WD40-repeat protein family that plays a critical role in multiple chromatin-centric processes. Overexpression of WDR5 correlates with poor clinical outcome in many human cancers, and WDR5 itself has emerged as an attractive target for therapy. Most drug-discovery efforts center on the WIN site of WDR5 that is responsible for recruitment of WDR5 to chromatin. Here, we describe discovery of a novel WDR5 WIN site antagonists containing a dihydroisoquinolinone bicyclic core using structure-based design. These compounds exhibit picomolar binding affinity and selective concentration-dependent anti-proliferative activities in sensitive MLL-fusion cell lines. Furthermore, these WDR5 WIN site binders inhibit proliferation in MYC–driven cancer cells and reduce MYC recruitment to chromatin at MYC/WDR5 co-bound genes. Thus, these molecules are useful probes to study the implication of WDR5 inhibition in cancers and serve as a potential starting point toward the discovery of anti-WDR5 therapeutics.
Keywords: WDR5, WIN site inhibitor, structure-based design, mixed-lineage leukemia
Graphical Abstract
INTRODUCTION
WD repeat domain 5 (WDR5) is a member of the WD40-repeat protein family1–3 and functions as a core scaffolding component in multiple protein complexes that control transcriptional regulation and histone code modification processes as a part of the epigenetic machinery.4 WDR5 maintains over 90% sequence homology among all vertebrates to form a characteristic circular barrel shaped 7-bladed β-propeller structure and provides two major interfaces, where a number of partner proteins can bind. The most prominent role of WDR5 is in the context of MLL/SET complexes that deposit histone H3 lysine 4 di- and tri-methylation (H3K4me2, me3),5–8 a mark of transcriptionally active chromatin. Binding interactions between WDR5 and members of the MLL/SET family of proteins (MLL1–4, SETd1A, and SETd1B) are well-characterized,9–12 and center on interaction of a conserved arginine in the MLL/SET proteins with an arginine-binding cavity on WDR5 known as the WIN site. In these complexes, WDR5 also interacts with at least three other proteins (RBBP5, ASH2L, and DPY30) through WIN-site independent mechanisms. Although all MLL/SET family members can engage the WIN site, complexes containing MLL1 are uniquely dependent on the WIN site of WDR5 for enzymatic activity.13
WDR5 is also involved in the association of MYC oncoprotein transcription factors with chromatin14. MYC does not bind WDR5 via the WIN site, but instead interacts with a distal cleft (the WBM site) that is also bound by RBBP5. WDR5 co-localizes with MYC on chromatin at key target genes, and mutations in MYC that disrupt the interaction with WDR5 reduce the binding of MYC to chromatin, and disable its pro-tumorigenic properties in vivo. WDR5 thus serves as a critical co-factor for MYC proteins to execute malignant gene expression programs. Furthermore, several cancers exhibit aberrant overexpression of WDR5 including bladder cancer,15 pancreatic cancer,16 breast cancer,5 colorectal cancer,17 gastric cancer,18 prostate cancer,19,20 neuroblastoma,21 head neck squamous cell carcinoma,22 and various leukemias.23 Often, overexpression of WDR5 is found in more aggressive cancers, and is associated with poor prognoses.15,17,24 Considering the multifaceted role of WDR5 in human malignancies, WDR5 has emerged as an attractive drug target for discovery of potent small molecule inhibitors.
Most drug discovery efforts targeting WDR5 center on the WIN site. The concept of targeting the WIN site of WDR5 as a therapeutic strategy first arose within the context of leukemias characterized by translocations of the MLL1 gene.11,25,26 In these cancers, translocation of a single allele in the 11q23 MLL1 gene to one of more than 70 different partner genes results in the development of leukemia.11,25,26 The most common fusion partners AF4, AF9, and ENL account for 70% of acute lymphoid leukemia (ALL) in infants and 5–10% of acute myeloid leukemia (AML) in adults.26–28 It was originally proposed29 that MLL-rearranged cancers would be sensitive to WIN site inhibitors because of a dependency on the remaining pristine MLL1 allele in these cancers, but this rationale has since been disproved.30 There is, however, a strong empirical sensitivity of MLL-fusion cancer cells to WIN site inhibitors,31 supporting the idea that they can be implemented for treatment of these malignancies.
Several structurally distinctive classes of WDR5 WIN site inhibitors have been reported, and Figure 1 depicts three representative classes of which their binding interactions at the site have been confirmed by X-ray co-crystal structures. Macrocyclic peptidomimetic compounds were designed to mimic the MLL peptide residues within the WIN site. MM-589 (Figure 1)32 was reported to exhibit sub-nanomolar affinity at the WIN site, selective inhibition of MLL1 HMT activity with low-nanomolar IC50, and anti-proliferative activities in MLL-fusion cancer cell lines MV4:11 and Molm13. OICR-9429 (Figure 1)33,34 is an example of non-peptidomimetic small-molecule WIN site inhibitor, wherein a basic methyl-piperazine moiety mimics the guanidine side-chain of R3765 in the MLL peptide. This compound has a reported Ki of 64 nM to WDR5 and has demonstrated reduced viability of primary human AML cells that express mutant forms of CCAAT-enhancer-binding protein α (C/EBPα)33 and gain-of-function p53 mutants.6
Figure 1.
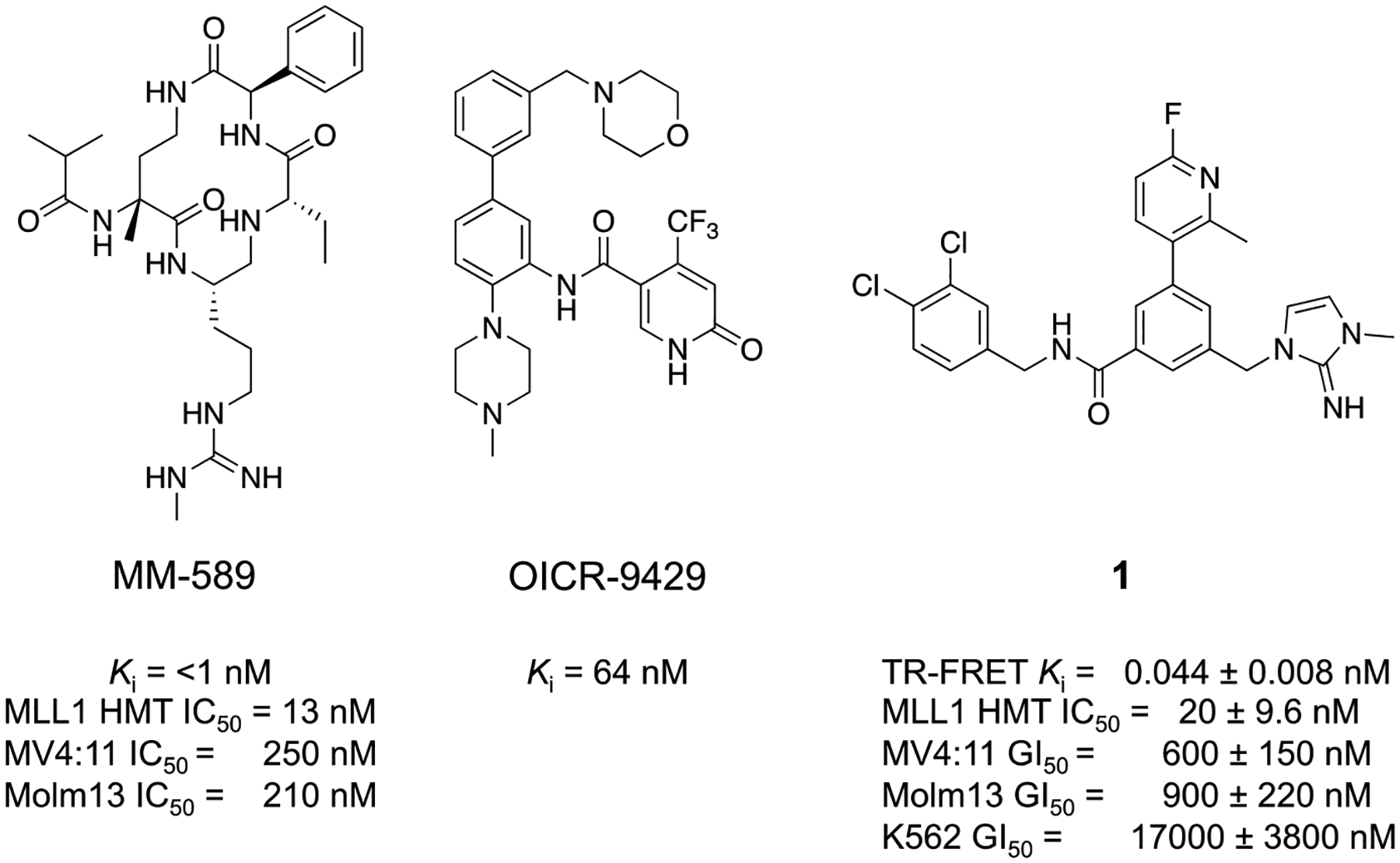
Chemical structures and in vitro profiles of three previously reported WDR5-WIN-site inhibitors.
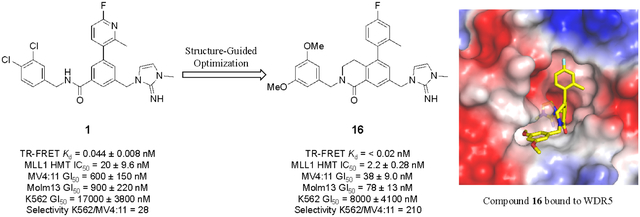
We have previously reported the discovery of WDR5 inhibitors using fragment-based methods and structure-based design.8,35 Our second-generation chemical probe was discovered using the imidazole-imine “warhead” moiety, which mimics the R3765 side-chain in the S2 pocket. The key binding interaction elements, such as a sandwiched π-π stacking interaction of the imidazole-imine in the S2 pocket, hydrogen-bond interaction of the carbonyl oxygen with the backbone NH of C261, and a hydrophobic biaryl moiety within the S4 pocket, mediated favorable binding of 1 (Figure 1)8 with a Ki of 44 pM to WDR5. Compound 1 also inhibited MLL1 HMT with low-nanomolar activity and proliferation of MLL-fusion cell lines MV4:11 and Molm13 with a GI50 values of 600 and 900 nM, respectively in a five-day cell proliferation assay. Compound 1 was then used as a chemical probe to investigate the implication of WDR5 WIN site inhibition on the interaction of WDR5 with chromatin, transcriptional alterations, and cellular functions that impact cell-fate. We found that blocking the WIN site of WDR5 with compound 1 caused displacement of WDR5 protein from its binding sites on chromatin and triggered a series of events including decreased expression of WDR5-bound ribosome protein genes, induction of nucleolar stress, increased p53 translation, and activation of p53-dependent apoptosis. Our previous results forecast a broad utility of potent WDR5 inhibitors as potential therapeutics against cancers.
Despite the picomolar binding affinity of compound 1, further improvements in cellular potency and pharmaceutical properties need to be made in order to demonstrate the therapeutic potential of WDR5 inhibitors. Here, we report our continued efforts to develop a potent and selective WDR5 inhibitor that builds upon our recently reported compound 1. Our efforts led to the discovery of a new class of potent WDR5 inhibitors that contain a novel dihydroisoquinolinone core unit, which was designed using X-ray structure-guided modifications. The new series of inhibitors offer significantly enhanced on-target mechanism-based anti-proliferative activity in WDR5 sensitive cells and drug-like properties. These improved compounds also produce consistent biomarker changes at physiologically relevant concentrations which support the primary mechanism of action.8
RESULTS AND DISCUSSION
Optimization of the Substituted Benzamide Series WDR5 Inhibitors Using Pharmacophore-based Approach.
The structure of compound 1 was divided into four pharmacophore units (P2, P4, P7, and core) based on their binding sub-site alignments in the WIN site of WDR5 with residues 3764–3773 (ARAEVHLRKS) in MLL1,36 and focused SAR efforts on each sub-unit were performed to optimize the potency and pharmaceutical properties of the series. The biochemical activity of each inhibitor was profiled using a time-resolved fluorescence energy transfer (TR-FRET) assay to determine the competitive binding affinity (Ki) against the MLL1 derived FITC-peptide probe followed by a H3K4 HMT inhibition assay as an orthogonal functional measurement of the effect on MLL1 mediated HMT activity.
As previously reported,8 WDR5 WIN site inhibitors display a robust antiproliferative response in MLL-fusion cancer cell lines that express wild-type p53. Therefore, WDR5 inhibition mediated cellular efficacy was assessed in MV4:11 and Molm13. The K562 leukemia cell line, which is null for p53, was used as an insensitive control. The half-maximal growth inhibition (GI50) ratio between K562 and MV4:11 serves as a cellular selectivity measure to ascertain primary target mediated activity.
The benzyl-3-methyl-1,3-dihydro-2H-imidazol-2-imine moiety of compound 1 bound to the deepest part of the WIN-site was incorporated into the new compound design for consistency in activity comparisons. The potentially reactive 4-fluoro-2-methylpyridin-3-yl P4 moiety of 1 was replaced with the chemically more stable 4-fluoro-2-methylphenyl isostere. We then set out to optimize the P7 moiety located in the hydrophobic S7 pocket of the WIN-site where H3769 and L3770 of MLL1 bind. A focused library of substituted benzamides was constructed by employing more than 50 benzylamines with varying substitution patterns and sizes to probe binding interactions at the site. Table 1 depicts representative examples to illustrate SAR trends.
Table 1.
SAR Profile of Substituted Benzamide WDR5 WIN site inhibitors
 |
|||||||
|---|---|---|---|---|---|---|---|
| compnd | R1 = | Kd (nM) TR-FRET | HMT IC50 (nM) | Cell Proliferation Assays GI50 (nM) | Selectivity | ||
| WDR5 | MLL1 | MV4:11 | Molml3 | K562 | K562/MV4:11 | ||
| 2 |  |
1.5 ±0.80 | 43.3 | 2300 ± 620 | 2000 ± 230 | 13000 ± 3000 | 5 |
| 3 | 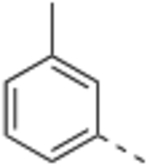 |
0.50 ±0.010 | 55.0 | 2400 ± 660 | 2700 ± 440 | 18000 ±6300 | 7 |
| 4 | 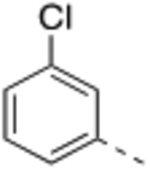 |
0.14 ±0.065 | 14.0 | 870 ± 320 | 840 ± 93 | 6400 ±1400 | 7 |
| 5 | 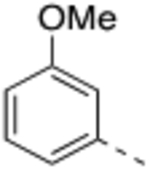 |
0.16 ±0.026 | 9.9 | 820 ± 400 | 970 ± 400 | 15000 ±850 | 18 |
| 6 | 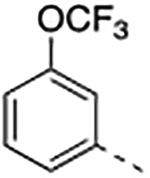 |
16 ± 14 | >3000 | 13000±1300 | 7600 ± 460 | 28000 ± 2400 | 2 |
| 7 |  |
0.030 ±0.014 | 3.4 | 390 ±150 | 390 ± 52 | 3600 ±1100 | 9 |
| 8 | 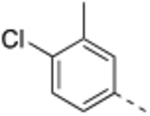 |
0.090 ± 0.002 | 28.4 | 1300±190 | 1500 ±220 | 10000 ± 3400 | 8 |
| 9 | 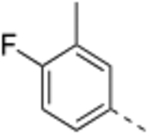 |
0.032 ± 0.005 | 11.6 | 360 ± 200 | 405 ± 83 | 7600±1500 | 21 |
| 10 |  |
0.035 ± 0.002 | 8.6 | 240 ± 28 | 300 ± 49 | 12000 ± 3800 | 50 |
| 11 | 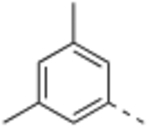 |
0.055 ± 0.021 | 23.9 | 340 ± 86 | 510 ±140 | 5900 ±1200 | 17 |
| 12 | 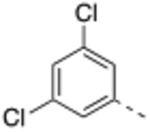 |
0.061 ± 0.023 | 3.4 | 490 ± 25 | 580 ±170 | 4900 ± 940 | 10 |
| 13 | 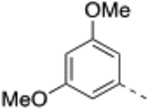 |
0.049 ± 0.008 | 3.4 | 470 ± 25 | 480 ±110 | 15000 ± 3600 | 32 |
TR-FRET Ki, and cell proliferation GI50 values represent four independent replicate determinations ± the standard deviation.
Histone methyltransferase inhibition (HMT) IC50 values represent two independent replicate determinations.
Selectivity is defined as GI50, K562/GI50, MV4:11.
Compound 2 was designed to establish the baseline activity profile of the unsubstituted phenyl moiety, and exhibited low-nanomolar binding affinity (Ki = 1.5 nM) to WDR5, inhibition of HMT with IC50 of 43 nM and low-micromolar anti-proliferative activity in the sensitive cell lines with only 5-fold selectivity over K562 cells. Early analogs indicated that the mono meta-substitution of phenyl group was more beneficial than other positions; therefore, a series of 3-substituted phenyl analogues were introduced to determine the optimal substituent in the S7 pocket. Compound 3 with a 3-methylphenyl demonstrated a marginal improvement in binding affinity, which was not translated in cellular potency in the sensitive cell lines. However, compounds 4 and 5 bearing either a 3-chloro or 3-methoxy groups displayed an order of magnitude enhanced binding affinities with sub-micromolar GI50’s in sensitive cell lines. In general, functional HMT IC50 values show good correlation and rank order with the cellular anti-proliferative activities (GI50) in the sensitive cell lines. For example 5 displayed a 4-fold improvement in HMT activity when compared to 2 and indeed 5 was consistently more potent in cells than 2. Interestingly, the 3-trifluoromethoxy of compound 6 exerted an undesirable binding interaction at the site to cause a large decrease in binding affinity and cellular activity. From this subset of mono-substituted compounds, 3-chloro and 3-methoxy groups provided beneficial binding interactions in the S7 pocket and were utilized in further optimization of the P7 unit.
We next explored either 3,4- or 3,5-disubstituted phenyl analogues for improved potency targeting the S7 sub-site in biochemical and cellular assays. Compound 7 containing 3,4-dichloro substitutions demonstrated a slight improvement in binding and cellular potency over its direct analog 1. This result also confirmed that replacement of the P4 group to the 4-fluoro-2-methylphenyl also proved to be beneficial to the in vitro potency of compounds. When compared to 4, the additional 4-chloro group in 7 also increased both binding affinity and HMT inhibition by 4 to 5-fold, and resulted in 2-fold enhanced cellular activity in the sensitive cell lines. As seen previously, compound 8 with 3-methyl-4-chloro substitution exhibited 3-fold reduced biological activities compared to 7, which is consistent with 3 and suggests that the 3-methyl group is a suboptimal phenyl substituent in the S7 sub-pocket for WDR5 potency. The 4-fluoro group in 9 and 10 were beneficial and added potency to existing 3-substituents of the phenyl group. Indeed, compound 10 with 4-fluoro-3-methoxyphenyl P7 moiety displayed the highest cellular potency in both MV4:11 and Molm-13 cells and selectivity over the insensitive K562 among examples in Table 1. The 3,5-disubstituted phenyl P7 compounds, represented by 11 – 13, were found to be equally effective by displaying similar in vitro potency compared to the 3,4-disubtituted series compounds. In summary, the 3-substituent of the phenyl P7 group is essential for the baseline affinity to the WIN site of WDR5, and the proper second substitutions at the 4- or 5-position are equally beneficial for enhancing potency further. Finally, compounds 5, 10 and 13 with the 3-methoxy substitution exhibited significantly improved GI50 ratios between MV4:11 and K562 compared to the 3-chloro or 3-methyl analogs. These results suggest that observed selective cytotoxicity in MV4:11 by the 3-methoxy series were mainly driven by the WDR5 inhibition mechanism, but they were generally less cytotoxic in insensitive cells. Based on the affinity, promising cellular activity, and selectivity index, compound 13 was chosen for further characterization and SAR expansion.
X-ray Co-crystal Structure of 13 Bound to WDR5.
An X-ray co-crystal structure of compound 13 bound to WDR5 (Figure 2) was obtained to determine the binding interactions at the WIN site for further structure-based design optimization. The binding mode of 13 bound to WDR5 (Figure 2A) was similar to the previously reported structure8 of 1.
Figure 2.

X-ray co-crystal structure of 13 bound to WDR5 (PDB ID: 6UFX). (A) Compound 13 (green-carbon capped sticks) bound to WDR5 represented as semi-transparent electrostatic potential surface with S2, S4, and S7 binding regions denoted. (B) Key H-bond (red dashed lines) and π–π stacking interaction residues of WDR5 that interact with 13. (C) Compound 13 with hydrogen atoms highlighting the design of the bicyclic dihydroisoquinolinone core.
The imidazole imine P2 group is positioned deeply in the S2 sub-pocket to engage double π-π stacking interactions with F133 and F263 and H-bond interaction to the carbonyl oxygen of C261 residue of WDR5. In addition, the imine NH formed two H-bond interactions with the backbone carbonyl oxygen of C261 with an interatomic distance of 2.8 Å and water bridged interaction to the side chains of S175 and S218 residues. The 4-fluoro-2-methylphenyl and 3,5-dimethoxyphenyl occupy the hydrophobic S4 and S7 sub-sites, respectively. The 3-methoxy unit the P7 group penetrates to the deepest part of S7 pocket to have hydrophobic interactions. Conversely, the 5-methoxy moiety is positioned toward the solvent accessible area where further chemical modifications are feasible for incorporation of property optimizing groups. An additional H-bond is also identified between the carbonyl of the benzamide core and the backbone NH of C261 with an interatomic distance of 2.8 Å, which is essential to stabilize the exit vector to the P7 group. The previously described key binding interaction elements of 13 (Figure 2B) contribute greatly to the overall binding affinity.
The X-ray structure of 13 revealed an opportunity for further structural modification of the core unit. Figure 2C highlights the bioactive conformation and positions on the core phenyl ring and the amide NH, where two hydrogen atoms provide appropriate vectors for a ring constraint design through a saturated bicyclic ring system to restrict the conformation. This modification may have several expected benefits. First, the new bicyclic core would preserve the binding conformation of the core unit and potentially increase the binding affinity by reducing conformational entropy. Second, the modification would effectively eliminate a potentially metabolically unstable secondary amide functional group, an H-bond donor, and two rotatable bonds that in principle may benefit pharmacokinetic and pharmaceutical properties of final target compounds.
Dihydroisoquinolinone Series WDR5 Inhibitors.
To further improve potency and pharmaceutical properties of the compounds, the tether design strategy was applied to form the dihydroisoquinoline core. Compounds 14 – 16 (Table 2) bearing optimized P7 units were synthesized, and they exhibited significantly enhanced biological activities compared to their direct open-chain analogs. Compound 14 had a 3 to 6-fold improvement in binding affinity, HMT inhibition and antiproliferative activity in the sensitive cell lines compared to 8. Binding affinities and HMT activities for both 15 and 16 could not be measured reliably because their IC50’s from our TR-FRET and HMT assays were below or at the theoretical limit of detection. The improved WDR5 affinities of 15 and 16 also impacted cellular potency of both compounds by lowering their GI50’s 6- to 12-fold only in the sensitive cell lines compared to 9 and 13. As expected, the cellular selectivity ratios assessed by GI50 differences between K562 and MV4:11 lines were significantly increased to 120 and 210 for 15 and 16, respectively. The results indicate that the structural modification on the core unit enhances the WDR5 affinity, which is translated to an increase in on-target mediated anti-proliferative activity in the WDR5 sensitive cells.
Table 2.
Biochemical and Cellular Activity of Compounds Containing Dihydroisoquinolinone Core
 |
|||||||
|---|---|---|---|---|---|---|---|
| compnd | R2 = | Kd (nM) TR-FRET | HMT IC50 (nM) | Cell Proliferation Assays GI50 (nM) | Selectivity | ||
| WDR5 | MLL1 | MV4:11 | Molml3 | K562 | K562/MV4:11 | ||
| 14 | 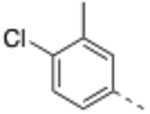 |
0.027 ± 0.008 | 4.7 | 240 ± 120 | 340 ± 68 | 8800 ±1300 | 37 |
| 15 | 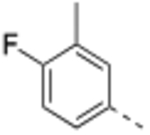 |
<0.02 | <2.0 | 44 ±25 | 55 ± 11 | 5400 ±1700 | 120 |
| 16 | 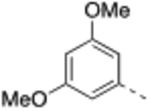 |
<0.02 | 2.2 | 38 ± 9.0 | 78 ±13 | 8000 ±4100 | 210 |
| R3 = | |||||||
| 17 |  |
0.043 ± 0.019 | 2.5 | 110 ± 37 | 200 ± 69 | 8600 ±1300 | 78 |
| 18 | 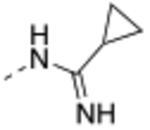 |
0.030 ±0.018 | 2.3 | 150 ±43 | 240 ±7.1 | 3700 ±1000 | 25 |
| 19 | 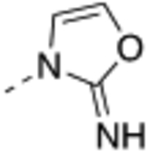 |
<0.02 | 4.0 | 180 ±56 | 330 ±61 | 3400 ± 930 | 19 |
| 20 | 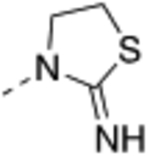 |
0.033 ± 0.009 | 15 | 290 ± 36 | 570 ± 39 | 2800 ± 490 | 10 |
| 21 | 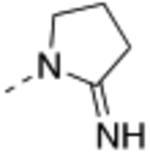 |
0.063 ± 0.020 | 14 | 400 ± 89 | 730 ±100 | 4900 ±1200 | 12 |
| 22 | 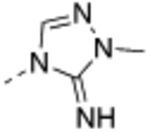 |
0.023 ± 0.010 | <2.0 | 84 ± 10 | 180 ±33 | 2900 ±190 | 35 |
TR-FRET Ki, and cell proliferation GI50 values represent four independent replicate determinations ± the standard deviation.
Histone methyltransferase inhibition (HMT) IC50 values represent two independent replicate determinations.
Selectivity is defined as GI50, K562/GI50, MV4:11.
Compound 17 – 22 (Table 2) bearing P2 variations on the compound 16 scaffold were synthesized and evaluated for their biological activities. P2 variants were selected on the basis of X-ray co-crystal structure of 16 bound to WDR5 (Figure 3) in which all beneficial interactions of the methyl imidazole imine moiety in the S2 pocket were preserved. The increased size and hydrophobicity of the P2 group using cyclopropyl in 17 and 18 caused the reduction in cellular potency in sensitive lines despite potent binding affinities and HMT inhibition activities. Compound 19 – 22 containing bioisosteres of the imidazole imine P2 group also exhibited reduced cellular potency in MV4:11 and Molm-13 cells compared to compound 16. Furthermore, 20 and 21 showed significantly diminished cellular selectivity, which may suggest an increase in cytotoxicity by off-target mechanisms. These results indicate the P2 moiety is a crucial component for cellular potency and selectivity. Finally, 16 with the methyl imidazole imine exhibited the best overall profile and was chosen as a chemical probe to interrogate the biological consequence of WDR5 inhibition.
Figure 3.

X-ray co-crystal structure of 16 bound to the WDR5 WIN-site (side-view, PDB ID: 6UCS). (A) Key H-bond (red dashed lines) and π−π stacking interaction residues of WDR5 that interact with 16. (B) Overlay of 13 (green sticks) and 16 (yellow sticks).
X-ray Co-crystal Structure of 16 Bound to WDR5.
To confirm the binding mode of the dihydroisoquinolinone core unit, the X-ray co-crystal structure of compound 16 bound to WDR5 (Figure 3) was obtained. The side view of the X-ray structure (Figure 3A) displays the same key binding interactions, such as the sandwich π−π stacking with side chain residues F133 and F263, bidentate H-bond with C261, and water mediated H-bond with side chain residues S175 and S218, as reported for 13 (Figure 2B). Overlays of the WDR5 bound conformations of 13 (green, Figure 3B) and 16 (yellow, Figure 3B) are nearly superimposable to further support the structure-based design. The ethylene tether of the dihydroisoquinolinone core is positioned in an unoccupied area of the WIN site and has no significant direct interactions within this region of the WIN-site. Therefore, the observed potency enhancement of compound 16 may be mainly mediated by reducing the conformational entropy by fixing the benzamide core conformation and the exit vector to the S7 sub-pocket.
Evidence of target engagement in cells.
We were interested in investigating whether inhibitor 16 is able to engage the WIN site of WDR5 in cells and specifically block interaction of WDR5 with proteins that bind the WIN site. A co-immunoprecipitation (co-IP) for WDR5 in MV4:11 cells treated with DMSO and 16 for 4 h demonstrated reduced binding of MLL1 with WDR5 upon 16 treatment but not DMSO treatment. RBBP5 was used a control for the specificity of WDR5 WIN site blockade, as RBBP5 is a member of the WRAD complex along with MLL1 but RBBP5 interacts with WDR5 on a binding surface distinct from the WIN site (WBN). Binding of RBBP5 with WDR5 was not affected by compound 16. Reduced IP of MLL1 but not RBBP5 with WDR5 after 16 treatment indicated that the chemical probe is able to engage the WIN site of WDR5 in live cells and specifically displace protein binding partners that engage at the site.
Detection of Displacement of WDR5 from Chromatin and Transcriptional Repression of WDR5 Bound Genes Leading to p53 Induction.
We previously determined that the primary mechanism of action of our early but moderately potent WDR5 WIN site inhibitor 1 is the rapid displacement of WDR5 from chromatin at a select set of ribosomal protein genes (RPGs) that is conserved across disparate cell types. The displacement of WDR5 from chromatin after compound 1 treatment (2 μM) is accompanied by a commensurate ~2-fold decrease in mRNA expression of WDR5-bound RPGs.8 Thus, we investigated whether our more potent inhibitor 16 can recapitulate the displacement of WDR5 from chromatin and repression of WDR5-bound RPGs as described for 1.
To investigate WDR5 binding to chromatin after compound 16 treatment, we used chromatin immunoprecipitation (ChIP) to quantify WDR5 enriched at three representative genes that we previously determined to be bound by WDR5 in MV4:11 cells (Figure 5A).8 Consistent with our previous report8 for 1, treatment of MV4:11 cells for 4 h with 600 nM 16 resulted in a roughly 90% reduction of WDR5 bound at all three WDR5-bound genes tested. To further assess whether 16 recapitulated the mode of action described for 1, we next sought to determine whether 16 selectively repressed expression of RPGs at which WDR5 displacement occurs. RT-qPCR analysis revealed that indeed compound 16 reduced expression of both representative WDR5-bound genes tested (RPS24 and RPL35) by roughly 2-fold (Figure 5B). However, expression of two ribosomal protein genes not bound by WDR5 (RPS11 and RPL14),8 was not reduced upon 16 treatment. Together, these data indicate that our WDR5 WIN site inhibitors 1 and 16 share a common primary molecular mode of action. Furthermore, compound 16 elicited similar level of cellular response at a significantly lower concentration to compound 1.
Figure 5.

Compound 16 displaced WDR5 from chromatin and repressed mRNA expression of WDR5-bound genes. (A) WDR5 or IgG control ChIP at three representative genes bound by WDR5 SNHG15 (E-box locus), RPS24 and RPL35 in MV4:11 cells after 4 h treatment with 0.1% DMSO or 600 nM 16. The SNHG15 gene body locus serves as a negative control for WDR5 binding, n = 3. (B) Relative mRNA expression of two-representative WDR5-bound ribosomal protein genes (RPS24 and RPL35) and two representative ribosomal genes not bound by WDR5 (RPS11 and RPL14) in MV4:11 cells after three-day treatment with 0.1% DMSO or 300 nM 16, as determined by RT-qPCR. Relative expression is normalized to DMSO treatment for each gene, n = 3.
The primary mode of action of our early WIN site inhibitor 1 at 2 μM (i.e. displacement of WDR5 from chromatin and repression of WDR5 bound genes) leads to induction of a p53-mediated cellular response in cells expressing an oncogenic MLL-fusion.8 Thus we sought to determine p53 and p21 protein level changes at 24h and 48h post treatment of compound 16 in MV4:11 cells. Figure 6A depicts the dose dependent elevation of both p53 and p21 proteins in a synchronized manner at both time points, and the effect was maximized at 300 nM concentration of compound 16 then plateaued. A time course analysis revealed that p53 and p21 protein levels started to increase from 8 h post treatment of compound 16 at 300 nM and continued to elevate up to 32 h (Figure 6B). Additionally, compound 16 caused 2.5-fold increase in caspase 3/7 activity in the MLL-fusion cell lines MV4:11 and Molm13 48 h post treatment (Figure 6C), consistent with induction of apoptosis. Cell cycle analysis of MV4:11 cells treated with 1μM compound 16 for 48 hours and subsequently stained with propidium iodide showed an approximate 4 fold increased SubG1 cells, which further supports WDR5 inhibitor mediated induction of apoptosis. G1 and S phase cells had similar levels in both treated and untreated cells, while an apparent decrease in G2M phase cells was observed in compound 16 treated cells (Figure 6D). These data support that all biomarker changes described above are mediated by the inhibition of WDR5 at the WIN site inhibitor by compound 16 and share a common mode of action with our less potent previously described compound 1.
Figure 6.

Compound 16 treatment induced p53 and p21 protein levels and apoptosis. (A) Western blot, showing the effects of DMSO or compound 16 (0.01 – 3.0 μM) on p53 and p21 protein levels in MV4:11 cells at 24h and 48h post treatment. GAPDH is a loading control. (B) Western blot, showing the effects of DMSO or compound 16 (0.3 μM) on p53 and p21 protein levels in MV4:11 cells in time course. GAPDH is a loading control. (C) Caspase 3/7 induction by compound 16 at 4, 24, 48, and 72 h time points in MV4:11 and Molm13 cells. (D) Cell cycle analysis of MV4:11 cells, showing 4-fold increased SubG1 cells by compound 16 treatment for 48 hours compared to DMSO control.
Displacement of MYC from Chromatin.
Several reports8,32 have empirically determined that MLL-fusion leukemia cell lines are sensitive to WIN site inhibition, but we reasoned that WDR5 WIN site inhibitors may have utility in other cancer types as well. We have previously reported that WDR5 facilitates the recruitment of the oncogenic transcription factor MYC to chromatin through a direct interaction.14 This protein–protein interaction occurs via MYC binding to a surface on WDR5 (the WBN site) that is distinct from the WIN site. Mutations in MYC that disrupt the interaction with WDR5 result in diminished recruitment of MYC to chromatin and impair the ability of MYC to drive tumorigenesis. Therefore, we reasoned that since the WIN site of WDR5 is required for WDR5 engagement with chromatin,8 displacement of WDR5 from chromatin by a WIN site inhibitor would also result in displacement of MYC from chromatin, and therefore WIN site inhibition would be effective against MYC driven tumors.
To test this hypothesis, we used ChIP to assess the ability of MYC to engage chromatin at genes that displayed WDR5 displacement upon 16 treatment in Figure 5A. We found that MYC binding to chromatin was also reduced at all three genes tested in response to 16 treatment (Figure 7), indicating that impeding WDR5 associated with chromatin through the WIN site also prevents MYC recruitment to MYC/WDR5 co-bound target genes.
Figure 7.

Compound 16 displaces MYC from chromatin at WDR5-displaced genes. MYC or IgG control ChIP at three representative genes, SNHG15 (E-box locus), RPS24 and RPL35, at which 16 displaces WDR5 (as shown in Figure 5A). in MV4:11 cells. MYC or IgG control ChIPs were performed in MV4:11 cells after 4 h treatment with 0.1% DMSO or 600 nM 16. The SNHG15 gene body locus serves as a negative control for MYC binding. n = 3.
Anti-proliferative Activity of Compound 16 in Other Cancers.
To further assess whether blocking the WIN site of WDR5 by a small molecule can inhibit proliferation in MYC-driven cancers, compound 1 and 16 were tested in cell lines listed in Table 3. Both compounds showed similar cellular selectivity profiles with 10–20 fold higher potency for compound 16 in sensitive cells suggesting the same mechanism of action. CHP-134 (neuroblastoma, wt p53) and Ramos (Burkitt’s lymphoma, mutant p53) lines show the highest vulnerability by the WDR5 inhibition at the WIN site among tested cells despite their different p53 status. Compound 16 also exhibited anti-proliferative activities against other Burkitt’s lymphoma lines with mutant p53 (Raji and Daudi). In addition, proliferation of the colon cancer line SW620 was moderately inhibited by compound 16 treatment, but SW480 cells were completely resistant. Above results support a potential utility of WDR5 WIN site inhibitors against MYC-driven cancers, and containing wild-type p53 may not be an absolute requirement for a mechanism-based sensitivity. We are currently focused on determining other factors that govern the WDR5 inhibition mediated cellular vulnerability in MYC-upregulated cancers.
Table 3.
Anti-proliferative Activity of Compound 16 in MYC-driven Cancers.
| Compnd | Cell line GI50 (μM)a | |||||
|---|---|---|---|---|---|---|
| CHP-134 | Ramos | Raji | Daudi | SW620 | SW480 | |
| 1 | 2.7 ± 0.8 | 7.1 ± 1.4 | 16 ± 2.2 | 6.8 ± 4.0 | 6.5 ± 3.4 | >29 |
| 16 | 0.26 ± 0.08 | 0.49 ± 0.24 | 1.2 ± 0.1 | 0.58 ± 0.26 | 3.2 ± 1.2 | >22 |
Cell proliferation GI50 values represent average of three to five independent replicate determinations ± the standard deviation.
WDR5 WIN-site inhibitors were then tested in cancer tissue-originated spheroids (CTOSs), which retain morphological characteristics of the original tumors. Five previously reported colorectal cancer CTOSs37,38 with different mutation profiles were treated with compound 1 and 16 (1 and 10 μM), and dose dependent inhibitions of cell growth were observed in all CTOS lines (Figure 8). Compound 16 exhibited significantly higher potency than 1 as expected. Intriguingly, the C45 line with multiple mutations including p53 was most sensitive by the compound 16 treatment. These results further support a broad utility of WDR5 WIN site inhibitors against solid cancers.
Figure 8.

Response of CTOSs to compound 1 and 16. (A) The sensitivity of CTOSs from five patients dosed with 1 and 10 μM compound 1 and 16. Relative growth ratios were calculated by comparing with 0.1% DMSO control. (B) Mutation profiles of the CTOSs. Gray block indicated presence of a mutation.
Chemical Synthesis.
Synthesis of compounds 2 – 13 (Scheme 1) began from 23, which was prepared from a previously reported protocol.39 Boronic pinacol ester 24 was prepared through Miyaura borylation of 23, and subsequent Suzuki-Miyaura cross-coupling reaction with 2-bromo-5-fluorotoluene gave the biphenyl methyl ester 25. Carboxylic acid 26 was obtained through hydrolysis of methyl ester 25. Intermediate 26 underwent amide bond formation with a variety of benzyl amines in a parallel manner to yield benzyl alcohols 27 – 38. The alcohol functional group of 27 – 38 was then brominated with phosphorus tribromide and then substituted with 1-methyl-2-aminoimidazole to yield compound 2 – 13.
Scheme 1.

Synthesis of the Substituted Benzamide Series Compounds 2 – 13a
aR1 is defined in Table 1. Conditions: (a) B2Pin2, KOAc, Pd(dppf)Cl2, 1,4-dioxane, 85 °C overnight, quant.; (b) 2-bromo-5-fluorotoluene, Pd(dppf)Cl2, K2CO3, 1,4-dioxane:H2O (3:1), 95 °C, 2 h, 68%; (c) 2 M LiOH, THF, rt, overnight, 94%; (d) R1-benzyl amine, TBTU, i-Pr2NEt, DMF, rt, 8 h, 37 – 84%; (e) PBr3, CH2Cl2, rt, 2 h; (f) 1-methyl-2-aminoimidazole, i-Pr2NEt, KI, MeCN, 80 °C, 4 h, 7 – 40%, over two steps.
Preparation of compound 14 – 22 (Scheme 2) began from hemiacetal 39, which was prepared in three steps from a previously reported protocol.40 Compound 39 underwent reductive amination with 2,4-dimethoxybenzylamine to construct the bicyclic core unit of lactam 40. Triflate 41 was synthesized using N-phenylbis(trifluoromethanesulfonimide) and subsequent deprotection of the 2,4-dimethoxybenzyl group gave intermediate 42. It was then subjected to Suzuki-Miyaura cross-coupling with 2-bromo-5-fluorotoluene to produce biphenyl lactam 43. Three selected substituted benzyl bromides were introduced at the lactam NH of 43 to yield 44 – 46. The methyl-2-imidazoleimine group of compound 14 – 16 was introduced from the corresponding intermediate methyl ester 44 – 46 employing three-steps sequence including reduction, bromination and nucleophilic substitution with 1-methyl-2-aminoimidazole. Finally, compound 17 – 22 bearing bioisosteric P2 units were prepared from bromide 52 by means of nucleophilic substitution with corresponding amines in a parallel manner.
Scheme 2.

Synthesis of Dihydroisoquinolinone Series Compounds 14 – 22a
aR2 and R3 are defined in Table 2. Conditions: (a) NaBH(OAc)3, CH2Cl2, rt, overnight, 65%; (b) phenyl triflimide, i-Pr2NEt, CH2Cl2, rt, overnight, 75%; (c) TFA, anisole, CH2Cl2, rt, overnight, 78%; (d) (4-fluoro-2-methylphenyl) boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane:H2O, 80 °C, overnight, 93%; (e) R2-alkyl bromide, NaH, DMF, 0 °C, 2 h, 57% - quant.; (f) LiBHEt3, THF, 0 °C, 1 h, 68% - quant.; (g) PBr3, CH2Cl2, 0 °C, 4 h, 54 – 93%; (h) R3-amine, i-Pr2NEt, KI, MeCN, 50 °C, overnight, 35 – 84%.
CONCLUSIONS
We describe a novel series of WDR5 WIN site inhibitors containing a bicyclic dihydroisoquinolinone core scaffold that was discovered by structure-guided cyclization of the linear benzamide. This modification fixed the bound conformation and removed a H-bond donor and two rotatable bonds, which resulted in improvements in both on-target potency and drug-like properties. The most potent WDR5 inhibitor compound 16 exhibited a picomolar binding affinity and clinically relevant anti-proliferative activities with enhanced selectivity in the MLL-fusion cell lines MV4:11 and Molm13. Compound 16 also effectively displaced WDR5 from chromatin, which led to the dose-dependent induction of p53 followed by caspase3/7 in the MLL-fusion cells.
A potential utility of WDR5 WIN site inhibitors in MYC-driven cancers was also assessed using compound 16 because displacement of WDR5 from chromatin also prevents MYC recruitment to MYC/WDR5 co-bound target genes. Indeed, compound 16 diminished MYC recruitment at WDR5-displaced genes and exhibited potent anti-proliferative effects in CHP-134 (neuroblastoma) and Ramos (Burkitt’s lymphoma) lines.
The observations that WDR5 WIN site inhibition impedes not only WDR5 but also MYC binding to chromatin, and prevents proliferation of MYC driven cell lines have tremendous therapeutic implications. MYC over-expression or dysregulation is found in the majority of cancers and contributes to an estimated 100,000 cancer-related deaths in the United States each year.41 Despite the known contributions of MYC function to cancer, MYC had long been considered “undruggable”. Our data presented here predicts that MYC can be indirectly therapeutically targeted in cancer via WDR5 WIN site inhibition at genes co-bound by WDR5 and MYC. Although the number of WDR5 target genes we recently identified in MV4:11 cells is fairly small, these genes are highly enriched in genes encoding components of the protein synthesis machinery.8 Enhanced protein synthesis is a core tumorigenic activity of MYC,42 and thus we predict that WDR5 WIN site inhibitors should effectively block this tumorigenic function of MYC by preventing MYC recruitment to a key set of protein synthesis genes.
In summary, this study demonstrates the successful targeting of WDR5 by structure-guided methods to produce a set of high-affinity WDR5 WIN-site inhibitors. Current efforts are focused on further optimization of the potency, pharmaceutical, and pharmacokinetic properties of this series to obtain candidates that are suitable for clinical development. In addition, we are expanding our understanding in mechanisms of action, cellular vulnerability and additional utility of WDR5 WIN site inhibitors.
EXPERMENTIAL SECTION
General Chemistry.
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on either a 400, 600 or 800 MHz Bruker spectrometer. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual nondeuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; comp, overlapping multiplets of nonmagnetically equivalent protons; br, broad. All final compounds were of 95% purity or higher as measured by analytical reversed-phase HPLC. Analytical HPLC was performed on an Agilent 1200 series system with UV detection at 214 and 254 nm, along with evaporative light scattering detection (ELSD). Low-resolution mass spectra were obtained on an Agilent 6140 mass spectrometer with electrospray ionization (ESI). LC-MS experiments were performed with the following parameters: Phenomenex Kinetex 2.6 μm XB-C18 100 Å LC column (50 mm × 2.1 mm); 2 min gradient, 5%−95% MeCN in H2O, and 0.1% TFA or 0.1% formic acid. Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and iodine. Silica gel chromatography was performed using a Teledyne ISCO Combiflash Rf system, eluting with varying concentrations of EtOAc in hexanes or MeOH in CH2Cl2. Preparative reversed-phase HPLC was performed on a Gilson HPLC equipped with a Phenomenex Kinetex C18 column, using varying concentrations of MeCN in H2O, and 0.1% TFA. Solvents for reactions, extraction, and washing were ACS grade, and solvents for chromatography were HPLC grade. Compounds 1, 23, and 39 were prepared according to previously reported protocols.8,39,40
General Procedure A: amide coupling.
The carboxylic acid (1.0 equiv.) was taken up in DMF (0.8 mL) and then N,N,N’,N’-Tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate (1.0 equiv.) and N,N-diisopropylethylamine (2.0 equiv.) were added, after stirring at room temperature for 10 min R1-benzyl amine (1.0 equiv.) was added. After stirring for 8 h at room temperature the reaction was diluted with EtOAc (5 mL) and washed with 1 M HCl (5 mL), sat. NaHCO3 (5 mL), water (5 mL) and brine (5 mL). The organic layer was then dried (MgSO4), filtered and concentrated. The crude residue was purified by silica gel chromatography (0 – 75% EtOAc:hexanes).
General Procedure B: bromination and 1-methyl-2-aminoimidazole addition.
The benzyl alcohol (1.0 equiv.) was taken up in CH2Cl2 (1.1 mL) and then PBr3 (3.0 equiv.) was added. The reaction was stirred at room temperature for 2 h and then sat. NaHCO3 (5 mL) was added. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine (5 mL), dried (MgSO4), filtered and concentrated. The residue was taken up in MeCN (0.75 mL) and then 1-methyl-2-aminoimidazole hydrochloride salt (3.0 equiv.), N,N-diisopropylethylamine (3.0 equiv.) and potassium iodide (0.1 equiv.) were added. The reaction was heated at 80 °C for 4 h and then cooled to room temperature and diluted with water (5 mL). The aqueous layer was extracted with EtOAc (3 × 5 mL) and the combined organic layers were washed with brine (5 mL), dried (MgSO4), filtered and concentrated. The residue was purified by reverse phase HPLC (Phenomenex Gemini C18, H2O:CH3CN gradient from 15–95% CH3CN, 0.1% TFA). The product was dissolved in CH2Cl2 and washed with sat. K2CO3 to remove TFA.
General procedure C: benzylation of methyl 5-(4-fluoro-2-methylphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate.
To a solution of lactam (1 equiv.) in DMF at 0 °C was added NaH (60% in mineral oil, 1.3 equiv.). After stirring for 15 min, the substituted R2-benzyl halide (1.3 equiv.) was added and the mixture was stirred for 1 h and quenched with saturated aqueous NH4Cl. The mixture was extracted with EtOAc, and the organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (0 – 100% EtOAc:hexanes).
General procedure D: reduction of methyl ester.
To a solution of methyl ester (1 equiv.) in THF at 0 °C was added dropwise 3 M solution of LiBHEt3 in THF (3 equiv.). The reaction was stirred for 30 min and quenched with sat. NaHCO3. The mixture was extracted with EtOAc, and the organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (0 – 100% EtOAc:hexanes).
General procedure E: bromination of benzyl alcohol.
To a solution of benzyl alcohol (1 equiv.) in CH2Cl2 at 0 °C was added a 1 M solution of PBr3 in CH2Cl2 (2 equiv.) dropwise. The reaction was stirred for 4 h, then quenched with sat. NaHCO3. The mixture was extracted with Et2O. The organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (0 – 100% EtOAc:hexanes).
General procedure F: displacement of benzyl bromide with amine.
A mixture of benzyl bromide (1 equiv.), R3-imidazole amine (3 equiv.), KI (1 equiv.), and N,N-diisopropylethylamine (3 equiv.) was stirred at 50 °C for 16 h. The solvent was removed under reduced pressure and the residue was purified by reverse phase HPLC (Phenomenex Gemini C18, H2O:CH3CN gradient from 15–95% CH3CN, 0.1% TFA). The product was dissolved in CH2Cl2 and washed with sat. aq. K2CO3 to remove TFA.
N-Benzyl-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (2).
General procedure B was followed using 27 (25 mg, 0.07 mmol) to obtain the title compound (7.1 mg, 23% yield). 1H NMR (400 MHz, MeOD) δ 7.76 (d, J = 4.8 Hz, 1H), 7.37 – 7.32 (m, 5H), 7.28 – 7.22 (m, 2H), 7.08 (dd, J = 10.0, 2.8 Hz, 1H), 7.02 – 6.99 (m, 1H), 6.63 (d, J = 8.4 Hz, 1H), 5.04 (s, 2H), 4.60 (s, 2H), 3.35 (s, 3H), 2.24 (s, 3H); 13C NMR (200 MHz, MeOD) δ 169.5, 163.9 (d, J = 244.0 Hz), 152.6, 143.6, 140.2, 139.4 (d, J = 8.0 Hz), 138.2 (d, J = 3.0 Hz), 138.1, 136.6, 132.5 (d, J = 8.0 Hz), 129.7, 128.9, 128.7, 128.4, 126.4, 118.0 (d, J = 20.0 Hz), 116.8, 114.9, 113.8 (d, J = 20.0 Hz), 49.6, 44.8, 32.6, 20.8; LCMS (ESI): m/z = 429.0 [M + H]+.
4’-Fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-N-(3-methylbenzyl)-[1,1’-biphenyl]-3-carboxamide (3).
General procedure B was followed using 28 (30 mg, 0.08 mmol) to obtain the title compound (8.9 mg, 24% yield). 1H NMR (400 MHz, MeOD) δ 7.86 (s, 1H), 7.77 (s, 1H), 7.70 (s, 1H), 7.49 (s, 1H), 7.27 – 7.20 (m, 4H), 7.17 (d, J = 6.4 Hz, 1H), 7.09 – 6.98 (m, 2H), 6.77 (d, J = 8.0 Hz, 1H), 4.73 (s, 2H), 4.56 (d, J = 2.8 Hz, 2H), 2.34 (s, 3H), 2.27 (s, 3H), 2.24 (s, 3H); 13C NMR (200 MHz, MeOD) δ 170.0, 163.8 (d, J = 244.0 Hz), 152.6, 143.8, 140.2, 139.4, 138.7 (d, J = 8.0 Hz), 136.9 (d, J = 3.0 Hz), 136.0, 132.5 (d, J = 8.0 Hz), 132.1, 129.6, 129.4, 129.0, 128.2, 125.8, 125.6, 118.4, 117.9 (d, J = 20.0 Hz), 116.5, 113.7 (d, J = 20.0 Hz), 64.8, 49.5, 44.7, 21.6, 20.8; LCMS (ESI): m/z = 443.1 [M + H]+.
N-(3-Chlorobenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (4).
General procedure B was followed using 29 (34 mg, 0.09 mmol) to obtain the title compound (10.8 mg, 26% yield). 1H NMR (400 MHz, MeOD) δ 7.77 (s, 1H), 7.76 (s, 1H), 7.38 (s, 1H), 7.36 – 7.23 (m, 5H), 7.08 – 6.97 (m, 2H), 6.67–6.63 (m, 2H), 5.07 (s, 2H), 4.58 (s, 2H), 3.37 (s, 3H), 2.25 (s, 3H); 13C NMR (200 MHz, MeOD) δ 169.5, 163.9 (d, J = 244.0 Hz), 152.3, 143.6, 142.7, 139.4 (d, J = 8.0 Hz), 138.1 (d, J = 3.0 Hz), 136.4, 135.5, 132.7, 132.5 (d, J = 8.0 Hz), 131.2, 128.9, 128.7, 128.5, 127.1, 126.4, 118.0 (d, J = 20.0 Hz), 116.9, 115.0, 113.9 (d, J = 20.0 Hz), 49.5, 44.2, 32.6, 20.8l LCMS (ESI): m/z = 463.0 [M + H]+.
N-(3-Methoxybenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (5).
General procedure B was followed using 30 (58 mg, 0.15 mmol) to obtain the title compound (21.0 mg, 30% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (t, J = 6.0 Hz, 1H), 7.80 (s, 1H), 7.74 (s, 1H), 7.38 (s, 1H), 7.29 – 7.17 (series of m, 3H), 7.11 (dt, J = 8.6, 2.9 Hz, 1H), 6.90 – 6.87 (m, 2H), 6.82 – 6.79 (m, 1H), 6.45 (d, J = 2.7 Hz, 1H), 6.35 (d, J = 2.7 Hz, 1H), 4.81 (s, 2H), 4.44 (d, J = 6.0 Hz, 2H), 3.72 (s, 3H), 3.06 (s, 3H), 2.21 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ166.3, 161.9 (d, J = 242.5 Hz), 159.7, 153.6, 141.6, 140.7, 139.0, 138.2 (d, J = 8.0 Hz), 137.3 (d, J = 3.2 Hz), 135.1, 131.9 (d, J = 8.0 Hz), 137.3 (d, J = 3.2 Hz), 135.1, 131.9 (d, J = 8.0 Hz), 131.4, 129.8, 126.8, 126.0, 119.9, 117.3 (d, J = 21.0 Hz), 113.6, 113.5, 113.2 (d, J = 21.0 Hz), 112.5, 111.9, 55.4, 47.1, 43.1, 31.5, 20.7; LCMS (ESI): m/z = 459.5 [M + H]+.
4’-Fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-N-(3-(trifluoromethoxy)benzyl)-[1,1’-biphenyl]-3-carboxamide (6).
General procedure B was followed using 31 (38 mg, 0.09 mmol) to obtain the title compound (10.4 mg, 23% yield). 1H NMR (400 MHz, MeOD) δ 7.87 (s, 1H), 7.71 (s, 1H), 7.50 (s, 1H), 7.47 – 7.38 (m, 2H), 7.29 – 7.24 (m, 2H), 7.19 (d, J = 7.6 Hz, 1H), 7.08 – 7.05 (m, 2H), 7.02 – 6.98 (m, 2H), 4.74 (s, 2H), 4.63 (s, 2H), 3.37 (s, 3H), 2.27 (s, 3H); 13C NMR (200 MHz, MeOD) δ 170.1, 163.8 (d, J = 244.0 Hz), 150.9, 143.9, 143.3, 143.1, 139.3 (d, J = 8.0 Hz), 138.7 (d, J = 3.0 Hz), 135.7, 132.5 (d, J = 8.0 Hz), 132.2, 131.4, 128.2, 127.4, 125.6, 122.7, 121.4, 121.2, 120.7, 117.9 (d, J = 20.0 Hz), 113.7 (d, J = 20.0 Hz), 64.8, 49.5, 44.1, 20.8; LCMS (ESI): m/z = 513.0 [M + H]+.
N-(3,4-Dichlorobenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (7).
General procedure B was followed using 32 (35 mg, 0.08 mmol) to obtain the title compound (8.8 mg, 21% yield). 1H NMR (400 MHz, MeOD) δ 7.81 (s, 1H), 7.78 (s, 1H), 7.53 (d, J = 1.6 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.41 (s, 1H), 7.31 – 7.24 (m, 2H), 7.10 (dd, J = 9.6, 2.4 Hz, 1H), 7.04 – 6.96 (m, 3H), 5.24 (s, 2H), 4.56 (s, 2H), 3.56 (s, 3H), 2.25 (s, 3H); 13C NMR (200 MHz, MeOD) δ 169.3, 163.9 (d, J = 244.0 Hz), 147.9, 143.9, 141.2, 139.4 (d, J = 8.0 Hz), 137.9 (d, J = 3.0 Hz), 136.8, 136.4, 132.7, 132.5 (d, J = 8.0 Hz), 132.2, 131.8, 130.8, 129.1, 128.7, 126.6, 118.9, 118.1 (d, J = 20.0 Hz), 116.9, 113.9 (d, J = 20.0 Hz), 49.5, 43.7, 33.4, 20.8; LCMS (ESI): m/z = 497.4 [M + H]+.
N-(4-Chloro-3-methylbenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (8).
General procedure B was followed using 33 (32 mg, 0.08 mmol) to obtain the title compound (9.6 mg, 25% yield). 1H NMR (400 MHz, MeOD) δ 7.80 (s, 1H), 7.78 (s, 1H), 7.40 (s, 1H), 7.32 – 7.23 (m, 2H), 7.18 (d, J = 8.0 Hz, 1H), 7.09 (d, J = 2.4 Hz, 1H), 7.07 (d, J = 2.4 Hz, 1H), 7.03–6.96 (m, 3H), 5.24 (s, 2H), 4.54 (s, 2H), 3.56 (s, 3H), 2.36 (s, 3H), 2.25 (s, 3H); 13C NMR (200 MHz, MeOD) δ 169.2, 164.0 (d, J = 244.0 Hz), 147.9, 143.8, 139.4 (d, J = 8.0 Hz), 139.1, 138.0 (d, J = 3.0 Hz), 137.3, 136.7, 136.6, 134.2, 132.6 (d, J = 8.0 Hz), 131.5, 130.2, 129.2, 127.8, 126.5, 118.8, 118.0 (d, J = 20.0 Hz), 116.9, 113.9 (d, J = 20.0 Hz), 49.8, 44.1, 33.4, 20.8, 20.2; LCMS: m/z = 477.4 [M + H]+.
N-(3-Methyl,−4-fluorobenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (9).
General procedure B was followed using 34 (83 mg, 0.22 mmol) to obtain the title compound (28.0 mg, 28% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (t, J = 6.0 Hz, 1H), 7.80 (s, 1H), 7.73 (s, 1H), 7.38 (s, 1H), 7.28 – 7.04 (series of m, 6H), 6.42 (d, J = 2.7 Hz, 1H), 6.32 (d, J = 2.7 Hz, 1H), 4.79 (s, 2H), 4.41 (d, J = 6.0 Hz, 2H), 3.04 (s, 3H), 2.21 (s, 3H), 2.20 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.2, 161.9 (d, J = 242.4 Hz), 160.1 (d, J = 240.0 Hz), 153.9, 140.7, 139.1, 138.2 (d, J = 7.9 Hz), 137.3 (d, J = 2.2 Hz), 135.9 (d, J = 3.1 Hz), 135.0, 131.9 (d, J = 8.3 Hz), 131.5, 131.0 (d, J = 4.8 Hz), 127.0 (d, J = 8.1 Hz), 126.8, 126.0, 124.3 (d, J = 17.0 Hz), 117.3 (d, J = 21.0 Hz), 115.1 (d, J = 22.0 Hz), 113.3, 113.2 (d, J = 21.0 Hz), 111.7, 47.1, 42.5, 31.4, 20.7, 14.6; LCMS (ESI): m/z = 461.4 [M + H]+.
N-(3-Methoxy,−4-fluorobenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (10).
General procedure B was followed using 35 (38 mg, 0.09 mmol) to obtain the title compound (18.0 mg, 40% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.04 (t, J = 5.7 Hz, 1H), 7.80 (s, 1H), 7.73 (s, 1H), 7.38 (s, 1H), 7.28 – 7.24 (m, 1H), 7.20 – 7.08 (series of m, 4H), 6.88 – 6.85 (m, 1H), 6.43 (d, J = 2.6 Hz, 1H), 6.34 (d, J = 2.6 Hz, 1H), 4.80 (s, 2H), 4.44 (d, J = 5.7 Hz, 2H), 3.81 (s, 3H), 3.05 (s, 3H), 2.21 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.3, 161.94 (d, J = 242.6 Hz), 153.9, 151.0 (d, J = 241.0 Hz), 147.3 (d, J = 10.6 Hz), 140.7, 139.2, 138.2 (d, J = 8.0 Hz), 137.3 (d, J = 2.2 Hz), 136.8 (d, J = 3.3 Hz), 135.1, 131.9 (d, J = 8.4 Hz), 131.5, 126.8, 126.0, 119.9 (d, J = 6.7 Hz), 117.3 (d, J = 21.0 Hz), 116.0 (d, J = 17.8 Hz), 113.5, 113.3, 113.2 (d, J = 21.0 Hz), 111.6, 56.3, 47.1, 42.8, 31.4, 20.7; LCMS (ESI): m/z = 477.0 [M + H]+.
N-(3,5-Dimethylbenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (11).
General procedure B was followed using 36 (94 mg, 0.25 mmol) to obtain the title compound (8 mg, 7% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.01 (t, J = 5.8 Hz, 1H), 7.81 (s, 1H), 7.74 (s, 1H), 7.38 (s, 1H), 7.29 – 7.25 (m, 1H), 7.18 (dd, J = 10.1, 2.5 Hz, 1H), 7.11 (dt, J = 8.6, 2.7 Hz, 1H), 6.91 (s, 2H), 6.86 (s, 1H), 6.44 (d, J = 2.8 Hz, 1H), 6.34 (d, J = 2.8 Hz, 1H), 4.80 (s, 2H), 4.40 (d, J = 5.8 Hz, 2H), 3.05 (s, 3H), 2.23 (s, 6H), 2.21 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 166.1, 161.9 (d, J = 242.6 Hz), 153.9, 140.6, 139.9, 139.1, 138.2 (d, J = 8.3 Hz), 137.7, 137.3 (d, J = 2.8 Hz), 135.1, 131.9 (d, J = 8.3 Hz), 131.4, 128.6, 126.8, 126.1, 125.6, 117.3 (d, J = 21.0 Hz), 113.3, 113.2 (d, J = 20.8 Hz), 111.7, 47.1, 43.1, 31.4, 21.4, 20.7; LCMS (ESI): m/z = 457.0 [M + H]+.
N-(3,5-Dichlorobenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (12).
General procedure B was followed using 37 (112 mg, 0.27 mmol) to obtain the title compound (12 mg, 9% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.01 (t, J = 5.4 Hz, 1H), 7.81 (s, 1H), 7.74 (s, 1H), 7.49 (s, 1H), 7.40 (s, 1H), 7.36 (s, 2H), 7.29 – 7.25 (m, 1H), 7.19 – 7.17 (m, 1H), 7.14 – 7.09 (m, 1H), 6.46 (d, J = 1.5 Hz, 1H), 6.36 (d, J = 1.5 Hz, 1H), 4.82 (s, 2H), 4.47 (d, J = 5.4 Hz, 2H), 3.06 (s, 3H), 2.21 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.5, 161.9 (d, J = 242.7 Hz), 153.6, 144.5, 140.8, 139.1, 138.2 (d, J = 8.2 Hz), 137.2 (d, J = 2.6 Hz), 134.7, 134.4, 131.9 (d, J = 8.4 Hz), 131.7, 126.9, 126.8, 126.6, 126.0, 117.3 (d, J = 21.0 Hz), 113.5, 113.2 (d, J = 21.0 Hz), 111.8, 47.1, 42.4, 31.4, 20.7; LCMS (ESI): m/z = 496.9 [M + H]+.
N-(3,5-Dimethoxybenzyl)-4’-fluoro-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (13).
General procedure B was followed using 38 (84 mg, 0.20 mmol) to obtain the title compound (12 mg, 12% yield). 1H NMR (400 MHz, CDCl3) δ 7.76 (s, 1H), 7.3 (s, 1H), 7.27 (s, 1H), 7.15 – 7.11 (m, 1H), 6.97 – 6.89 (m, 2H), 6.79 – 6.74 (m, 1H), 6.97 – 6.89 (m, 2H), 6.79 – 6.74 (m, 1H), 6.50 (d, J = 7.4 Hz, 2H), 6.36 (t, J = 2.2 Hz, 1H), 6.14–6.12 (m, 2H), 4.86 (s, 2H), 4.57 (d, J = 5.4 Hz, 2H), 3.77 (s, 6H), 3.24 (s, 3H), 2.20 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 166.7, 162.2 (d, J = 244 Hz), 161.0, 142.1, 140.8, 137.7 (d, J = 7.9 Hz), 136.4 (d, J = 3.3 Hz), 135.5, 135.2, 131.3, 131.2 (d, J = 7.9 Hz), 128.4, 125.7, 116.9 (d, J = 21.0 Hz), 115.1, 113.1, 112.8 (d, J = 21.0 Hz), 105.9, 105.7, 99.3, 55.4, 48.9, 43.9, 33.0, 20.6; LCMS: m/z = 489.5 [M + H]+.
2-(4-Chloro-3-methylbenzyl)-5-(4-fluoro-2-methylphenyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (14).
General procedure F was followed using 50 (30 mg, 0.06 mmol) and 1-methyl-1H-imidazol-2-amine hydrochloride (25 mg, 0.18 mmol) to obtain the title compound (25 mg, 81% yield). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 1.6 Hz, 1H), 7.29 (m, 1H), 7.25 (d, J = 1.6 Hz, 1H), 7.20 (s, 1H), 7.09 (dd, J = 2.0, 8.0 Hz, 1H), 7.03 (dd, J = 6.4, 8.4 Hz, 1H), 6.97 (dd, J = 2.4, 9.6 hz, 1H), 6.91 (dt, J = 2.4, 8.4 Hz, 1H), 6.20 (m, 2H), 4.97 (s, 2H), 4.72 (ABq, JAB = 14.8 Hz, 2H), 3.37 (t, J = 6.4 Hz, 2H), 3.33 (s, 3H), 2.56 (m, 2H), 2.36 9s, 3H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.4, 162.4 (d, J = 244.0 Hz), 153.0, 139.7, 138.5 (d, J = 8.0 Hz), 136.5, 136.1, 135.9, 135.2, 135.0, 133.7, 132.5, 131.0 (d, J = 8.0 Hz), 130.8, 130.3, 129.4, 127.0 (d, J = 3.0 Hz), 116.8 (d, J = 21.0 Hz), 114.0, 112.8 (d, J = 21.0 Hz), 112.1, 50.1, 48.4, 45.3, 32.4, 25.7, 20.3, 20.2; LCMS (ESI): >95%, m/z = 503.4 [M + H]+.
5-(4-Fluoro-2-methylphenyl)-2-(4-fluoro-3-methylbenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (15).
General procedure F was followed using 51 (35 mg, 0.07 mmol) and 1-methyl-1H-imidazol-2-amine hydrochloride (30 mg, 0.22 mmol) to obtain the title compound (18 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 1.3 Hz, 1H), 7.16 (d, J = 1.5 Hz, 1H), 7.13 (d, J = 7.3 Hz, 1H), 7.10 – 7.07 (m, 1H), 7.01 – 6.98 (m, 1H), 6.95 – 6.86 (m, 3H), 6.09 (s, 2H), 4.81 (s, 2H), 4.68 (ABq, JAB = 14.6 Hz, 2H), 3.72 (brs, 1H), 3.33 (t, J = 6.6 Hz, 1H), 3.20 (s, 3H), 2.62 – 2.45 (m, 2H), 2.22 (s, 3H), 2.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.4, 162.2 (d, J = 244.5 Hz), 160.9 (d, J = 243.0 Hz), 154.6, 139.6, 138.5 (d, J = 7.8 Hz), 135.8 (d, J = 6.4 Hz), 135.1 (d, J = 3.1 Hz), 132.8 (d, J = 3.5 Hz), 132.1, 131.4 (d, J = 5.2 Hz), 131.0 (d, J = 8.2 Hz), 130.3, 127.1 (d, J = 8.1 Hz), 126.8, 125.3 (d, J = 17.5 Hz), 116.8 (d, J = 21.0 Hz), 115.8 (d, J = 22.5 Hz), 113.2, 112.8 (d, J = 21.1 Hz), 111.2, 50.0, 47.9, 45.2, 31.7, 25.6, 20.2, 14.6 (d, J = 3.5 Hz); LCMS (ESI): >95%, m/z = 487.6 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (16).
General procedure F was followed using 52 (30 mg, 0.06 mmol) and 1-methyl-1H-imidazol-2-amine hydrochloride (24 mg, 0.18 mmol) to obtain the title compound (20 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 2.0 Hz, 1H), 7.31 (d, J = 2.0 Hz, 1H), 7.91 (m, 3H), 6.48 (dd, J = 4.8, 2.8 Hz, 2H), 6.44 (d, J = 2.0 Hz, 2H), 6.37 (t, J = 2.0 Hz, 1H), 5.28 (s, 2H), 4.66 (ABq, JAB = 14.8 Hz, 2H), 3.76 (s, 6H), 3.60 (s, 3H), 3.38 (t, J = 6.4 Hz, 2H), 2.54 (m, 2H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.4, 162.3 (d, J = 244.8 Hz), 161.2, 152.7, 139.7, 139.6, 138.5 (d, J = 7.9 Hz), 136.1, 135.1, 135.0 (d, J = 3.1 Hz), 132.6, 131.0 (d, J = 8.4 Hz), 130.3, 129.4, 127.2, 116.7 (d, J = 20.9 Hz), 114.1, 112.7 (d, J = 21.0 Hz), 112.0, 106.2, 99.4, 55.5, 50.6, 48.3, 45.1, 32.3, 25.7, 20.2; LCMS (ESI): >95%, m/z = 515.0 [M + H]+.
7-((3-Cyclopropyl-2-imino-2,3-dihydro-1H-imidazol-1-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-3,4-dihydroisoquinolin-1(2H)-one (17).
General procedure F was followed using 52 (28 mg, 0.06 mmol) and 1-cyclopropyl-1H-imidazol-2-amine (17.3 mg, 0.14 mmol) to obtain the title compound (24 mg, 79% yield). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 2.0 Hz, 1H), 7.26 (d, J = 2.0 Hz, 1H), 7.02 (dd, J = 6.0, 8.4 Hz, 1H), 6.95 (dd, J = 2.8, 10.0 Hz, 1H), 6.91 (m, 1H), 6.46 (d, J = 2.0 Hz, 2H), 6.36 (m, 1H), 6.15 (d, J = 2.8 Hz, 1H), 6.09 (d, J = 2.8 Hz, 1H), 5.00 (s, 2H), 4.70 (ABq, JAB = 14.8 Hz, 2H), 3.76 (s, 6H), 3.36 (t, J = 6.4 Hz, 2H), 2.84 (m, 1H), 2.53 (m, 2H), 2.01 (s, 3H), 1.01 (m, 2H), 0.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 164.5, 162.4 (d, J = 245.0 Hz), 161.2, 154.0, 139.8, 139.6, 138.5 (d, J = 8.0 Hz), 136.1, 135.4, 135.1, 132.7, 131.0 (d, J = 8.0 Hz), 130.3, 127.1, 116.8 (d, J = 21.0 Hz), 113.0, 112.8 (d, J = 21.0 Hz), 111.8, 106.2, 99.5, 55.5, 50.7, 48.1, 45.1, 25.9, 25.7, 20.3, 6.4; LCMS (ESI): >95%, m/z = 541.6 [M+H]+.
N-((2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinolin-7-yl)methyl)cyclopropanecarboximidamide (18).
General procedure F was followed using 52 (35 mg, 0.07 mmol) and cyclopropanecarboximidamide hydrochloride (25 mg, 0.21 mmol) to obtain the title compound (12 mg, 35% yield). 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J =1.6 Hz, 1H), 7.26 (m, 1H), 7.03 (dd, J = 6.0, 8.4 Hz, 1H), 6.97 (dd, J = 2.4, 9.6 Hz, 1H), 6.91 (dt, J = 2.4, 8.4 Hz, 1H), 6.47 (d, J = 2.0 Hz, 2H), 6.37 (t, J = 2.4 Hz, 1H), 4.72 (ABq, JAB = 14.8 Hz, 2H), 4.48 (brs, 2H), 3.77 (s, 6H), 3.37 (t, J = 6.4 Hz, 2H), 2.56 (m, 2H), 2.05 (s, 3H), 1.44 (m, 1H), 0.8 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 164.7, 162.9, (d, J = 245.0 Hz), 161.2, 139.9, 139.3, 138.5 (d, J = 7.5 Hz), 135.4, 132.4, 131.0 (d, J = 7.5 Hz), 130.2, 126.8, 116.8 (d, J = 21.0 Hz), 112.8 (J = 21.0 Hz), 106.2, 99.5, 55.5, 50.7, 45.3, 29.9, 25.7, 20.3, 15.7, 6.7; LCMS (ESI): >95%, m/z = 502.6 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-7-((2-iminooxazol-3(2H)-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (19).
General procedure F was followed using 52 (30 mg, 0.06 mmol) and oxazol-2-amine (15 mg, 0.18 mmol) to obtain the title compound (19 mg, 63% yield). 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 1.9 Hz, 1H), 7.24 (d, J = 1.9 Hz, 1H), 7.01 (dd, J = 8.6, 6.0 Hz, 1H), 6.96 (dd, J = 9.7, 2.6 Hz, 1H), 6.90 (td, J = 8.4, 2.6 Hz, 1H), 6.62 (d, J = 1.9 Hz, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.37 – 6.34 (m, 2H), 4.77 (s, 2H), 4.71 (ABq, JAB = 14.8 Hz, 2H), 3.76 (s, 6H), 3.37 (d, J = 6.6 Hz, 2H), 2.65 – 2.57 (m, 1H), 2.55 – 2.47 (m, 1H), 2.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.3, 162.4 (d, J = 244.1 Hz), 161.2, 158.9, 139.7 (d, J = 7.2 Hz), 138.4 (d, J = 7.9 Hz), 136.3, 135.0, 132.4, 130.9 (d, J = 8.3 Hz), 130.4, 127.8, 127.0, 116.8 (d, J = 21.0 Hz), 116.2, 112.8 (d, J = 21.0 Hz), 106.2, 99.4, 55.5, 50.7, 47.9, 45.1, 25.7, 20.2; LCMS (ESI): >95%, m/z = 502.0 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-7-((2-iminothiazolidin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (20).
General procedure F was followed using 52 (35 mg, 0.07 mmol) and thiazolidin-2-imine (35.9 mg, 0.35mmol) to obtain the title compound (22 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.28 (s, 1H), 6.97 (m, 3H), 6.47 (d, J = 2.0 Hz, 2H), 6.37 (m, 1H), 4.72 (ABq, JAB = 14.8 Hz, 2H), 4.66 (s, 2H), 3.77 (s, 6H), 3.57 (t, J = 6.8 Hz, 2H), 3.38 (t, J = 6.8 Hz, 2H), 3.16 (t, J = 6.8 Hz, 2H), 2.56 (m, 2H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.6, 162.4 (d, J = 244.0 Hz), 161.2, 139.8, 139.6, 138.5 (d, J = 7.0 Hz), 136.1, 135.7, 135.3, 132.7, 131.0 (d, J = 8.0 Hz), 130.2, 127.3, 116.8 (d, J = 21.0 Hz), 112.8 (d, J = 21.0 Hz), 106.3, 99.4, 55.5, 51.7, 50.7, 49.2, 45.2, 27.2, 25.7, 20.3; LCMS (ESI): >95%, m/z = 520.4 [M+H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-7-((2-iminopyrrolidin-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (21).
General procedure F was followed using 52 (35 mg, 0.07 mmol) and pyrrolidin-2-imine hydrochloride (17 mg, 0.14 mmol) to obtain the title compound (21 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.21 (s, 1H), 7.02 (dd, J = 6.0, 8.4 Hz, 1H), 6.92 (m, 2H), 6.47 (d, J = 2.0 Hz, 2H), 6.36 (m, 1H), 4.71 (ABq, JAB = 14.8 Hz, 2H), 4.62 (s, 2H), 3.77 (s, 6H), 3.35 (m, 4H), 2.58 (m, 4H), 2.05 (s, 3H), 1.97 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.2, 164.6, 162.4 (d, J = 244.0 Hz), 161.2, 139.8, 139.5, 138.5 (d, J = 8.0 Hz), 136.1, 135.6, 135.4 (d, J = 3.0 Hz), 132.5, 131.1 (d, J = 8.0 Hz), 130.2, 127.1, 116.8 (d, J = 21.0 Hz), 112.8 (d, J = 21.0 Hz), 106.2, 99.4, 55.5, 50.7, 50.1, 47.6, 45.2, 32.8, 25.7, 20.3, 19.7; LCMS (ESI): >95%, m/z = 502.5 [M+H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-7-((5-imino-1-methyl-1,5-dihydro-4H-1,2,4-triazol-4-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (22).
General procedure F was followed using 52 (30 mg, 0.06 mmol) and 1-methyl-1H-1,2,4-triazol-5-amine (18 mg, 0.18 mmol) to obtain the title compound (26 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 1.8 Hz, 1H), 7.20 (d, J = 1.9 Hz, 1H), 7.18 (s, 1H), 7.00 (dd, J = 8.5, 4.7 Hz, 1H), 6.95 (dd, J = 9.6, 4.1 Hz, 1H), 6.90 (td, J = 8.4, 3.9 Hz, 1H), 6.45 (d, J = 2.2 Hz, 2H), 6.36 (t, J = 2.2 Hz, 1H), 4.83 (s, 2H), 4.70 (ABq, JAB = 14.7 Hz, 2H), 3.75 (s, 6H), 3.40 (s, 3H), 3.37 (t, J = 6.7 Hz, 1H), 2.64 – 2.57 (m, 1H), 2.55 – 2.49 (m, 1H), 2.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.2, 162.4 (d, J = 244.1 Hz), 161.2, 154.0, 140.0, 139.6, 138.4 (d, J = 7.8 Hz), 136.6, 135.0, 134.8 (d, J = 3.0 Hz), 134.0, 132.2, 130.0 (d, J = 8.3 Hz), 130.7, 127.1, 116.9 (d, J = 21.1 Hz), 112.9 (d, J = 21.0 Hz), 106.2, 99.4, 55.5, 50.7, 46.5, 45.1, 33.5, 25.7, 20.2; LCMS (ESI): >95%, m/z = 516.0 [M + H]+.
Methyl 3-(hydroxymethyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (24).
Bis(pinacolato)diboron (358 mg, 1.41 mmol), KOAc (276 mg, 2.82 mmol), and Pd(dppf)Cl2 (69.0 mg, 0.094 mmol) were added to a solution of methyl 3-bromo-5-(hydroxymethyl)benzoate (23) (230 mg, 0.940 mmol) in 1,4-dioxane (9.4 mL). The reaction mixture was stirred at 85 °C overnight. The reaction mixture was concentrated and the residue was purified by flash column chromatography on silica gel (0 – 40% EtOAc:hexanes) to provide the title compound (264 mg, quantitative). 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H), 8.15 (s, 1H), 7.99 (s, 1H), 4.75 (d, J = 2.7 Hz, 2H), 3.92 (s, 3H), 1.35 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 167.2, 140.7, 137.8, 135.7, 130.9, 130.1, 127.9, 84.3, 64.9, 52.2, 25.0; LCMS (ESI): m/z = 293.4 [M + H]+.
Methyl 4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxylate (25).
To 24 (5.00 g, 30.3 mmol) in 1,4-dioxane:H2O (3:1, 120 mL) was added 2-bromo-5-fluorotoluene (8.54 g, 45.4 mmol), K2CO3 (12.6 g, 90.9 mmol) and Pd(dppf)Cl2 (1.11 g, 1.51 mmol). The mixture was degassed and then heated to 95 °C for 2 h. The reaction was cooled to room temperature, filtered through a pad of celite and washed with EtOAc. The filtrate was diluted with water (40 mL) and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 40 mL) and the combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated. The residue was purified by flash column chromatography on silica gel (0 – 30% EtOAc:hexanes) to provide the title compound (5.67 g, 68% yield). 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.89 (t, J = 1.2 Hz, 1H), 7.49 (s, 1H), 7.18 (dd, J = 8.4, 6.0 Hz, 1H), 6.99 – 6.90 (m, 2H), 4.79 (s, 2H), 3.92 (s, 3H), 2.23 (s, 3H), 2.06 (bs, 1H); 13C NMR (100 MHz, CDCl3) δ 167.1, 162.4 (d, J = 245.0 Hz), 141.7, 141.5, 137.9 (d, J = 8.0 Hz), 136.8 (d, J = 3.0 Hz), 132.3, 131.3 (d, J = 8.0 Hz), 130.5, 129.7, 126.6, 117.2 (d, J = 21.0 Hz), 112.9 (d, J = 21.0 Hz), 64.8, 52.4, 20.7; LCMS (ESI): m/z = 275.1 [M + H]+.
4’-Fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxylic acid (26).
To 25 (4.67 g, 17.0 mmol) in THF (68 mL) was added LiOH (2 M, 17 mL). The reaction was stirred at room temperature overnight. The reaction was then diluted with water (15 mL) and washed with EtOAc (15 mL). The aqueous layer was then acidified to pH ~ 2 using 1 M HCl and then extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated to provide the title compound (4.18 g, 94% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (s, 1H), 7.70 (s, 1H), 7.49 (s, 1H), 7.27 (dd, J = 8.4, 6.0 Hz, 1H), 7.20 (dd, J = 10.0, 2.4 Hz, 1H), 7.12 – 7.07 (m, 1H), 5.36 (bs, 1H), 4.61 (s, 2H), 2.22 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.3, 161.5 (d, J = 242.0 Hz), 143.3, 140.3, 137.6 (d, J = 8.0 Hz), 136.9 (d, J = 3.0 Hz), 131.4, 131.3 (d, J = 8.0 Hz), 130.8, 128.1, 125.9, 116.9 (d, J = 20.0 Hz), 112.9 (d, J = 20.0 Hz), 62.3, 20.1; LCMS (ESI): m/z = 261.1 [M + H]+.
N-Benzyl-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (27).
General procedure A was followed using 26 (50 mg, 0.19 mmol) and benzylamine (20 mg, 0.19 mmol) to obtain the title compound (51.2 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.62 (s, 1H), 7.42 (s, 1H), 7.37–7.29 (m, 5H), 7.17 (dd, J = 8.4, 6.0 Hz, 1H), 6.99 – 6.90 (m, 2H), 6.42 (bs, 1H), 4.79 (s, 2H), 4.67 (d, J = 5.6 Hz, 2H), 2.23 (s, 3H), 1.92 (bs, 1H); 13C NMR (200 MHz, CDCl3) δ 167.1, 161.6 (d, J = 244.0 Hz), 141.9, 141.5, 138.0, 137.8 (d, J = 8.0 Hz), 136.7 (d, J = 3.0 Hz), 134.8, 131.2 (d, J = 8.0 Hz), 130.8, 128.9, 128.0, 127.8, 127.1, 123.9, 117.0 (d, J = 20.0 Hz), 112.8 (d, J = 20.0 Hz), 64.7, 44.3, 20.6; LCMS (ESI): m/z = 350.1 [M + H]+.
4’-Fluoro-5-(hydroxymethyl)-2’-methyl-N-(3-methylbenzyl)-[1,1’-biphenyl]-3-carboxamide (28).
General procedure A was followed using 26 (50 mg, 0.19 mmol) and 3-methylbenzylamine (23 mg, 0.19 mmol) to obtain the title compound (58.6 mg, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.62 (s, 1H), 7.42 (s, 1H), 7.25 – 7.22 (m, 1H), 7.17 – 7.11 (m, 3H), 6.99 – 6.90 (m, 2H), 6.40 (bs, 1H), 4.79 (s, 2H), 4.62 (d, J = 5.6 Hz, 2H), 2.35 (s, 3H), 2.23 (s, 3H), 1.92 (bs, 1H); 13C NMR (100 MHz, CDCl3) δ 166.9, 160.9 (d, J = 244.0 Hz), 141.8, 141.4, 138.5, 137.8, 137.7 (d, J = 8.0 Hz), 136.6 (d, J = 3.0 Hz), 134.7, 131.1 (d, J = 8.0 Hz), 130.6, 128.8, 128.7, 128.4, 127.0, 125.0, 123.8, 116.9 (d, J = 21.0 Hz), 112.7 (d, J = 21.0 Hz), 64.6, 44.2, 21.3, 20.5; LCMS (ESI): m/z = 364.0 [M + H]+.
N-(3-Chlorobenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (29).
General procedure A was followed using 26 (50 mg, 0.19 mmol) and 3-chlorobenzylamine (27 mg, 0.19 mmol) to obtain the title compound (60.3 mg, 82% yield). 1H NMR (400 MHz, MeOD) δ 7.87 (s, 1H), 7.71 (s, 1H), 7.50 (s, 1H), 7.39 (s, 1H), 7.36 – 7.24 (m, 4H), 7.08 – 6.97 (m, 2H), 4.74 (s, 2H), 4.59 (s, 2H), 2.27 (s, 3H); 13C NMR (200 MHz, MeOD) δ 170.1, 163.8 (d, J = 244.0 Hz), 143.8, 143.1, 142.9, 139.4 (d, J = 8.0 Hz), 138.7 (d, J = 3.0 Hz), 135.7, 135.5, 132.5 (d, J = 8.0 Hz), 132.2, 131.2, 128.7, 128.4, 128.2, 127.0, 125.6, 117.9 (d, J = 20.0 Hz), 113.7 (d, J = 20.0 Hz), 64.8, 44.2, 20.8; LCMS (ESI): m/z = 384.0 [M + H]+.
N-(3-Methoxybenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (30).
General procedure A was followed using 26 (101 mg, 0.39 mmol) and 3-methoxybenzylamine (53 mg, 0.39 mmol) to obtain the title compound (93 mg, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.61 (s, 1H), 7.41 (s, 1H), 7.28 – 7.24 (m, 1H), 7.16–7.12 (m, 1H), 6.98 – 6.91 (m, 4H), 6.84–6.82 (m, 1H), 6.51 – 6.48 (m, 1H), 4.76 (s, 2H), 4.60 (d, J=5.6 Hz, 2H), 3.80 (s, 3H), 2.22 (s, 3H); 13C NMR (150 MHz, CDCl3) δ167.1, 162.2 (d, J = 246.3 Hz), 160.0, 141.8, 141.5, 139.6, 137.7 (d, J = 8.1 Hz), 136.7, (d, J = 2.4 Hz), 134.7, 131.1 (d, J = 8.1 Hz),130.8, 129.9, 127.0, 123.8, 120.2, 116.9 (d, J = 21.8 Hz), 113.7, 113.0, 112.7 (d, J = 21.8 Hz), 66.5, 55.3, 44.2, 20.6; LCMS (ESI): m/z = 380.4 [M + H]+.
4’-Fluoro-5-(hydroxymethyl)-2’-methyl-N-(3-(trifluoromethoxy)benzyl)-[1,1’-biphenyl]-3-carboxamide (31).
General procedure A was followed using 26 (50 mg, 0.19 mmol) and 3-(trifluoromethoxy)benzylamine (37 mg, 0.19 mmol) to obtain the title compound (63.7 mg, 76% yield.). 1H NMR (400 MHz, MeOD) δ 7.87 (s, 1H), 7.71 (s, 1H), 7.50 (s, 1H), 7.47 – 7.43 (m, 1H), 7.39 – 7.37 (m, 1H), 7.29 (s, 1H), 7.27 – 7.23 (m, 1H), 7.19 (d, J = 8.0 Hz, 1H), 7.08 – 7.05 (m, 1H), 7.02–6.97 (m, 1H), 4.74 (s, 2H), 4.63 (s, 2H), 2.27 (s, 3H); 13C NMR (200 MHz, MeOD) δ 170.1, 163.8 (d, J = 244.0 Hz), 150.9, 143.9, 143.3, 143.1, 139.3 (d, J = 8.0 Hz), 138.7 (d, J = 3.0 Hz), 135.7, 132.5 (d, J = 8.0 Hz), 132.2, 131.4, 128.2, 127.4, 125.6, 121.2, 120.7, 117.9 (d, J = 20.0 Hz), 113.7 (d, J = 20.0 Hz), 64.8, 44.1, 20.8; LCMS (ESI): m/z = 433.9 [M + H]+.
N-(3,4-Dichlorobenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (32).
General Procedure A was followed using 26 (50 mg, 0.19 mmol) and 3,4-dichlorobenzylamine (34 mg, 0.19 mmol) to obtain the title compound (67.1 mg, 84% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.89 (s, 1H), 7.72 (s, 1H), 7.59 (s, 1H), 7.57 (s, 1H), 7.44 (s, 1H), 7.33 – 7.25 (m, 2H), 7.19 – 7.07 (m, 2H), 4.63 (d, J = 5.6 Hz, 2H), 4.48 (d, J = 5.6 Hz, 2H), 2.24 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.6, 163.0 (d, J = 243.0 Hz), 143.4, 141.3, 140.5, 138.1 (d, J = 8.0 Hz), 137.6 (d, J = 3.0 Hz), 134.3, 131.8 (d, J = 8.0 Hz), 131.2, 130.9, 130.3, 129.7, 129.6, 128.1, 126.5, 124.7, 117.2 (d, J = 21.0 Hz), 113.1 (d, J = 21.0 Hz), 63.0, 42.2, 20.6; LCMS (ESI): m/z = 417.9 [M + H]+.
N-(4-Chloro-3-methylbenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (33).
General procedure A was followed using 26 (50 mg, 0.19 mmol) and 4-chloro-3-methylbenzylamine (30 mg, 0.19 mmol) to obtain the title compound (61.1 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.88 (s, 1H), 7.71 (s, 1H), 7.42 (s, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.29 – 7.25 (m, 2H), 7.20 – 7.16 (m, 2H), 7.13 – 7.08 (m, 1H), 4.61 (d, J = 5.6 Hz, 2H), 4.44 (d, J = 5.6 Hz, 2H), 2.30 (s, 3H), 2.24 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.4, 161.8 (d, J = 242.0 Hz), 143.4, 140.5, 139.1, 138.1 (d, J = 8.0 Hz), 137.6 (d, J = 3.0 Hz), 135.5, 134.5, 131.8 (d, J = 8.0 Hz), 130.6, 130.2, 129.1, 127.0, 126.4, 124.7, 117.2 (d, J = 20.0 Hz), 113.1 (d, J = 20.0 Hz), 63.0, 42.5, 20.6, 20.0; LCMS (ESI): m/z = 397.9 [M + H]+.
N-(4-Fluoro-3-methylbenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (34).
General procedure A was followed using 26 (75 mg, 0.29 mmol) and 4-fluoro-3-methylbenzylamine (40 mg, 0.29 mmol) to obtain the title compound (92 mg, 84% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.06 (t, J = 5.9 Hz, 1H), 7.87 (s, 1H), 7.71 (s, 1H), 7.42 (s, 1H), 7.29–7.06 (series of m, 6H), 5.33 (t, J = 5.7 Hz, 1H), 4.61 (d, J = 5.9 Hz, 2H), 4.43 (d, J = 5.7 Hz, 2H), 2.24 (s, 3H), 2,22 (S, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.4, 161.9 (d, J = 242.0 Hz), 160.1 (d, J = 240.0 Hz), 159.3, 143.4, 140.5, 138.1 (d, J = 8.0 Hz), 137.7 (d, J = 2.5 Hz), 136.0 (d, J = 3.0 Hz), 134.6, 131.8 (d, J = 8.4 Hz), 131.0 (d, J = 5.1Hz), 130.2, 127.0 (d, J = 8.4 Hz), 126.5, 124.7, 124.3 (d, J = 17.1 Hz), 117.2 (d, J = 20.9 Hz), 115.1 (d, J = 22.0 Hz), 113.1 (d, J = 20.8 Hz), 63.0, 42.5, 20.7, 14.6; LCMS (ESI): m/z = 382.3 [M + H]+.
N-(4-Fluoro-3-methoxylbenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (35).
General procedure A was followed using 26 (76 mg, 0.29 mmol) and 4-fluoro-3-methoxybenzylamine (45 mg, 0.29 mmol) to obtain the title compound (43 mg, 37% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (t, J = 6.0 Hz, 1H), 7.86 (s, 1H), 7.70 (s, 1H), 7.41 (s, 1H), 7.28 – 7.25 (m, 1H), 7.20 – 7.08 (series of m, 4H), 6.88 – 6.85 (m, 1H), 5.32 (t, J = 5.7 Hz, 1H), 4.59 (d, J = 5.7 Hz, 2H), 4.44 (d, J = 6.0 Hz, 2H), 3.81 (s, 3H), 2.22 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.5, 161.9 (d, J = 242.3 Hz), 151.0 (d, J = 240.8 Hz), 147.3 (d, J = 10.7 Hz), 143.5, 140.5, 138.2 (d, J = 8.1 Hz), 137.7 (d, J = 2.3 Hz), 137.0 (d, J = 3.3 Hz), 135.7, 131.9 (d, J = 8.2 Hz), 130.3, 126.5, 124.8, 120.0 (d, J = 6.7 Hz), 117.2 (d, J = 20.9 Hz), 116.0 (d, J = 18.03 Hz), 113.5, 113.1 (d, J = 20.7 Hz), 63.0, 56.3, 42.8, 20.7; LCMS (ESI): m/z = 398.3 [M + H]+.
N-(3,5-Dimethylbenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (36)
General procedure A was followed using 26 (75 mg, 0.29 mmol) and 3,5-dimethylbenzylamine (39 mg, 0.29 mmol) to obtain the title compound (87 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.01 (t, J = 5.7 Hz, 1H), 7.88 (s, 1H), 7.72 (s, 1H), 7.42 (s, 1H), 7.29–7.26 (m, 1H), 7.19 (dd, J = 10.1, 2.7 Hz, 1H), 7.11 (dt, J = 8.8, 2.9 Hz, 1H), 6.92 (s, 2H), 6.87 (s, 1H), 5.33 (t, J = 5.7 Hz, 1H), 4.60 (d, J = 5.7 Hz, 2H), 4.41 (d, J = 5.9 Hz, 2H), 2.24 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.3, 161.9 (d, J = 242.1 Hz), 143.4, 140.5, 140.0, 138.1 (d, J = 7.9 Hz), 137.7 (d, J = 2.4 Hz), 137.6, 134.7, 131.8 (d, J = 8.3 Hz), 130.2, 128.6, 126.5, 125.6, 124.8, 117.2 (d, J = 20.9 Hz), 113.1 (d, J = 20.8 Hz), 63.0, 43.1, 21.4, 20.7; LCMS (ESI): m/z = 378.4 [M + H]+.
N-(3,5-Dichlorobenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (37).
General procedure A was followed using 26 (68 mg, 0.26 mmol) and 3,5-dichlorobenzylamine (46 mg, 0.26 mmol) to obtain the title compound (87 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.14 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.72 (s, 1H), 7.49 (t, J = 1.9 Hz, 1H), 7.44 (s, 1H), 7.38 (d, J = 1.9 Hz, 2H), 7.30 – 7.26 (m, 1H), 7.20 (dd, J = 7.5, 2.8 Hz, 1H), 7.12 (dt, J = 8.5, 2.7 Hz, 1H), 5.34 (t, J = 5.9 Hz, 1H), 4.61 (d, J = 5.8 Hz, 2H), 4.48 (d, J = 5.9 Hz, 2H), 2.25 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 166.7, 161.9 (d, J = 242.3 Hz), 144.6, 143.6, 140.6, 138.2 (d, J = 8.1 Hz), 137.6 (d, J = 2.7 Hz), 134.4, 134.2, 131.8 (d, J = 8.5 Hz), 130.5, 126.9, 126.6, 126.5, 124.7, 117.2 (d, J = 20.9 Hz), 113.2 (d, J = 20.9 Hz), 63.0, 42.4, 20.7; LCMS (ESI): m/z = 418.3 [M + H]+.
N-(3,5-Dimethoxybenzyl)-4’-fluoro-5-(hydroxymethyl)-2’-methyl-[1,1’-biphenyl]-3-carboxamide (38).
General procedure A was followed using 26 (100 mg, 0.38 mmol) and 3,5-dimethoxybenzylamine (64 mg, 0.38 mmol) to obtain the title compound (93 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ 7.76 (s, 1H), 7.61 (s, 1H), 7.41 (s, 1H), 7.17 – 7.13 (m, 1H), 6.98 – 6.89 (m, 2H), 6.49 (d, J = 2.3 Hz, 2H), 6.38 (t, J = 2.2 Hz, 1H), 4.77 (s, 2H), 4.57 (d, J = 5.8 Hz, 2H), 3.77 (s, 6H), 2.22 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 167.1, 162.2 (d, J = 244.7 Hz), 161.2, 141.9, 141.5, 140.3, 137.7 (d, J = 7.7 Hz), 136.7 (d, J = 2.7 Hz), 134.7, 131.1 (d, J = 7.7 Hz), 13.8, 127.1, 123.9, 116.9 (d, J = 21.2 Hz), 112.7 (d, J = 21.2 Hz), 106.0, 99.5, 64.7, 55.4, 44.4, 20.6; LCMS (ESI): m/z = 410.4 [M + H]+.
Methyl 2-(2,4-dimethoxybenzyl)-5-hydroxy-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (40).
To a solution of dimethyl 2-hydroxy-2,3-dihydrobenzofuran-4,6-dicarboxylate (39) (518.7 mg, 2.05 mmol, 1 equiv.) in CH2Cl2 (40 mL) was added 2,4-dimethoxy benzylamine (515.8 mg, 3.08 mmol, 1.5 equiv.). The reaction was stirred at room temperature for 10 min, then NaBH(OAc)3 (653.8 mg, 3.08 mmol, 1.5 equiv) was added. The resulting mixture was stirred at room temperature for 16 h and followed by LC-MS analysis. The solvent was removed under reduced pressure and toluene (8 mL) was added to the residue. The mixture was heated at 110 °C for 1 h, then cooled to room temperature. The mixture was extracted with EtOAc and water. The organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography (0 – 70% EtOAc:hexanes) to provide 40 (503.0 mg, 65% yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ 8.37 (s,1H), 7.67 (s, 1H), 7.29 (s, 1H), 6.44 (m, 2H), 4.75 (s, 2H), 3.88 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.54 (t, J = 6.8 Hz, 2H), 2.96 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 167.0, 164.3, 160.5, 158.80, 152.8, 131.1, 130.9, 130.7, 129.3, 121.6, 118.8, 117.6, 104.4, 98.5, 55.5, 52.3, 45.4, 45.2, 21.9; LCMS (ESI): m/z = 372.4 [M + H]+.
Methyl 2-(2,4-dimethoxybenzyl)-1-oxo-5-(((trifluoromethyl)sulfonyl)oxy)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (41).
To a solution of 40 (2.1 g, 5.65 mmol, equiv.) in CH2Cl2 (25 mL) was added N,N-diisopropylethylamine (1.48 mL, 8.5 mmol, 1.5 equiv.), N-Phenyl-bis(trifluoromethanesulfonimide) (2.42 g, 6.78 mmol, 1.2 equiv.). The reaction mixture was stirred at room temperature for 16 h, then diluted with CH2Cl2 and washed with water. The organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography (0 – 50% EtOAc:hexanes) to provide 41 (2.2 g, 75% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 8.02 (s, 1H), 7.30 (m, 1H), 6.48 (m, 2H), 4.75 (s, 2H), 3.96 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 3.60 (t, J = 6.4 Hz, 2H), 3.05 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 165.0, 162.1, 160.8, 158.9, 145.9, 136.2, 133.0, 131.5, 131.05, 129.6, 125.1, 118.8 (q, J = 318.0 Hz), 117.3, 104.6, 98.7, 55.6, 52.9, 45.5, 44.6, 23.2; LCMS (ESI): m/z = 504.4 [M + H]+.
Methyl 1-oxo-5-(((trifluoromethyl)sulfonyl)oxy)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (42).
To a solution of 41 (1.8 g, 3.6 mmol, 1 equiv.) in CH2Cl2 (10 mL) was added anisole (1.96 mL, 18 mmol, 5 equiv.) and TFA (20 mL). The reaction was stirred at room temperature overnight. The solvent was removed under reduced pressure. The residue was dissolved in EtOAc, and washed with sat. NaHCO3. The organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography (0 – 50% EtOAc:hexanes) to provide 42 (1.0 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.77 (s, 1H), 8.09 (s, 1H), 6.74 (brs, 1H), 3.97 (s, 3H), 3.65 (m, 2H), 3.16 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 164.8, 163.7, 146.2, 136.8, 132.1, 131.2, 129.3, 125.9, 118.7 (q, J = 319.0 Hz), 53.0, 39.4, 23.4; LCMS (ESI): m/z = 354.2 [M + H]+.
Methyl 5-(4-fluoro-2-methylphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (43).
To a solution of 42 (2.53 g, 7.16 mmol, 1 equiv.) in 1,4-dioxane (72 mL) was added (4-fluoro-2-methylphenyl) boronic acid (1.43 g, 9.31 mmol, 1.3 equiv.), Na2CO3 (1.9 g, 17.9 mmol, 2.5 equiv.), water (7.2 mL), and Pd(PPh3)4 (497 mg, 0.43 mmol, 0.06 equiv.). The mixture was degassed and back filled with Argon gas, then heated at 80 °C for 16 h. The mixture was diluted with EtOAc, and washed with water. The organic layer was dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography (0 – 80% EtOAc:hexanes) to provide 43 (2.1 g, 93% yield) as an off-white foam. 1H NMR δ 8.78 (s, 1H), 7.99 (s, 1H), 7.00 (m, 3H), 6.21 (brs, 1H), 3.94 (s, 3H), 3.50 (m, 2H), 2.69 (m, 2H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.4, 165.7, 162.5 (d, J = 245.0 Hz), 142.1, 139.8, 138.4 (d, J = 8.0 Hz), 134.6 (d, J = 5.0 Hz), 134.2, 130.9 (d, J = 8.0 Hz), 129.9, 129.1, 128.7, 117.0 (d, J = 21.0 Hz), 113.04 (d, J = 21.0 Hz), 52.4, 39.8, 26.3, 20.1; LCMS (ESI): m/z = 314.4 [M + H]+.
Methyl 2-(4-chloro-3-methylbenzyl)-5-(4-fluoro-2-methylphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (44).
General procedure C was followed using 43 (316 mg, 1.0 mmol) and 1-chloro-4-(chloromethyl)-2-methylbenzene (230 mg, 1.3 mmol) to obtain the title compound (260.0 mg, 57% yield). 1H NMR (400 MHz, CDCl3) δ 8.84 (d, J =1.6 Hz, 1H), 7.97 (d, J = 1.6 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 7.20 (s, 1H), 7.10 (m, 1H), 6.99 (m, 4H), 4.73 (ABq, JAB = 14.8 Hz, 2H), 3.95 (s, 3H), 3.39 (t, J = 6.4 Hz, 2H), 2.64 (m, 2H), 2.35 (s, 3H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.6, 163.9, 162.6, (d, J = 245.0 Hz), 141.3, 139.5, 138.5 (d, J = 7.0 Hz), 136.6, 135.8, 134.5, 133.9, 133.8, 131.1 (d, J = 5.0 Hz), 131.0, 130.3, 129.5, 129.3, 127.1, 117.0 (d, J = 22.0 Hz), 113.0 (d, J = 22.0 Hz), 52.5, 50.2, 45.0, 26.2, 20.2; LCMS (ESI): m/z = 452.4 [M + H]+.
Methyl 5-(4-fluoro-2-methylphenyl)-2-(4-fluoro-3-methylbenzyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (45).
General procedure C was followed using 43 (219 mg, 0.70 mmol) and 4-(bromomethyl)-1-fluoro-2-methylbenzene (284 mg, 1.4 mmol) to obtain the title compound (357.0 mg, quant.). 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 1.4 Hz, 1H), 8.00 (s, 1H), 7.93 (d, J = 1.5 Hz, 1H), 7.14 (d, J = 7.2 Hz, 1H), 7.11 – 7.08 (m, 1H), 7.02 – 6.88 (m, 3H), 4.70 (ABq, JAB = 14.6 Hz, 2H), 3.91 (s, 3H), 3.37 (t, J = 6.6 Hz, 2H), 2.69 – 2.52 (m, 2H), 2.22 (s, 3H), 2.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.5, 163.8, 162.6, 162.5 (d, J = 245.3 Hz), 160.9 (d, J = 243.2 Hz), 141.3, 139.5, 138.4 (d, J = 8.0 Hz), 134.5 (d, J = 3.1 Hz), 133.8, 132.6 (d, J = 3.5 Hz), 131.5 (d, J = 5.1 Hz), 130.9 (d, J = 8.3 Hz), 130.3, 129.2 (d, J = 2.3 Hz), 127.2 (d, J = 8.0 Hz), 125.3 (d, J = 17.6 Hz), 116.9 (d, J = 21.2 Hz), 115.2 (d, J = 22.5 Hz), 112.9 (d, J = 21.3 Hz), 52.4, 50.0, 44.8, 36.6, 31.5, 26.1, 20.1, 14.6 (d, J = 3.4 Hz); LCMS (ESI): m/z = 436.0 [M + H]+.
Methyl 2-(3,5-dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (46).
General procedure C was followed using 43 (0.68 g, 2.1 mmol) and 3,5-dimethoxybenzyl bromide (0.64 g, 2.8 mmol) to obtain the title compound (0.77 g, 78% yield). 1H NMR (400 MHz, CDCl3) δ 8.84 (d, J = 1.6 Hz, 1H), 7.96 (d, J = 1.6 Hz, 1H), 7.00 (m, 3H), 6.48 (m, 2H), 6.37 (m, 1H), 4.74 (ABq, JAB = 14.8 Hz, 2H), 3.94 (s, 3H), 3.76 (s, 6H), 3.40 (t, J = 6.4 Hz, 2H), 2.63 (m, 2H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.5, 163.8, 162.5 (d, J = 246.0 Hz), 161.2, 141.3, 139.5, 139.4, 138.4 (d, J = 8.0 Hz), 134.5 (d, J = 3.0 Hz, 133.8, 130.9 (d, J = 8.0 Hz), 130.3, 129.2, 116.9 (d, J = 21.0 Hz), 112.9 (d, J = 21.0 Hz), 106.3, 99.4, 55.5, 52.4, 50.7, 44.8, 26.1, 20.2; LCMS (ESI): m/z = 464.4 [M + H]+.
2-(4-Chloro-3-methylbenzyl)-5-(4-fluoro-2-methylphenyl)-7-(hydroxymethyl)-3,4-dihydroisoquinolin-1(2H)-one (47).
General procedure D was followed using 44 (263 mg, 0.58 mmol) to obtain the title compound (168.0 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 7.32 (s, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.07 (m, 1H), 7.01 (dd, J = 6.0, 8.4 Hz, 1H), 6.89 (dt, J = 2.4, 8.4 Hz, 1H), 4.75 (s, 2H), 4.70 (ABq, JAB = 14.8 Hz, 2H), 3.35 (t, J = 6.4 Hz, 2H), 3.18 (brs, 1H), 2.55 (m, 2H), 2.33 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.9, 162.3 (d, J = 245.0 Hz), 140.2, 139.2, 138.4 (d, J = 8.0 Hz), 136.4, 135.9, 135.5, 135.4 (d, J = 3.0 Hz), 133.6, 131.8, 131.0 (d, J = 8.0 Hz), 130.8, 129.8, 129.3, 126.9, 126.4, 116.7 (d, J = 21.0 Hz), 112.7 (d, J = 21.0 Hz), 64.6, 50.1, 45.3, 25.6, 20.2; LCMS (ESI): m/z = 424.4 [M + H]+.
5-(4-Fluoro-2-methylphenyl)-2-(4-fluoro-3-methylbenzyl)-7-(hydroxymethyl)-3,4-dihydroisoquinolin-1(2H)-one (48).
General procedure D was followed using 45 (327 mg, 0.7 mmol) to obtain the title compound (306 mg, quant.). 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 7.31 (s, 1H), 7.15 (d, J = 7.3 Hz, 1H), 7.11 – 7.08 (m, 1H), 7.04 – 7.01 (m, 1H), 6.97 – 6.89 (m, 3H), 5.00 (brs, 1H), 4.70 (ABq, JAB = 14.7 Hz, 2H), 3.91 – 3.72 (m, 2H), 3.35 (t, J = 6.5 Hz, 2H), 2.62 – 2.49 (m, 2H), 2.24 (s, 3H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.7, 162.3 (d, J = 244.0 Hz), 160.9 (d, J = 243.0 Hz), 138.5 (d, J = 8.8 Hz), 135.5, 132.9, 131.4 (d, J = 5.2 Hz), 131.0 (d, J = 8.2 Hz), 129.9, 127.2 (d, J = 8.1 Hz), 126.3, 126.1, 125.3, 125.2, 116.8 (d, J = 21.2 Hz), 115.2 (d, J = 22.5 Hz), 112.8 (d, J = 20.8 Hz), 50.0, 45.3, 37.1, 25.7, 20.2, 14.7 (d, J = 3.4 Hz); LCMS (ESI): m/z = 408.0 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-7-(hydroxymethyl)-3,4-dihydroisoquinolin-1(2H)-one (49).
General procedure D was followed using 46 (0.77 g, 1.7 mmol) to obtain the title compound (0.60 g, 83% yield). 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 1.6 Hz, 1H), 7.32 (d, J = 1.6 Hz, 1H), 7.03 (dd, J = 6.0, 8.4 Hz, 1H), 6.97 (dd, J = 2.8, 10.0 Hz, 1H), 6.91 (m, 1H), 6.47 (d, J = 2.0 Hz, 2H), 6.36 (m, 1H), 4.73 (m, 4H), 3.76 (s, 6H), 3.38 (t, J = 6.4 Hz, 2H), 2.58 (m, 2H), 2.15 (m, 1H), 2.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.8, 162.4 (d, J = 245.0 Hz), 161.2, 139.9, 139.8, 139.3, 138.5 (d, J = 7.0 Hz), 135.7, 135.4 (d, J = 3.0 Hz), 131.7, 131.0 (d, J = 9.0 Hz), 130.1, 126.4, 116.8 (d, J = 21.0 Hz), 112.8 (d, J = 21.0 Hz), 106.2, 99.5, 64.9, 55.5, 50.7, 45.3, 25.7, 20.2; LCMS (ESI): m/z = 436.4 [M + H]+.
7-(Bromomethyl)-2-(4-chloro-3-methylbenzyl)-5-(4-fluoro-2-methylphenyl)-3,4-dihydroisoquinolin-1(2H)-one (50).
General procedure E was followed using 47 (90 mg, 0.21 mmol) to obtain the title compound (94.0 mg, 91% yield). 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J =1.6 Hz, 1H), 7.31 (d, J = 1.6 Hz, 1H), 7.24 (s, 1H), 7.17 (s, 1H), 7.03 (m, 4H), 6.94 (m, 2H), 4.69 (ABq, JAB = 14.8 Hz, 2H), 4.52 (s, 3H), 3.34 (t, J = 6.4 Hz, 2H), 2.53 (m, 2H), 2.33 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.2, 162.5 (d, J = 245.0 Hz), 139.8, 138.5 (d, J = 8.0 Hz), 136.8, 136.7, 136.6, 135.9, 134.9 (d, J = 3.0 Hz), 133.7, 131.0, 130.9, 130.4, 129.4, 128.4, 127.0, 116.9 (d, J = 21.0 Hz), 113.0 (d, J = 21.0 Hz), 50.1, 45.2, 32.7, 25.8, 20.2; LCMS (ESI): m/z = 486.4 [M + H]+.
7-(Bromomethyl)-5-(4-fluoro-2-methylphenyl)-2-(4-fluoro-3-methylbenzyl)-3,4-dihydroisoquinolin-1(2H)-one (51).
General procedure E was followed using 48 (320 mg, 0.78 mmol) to obtain the title compound (197.8 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 1.6 Hz, 1H), 7.32 (d, J = 1.6 Hz, 1H), 7.15 (d, J = 7.2 Hz, 1H), 7.12 – 7.08 (m, 1H), 7.05 – 7.02 (m, 1H), 6.99 – 6.89 (m, 3H), 4.70 (ABq, JAB = 14.5 Hz, 2H), 4.54 (s, 2H), 3.36 (t, J = 6.6 Hz, 2H), 2.64 – 2.47 (m, 2H), 2.24 (s, 3H), 2.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.1, 162.4 (d, J = 244.9 Hz), 160.9 (d, J = 147.4 Hz), 139.7, 138.5 (d, J = 8.0 Hz), 136.7 (d, J = 8.7 Hz), 134.9 (d, J = 3.0 Hz), 133.7, 132.7 (d, J = 3.4 Hz), 131.4 (d, J = 5.2 Hz), 130.9 (d, J = 8.3 Hz), 130.4, 128.4, 127.2 (d, J = 8.0 Hz), 125.3 (d, J = 17.4 Hz), 116.9 (d, J = 21.0 Hz), 115.2 (d, J = 22.5 Hz), 112.9 (d, J = 21.1 Hz), 50.0, 45.1, 32.7, 25.7, 20.2, 14.7 (d, J = 3.4 Hz); LCMS (ESI): m/z = 470.0 [M + H]+.
7-(Bromomethyl)-2-(3,5-dimethoxybenzyl)-5-(4-fluoro-2-methylphenyl)-3,4-dihydroisoquinolin-1(2H)-one (52).
General procedure E was followed using 49 (0.60 g, 1.4 mmol) to obtain the title compound (0.64 g, 93% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 1.9 Hz, 1H), 7.40 (d, J = 1.9 Hz, 1H), 7.21 – 7.16 (m, 2H), 7.08 (td, J = 8.7, 4.0 Hz, 1H), 6.45 (d, J = 2.2 Hz, 2H), 6.40 (t, J = 2.2 Hz, 1H), 4.81 (s, 2H), 4.63 (ABq, JAB = 14.9 Hz, 2H), 3.71 (s, 3H), 3.40 (t, J = 6.6 Hz, 2H), 2.68 – 2.60 (m, 1H), 2.50 – 2.43 (m, 1H), 2.03 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 163.0, 161.5 (d, J = 242.4 Hz), 160.6, 139.9, 138.9, 138.3 (d, J = 8.0 Hz), 136.8, 136.5, 134.9 (d, J = 2.7 Hz), 133.4, 131.1 (d, J = 8.6 Hz), 129.9, 127.8, 116.5 (d, J = 21.0 Hz), 112.5 (d, J = 20.9 Hz), 105.6, 98.7, 55.1, 49.8, 44.8, 33.7, 25.1, 19.6; LCMS (ESI): m/z = 497.8 [M + H]+.
Protein Expression and Purification.
Human WDR5 (aa: 22–334) was cloned into a modified pET27 vector (pBG104) with a 6xHis-SUMO tag present at the N terminus. The plasmid was then transformed into E. coli BL21-Gold (DE3) cells. One hundred milliliters of LB starter was used to inoculate a 10l fermentation culture (BioFlo 415, New Brunswick Scientific), grown at 37 °C. Fermentation growth media contained KH2PO4 (4 g/L), K2HPO4 (6 g/L), Na2SO4 (2 g/L), K2SO4 (1 g/L), NaCl (0.5 g/L), Yeast Extract (5 g/L), glycerol (2 ml/L), Antifoam (0.2 ml/L), 5% LB medium, glucose (25 g/L), MgCl2 (2 mM), CaCl2 (0.1 mM), NH4Cl (2.5 g/L), and Kanamycin (50 μg/ml). When the cell density reached OD600 = 2.0, the temperature was lowered to 25 °C, and WDR5 expression induced by treatment with 1 mM isopropyl-β-D-thioga-lactoside (IPTG) overnight. Cell pellets were collected, dissolved in lysis buffer containing 1XPBS plus 300 mM NaCl, 20 mM imidazole, 5 mM BME, and 10% glycerol, and lysed by homogenization (APV-2000, APV). The lysate was cleared by centrifugation, filtered, and then applied to the Ni-column (140 mL, ProBond, Invitrogen). Bound protein was eluted using an imidazole gradient (0–500 mM). The His-SUMO-tag was cleaved by SUMO protease during dialysis and subsequently eliminated through a second Ni-column. WDR5 protein was then purified by size-exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) using crystallization buffer consisting of 20 mM HEPES, pH 7.0, 250 mM NaCl, and 5 mM DTT. The purity of protein was checked using SDS–PAGE. Purified WDR5 was then concentrated to 10 mg/mL, and was stored at −80 °C.
Protein Crystallization, Data Collection, and Structure Refinement.
WDR5 apo- and co-crystals were obtained at 18 °C using the sitting drop vapor diffusion method. The crystallization condition was 0.1 M Bis-Tris or HEPES or HCl-Tris pH 6.0–8.0, 0.2 M ammonium acetate, 20% to 30% PEG3350. A soaking method was applied for some of the compounds using WDR5 apo-crystals. Crystals were flash frozen directly in liquid nitrogen. Diffraction data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-D and G beamlines at the Advanced Photon Source (APS) at Argonne National Laboratory. Diffraction data were indexed, integrated, and scaled using HKL2000.43 Molecular replacement was applied using Phaser as implemented in CCP444 using a published structure (PDB code 3EG6). Refinement of the structural models was performed using PHENIX45 along with rounds of manual model building in COOT46. All structure images were prepared with PyMOL. A summary of the final refinement statistics for structures including compounds 13 and 16 can be found in Table S1.
TR-FRET Competition Assay.
The 10-mer-Thr-FAM (ARTEVHLRKS-(Ahx-Ahx)(Lys-(5-FAM)))36 peptide was purchased from GeneScript and used without additional purification. TR-FRET emissions were recorded on a BioTek Cytation 3 instrument.
For the 10-mer-Thr-FAM peptide TR-FRET assay, LanthaScreen Elite Tb-anti His antibody (Tb-Ab) was purchased from Thermo-Fisher and used at 1 nM. The 10-mer-Thr-FAM peptide was used at 150 nM, while WDR5-His-SUMO tag protein was used at 2 nM. The working assay buffer composition was modified to pH 7.2 (1X Phosphate Buffered Saline, 300 mM NaCl, 0.5 mM TCEP, 0.1%CHAPS). Stock compounds were dispensed to a white, flat-bottom OptiPlate plate (PerkinElmer) using an Echo Liquid Handler. An 11-point, Semi-log dilution scheme with a top concentration of 1 μM (0.010 nM low concentration) was used with a final volume of 20 μl. Both the top concentration and the dilution scheme was adjusted to fit the anticipated potency of the compounds. Using the above probe concentration and assay conditions, the calculated lower Ki limit was ~ 0.020 ± 0.010 nM. Positive control wells (0% displacement) consisted of His-SUMO-WDR5 and 10-mer-Thr-FAM probe/Tb antibody mix occupying column 24, while negative control wells (100% displacement) consisting of the 10-mer-Thr-FAM probe/terbium antibody mix alone occupy column 1. The assay performed with an average Z’ value of 0.7 and was found to be tolerant to up to 5% DMSO, but routinely screened at 1% DMSO.
For IC50 determinations, plates were covered, shielded from light, and incubated for 1h at room temperature with rocking. For the TR-FRET assay, measurement plates were excited at a wavelength of 340 nm, and emission wavelengths of 495 and 520 nm were used. The ratio of the 520/495 wavelengths were used to assess the degree of the FRET signal and resulting peptide displacement. TR-FRET plate positive control wells include column 24 containing 10-mer-Thr-FAM peptide, His-SUMO-WDR5, and Tb-anti-His antibody to measure maximum signal from the FRET response. The 520/495 emission ratio (TR-FRET) was used to calculate an IC50 (inhibitor concentration at which 50% of the bound peptide is displaced) by fitting the inhibition data using XLFit software (Guilford, UK) to single-site binding model. This was converted into a binding inhibition/displacement constant (Ki) using the formula:47
where [I]50 is the concentration of the free inhibitor at 50% inhibition, [L]50 is the concentration of the free labeled ligand at 50% inhibition, [P]0 is the concentration of the free protein at 0% inhibition, and Kdpep represents the dissociation constant of the 10-mer-Thr-FAM probe.
MLL1 Histone Methyltransferase Assay.
HMT inhibition activity assays (assay catalog #HMT-15–105, MLL1 complex no extra WRAD2) were performed at Reaction Biology Corporation in a HotSpot radioisotope based filter-binding format. The assay was performed using purified recombinant human MLL1complex (total MW = 212 kDa): MLL1 (aa 3745–3969, GenBank Accession No. NM_005933), WDR5 (aa 22–334, GenBank Accession No. NM_017588), RbBP5 (aa 1–538, GenBank Accession No. NM_005057), Ash2L (aa 2–534, GenBank Accession No. NM_001105214), DPY-30 (aa 1–99, GenBank Accession No. NM_0325742), all with N-terminal His tag were expressed in E. coli, and mixed molar ratio of 1:1:1:1:2, respectively, in a reaction buffer (50 mM Tris-HCl (pH 8.5), 5 mM MgCl2, 50 mM NaCl, 0.01% Brij35, 1 mM DTT, 1% DMSO. Purified oligonucleosomes were obtained from HeLa cells (primarily oligomers of 3–6 units, 600–1200 bp DNA), and S-adenosyl-L-[methyl-3H]methionine (SAM) was used as the methylation cofactor.48 S-Adenosylhomocysteine (SAH) was used as a positive control. Reaction conditions consisted of 7–10 nM of MLL1 complex, 0.05 mg/mL HeLa oligonucleosomes, and 1 μM of SAM substrate. Compounds were tested in duplicate using a 10-point CRC 3-fold serial dilution from a 3 μM top concentration. See the SI and www.reactionbiology.com for further details.
Cellular Proliferation Assays.
Cell proliferation was assayed using the Promega CellTiter-Glo Luminescent Kit. White, opaque, flat-bottomed 384-well plates were used. Two hundred cells were seeded per well for all cell lines. Cells were treated with 0.3% DMSO vehicle only or 22 two-fold dilutions of WDR5 inhibitors with a top concentration of 30 μM. Final DMSO concentration was 0.3% in all compound treatment experiments and at least two biological replicates were performed. The total volume of cells with inhibitor was 32 μL per well. Thirty-two μL of sterile media was added to all of the empty wells around the edge of the plate to prevent evaporation. Plates were incubated at 37 °C, 5% CO2 for 5 days. At the end of incubation, the plates were allowed to reach room temperature before adding 16 μL of CellTiter-Glo reagent per well. Plates were incubated at room temperature, covered from light, for 15 min before the luminescence was measured using the CellTiter-Glo protocol on a Cytation3 plate reader (BioTek, Winooski, VT). The raw luminescence values were normalized to the DMSO vehicle only wells and PRISM software or XLfit (IDBS, Guildford, United Kingdom) was used to generate GI50 values. Error bars on proliferation curves represent standard errors of the mean.
WDR5 Immunoprecipitation.
40 × 106 MV4:11 cells were resuspended in 8 mL of media and treated for 4 h with DMSO or 1 μM compound 16. Cell lysates were prepared in IP buffer [50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100] plus Complete Protease Inhibitor Cocktail (Roche), briefly sonicated, and cleared by centrifugation. Immune complexes were precipitated overnight at 4 °C with the normal rabbit IgG or anti-WDR5 antibody (Bethyl), captured for 2 h at 4 °C with Protein-A Agarose (Sigma), washed four times in IP buffer, and resolved by SDS-PAGE. Antibodies used for immunoblotting were MLL1 and WDR5 from Cell Signaling Technologies, and RBBP5 from Bethyl.
Chromatin Immunoprecipitation.
Chromatin immunoprecipitation (ChIP) assays were performed as previously described.14 Cells were treated with 0.1% DMSO or 600 nM compound 16 for 4 h at 37 °C, then washed in PBS and cross-linked with 1% formaldehyde at room temperature for 10 min. The reaction was quenched with 125 mM glycine for 10 min at room temperature, then cells were washed with ice cold PBS. Cells were lysed in Formaldehyde Lysis Buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton, 1% SDS, and Complete Protease Inhibitor cocktail) using 250 μL of buffer per 1 × 107 cells, on ice, for 10 min. Chromatin was sheared by 25-min sonication (BioRuptor) to yield a mean chromatin size of ~250 bp, and debris cleared by centrifugation. Sheared chromatin was diluted 10-fold in Formaldehyde Lysis Buffer without SDS before immunoprecipitation overnight at 4 °C using the appropriate antibody and Protein A agarose. Chromatin from 6 million cells was used per reaction. Immune complexes were washed sequentially with Low Salt Wash buffer (20 mM Tris pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton), High Salt Wash Buffer (20 mM Tris pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton), LiCl Wash buffer (25 mM LiCl, 1 mM EDTA, 1% Triton) and twice with TE (pH 8.0). Protein–DNA complexes were de-crosslinked overnight at 65 °C in Elution Buffer (TE, 0.1% SDS, 40 μg Proteinase K). Proteinase K was heat inactivated for 20 min at 95 °C then 150 μL of TE was added. 1 μL of DNA was used in a 15 μL PCR reaction using KAPA SYBR FAST qPCR Master Mix 2X Universal and quantified on an Eppendorf Realplex2 Mastercycler in triplicate. ChIP signals were calculated as percent input. ChIP experiments were completed in biological triplicate with error bars representing the standard error of the mean.
RT-qPCR Quantification of mRNA Expression.
MV4:11 cells were treated for three days with 0.1% DMSO or 300 nM compound 16. Cells were then lysed in 500 mL Trizol, and total RNA was extracted using the Zymo Research Direct-zol RNA MiniPrep kit with on-column DNase digestion. After extraction, 1 μg of RNA was reverse transcribed using MuLV reverse transcriptase (Life Tech N8080018) in 20 μL reaction, then diluted three-fold with nuclease-free water. 1 μL of cDNA was used in a 15 μL qPCR reaction using KAPA SYBR FAST qPCR Master Mix 2X Universal and quantified on an Eppendorf Realplex2 Mastercycler in triplicate. Relative mRNA expression of genes of interest was quantified using the CT method, normalizing to the signal from GAPDH. mRNA expression studies were completed in triplicate with error bars representing the standard error of the mean.
Caspase Assay.
MV-4–11 cells were incubated with compounds in 384 well plates at in a 32 μL volume containing 1600 cells. A 16 μL volume of Caspase-Glo® 3/7 reagent (Promega Biosciences, Madison WI) was added and luminescence was measured using a Cytation 3 plate reader.
Cell Cycle Analysis.
4 million cells were treated for three days with the indicated amount of compound. Cell pellets were permeabilized in 70% Ethanol for 1 hour at 4 °C, washed twice with PBS, incubated for 5 minutes in 50 μL PBS containing 5 μg RNAse A (Qiagen), then stained by adding 450 μL PBS and propidium iodide (Sigma) to 1 μg/mL. Cell cycle analysis was then carried out using an LSRII Flow Cytometer and data was analyzed with FACSDiva software (BD Biosciences).
Western Blot.
Compound treated cells were lysed in RIPA containing protease and phosphatase inhibitor cocktail. Changes in protein levels were measured by western blot using monoclonal antibodies against p53 (DO-1, Santa Cruz Biotechnology), p21 (12D1, Cell Signaling Technology, Danvers, MA), and GAPDH (14C10, Cell Signaling Technology.
Colorectal CTOSs Assays.
CTOSs were prepared and cultured as described previously.36, 37 CTOSs were cultured with StemPro hESC medium (Invitrogen) at 37 °C with 5% CO2. For the sensitivity assays for compound 1 and 16, CTOSs were dissociated into single cells using TrypLE express (Invitrogen) and filtered through a 35 μm cell strainer (BD Falcon). Cell suspension in Matrigel (Corning) at 200–400 cells/μl were prepared, and 5μl of Matrigel drop containing cancer cells were put in wells of 96-well plates. After the gels solidified, StemPro medium with indicated concentration of drugs were applied. Following 7 to 9 days of culture, viability of the cultured cells was evaluated by Celltiter-Glo (Promega).
Supplementary Material
Figure 4.

Compound 16 displaced MLL1 from the WRAD protein complex. Endogenous immunoprecipitation (IP) using anti-WDR5 antibody or control IgG in MV4:11 cells treated for 4 h with DMSO or 1 μM 16. IP’ed material and whole cell lysates were probed with MLL1, RBBP5 and WDR5 antibodies.
ACKNOWLEDGMENT
We thank the Vanderbilt High-Throughput Screening Core facility for compound management and Vanderbilt University Biomolecular NMR Facility for use of Bruker NMR spectrometers. This facility receives support from an NIH SIG Grant (1S-10RR025677-01) and Vanderbilt University matching funds. The authors also thank contributing and supporting members of Leidos Biomedical Research at Frederick National Labs for Cancer Research, Reaction Biology Corp. and the NCI Chemical Biology Consortium. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy Office of Science by the Argonne National Laboratory, was supported by US Department of Energy [Contract No. DE-AC02-06CH11357]. Use of the Life Sciences Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri- Corridor Grant [grant number 085P1000817]. This work was also funded by NCI/NIH CA211305 to L.R.T.
Funding Sources
This work was generously supported in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E (to S.W.F), grant CA200709 from the National Institutes of Health (to W.P.T), The Vanderbilt Ingram Cancer Center Support grant (NIH: CA68485), GI Special Programs in Research Excellence P50 CA236733 and startup funds to S.W.F. provided by Vanderbilt University. This work was also supported by grants from the Robert J. Kleberg, Jr., and Helen C. Kleberg Foundation (to WPT and SWF), The TJ Martell Foundation (to W.P.T. and S.W.F.), St. Baldrick’s Foundation (W.P.T.), Alex’s Lemonade Stand Foundation (W.P.T.), Edward P. Evans Foundation (W.P.T.), the NCI/NIH (CA211305; L.R.T.), and the IBSTO Training Program (T32 CA119925; E.R.A./W.P.T.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
ABBREVIATIONS USED
- ALL
acute lymphoid leukemia
- AML
acute myeloid leukemia
- C/EBPα
CCAAT-enhancer-binding protein α
- ChIP
chromatin immunoprecipitation
- CTOSs
cancer tissue-originated spheroids
- GI50
half-maximal growth inhibition
- HMT
histone methyltransferases
- Ki
binding inhibition constant
- MLL
Mixed-Lineage Leukemia
- TR-FRET
time-resolved fluorescence energy transfer
- WDR5
WD repeat domain 5 protein
- WIN
WDR5 interaction motif
Footnotes
Supporting Information. X-ray refinement statistics, MLL1 HMT assay details and titration curves of compound 16. This material is available free of charge via the internet at http://pubs.acs.org.
Accession Codes. Atom coordinates and structure factors for WDR5-ligand complexes can be accessed in the PDB via the following accession codes: Compound 13/WDR5 complex (6UFX), Compound 16/WDR5 complex (6UCS). Authors will release the atomic coordinates upon article publication.
REFERENCES
- (1).Smith TF; Gaitatzes C; Saxena K; Neer EJ The WD Repeat: A Common Architecture for Diverse Functions. Trends Biochem. Sci 1999, 24 (5), 181–185. [DOI] [PubMed] [Google Scholar]
- (2).Stirnimann CU; Petsalaki E; Russell RB; Müller CW WD40 Proteins Propel Cellular Networks. Trends Biochem. Sci 2010, 35 (10), 565–574. [DOI] [PubMed] [Google Scholar]
- (3).Xu C; Min J Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2 (3), 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Guarnaccia A; Tansey W; Guarnaccia AD; Tansey WP Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med 2018, 7 (2), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dai X; Guo W; Zhan C; Liu X; Bai Z; Yang Y WDR5 Expression Is Prognostic of Breast Cancer Outcome. PLoS One 2015, 10 (9), e0124964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhu J; Sammons MA; Donahue G; Dou Z; Vedadi M; Getlik M; Barsyte-Lovejoy D; Al-awar R; Katona BW; Shilatifard A; Huang J; Hua X; Arrowsmith CH; Berger SL Gain-of-Function P53 Mutants Co-Opt Chromatin Pathways to Drive Cancer Growth. Nature 2015, 525 (7568), 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang F; Jeon KO; Salovich JM; Macdonald JD; Alvarado J; Gogliotti RD; Phan J; Olejniczak ET; Sun Q; Wang S; Camper D; Yuh JP; Shaw JG; Sai J; Rossanese OW; Tansey WP; Stauffer SR; Fesik SW Discovery of Potent 2-Aryl-6,7-Dihydro-5 H-Pyrrolo[1,2- a]Imidazoles as WDR5-WIN-Site Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem 2018, 61 (13), 5623–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Aho ER; Wang J; Gogliotti RD; Howard GC; Phan J; Acharya P; Macdonald JD; Cheng K; Lorey SL; Lu B; Wenzel S; Foshage AM; Alvarado J; Wang F; Shaw JG; Zhao B; Weissmiller AM; Thomas LR; Vakoc CR; Hall MD; Hiebert SW; Liu Q; Stauffer SR; Fesik SW; Tansey WP Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019, 26 (11), 2916–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Patel A; Dharmarajan V; Cosgrove MS Structure of WDR5 Bound to Mixed Lineage Leukemia Protein-1 Peptide. J. Biol. Chem 2008, 283 (47), 32158–32161. [DOI] [PubMed] [Google Scholar]
- (10).Patel A; Vought VE; Dharmarajan V; Cosgrove MS A Conserved Arginine-Containing Motif Crucial for the Assembly and Enzymatic Activity of the Mixed Lineage Leukemia Protein-1 Core Complex. J. Biol. Chem 2008, 283 (47), 32162–32175. [DOI] [PubMed] [Google Scholar]
- (11).Hess JL MLL: A Histone Methyltransferase Disrupted in Leukemia. Trends Mol. Med 2004, 10 (10), 500–507. [DOI] [PubMed] [Google Scholar]
- (12).Dharmarajan V; Lee J-H; Patel A; Skalnik DG; Cosgrove MS Structural Basis for WDR5 Interaction (Win) Motif Recognition in Human SET1 Family Histone Methyltransferases. J. Biol. Chem 2012, 287 (33), 27275–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Alicea-Velázquez NL; Shinsky SA; Loh DM; Lee JH; Skalnik DG; Cosgrove MS Targeted Disruption of the Interaction between WD-40 Repeat Protein 5 (WDR5) and Mixed Lineage Leukemia (MLL)/SET1 Family Proteins Specifically Inhibits MLL1 and SETd1A Methyltransferase Complexes. J. Biol. Chem 2016, 291 (43), 22357–22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Thomas LR; Wang Q; Grieb BC; Phan J; Foshage AM; Sun Q; Olejniczak ET; Clark T; Dey S; Lorey S; Alicie B; Howard GC; Cawthon B; Ess KC; Eischen CM; Zhao Z; Fesik SW; Tansey WP Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58 (3), 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen X; Xie W; Gu P; Cai Q; Wang B; Xie Y; Dong W; He W; Zhong G; Lin T; Huang J Upregulated WDR5 Promotes Proliferation, Self-Renewal and Chemoresistance in Bladder Cancer via Mediating H3K4 Trimethylation. Sci. Rep 2015, 5 (1), 8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Carugo A; Genovese G; Seth S; Nezi L; Rose JL; Bossi D; Cicalese A; Shah PK; Viale A; Pettazzoni PF; Akdemir KC; Bristow CA; Robinson FS; Tepper J; Sanchez N; Gupta S; Estecio MR; Giuliani V; Dellino GI; Riva L; Yao W; Di Francesco ME; Green T; D’Alesio C; Corti D; Kang Y; Jones P; Wang H; Fleming JB; Maitra A; Pelicci PG; Chin L; DePinho RA; Lanfrancone L; Heffernan TP; Draetta GF In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16 (1), 133–147. [DOI] [PubMed] [Google Scholar]
- (17).Tan X; Chen S; Wu J; Lin J; Pan C; Ying X; Pan Z; Qiu L; Liu R; Geng R; Huang W PI3K/AKT-Mediated Upregulation of WDR5 Promotes Colorectal Cancer Metastasis by Directly Targeting ZNF407. Cell Death Dis. 2017, 8 (3), e2686–e2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sun W; Guo F; Liu M Up-Regulated WDR5 Promotes Gastric Cancer Formation by Induced Cyclin D1 Expression. J. Cell. Biochem 2018, 119 (4), 3304–3316. [DOI] [PubMed] [Google Scholar]
- (19).Malek R; Gajula RP; Williams RD; Nghiem B; Simons BW; Nugent K; Wang H; Taparra K; Lemtiri-Chlieh G; Yoon AR; True L; An SS; DeWeese TL; Ross AE; Schaeffer EM; Pienta KJ; Hurley PJ; Morrissey C; Tran PT TWIST1-WDR5- Hottip Regulates Hoxa9 Chromatin to Facilitate Prostate Cancer Metastasis. Cancer Res. 2017, 77 (12), 3181–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tran PT; Gajula R; Williams R; Malek R; Nugent K; Walker A; Chettiar S; Wang H; Taparra K; Cades J; Herman J A Twist1-MLL-WDR5-HOTTIP Complex Regulates HOXA9 Chromatin to Facilitate Metastasis of Prostate Cancer. Int. J. Radiat. Oncol 2014, 90 (1), S177. [Google Scholar]
- (21).Sun Y; Bell JL; Carter D; Gherardi S; Poulos RC; Milazzo G; Wong JWH; Al-Awar R; Tee AE; Liu PY; Liu B; Atmadibrata B; Wong M; Trahair T; Zhao Q; Shohet JM; Haupt Y; Schulte JH; Brown PJ; Arrowsmith CH; Vedadi M; MacKenzie KL; Huttelmaier S; Perini G; Marshall GM; Braithwaite A; Liu T WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 2015, 75 (23), 5143–5154. [DOI] [PubMed] [Google Scholar]
- (22).Wu Y; Diao P; Li Z; Zhang W; Wang D; Wang Y; Cheng J Overexpression of WD Repeat Domain 5 Associates with Aggressive Clinicopathological Features and Unfavorable Prognosis in Head Neck Squamous Cell Carcinoma. J. Oral Pathol. Med 2018, 47 (5), 502–510. [DOI] [PubMed] [Google Scholar]
- (23).Ge Z; Song EJ; Kawasawa YI; Li J; Dovat S; Song C; Ge Z; Song EJ; Kawasawa YI; Li J; Dovat S; Song C WDR5 High Expression and Its Effect on Tumorigenesis in Leukemia. Oncotarget 2016, 7 (25), 37740–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lu K; Tao H; Si X; Chen Q The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front. Oncol 2018, 8, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Meyer C; Hofmann J; Burmeister T; Gröger D; Park TS; Emerenciano M; Pombo de Oliveira M; Renneville A; Villarese P; Macintyre E; Cavé H; Clappier E; Mass-Malo K; Zuna J; Trka J; De Braekeleer E; De Braekeleer M; Oh SH; Tsaur G; Fechina L; van der Velden VHJ; van Dongen JJM; Delabesse E; Binato R; Silva MLM; Kustanovich A; Aleinikova O; Harris MH; Lund-Aho T; Juvonen V; Heidenreich O; Vormoor J; Choi WWL; Jarosova M; Kolenova A; Bueno C; Menendez P; Wehner S; Eckert C; Talmant P; Tondeur S; Lippert E; Launay E; Henry C; Ballerini P; Lapillone H; Callanan MB; Cayuela JM; Herbaux C; Cazzaniga G; Kakadiya PM; Bohlander S; Ahlmann M; Choi JR; Gameiro P; Lee DS; Krauter J; Cornillet-Lefebvre P; Te Kronnie G; Schäfer BW; Kubetzko S; Alonso CN; zur Stadt U; Sutton R; Venn NC; Izraeli S; Trakhtenbrot L; Madsen HO; Archer P; Hancock J; Cerveira N; Teixeira MR; Lo Nigro L; Möricke A; Stanulla M; Schrappe M; Sedék L; Szczepański T; Zwaan CM; Coenen EA; van den Heuvel-Eibrink MM; Strehl S; Dworzak M; Panzer-Grümayer R; Dingermann T; Klingebiel T; Marschalek R The MLL Recombinome of Acute Leukemias in 2013. Leukemia 2013, 27 (11), 2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Muntean AG; Hess JL The Pathogenesis of Mixed-Lineage Leukemia. Annu. Rev. Pathol. Mech. Dis 2012, 7 (1), 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Winters AC; Bernt KM MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr 2017, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krivtsov AV; Armstrong SA MLL Translocations, Histone Modifications and Leukaemia Stem-Cell Development. Nat. Rev. Cancer 2007, 7 (11), 823–833. [DOI] [PubMed] [Google Scholar]
- (29).Cao F; Townsend EC; Karatas H; Xu J; Li L; Lee S; Liu L; Chen Y; Ouillette P; Zhu J; Hess JL; Atadja P; Lei M; Qin ZS; Malek S; Wang S; Dou Y Targeting MLL1 H3K4 Methyltransferase Activity in Mixed-Lineage Leukemia. Mol. Cell 2014, 53 (2), 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen Y; Anastassiadis K; Kranz A; Stewart AF; Arndt K; Waskow C; Yokoyama A; Jones K; Neff T; Lee Y; Ernst P MLL2, Not MLL1, Plays a Major Role in Sustaining MLL-Rearranged Acute Myeloid Leukemia. Cancer Cell 2017, 31 (6), 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Aho ER; Weissmiller AM; Fesik SW; Tansey WP Targeting WDR5: A WINning Anti-Cancer Strategy? Epigenetics insights 2019, 12, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Karatas H; Li Y; Liu L; Ji J; Lee S; Chen Y; Yang J; Huang L; Bernard D; Xu J; Townsend EC; Cao F; Ran X; Li X; Wen B; Sun D; Stuckey JA; Lei M; Dou Y; Wang S Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)–Mixed Lineage Leukemia (MLL) Protein–Protein Interaction. J. Med. Chem 2017, 60 (12), 4818–4839. [DOI] [PubMed] [Google Scholar]
- (33).Grebien F; Vedadi M; Getlik M; Giambruno R; Grover A; Avellino R; Skucha A; Vittori S; Kuznetsova E; Smil D; Barsyte-Lovejoy D; Li F; Poda G; Schapira M; Wu H; Dong A; Senisterra G; Stukalov A; Huber KVM; Schönegger A; Marcellus R; Bilban M; Bock C; Brown PJ; Zuber J; Bennett KL; Al-awar R; Delwel R; Nerlov C; Arrowsmith CH; Superti-Furga G Pharmacological Targeting of the Wdr5-MLL Interaction in C/EBPα N-Terminal Leukemia. Nat. Chem. Biol 2015, 11 (8), 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Getlik M; Smil D; Zepeda-Velázquez C; Bolshan Y; Poda G; Wu H; Dong A; Kuznetsova E; Marcellus R; Senisterra G; Dombrovski L; Hajian T; Kiyota T; Schapira M; Arrowsmith CH; Brown PJ; Vedadi M; Al-awar R Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J. Med. Chem 2016, 59 (6), 2478–2496. [DOI] [PubMed] [Google Scholar]
- (35).Wang F; Jeon KO; Salovich JM; Macdonald JD; Alvarado J; Gogliotti RD; Phan J; Olejniczak ET; Sun Q; Wang S; Camper D; Yuh JP; Shaw JG; Sai J; Rossanese OW; Tansey WP; Stauffer SR; Fesik SW Discovery of Potent 2-Aryl-6,7-Dihydro-5 H -Pyrrolo[1,2- a]Imidazoles as WDR5-WIN-Site Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem 2018, 61 (13), 5623–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Karatas H; Townsend EC; Bernard D; Dou Y; Wang S Analysis of the Binding of Mixed Lineage Leukemia 1 (MLL1) and Histone 3 Peptides to WD Repeat Domain 5 (WDR5) for the Design of Inhibitors of the MLL1–WDR5 Interaction. J. Med. Chem 2010, 53 (14), 5179–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kondo J; Endo H; Okuyama H; Ishikawa O; Iishi H; Tsujii M; Ohue M; Inoue M Retaining Cell-Cell Contact Enables Preparation and Culture of Spheroids Composed of Pure Primary Cancer Cells from Colorectal Cancer. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (15), 6235–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kondo J; Ekawa T; Endo H; Yamazaki K; Tanaka N; Kukita Y; Okuyama H; Okami J; Imamura F; Ohue M; Kato K; Nomura T; Kohara A; Mori S; Dan S; Inoue M High-Throughput Screening in Colorectal Cancer Tissue-Originated Spheroids. Cancer Sci. 2019, 110 (1), 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yang H; Medeiros PF; Raha K; Elkins P; Lind KE; Lehr R; Adams ND; Burgess JL; Schmidt SJ; Knight SD; Auger KR; Schaber MD; Franklin GJ; Ding Y; DeLorey JL; Centrella PA; Mataruse S; Skinner SR; Clark MA; Cuozzo JW; Evindar G Discovery of a Potent Class of PI3Kα Inhibitors with Unique Binding Mode via Encoded Library Technology (ELT). ACS Med. Chem. Lett 2015, 6 (5), 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bia H; Bailey S; Bhumralkar DR; Bi FC; Guo F; He M; Humphries PS; Ling AL; Lou J; Nukul S; Zhou R Fused Phenyl Amido Heterocyclic Compounds. US Patent 7,842,713, November 30, 2010.
- (41).Vita M; Henriksson M The Myc Oncoprotein as a Therapeutic Target for Human Cancer. Semin. Cancer Biol. 2006, 16 (4), 318–330. [DOI] [PubMed] [Google Scholar]
- (42).van Riggelen J; Yetil A; Felsher DW MYC as a Regulator of Ribosome Biogenesis and Protein Synthesis. Nat. Rev. Cancer 2010, 10 (4), 301–309. [DOI] [PubMed] [Google Scholar]
- (43).Otwinowski Z; Minor W [20] Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (44).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP 4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr 2011, 67 (4), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Adams PD; Grosse-Kunstleve RW; Hung L-W; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX : Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr. Sect. D Biol. Crystallogr 2002, 58 (11), 1948–1954. [DOI] [PubMed] [Google Scholar]
- (46).Emsley P; Cowtan K Coot : Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr 2004, 60 (12), 2126–2132. [DOI] [PubMed] [Google Scholar]
- (47).Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S Development and Optimization of a Binding Assay for the XIAP BIR3 Domain Using Fluorescence Polarization. Anal. Biochem 2004, 332 (2), 261–273. [DOI] [PubMed] [Google Scholar]
- (48).Ferry JJ; Smith RF; Denney N; Walsh CP; McCauley L; Qian J; Ma H; Horiuchi KY; Howitz KT Development and Use of Assay Conditions Suited to Screening for and Profiling of SET-Domain-Targeted Inhibitors of the MLL/SET1 Family of Lysine Methyltransferases. Assay Drug Dev. Technol 2015, 13 (4), 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.