

ABSTRACT
Kv12.1 K+ channels are expressed in several brain areas, but no physiological function could be attributed to these subunits so far. As genetically-modified animal models are not available, identification of native Kv12.1 currents must rely on characterization of distinct channel properties. Recently, it was shown in Xenopus laevis oocytes that Kv12.1 channels were modulated by membrane PI(4,5)P2. However, it is not known whether these channels are also sensitive to physiologically-relevant PI(4,5)P2 dynamics. We thus studied whether Kv12.1 channels were modulated by activation of phospholipase C β (PLCβ) and found that they were insensitive to receptor-triggered depletion of PI(4,5)P2. Thus, Kv12.1 channels add to the growing list of K+ channels that are insensitive to PLCβ signaling, although modulated by PI(4,5)P2 in Xenopus laevis oocytes.
KEYWORDS: Kv12; Kv7; KCNQ; M current; mode shift; PI(4,5)P2; Kv7; PI(3,4,5)P3; phospholipase C; GqPCR; Ci-VSP
Introduction
The ether-à-go-go-gene-like (Elk; Kv12) family of voltage-gated potassium (Kv) channels comprises three members (Kv12.1-Kv12.3) that are predominantly expressed in neurons of various brain regions [1-6]. Despite recent significant progress in understanding biophysical properties and characteristics of these channels [7–10], at present only little information is available on physiological functions of the family members. Genetic deletion and pharmacological inhibition revealed that Kv12.2 channels constitute subthreshold K+ conductance regulating excitability of pyramidal neurons in hippocampus [11], but no native physiological relevance could be attributed to Kv12.1 and Kv12.3 subunits so far. As Kv12.1 channels activate at hyperpolarized membrane potentials [7,8,10], it is reasonable to assume that these channels also modulate neuronal excitability.
A prominent and characteristic feature of Kv12.1 channels is a mode shift of activation (also referred to as pre-pulse facilitation or voltage-dependent potentiation) [7,8,12]. This biophysical phenomenon describes hysteresis of voltage-dependent channel activation that most probably is caused by time-dependent stabilization of the channel´s voltage sensor in a “relaxed” open state in response to depolarized (conditioning) holding potentials [13,14]. It was shown recently that prolonged depolarization of the membrane potential induces slow rearrangement of a structural interaction between domains in the C- and N-terminus of Kv12.1 channels [8]. This rearrangement apparently is coupled to channel gating and necessary for transition of Kv12.1 channels into the more stable gating mode that favors channel opening [8]. Accordingly, conditioning depolarisation of the membrane potential causes a large shift in voltage dependence of activation to hyperpolarized potentials and a slowing of deactivation of Kv12.1 channels [7,8,10]. This mode shift may constitute a biophysical adaption to dampen excitability of neurons upon ongoing stimulation possibly also to prevent hyperexcitability in nervous tissue. Analogous mode shift of related Kv11.1 channels might contribute to repolarization of cardiac action potentials through slowing of channel deactivation [15,16], but to our knowledge physiological relevance of this mode shift in neurons has not been demonstrated.
Recently, it was shown that Kv12.1 channel activity was regulated through membrane-associated phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) [7] that is a minor component of the inner leaflet of the plasma membrane and a well-known co-factor of many ion channels [17–19]. Li and colleagues showed that loss of phosphoinositides (PI) induced by excision of membrane patches from Xenopus laevis oocytes caused acceleration of Kv12.1 channel activation and deactivation and a potentiation of steady-state K+ currents. Furthermore, excision of membrane patches expressing Kv12.1 channels caused a shift of voltage dependence of channel activation to hyperpolarized potentials and almost completely eliminated voltage-dependent mode shift [7]. As application of a water soluble PI(4,5)P2 analogue restored Kv12.1 channel kinetics as well as voltage dependence, and partially brought-back mode shift, the authors consistently concluded that PI(4,5)P2 in a bimodal way of action stabilized the open state, but at the same time inhibited voltage dependent activation of Kv12.1 channels [7]. Accordingly, variations of membrane PI(4,5)P2 levels might control excitability of neurons through modulating Kv12.1 channel activity. Most interestingly, impact of PI(4,5)P2 level changes on neuronal excitability might vary considerably with the general excitation status of the neuron, as the extent of mode shift apparently also was PI-dependent [7]. Unfortunately, it has not been shown yet whether Kv12.1 channels were sensitive to physiologically-relevant PI(4,5)P2 depletion at all. As activation of phospholipase C β (PLCβ) through Gq protein-coupled receptors (GqPCR) constitutes an important signaling pathway to deplete PI(4,5)P2 in neurons [20,21], we studied whether Kv12.1 channels were modulated by activation of Gq protein-coupled muscarinic receptors.
Results
Analyzing voltage-dependent mode shift of Kv12.1 channels
We analyzed sensitivity to GqPCR signaling of human Kv12.1 channels heterologously expressed in CHO cells. To be able to analyze whether voltage-dependent activation of Kv12.1 channels was sensitive to activation of the GqPCR pathway, we first established appropriate voltage protocols to study voltage dependence of these channels. As recently reported [10], we applied depolarizing holding potentials (conditioning voltage; 200 ms) before a series of activating voltage steps (pulse potentials from −140 mV to +10 mV; 600 ms) (Figure 1(A,B)). We activated Kv12.1 channels for as short as 600 ms, as we previously found that 600 ms allowed for steady-state activation of Kv12.1 channels, whereas longer activating pulses attenuated mode shift due to longer time intervals at hyperpolarized potentials [10]. Accordingly, we also recorded tail currents at correspondingly depolarized potentials to minimize time at hyperpolarized voltages (Figure 1(B)). These voltage protocols elicited robust, outwardly-rectifying and voltage-dependent Kv12.1 currents in CHO cells (Figure 1(A)). For conditioning voltages of −60 mV, half-maximal voltage (Vh) and slope factor of Kv12.1 channel activation were −21.3 ± 2.1 mV and −14.6 ± 1.3 mV, respectively (n = 6; Figure 1(C–E)). When cells were held at a depolarized conditioning potential of 0 mV, Vh of channel activation was
Figure 1.
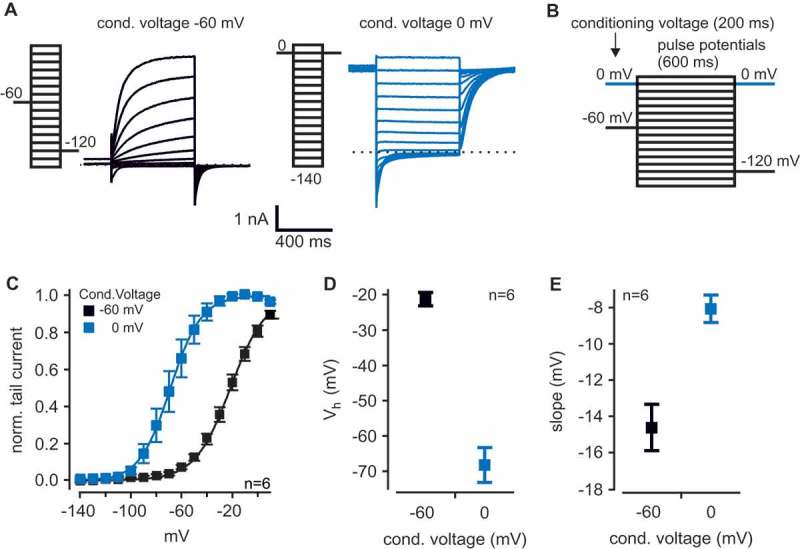
Mode shift of recombinant human Kv12.1 channels.
(A) Representative patch clamp recording of a CHO cell transiently transfected with human Kv12.1 channels activated by the voltage steps as indicated. (B) Voltage protocols consisted of a 200 ms conditioning potential step to −60 mV (black) or 0 mV (blue) followed by 600 ms activating pulse potentials from −140 mV to +10 mV (10 mV increments). Tail currents were elicited either at −120 mV or at 0 mV to minimize time at hyperpolarized membrane potentials (voltage protocols were established in a recent publication [10]). (C-E) Summary of voltage dependence of human Kv12.1 channels. (C) Voltage dependence was analyzed with Boltzmann fits to individual recordings as shown in (A). Solid lines in (C) represent Boltzmann fits to averaged data. Depolarized conditioning potential of 0 mV induced a large shift of voltage dependence to hyperpolarized potentials, as recently reported [10]. (D) shows mean Vh and (E) displays mean slope factor of channel activation in dependence of conditioning potentials of −60 mV and 0 mV (values were derived from Boltzmann fits shown in (C).
-68.2 ± 5.2 mV and the slope factor was −8.1 ± 0.8 mV (n = 6; Figure 1(C–E)). Accordingly, as reported by us and others [7,8,10] depolarizing conditioning potentials induced a large shift of voltage dependence to hyperpolarized potentials by about −50 mV. This shift of voltage dependence in response to depolarized holding potentials is generally referred to as mode shift of activation [12].
Kv12.1 channels are insensitive to physiological PI(4,5)P2 depletion through PLCβ
Recently, it was shown that activity of Kv12.1 channels was modulated by PI in membrane patches excised from Xenopus laevis oocytes [7,8]. We thus set out to analyze in CHO cells whether these channels were also sensitive to stimulation of GqPCR signaling that physiologically leads to depletion of membrane PI(4,5)P2 through activation of PLCβ. To this end, we overexpressed Gq-coupled muscarinic receptor type 1 (m1R) in CHO cells and activated the receptor by extracellular application of oxotremorine-M (Oxo-M; 10 µM). In this expression system, activation of recombinant m1R reconstitutes GqPCR signaling and induces depletion of PI(4,5)P2 through endogenous PLCβ thereby producing second messengers diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (I(1,4,5)P3) [e.g. 20,22–25]. At start, we co-expressed bona fide PI(4,5)P2-sensitive homomeric Kv7.4 or Kv7.3 channels together with m1R. We utilized Kv7.3 channels carrying a A315T mutation in the pore region to increase whole cell currents without however affecting the channel´s sensitivity to PI(4,5)P2 depletion (Kv7.3T [26,27]). We found that stimulation of m1R strongly reduced Kv7.4- and Kv7.3T-mediated currents (Figure 2(A,B)). It is well established that m1R-dependent inhibition of Kv7 channels mainly depends on PLCβ-dependent depletion of PI(4,5)P2 [22,23,28,29] and that degree of inhibition corresponds to PI(4,5)P2 affinity of the respective Kv7 isoform [30]. In line, activation of m1R caused stronger inhibition of Kv7.4 that exhibit lower PI(4,5)P2 affinity than Kv7.3T channels [29–31]. These recordings demonstrated robust activation of GqPCR signaling and substantial PLCβ-dependent PI(4,5)P2 depletion in CHO cells upon stimulation of m1R. In contrast, Kv12.1-mediated steady-state currents elicited by voltages between -140 mV and + 10 mV were not affected by stimulation of m1R through Oxo-M (Figure 2(A–E)). GqPCR-dependent activation of PLCβ also did not change kinetics of activation (Figure 2(F)) and deactivation (Figure 2(G)) of human Kv12.1 channels in CHO cells. After 60 s of application of Oxo-M (10 µM), Vh of activation was −23.7 ± 2.1 mV and −67.3 ± 5.6 mV for conditioning voltages of −60 mV and 0 mV, respectively (Figure 2(H,I)). Thus, also voltage dependence of human Kv12.1 channels was not affected by stimulation of PLCβ demonstrating that mode shift of Kv12.1 channels was not sensitive to activation of the pathway in CHO cells. To rule out that for some reason depletion of PI(4,5)P2 was reduced in CHO cells expressing Kv12.1 channels, we then directly assessed activity of PLCβ under our experimental conditions. We quantified putative PLCβ activity using pleckstrin homology (PH) domain of PLCδ1 fused to RFP (PLCδ1-PH-RFP) as genetically-encoded, optical biosensor that binds to membrane PI(4,5)P2, but also to cytoplasmic I(1,4,5)P3 [32]. PLCδ1-PH-RFP associates to the plasma membrane, when resting PI(4,5)P2 levels are high, and translocates into the cytoplasm in response to PLCβ-dependent decrease of membrane PI(4,5)P2 and a corresponding increase of cytoplasmic I(1,4,5)P3 [32,33]. Accordingly, changes of the fluorescence intensity at the membrane or in the cytoplasm are a measure for the activity of PLCβ during stimulation of GqPCR [17,34]. Here, we used confocal imaging to monitor translocation of PLCδ1-PH-RFP from membrane to the cytoplasm of transiently transfected CHO cells during application of Oxo-M (10 µM). In cells, coexpressing m1R together with PLCδ1-PH-RFP and Kv7.4 channels, RFP-associated fluorescence in the cytoplasm strongly increased during activation of the GqPCR. This increase of fluorescence indicated translocation of the sensor from membrane into cytoplasm and thus PLCβ-mediated PI(4,5)P2 depletion (Figure 2(J)). Upon wash-out of Oxo-M, the signal recovered within a couple of minutes demonstrating reversibility of PI(4,5)P2 depletion. When we activated m1R in cells coexpressing Kv12.1 channels, sensor translocation into the cytoplasm and its return to the plasma membrane were indistinguishable from cells expressing Kv7.4 (Figure 2(J,K)). These experiments demonstrated that PLCβ was strongly activated under our experimental conditions and that PLCβ activity and thus PI(4,5)P2 depletion were the same in cells expressing Kv7.4 and Kv12.1 channels. Taking together these data showed that human Kv12.1 channels were not sensitive to activation of GqPCR signaling and more importantly PLCβ-mediated PI(4,5)P2 depletion in CHO cells.
Figure 2.
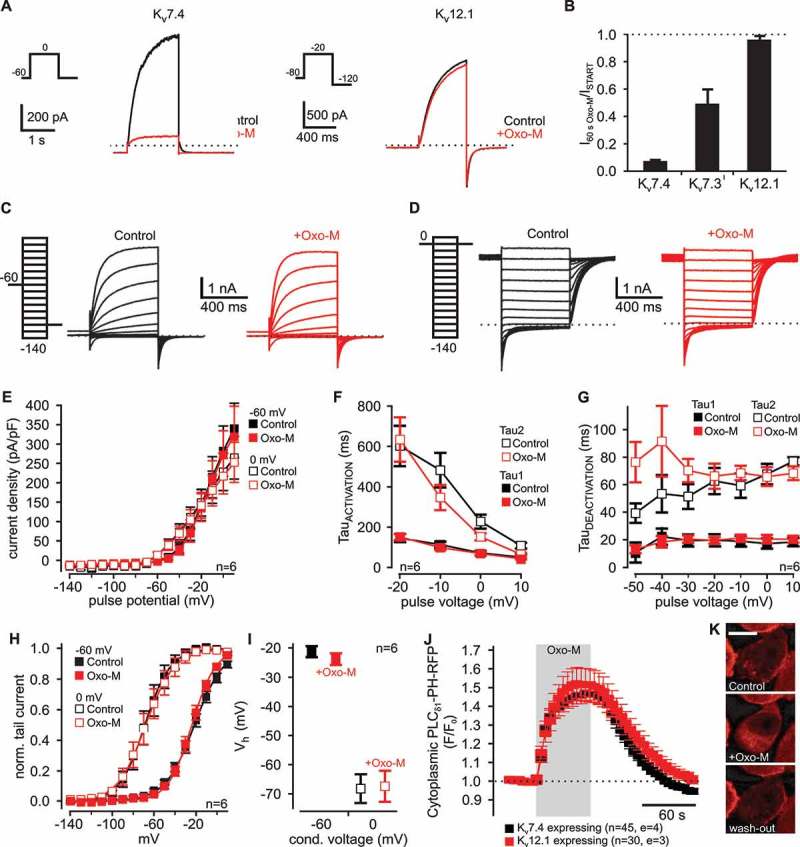
Human Kv12.1 channels are insensitive to GqPCR/PLCβ signaling.
(A + B) Activation of muscarinic Gq protein-coupled receptor type 1 (m1R) by Oxo-M (10 µM) strongly inhibited bona fide PI(4,5)P2-sensitive Kv7.4 and Kv7.3T channels. In contrast, Kv12.1-mediated currents were resistant to stimulation of m1R. (A) shows representative Kv7.4- and Kv12.1-mediated whole cell currents before (black) and at the end of a 60 s application of 10 µM Oxo-M (red) (voltage protocol and scale bars as indicated). (B) shows a summary of recordings as presented in (A). (C-E) Steady-state Kv12.1 currents elicited by voltage steps between −140 mV and + 10 mV were not sensitive to activation of m1R through Oxo-M (10 µM). (F) Neither activating kinetics nor (G) deactivating kinetics of Kv12.1 channels were altered upon activation of m1R (time constants were calculated from double-exponential fits to the activating current component and to deactivating tail currents). (C + D) show representative whole cell Kv12.1 currents elicited by the voltage protocols indicated. To induce voltage-dependent mode shift of Kv12.1 channels, voltage protocols consisted of a 200 ms conditioning potential step to (C) −60 mV or (D) 0 mV followed by 600 ms activating pulse potentials from −140 mV to + 10 mV (10 mV increments). (H + I) Oxo-M-dependent activation of m1R did not change voltage-dependence or mode-shift of Kv12.1 channels expressed in CHO cells. (H) shows a summary of the voltage dependence of human Kv12.1 channels and (I) displays mean Vh derived from Boltzmann fits to individual recordings as shown in (C) an (D) (solid lines in (H) represent Boltzmann fits to averaged data; Data with Oxo-M were analyzed at the end of 60 s Oxo-M application; control recordings are also shown in Figure 1). (J + K) In CHO cells coexpressing m1R together with Kv7.4, application of Oxo-M (10 µM) induced robust and reversible translocation of the optical PI(4,5)P2/I(1,4,5)P3 biosensor PLCδ1-PH-mRFP from the membrane to the cytoplasm. PLCδ1-PH-RFP translocation was indistinguishable between cells coexpressing Kv12.1 or Kv7.4 channels together with m1R. Thus, PLCβ activation was comparable in cells expressing Kv7.4 or Kv12.1. (J) shows mean RFP fluorescence intensities measured in confocal sections averaged over a region of interest in the cytoplasm of CHO cells coexpressing PLCδ1-PH-RFP together with either Kv12.1 or Kv7.4. (K) shows representative images of a CHO cell expressing Kv12.1 together with m1R before (top), after 60 s application of Oxo-M (middle) and after wash out of the agonist (bottom). The scale bar represents 10 µm.
Recombinant Kv12.1 channels are insensitive to Ci-VSP-dependent PI(4,5)P2 depletion
To assess in more detail whether Kv12.1 channels were sensitive to PI(4,5)P2 depletion in CHO cells, we utilized voltage-sensitive PI 5-phosphatase from ciona intestinalis (Ci-VSP). Upon depolarization of the membrane potential, Ci-VSP rapidly removes PI(4,5)P2 and PI(3,4,5)P3 from the membrane by dephosphorylation to PI(4)P and PI(3,4)P2, respectively [35–39]. In these experiments, we selected cells for clear membrane localization of Ci-VSP-RFP-associated fluorescence and recorded Kv7 and Kv12.1 currents activated by depolarizing voltage steps from the holding potential of −80 mV to 0 mV or −20 mV, respectively (Figure 3(A)). After recording stable control current amplitudes for at least 30 s, the holding potential was depolarized for another 30 s to +80 mV to activate Ci-VSP. As shown in Figure 3(A,B), currents through homomeric Kv7.2 and Kv7.3T channels were strongly inhibited by activation of Ci-VSP. After 30 s of Ci-VSP activation at +80 mV, Kv7.2 and Kv7.3T currents were reduced to 27.8 ± 4.6% and 40.6 ± 1.9% of baseline current amplitudes, respectively (Figure 3(A,B)). This inhibition of Kv7-mediated currents, especially inhibition of Kv7.3T channels that exhibit higher PI(4,5)P2 affinity than Kv7.2, demonstrated substantial depletion of membrane PI(4,5)P2 upon activation of Ci-VSP, in line with many previous reports [e.g. 29, 38]. In these experiments, Kv7 currents returned to baseline within a minute after deactivation of Ci-VSP at hyperpolarized potentials demonstrating reversible PI(4,5)P2 depletion and its resynthesis through endogenous PI kinases (Figure 3(B)) [38]. Interestingly after deactivation of Ci-VSP, Kv7.2-mediated currents transiently over-recovered before returning to baseline amplitudes indicating some kind of over-recovery of PI(4,5)P2 levels possibly stimulated by Ci-VSP-induced PI(4,5)P2 depletion [c.f. 38].
Figure 3.
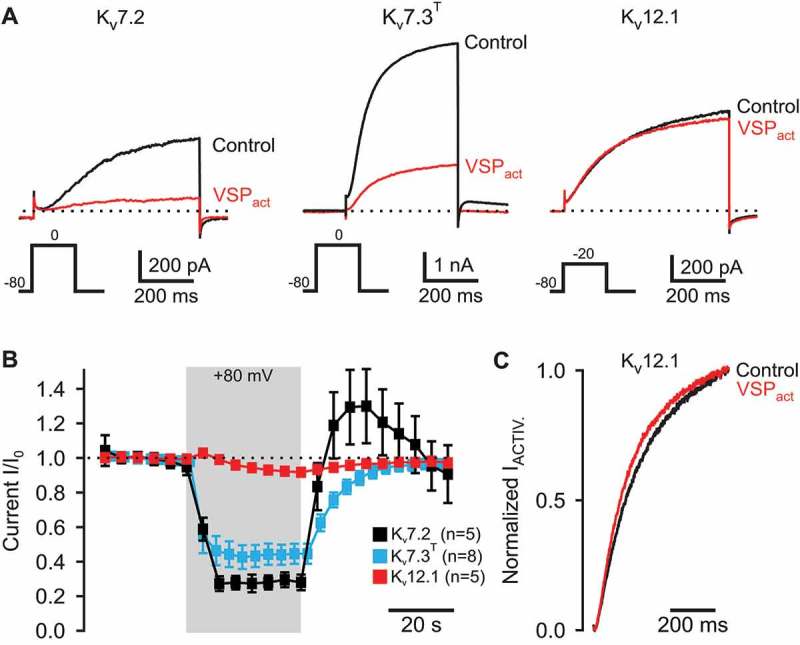
In CHO cells, human Kv12.1 channels are insensitive to activation of a voltage-sensitive PI(4,5)P2/PI(3,4,5)P3 5-phosphatase from ciona intestinalis.
(A + B) Activation of Ci-VSP through 30 s depolarization of the holding potential to +80 mV reversibly inhibited recombinant Kv7.2 and Kv7.3T channels. In contrast, Kv12.1-mediated current amplitudes were largely insensitive to voltage-dependent activation of Ci-VSP. (A) shows representative recordings of Kv7.2, Kv7.3T or Kv12.1 currents before (black) and at the end of Ci-VSP activation for 30 s (red) (voltage steps to activate K+ channel were applied every 5 s; Ci-VSP was activated by depolarization to + 80 mV between these voltage steps). (B) displays averaged time course of recordings as shown in (A). (C) In only three out of five cells, we found slight acceleration of Kv12.1 activating kinetics upon voltage-dependent activation of Ci-VSP. (C) shows exemplary normalized Kv12.1 currents activating upon a voltage step from −80 mV to −20 mV before (black) and after activation of Ci-VSP at +80 mV for 30 s (red).
In contrast to Kv7 currents, Kv12.1 channels were almost completely resistant to activation of co-expressed Ci-VSP utilizing the same voltage protocol to activate the phosphatase. After 30 s at +80 mV, Kv12.1 current amplitudes were 92.7 ± 1.3% of control currents before voltage-dependent activation of Ci-VSP (n = 5; Figure 3(A,B)). In three out of five cells tested, we detected slight acceleration of the activating kinetics of Kv12.1 channels in response to stimulation of Ci-VSP (Figure 3(C)). As these changes of kinetics were small and not detectable in all cells, we did not examine the phenomenon any further.
In summary, Kv12.1 channels were not affected by Ci-VSP-dependent depletion of PI(4,5)P2 in CHO cells. Thus, we conclude that Kv12.1 channels are not relevantly modulated by membrane PI(4,5)P2 in this cell line. As Ci-VSP stoichiometrically converts PI(4,5)P2 into PI(4)P thereby substantially increasing PI(4)P levels in the membrane [39], these data also demonstrated that Kv12.1 channels are not modulated by membrane-associated PI(4)P.
Discussion
Activation of PLCβ through Gq/11-coupled receptors is an important intercellular signaling pathway that induces PI dynamics in neurons [17,18,21]. Although PLCβ possibly also hydrolyses PI(4)P [20,40,41], it is well known that PI(4,5)P2 is the prime substrate of its enzymatic activity in living cells [21]. Whereas actual changes in membrane-associated PI(4,5)P2 might vary significantly depending on the receptor type, as well as on the activity of PLCβ and PI(4,5)P2 resynthesis pathways, it is generally believed that the signaling cascade affects cellular physiology also through depletion of PI(4,5)P2 [20,21,32]. Thus, as PI(4,5)P2 is an important cofactor for many ion channels, activation of PLCβ is directly linked to neuronal excitability through PI(4,5)P2 depletion and PI(4,5)P2-dependent modulation of ion channel activity. As one prominent example, stimulation of endogenous Gq-coupled muscarinic receptors induces significant depolarization of neuronal membrane potentials through PI(4,5)P2-dependent inhibition of Kv7 potassium channels [22,23,42].
Here, we evaluated whether human Kv12.1 channels were sensitive to physiological changes of PI(4,5)P2 for two reasons: (i) As no physiological function could be assigned to Kv12.1 channels yet, novel properties of these channels might be useful to identify native Kv12.1-mediated currents in neurons [c.f. 10]. We thus considered PLCβ sensitivity of Kv12.1 channels a potentially interesting feature fostering future attribution of neuronal K+ currents to Kv12.1 channels. And (ii), recently it was shown for Kv12.1 channels that excision of membrane patches from Xenopus laevis oocytes speeded activation and deactivation, shifted voltage dependence to hyperpolarized potentials and significantly attenuated mode shift [7]. Importantly, such excision of membrane patches into solution without ATP causes depletion of PI(4,5)P2 (and possibly other PI species) through irreversible activation of phosphatases [19,24,28,31,43,44]. As application of a water-soluble PI(4,5)P2 analogue restored Kv12.1 channel properties in the excised inside-out patches, these findings demonstrated PI(4,5)P2 sensitivity of Kv12.1 channels [7]. We considered this PI(4,5)P2 sensitivity especially interesting, as by attenuating mode shift and by affecting voltage dependence of these channels, impact of PLCβ activation on the excitability of Kv12.1 expressing neurons might significantly vary depending on synaptic input and thus excitation status of the respective neuron. Utilizing m1R as G protein-coupled receptor to activate PLCβ in an expression system, we studied sensitivity of human Kv12.1 channels to GqPCR signaling. It has been shown in many studies that overexpression of a GqPCR (such as m1R) adequately reconstitutes PLCβ signaling in expression systems (including CHO cells used in our report). Through activation of endogenous PLCβ, stimulation of recombinant m1R substantially depletes PI(4,5)P2 [20,22,23,24,25,30,34], produces reasonable amounts of DAG and I(1,4,5)P3, induces I(1,4,5)P3-dependent Ca2+ release and activates downstream effectors of the cascade (e.g. protein kinase C, PKC) [20,22,30,34,45,46]. This has been used successfully to explore sensitivity of ion channels to physiological PI(4,5)P2 depletion [20,22,29,30,47], but also the modulation of ion channels through second messengers downstream of PLCβ activation [48–51]. Importantly, heterologous systems can even be utilized to dissect PI(4,5)P2 dependence of ion channels from their sensitivity to second messengers produced during activation of PLCβ [47,49,52]. Heterologous expression systems accordingly constitute a well-accepted model to study the GqPCR pathway and PI(4,5)P2 dependence of ion channels [17,18]. The stimulation of PLCβ as well as voltage-dependent activation of Ci-VSP strongly inhibited bona-fide PI(4,5)P2-dependent Kv7 channels (c.f. Figures 2 and 3). Especially, the significant inhibition of Kv7.3T channels that exhibit considerably higher PI(4,5)P2 affinity than Kv7.2 or Kv7.4 channels [29,30,38] demonstrated substantial PI(4,5)P2 depletion under our experimental conditions. Translocation of the optical PI(4,5)P2 biosensor PLCδ1-PH-RFP from the membrane during stimulation of m1R additionally showed strong reduction of PI(4,5)P2 levels in these experiments. However, Kv12.1 channels were not sensitive to this PLCβ-mediated PI(4,5)P2 depletion at all. Theoretically, a rise in DAG, I(1,4,5)P3 and intracellular Ca2+ or activation of PKC could stimulate Kv12.1, which might counterbalance PI depletion thereby masking PI(4,5)P2 sensitivity of these channels. However, although we consider such a mechanism rather unlikely, we want to point out that we cannot exclude sensitivity of Kv12.1 to these second messengers completely at present. Kv7-mediated currents are indeed reduced by PKC-dependent phosphorylation [48] and Ca2+/calmodulin signaling [53], but importantly GqPCR-dependent inhibition of these channels almost exclusively depends on PI(4,5)P2 depletion and not on messengers produced by PLCβ [23,29]. Based on these considerations, we conclude that Kv12.1 channels are resistant to stimulation of GqPCR and importantly insensitive to physiologically-relevant PI(4,5)P2 dynamics. This insensitivity of Kv12.1 channels to stimulation of PLCβ is not surprising, as several other K+ channels, including members of the Kv1, Kv2, Kv3, and Kv4 families, have been described to be not affected by stimulation of the PLCβ pathway and of Ci-VSP [24,25]. However, it is surprising that for some of these Kv channels (Kv1.1, Kv1.4, Kv1.5, Kv3.4), just as for Kv12.1 channels, PI(4,5)P2 sensitivity was reported using excised patches from Xenopus laevis oocytes as experimental model [54,55]. Importantly however, like Kv12.1 these channels were resistant to activation of m1R and Ci-VSP in mammalian cell lines [24,25]. As comprehensively discussed by the Hille group [24,25], we do not think that our findings represent a discrepancy to results obtained in the frog oocytes (presented by Li and colleagues [7]). First, upon excision of inside-out patches levels of membrane PI(4,5)P2 might fall well below levels reached by physiological stimulation of PLCβ (and possibly even by activation of Ci-VSP) and other PI species may also be depleted during the patch excision. Conversely, perfusion of membrane patches with water soluble PI analogues might introduce super-physiological PI(4,5)P2 levels within or close to the membrane [25]. Therefore, on the one hand PI sensitivity of ion channels might be overestimated upon PI(4,5)P2 depletion through patch excision and subsequent application of exogenous PI(4,5)P2. On the other hand, the degree of PLCβ-dependent PI(4,5)P2 depletion might be just too low to induce relevant modulation of Kv12.1 channel activity. And second, as comprehensively discussed and pointed-out by Li and colleagues [7], Kv12.1 channels might exhibit low PI affinity and low selectivity between different PI species. This conclusion is supported by the fact that not only PI(4,5)P2 but probably also other PI species might be depleted after excision of the membrane patches from oocytes. This depletion of several PI species upon patch excision might affect ion channels with unselective PI specificity even stronger than a highly selective channel that recognizes only a certain PI species. Supporting this notion, in membrane patches excised from Xenopus oocytes mode shift of Kv12.1 channels was sensitive to application of PI(4)P, PI(4,5)P2 and PI(3,4,5)P3 [7]. This strongly indicated that Kv12.1 channels (in contrast to e.g. Kv7 channels) do not exhibit highly selective binding to a certain PI species, but rather general electrostatic interactions with several PI [7]. Completely in line, Kv12.1 channels were resistant to activation of Ci-VSP in CHO cells, which stoichiometrically converts PI(4,5)P2 and PI(3,4,5)P3 into PI(4)P and PI(3,4)P2, respectively [35–37,39]. Whilst depleting PI(4,5)P2 from the membrane, activation of Ci-VSP thus leaves constant the total PI concentration in the plasma membrane. At the same time however, Ci-VSP substantially increases PI(4)P levels [38,39], which revealed that activity of Kv12.1 channels is not relevantly modulated by PI(4)P. This low sensitivity to Ci-VSP activation (and PLCβ signaling) may as well point to lack of selectivity towards PI(4,5)P2 thereby indicating a rather general PI sensitivity of Kv12.1 channels. In line, EC50 values of Kv12.1 channels for application of exogenous PI(4,5)P2 expressed in Xenopus oocytes (approx. 10 µM [7]) were well in the range previously demonstrated for Kv7.3 channels [28]. However, most probably due to high PI(4,5)P2 specificity and selectivity, Kv7.3 channels are readily inhibited by activation of m1R and Ci-VSP, whereas Kv12.1 channels are not. In line, sensitivity of K+ channels to activation of Ci-VSP generally correlates well with their sensitivity towards receptor-triggered activation of PLC [29], which apparently is also true for Kv12.1 channels.
Thus, in summary, human Kv12.1 channels most probably exhibit unselective PI sensitivity thereby adding to the growing list of Kv channels resistant to stimulation of GqPCR/PLCβ-signaling and Ci-VSP-dependent PI(4,5)P2 depletion [c.f. 24, 25].
Methods
Cell culture and transfection
Chinese hamster ovary (CHO) dhFR− cells were maintained as previously reported [56]. In brief, cells were kept in MEM Alpha Medium supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (pen/strep) (all Invitrogen GmbH, Darmstadt, Germany) in a humidified atmosphere at 5% CO2 and 37°C. Transient transfection of CHO cells in culture was done with jetPEI transfection reagent (Polyplus Transfection, Illkirch, France) and all experiments were performed at room temperature (22°C-25°C) approx. 24–48 h after transfection. The following vectors for transient expression in CHO cells were used: Kv7.2-pBK-CMV (gene: human KCNQ2; UniProt accession number: O43526), Kv7.3(A315T)-pBK-CMV (human KCNQ3(A315T); O43525), Kv7.4-pBK-CMV (human KCNQ4; P56696), Kv12.1(Elk1)-pcDNA3.1-IRES-eGFP (human KCNH8; Q96L42), human muscarinic receptor 1 (human M1R)-pSGHV0 (Q96RH1), Ci-VSP-mRFP-C1 (Q4W8A1), PLCδ1-PH-mRFP-C1 (amino acids 1–70; P51178) and pEGFP-C1 (transfection control; Addgene, Teddington, UK).
Electrophysiological recordings
Electrophysiological recordings (in the whole cell configuration) were performed with an Axopatch 200B amplifier (Molecular Devices, Union City, CA) in voltage-clamp mode [57]. Recordings were low-pass filtered at 2 kHz and sampled at 5 kHz. In the figures, voltage protocols are indicated and the dashed lines highlight zero current. Borosilicate glass patch pipettes (Sutter Instrument Company, Novato, CA, USA) used had an open pipette resistance of 2–3 MΩ after back-filling with intracellular solution containing (in mM) 135 KCl, 2.41 CaCl2 (100 nM free Ca2+), 3.5 MgCl2, 5 HEPES, 5 EGTA, 2.5 Na2ATP, 0.1 Na3GTP, pH 7.3 (with KOH), 290–295 mOsm/kg. Series resistance (Rs) typically was below 6 MΩ and compensated throughout the recordings (80–90%), and liquid junction potentials were not compensated (approx. −4 mV). For presentation whole cell currents were normalized to the cell capacitance (current density; pA/pF) or to baseline current amplitude (I/I0). The extracellular solution contained (in mM) 144 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 10 HEPES and 5.6 D-glucose, pH 7.4 (with NaOH), 305–310 mOsm/kg.
Voltage-dependent activation of Ci-VSP
For coexpression of ion channels with Ci-VSP-RFP, cells were selected for clear membrane localization of RFP (attached to Ci-VSP). Kv channel-mediated currents were elicited every 5s with the voltage protocol indicated in the figure and Ci-VSP was activated in between those voltage steps by depolarizing the holding potential to +80 mV for a total of 30 s, as previously reported [47].
Confocal microscopy
Confocal imaging was performed with an upright LSM 710 – Axio Examiner.Z1 microscope equipped with a W Plan/Apochromat 20x/1.0 DIC M27 75 mm water immersion objective (Zeiss, Jena, Germany) [58]. Red fluorescent protein (RFP) was excited at 561 nm with a DPSS 561–10 laser (Zeiss) and fluorescence emission was sampled at 582–754 nm. Green fluorescent protein (GFP) was excited at 488 nm with an argon laser and fluorescence emission was recorded at 493–597 nm. The sample interval for time series was 5 s. In these experiments, Kv7.4 or Kv12.1 expressing cells were identified through coexpression of GFP or the GFP expression associated with the pcDNA3.1-IRES-eGFP plasmid, respectively.
Solutions and substances
Oxotremorine-M (Oxo-M) was purchased from Biotrend Chemikalien GmbH (Cologne, Germany) and was diluted to a concentration of 10 µM in extracellular solution. Oxo-M was applied locally through a custom-made application system via a glass capillary brought into close proximity to cells under investigation.
Data analysis
Patch clamp recordings were analyzed with IgorPro (Wavemetrics, Lake Oswego, OR) and the PatchMaster (HEKA) software. Voltage dependence of activation was derived from tail current amplitudes using voltage protocols indicated: Tail currents were fitted with a two-state Boltzmann function with I = Imin + (Imax-Imin)/(1 + exp((V-Vh)/s)), where I is current, V is the membrane voltage, Vh is the voltage at half maximal activation, and s describes the steepness of the curve [59]. Results are shown as conductance-voltage curves, obtained by normalizing to (Imax-Imin), obtained from fits to data of individual experiments. Time constants of activation and deactivation were derived from double-exponential fits to deactivating current components at indicated potentials. Imaging time series were analyzed measured with confocal microscopy with Zen2009 (Zeiss) and IgorPro (Wavemetrics). PLCδ1-PH-mRFP fluorescent intensities were derived after background subtraction from averages over a region of interest (ROI) in the cytoplasm of transfected cells and are presented as cytoplasmic F/F0.
Data presentation
In electrophysiological experiments, n represents the number of individual cells and accordingly the number of independent experiments (no pseudo-replication). In imaging experiments (c.f. Figure 2(J)), n represents the number of individual cells and e denotes the number of independent experiments.
Funding Statement
This work was funded by Research Grants of the University Medical Center Giessen and Marburg (UKGM 17/2013; UKGM 13/2016 to M.G.L.) and by the German Research Foundation (DFG Priority Program 1608:”Ultrafast and temporally precise information processing: Normal and dysfunctional hearing”, [LE 3600/1-1 to MGL]).
Acknowledgments
The authors thank Dr. T. Jegla for constructive comments on the manuscript and helpful discussion of data. We acknowledge the kind gift of plasmids for Kv7 channels from Dr T. Jentsch, Kv12.1 (Elk) from Dr T. Jegla, Ci-VSP from Dr Y. Okamura, and PLCδ1-PH from Dr T. Meyer. We thank Olga Ebers and Neslihan Özen for superb technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Engeland B, Neu A, Ludwig J, et al. Cloning and functional expression of rat ether-a-go-go-like K+ channel genes. J Physiol. 1998. December 15;513(Pt 3):647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Miyake A, Mochizuki S, Yokoi H, et al. New ether-a-go-go K(+) channel family members localized in human telencephalon. J Biol Chem. 1999. August 27;274(35):25018–25025. [DOI] [PubMed] [Google Scholar]
- [3].Shi W, Wang HS, Pan Z, et al. Cloning of a mammalian elk potassium channel gene and EAG mRNA distribution in rat sympathetic ganglia. J Physiol. 1998. September 15;511(Pt 3):675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zou A, Lin Z, Humble M, et al. Distribution and functional properties of human KCNH8 (Elk1) potassium channels. Am J Physiol Cell Physiol. 2003. December;285(6):C1356–66. [DOI] [PubMed] [Google Scholar]
- [5].Trudeau MC, Titus SA, Branchaw JL, et al. Functional analysis of a mouse brain Elk-type K+ channel. J Neurosci. 1999. April 15;19(8):2906–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Saganich MJ, Machado E, Rudy B.. Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci. 2001. July 1;21(13):4609–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li X, Anishkin A, Liu H, et al. Bimodal regulation of an Elk subfamily K+ channel by phosphatidylinositol 4,5-bisphosphate. J Gen Physiol. 2015. November;146(5):357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dai G, Zagotta WN. Molecular mechanism of voltage-dependent potentiation of KCNH potassium channels. eLife. 2017. April 27;6 . DOI: 10.7554/eLife.26355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kazmierczak M, Zhang X, Chen B, et al. External pH modulates EAG superfamily K+ channels through EAG-specific acidic residues in the voltage sensor. J Gen Physiol. 2013. June;141(6):721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dierich M, Evers S, Wilke BU, et al. Inverse modulation of neuronal Kv12.1 and Kv11.1 channels by 4-aminopyridine and NS1643. Front Neurosci. 2018;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang X, Bertaso F, Yoo JW, et al. Deletion of the potassium channel Kv12.2 causes hippocampal hyperexcitability and epilepsy. Nat Neurosci. 2010. September;13(9):1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Villalba-Galea CA. Hysteresis in voltage-gated channels. Channels (Austin). 2017. March 4;11(2):140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bezanilla F, Taylor RE, Fernandez JM. Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J Gen Physiol. 1982. January;79(1):21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Villalba-Galea CA, Sandtner W, Starace DM, et al. S4-based voltage sensors have three major conformations. Proc Natl Acad Sci U S A. 2008. November 18;105(46):17600–17607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Trudeau MC, Warmke JW, Ganetzky B, et al. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995. July 7;269(5220):92–95. [DOI] [PubMed] [Google Scholar]
- [16].Sanguinetti MC, Jiang C, Curran ME, et al. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995. April 21;81(2):299–307. [DOI] [PubMed] [Google Scholar]
- [17].Leitner MG, Halaszovich CR, Ivanova O, et al. Phosphoinositide dynamics in the postsynaptic membrane compartment: mechanisms and experimental approach. Eur J Cell Biol. 2015. Jul-Sep;94(7–9):401–414. [DOI] [PubMed] [Google Scholar]
- [18].Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013. July;93(3):1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Horowitz LF, Hirdes W, Suh BC, et al. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005. September;126(3):243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kadamur G, Ross EM. Mammalian phospholipase C. Annu Rev Physiol. 2013;75:127–154. [DOI] [PubMed] [Google Scholar]
- [22].Shapiro MS, Roche JP, Kaftan EJ, et al. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels that underlie the neuronal M current. J Neurosci. 2000. Mar 1;20(5):1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gomez-Posada JC, Etxeberria A, Roura-Ferrer M, et al. A pore residue of the KCNQ3 potassium M-channel subunit controls surface expression. J Neurosci. 2010. July 7;30(27):9316–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shapiro MS, Roche JP, Kaftan EJ, et al. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels that underlie the neuronal M current. J Neurosci. 2000. March 1;20(5):1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002. August 1;35(3):507–520. S0896627302007900 [pii]. [DOI] [PubMed] [Google Scholar]
- [26].Zhang H, Craciun LC, Mirshahi T, et al. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003. March 27;37(6):963–975. [DOI] [PubMed] [Google Scholar]
- [27].Rjasanow A, Leitner MG, Thallmair V, et al. Ion channel regulation by phosphoinositides analyzed with VSPs-PI(4,5)P2 affinity, phosphoinositide selectivity, and PI(4,5)P2 pool accessibility. Front Pharmacol. 2015;6:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hernandez CC, Falkenburger B, Shapiro MS. Affinity for phosphatidylinositol 4,5-bisphosphate determines muscarinic agonist sensitivity of Kv7 K+ channels. J Gen Physiol. 2009. November;134(5):437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li Y, Gamper N, Hilgemann DW, et al. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2005. October 26;25(43):9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998. March 12;8(6):343–346. [DOI] [PubMed] [Google Scholar]
- [31].Hirose K, Kadowaki S, Tanabe M, et al. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 1999. May 28;284(5419):1527–1530. [DOI] [PubMed] [Google Scholar]
- [32].Varnai P, Balla T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta. 2006. August;1761(8):957–967. [DOI] [PubMed] [Google Scholar]
- [33].Murata Y, Iwasaki H, Sasaki M, et al. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005. June 30;435(7046):1239–1243. [DOI] [PubMed] [Google Scholar]
- [34].Murata Y, Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J Physiol. 2007. September 15;583(Pt 3):875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Iwasaki H, Murata Y, Kim Y, et al. A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A. 2008. June 10;105(23):7970–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Falkenburger BH, Jensen JB, Hille B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol. 2010. February;135(2):99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Halaszovich CR, Schreiber DN, Oliver D. Ci-VSP is a depolarization-activated phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate 5ʹ-phosphatase. J Biol Chem. 2009. January 23;284(4):2106–2113. [DOI] [PubMed] [Google Scholar]
- [38].Wilson DB, Bross TE, Hofmann SL, et al. Hydrolysis of polyphosphoinositides by purified sheep seminal vesicle phospholipase C enzymes. J Biol Chem. 1984. October 10;259(19):11718–11724. [PubMed] [Google Scholar]
- [39].Ryu SH, Cho KS, Lee KY, et al. Purification and characterization of two immunologically distinct phosphoinositide-specific phospholipases C from bovine brain. J Biol Chem. 1987. September 15;262(26):12511–12518. [PubMed] [Google Scholar]
- [40].Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980. February 14;283(5748):673–676. [DOI] [PubMed] [Google Scholar]
- [41].Kruse M, Hammond GR, Hille B. Regulation of voltage-gated potassium channels by PI(4,5)P2. J Gen Physiol. 2012. August;140(2):189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zaydman MA, Silva JR, Delaloye K, et al. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci U S A. 2013. August 6;110(32):13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li Y, Gamper N, Shapiro MS. Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J Neurosci. 2004. June 2;24(22):5079–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kruse M, Hille B. The phosphoinositide sensitivity of the K(v) channel family. Channels (Austin). 2013. Nov-Dec;7(6):530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Falkenburger BH, Dickson EJ, Hille B. Quantitative properties and receptor reserve of the DAG and PKC branch of G(q)-coupled receptor signaling. J Gen Physiol. 2013. May;141(5):537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dickson EJ, Falkenburger BH, Hille B. Quantitative properties and receptor reserve of the IP(3) and calcium branch of G(q)-coupled receptor signaling. J Gen Physiol. 2013. May;141(5):521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lindner M, Leitner MG, Halaszovich CR, et al. Probing the regulation of TASK potassium channels by PI(4,5)P-2 with switchable phosphoinositide phosphatases. J Physiol London. 2011. July 1 2011;589(13):3149–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kosenko A, Kang S, Smith IM, et al. Coordinated signal integration at the M-type potassium channel upon muscarinic stimulation. EMBO J. 2012. May 29;31(14):3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wilke BU, Lindner M, Greifenberg L, et al. Diacylglycerol mediates regulation of TASK potassium channels by Gq-coupled receptors. Nat Commun. 2014;5:5540. [DOI] [PubMed] [Google Scholar]
- [50].Gamper N, Li Y, Shapiro MS. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell. 2005. August;16(8):3538–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Storch U, Forst AL, Pardatscher F, et al. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc Natl Acad Sci U S A. 2017. January 3;114(1):E37–E46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hofmann T, Obukhov AG, Schaefer M, et al. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999. January 21;397(6716):259–263. [DOI] [PubMed] [Google Scholar]
- [53].Chang A, Abderemane-Ali F, Hura GL, et al. A Calmodulin C-Lobe Ca(2+)-dependent switch governs Kv7 channel function. Neuron. 2018. February 21;97(4):836–852 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Decher N, Gonzalez T, Streit AK, et al. Structural determinants of Kvbeta1.3-induced channel inactivation: a hairpin modulated by PIP2. EMBO J. 2008. December 3;27(23):3164–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Oliver D, Lien CC, Soom M, et al. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004. April 9;304(5668):265–270. [DOI] [PubMed] [Google Scholar]
- [56].Leitner MG, Michel N, Behrendt M, et al. Direct modulation of TRPM4 and TRPM3 channels by the phospholipase C inhibitor U73122. Br J Pharmacol. 2016. August;173(16):2555–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Leitner MG, Halaszovich CR, Oliver D. Aminoglycosides inhibit KCNQ4 channels in cochlear outer hair cells via depletion of phosphatidylinositol(4,5)bisphosphate. Mol Pharmacol. 2011. January;79(1)51–60. [DOI] [PubMed] [Google Scholar]
- [58].Halaszovich CR, Leitner MG, Mavrantoni A, et al. A human phospholipid phosphatase activated by a transmembrane control module. J Lipid Res. 2012. November;53(11):2266–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Leitner MG, Feuer A, Ebers O, et al. Restoration of ion channel function in deafness-causing KCNQ4 mutants by synthetic channel openers. Br J Pharmacol. 2012. April;165(7):2244–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]