

ABSTRACT
Little is known about the reproductive effects of paternal cannabis exposure. We evaluated associations between cannabis or tetrahydrocannabinol (THC) exposure and altered DNA methylation in sperm from humans and rats, respectively. DNA methylation, measured by reduced representation bisulfite sequencing, differed in the sperm of human users from non-users by at least 10% at 3,979 CpG sites. Pathway analyses indicated Hippo Signaling and Pathways in Cancer as enriched with altered genes (Bonferroni p < 0.02). These same two pathways were also enriched with genes having altered methylation in sperm from THC-exposed versus vehicle-exposed rats (p < 0.01). Data validity is supported by significant correlations between THC exposure levels in humans and methylation for 177 genes, and substantial overlap in THC target genes in rat sperm (this study) and genes previously reported as having altered methylation in the brain of rat offspring born to parents both exposed to THC during adolescence. In humans, cannabis use was also associated with significantly lower sperm concentration. Findings point to possible pre-conception paternal reproductive risks associated with cannabis use.
KEYWORDS: Cannabis, tetrahydrocannabinol, sperm, DNA methylation, epigenetic reprogramming, rat, human
Introduction
Cannabis is one of the most widely used psychoactive drugs, with more than 180 million users globally [1]. In the United States, the landscape of cannabis use and availability has changed dramatically in the past 20 years. More than half of US states have legalized cannabis use in some form, ten of which have legalized recreational use along with the District of Columbia [2]. The National Survey on Drug Use and Health (NSDUH) reported that in 2015 among 18–25 year olds, 32.2% and 19.8% report past year and past month use, respectively. Among males, these rates are higher, with 36.0% and 23.4% reporting past year and past month use, respectively. Although rates decline across the lifespan, cannabis use rates were still 20.6% and 12.9%, respectively, among 26–34 year old men [3]. Since mean paternal age for first child in the United States is 27.4 years, a substantial number of males of child-bearing age may have recent exposure to cannabis at or around the time they conceive.
A large number of studies have assessed the deleterious effects of cannabis use on a range of outcomes [4], including adverse effects of prenatal exposure during pregnancy via maternal use [1]. A growing body of literature has begun to focus on the potential heritability of effects resulting from pre-conception cannabis exposure [5]. At least two studies in rodents have reported that adolescent exposure to cannabinoid agonists (prior to conception) results in different drug seeking behavioral phenotypes in adult offspring [6,7]. There is a currently a gap in knowledge about cannabis use effects on paternal reproductive factors. Identifying the sources (pre- versus post-conception) and their differential impacts on offspring is necessary for setting prevention effort priorities.
Epigenetic modifications to the genome, including DNA methylation, play an essential role in regulating gene activity over the life course and have been implicated as a potential mechanism underlying the heritable effects of pre-conception cannabis exposure [5]. DNA methylation is erased and established anew with each generation, first just after fertilization, then again in the developing embryo wherein the methylation present in the primordial germ cells is erased, and after sex specification, is re-established in a sex-specific manner. The physical act of reprogramming of the methylome may render the fidelity of this process vulnerable to endogenous or exogenous influences, including for example obesity [8]. In post-pubertal males, the DNA methylation patterns in maturing sperm are finalized and maintained during the post-spermatogonial stages of spermatogenesis [9]. Supporting this, Donkin et al. concluded that DNA methylation changes do indeed occur during the final stages of sperm maturation in humans from their comparison of sperm DNA methylation from men one week before and one week after they underwent bariatric surgery [10]. Because sperm maturation is a continual process throughout the adult male’s life, exposures like cannabis could have an impact on the integrity of the sperm methylome, with implications for heritability of such alterations by subsequent generations.
It is currently unknown whether cannabis use alters the epigenetic profile of sperm in male human users. The objective of the present study was to explore differences in sperm methylation profiles in well characterized and biologically verified recreational cannabis users compared to non-users using reduced representation bisulfite sequencing, a quantitative, genome-scale approach [11]. We compared these results to those obtained using mature sperm from rats exposed to tetrahydrocannabinol (THC), the major psychoactive component of cannabis.
Results
Figure 1 provides an overview of recruitment and enrollment for the study. A total of 37 men were evaluated to yield the 24 who enrolled and completed the study. Participants in the cannabis user and non-user groups were similar in terms of age, ethnicity, and other baseline characteristics (see Table 1). On a screening test for alcohol use disorders, the cannabis users reported somewhat higher weekly alcohol intake, but neither group reported alcohol use that was clinically significant. Among cannabis users, levels of THC ranged from 50 ng/mL to 739 ng/mL (mean = 260.8 ng/mL, SD = 228.9 ng/mL). Creatinine adjusted THC levels ranged from 38 ng/mL to 1,628 ng/mL (mean = 329.8 ng/mL, SD = 460.9 ng/mL).
Figure 1.
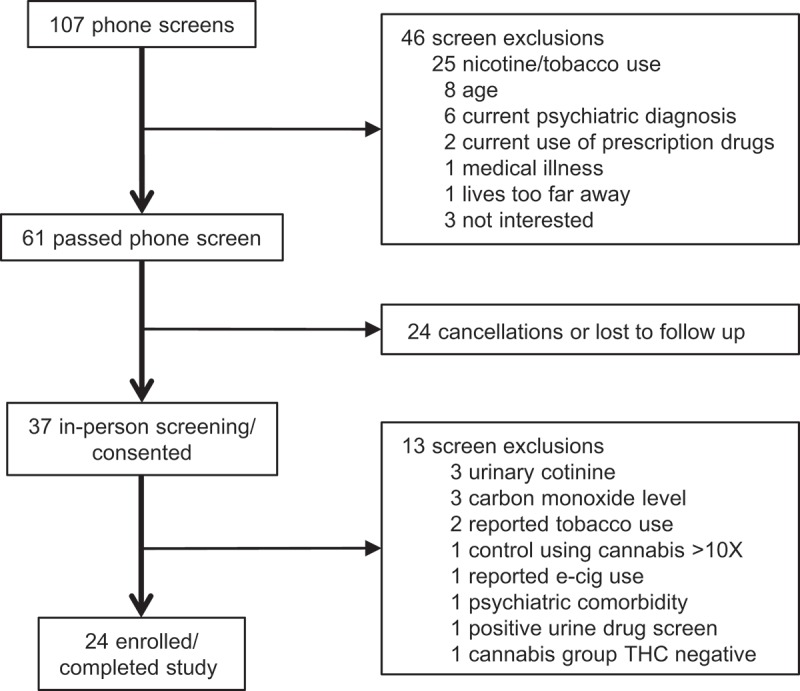
Study procedures for screening and consent. Twenty-four men, including 12 cannabis users and 12 non-users, were enrolled and participated in the study.
Table 1.
Participant characteristics.
| Cannabis Users | Non-Users | p | |
|---|---|---|---|
| Demographics/Physical/Cognitive | |||
| Age in years (SD) | 21.8 (3.8) | 25.8 (6.7) | NS |
| Race (% Caucasian) | 58.3 | 75 | NS |
| Height in inches (SD) | 69.1 (2.4) | 69.7 (2.5) | NS |
| Weight in pounds (SD) | 164.7 (30.2) | 176.1 (39.7) | NS |
| IQ Est (SD) | 116.3 (14.6) | 115.5 (11.5) | NS |
| THC concentration – unadjusted ng/mL (SD) | 260.8 (228.9) | 0.0 (0.0) | p < 0.01 |
| THC concentration – Creatinine adjusted ng/mL (SD) | 329.8 (460.9) | 0.0 (0.0) | p < 0.05 |
| Semen Analysis | |||
| Duration of abstinence before sample in days (SD) | 3.8 (1.2) | 4.3 (1.3) | NS |
| Interval between ejaculation and analysis in minutes (SD) | 19.6 (9.9) | 25.0 (10.0) | NS |
| Volume in mL (SD) | 3.2 (1.4) | 3.4 (1.2) | NS |
| Semen pH (SD) | 8.4 (0.2) | 8.4 (0.1) | NS |
| Sperm Concentration (SD) | 58.1 (26.5) | 96.3 (49.7) | p < 0.05 |
| % Motile (SD) | 63.3 (8.5) | 63.8 (16.0) | NS |
| % Normal Morphology (SD) | 3.3 (3.3) | 3.5 (2.0) | NS |
There were no differences between the cannabis users and non-users with respect to time since last ejaculation, interval between ejaculation and analysis, semen volume, semen pH, percent motility, or percent normal morphology (Table 1). Cannabis users had significantly lower sperm concentrations compared to non-users (t = −2.4; p < 0.05, Figure. S1).
Reduced Representation Bisulfite Sequencing (RRBS) is a quantitative method commonly used to define and compare DNA methylation profiles on a genomic scale [11] and was used to generate quantitative sperm DNA methylation data for the user and non-user groups. Data for more than five million CpG sites were generated using this method for each individual. Likely due to the relatively small sample size, none of the CpG site comparisons resulted in a p value that was less than the Bonferroni corrected p value of <10−8 for significance, as has been observed in other studies [12–14]. We therefore increased the stringency for inclusion of CpGs in our final set of differentially methylated sites using additional criteria. There were 6,640 CpG sites that differed (p < 0.05) between the cannabis users and non-users after data preprocessing and filtering. This included retention of sites with measured values for all samples, ≥5X coverage, a > 10% difference in means between groups, and removal of CpG-altering SNPs with >5% heterozygosity frequency as reported in dbSNP (see Supplementary Information). The majority of the CpGs (78.3%) had lower levels of methylation in the user group, indicating a non-random distribution of methylation differences.
We further limited CpG sites to those occurring within a RefSeq gene, inclusive of the 5,000 bp region upstream, and for which there were ten or more CpG sites with a significant difference between the user and non-user groups. While this approach might overlook intergenic sites for which the methylation status of a single CpG could regulate transcriptional activity, it improves the probability that the identified regions are bona fide results as opposed to chance findings given the overall large number of individual comparisons made. There were 46 genes (708 CpG sites) meeting these criteria, the vast majority of which were less methylated in the user group (Figure 2(a); also see Table S1). The maximum number of CpG sites associated with any given gene by RRBS was for the Aryl Hydrocarbon Receptor Repressor (AHRR), with 94 altered CpGs, all hypomethylated by 10% or greater in the users.
Figure 2.
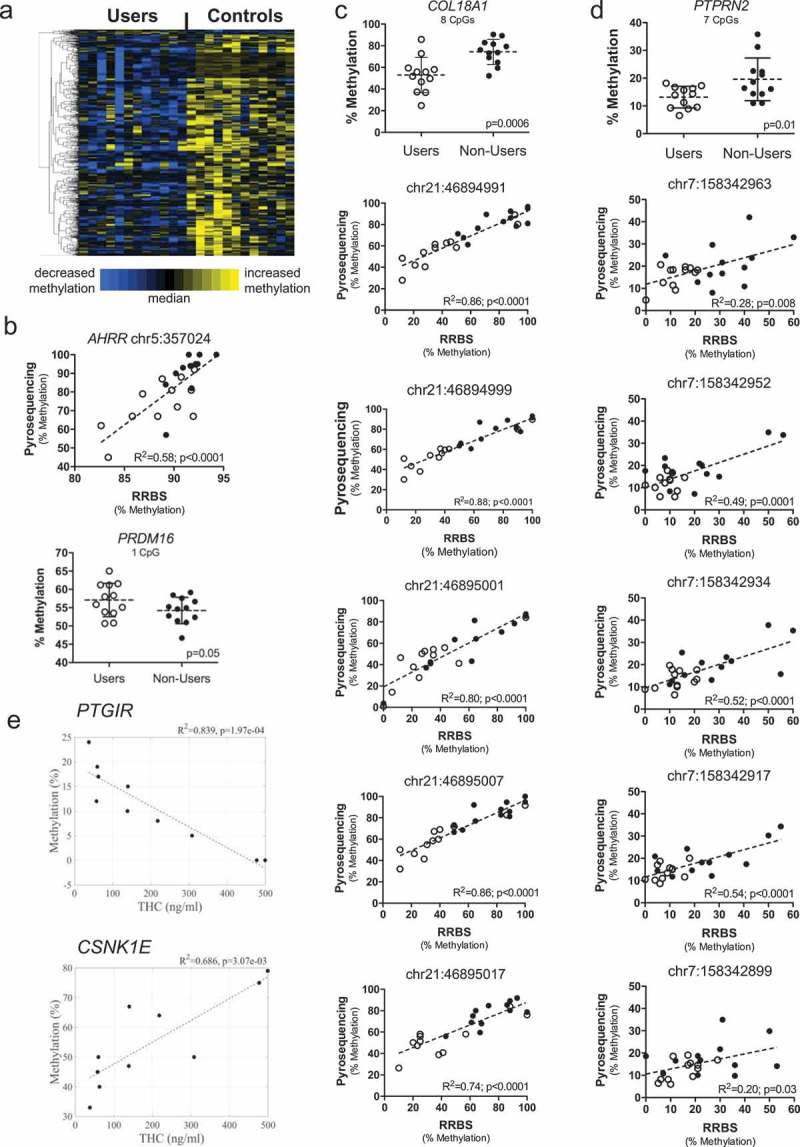
Cannabis use is associated with DNA methylation in sperm. (a) Heat map showing DNA methylation relative to the median for 708 CpG sites (rows) associated with 46 genes with differential methylation between cannabis users (left columns) and non-user controls (right columns). Each column represents one participant, while each row represents one CpG site. Row clustering was unsupervised. Methylation levels are median-centered. (b) Top, bisulfite pyrosequencing data compared to RRBS data from users to non-users for a CpG within the intragenic repeated sequence of AHRR by linear regression. Bottom, bisulfite pyrosequencing data for one of the few genes, PRDM16, showing increased methylation in the user group. Each data point is the average of replicate measures. The mean is shown by the center dashed bar with error bars representing standard deviation. Unpaired t test, one-tailed, F test to compare variances (p = 0.44). D’Agostino & Pearson normality test showed data was normally distributed in each group. (c) Top graph: bisulfite pyrosequencing data for COL18A1 showing all eight CpG sites analyzed together discriminate users from non-users. Each data point is the average of replicate measures. The mean is shown by the center dashed bar with error bars representing standard deviation. Unpaired t test, one-tailed, F test to compare variances (p = 0.27). D’Agostino & Pearson normality test showed data was normally distributed in each group. Bottom five graphs: Comparison of methylation values from RRBS versus bisulfite pyrosequencing for the five CpG sites identified as differentially methylated by RRBS, analyzed using linear regression. (d) Top graph: bisulfite pyrosequencing data for PTPRN2 showing all seven CpG sites analyzed together discriminate users from non-users. Each data point is the average of replicate measures. The mean is shown by the center bar with error bars representing standard deviation. Unpaired t test with Welch’s correction, one-tailed, F test to compare variances (p = 0.03). D’Agostino & Pearson normality test showed data was normally distributed in each group. Bottom five graphs: Comparison of methylation values from RRBS versus bisulfite pyrosequencing for the five CpG sites identified as differentially methylated by RRBS, analyzed by linear regression. (e) Correlations between urinary THC concentration in the human cannabis user group and sperm DNA methylation levels for CpGs identified as differentially methylated in PTGIR (top) and CSNK1E (bottom), analyzed by linear regression. Panels B-D: Users, open circles; non-users, closed circles.
Two of the 94 CpGs are located closer to the 3ʹ end of AHRR, very near to but not identical with the CpGs that have been identified as hypomethylated in prior studies of infants born to smoking versus non-smoking mothers [15]. The remaining 92 are located within an intronic CpG island that is comprised of a 62-nucleotide sequence repeated 48 times that is unique to AHRR. With one PCR primer anchored upstream, outside of the repetitive region, and the other within, we used bisulfite pyrosequencing to quantify three CpG sites in this sequence and compared this to the results from RRBS. We found good correlation between these two methods for one of three CpGs analyzed (Figure 2(b), top). In spite of the weak correlation between methods for the other two CpGs (perhaps due to the repetitive nature of the sequence), the average methylation of all three AHRR sites distinguished users from non-users (p = 0.03; not shown). We also validated increased methylation by pyrosequencing for one of the few hypermethylated CpGs in the user group associated with the gene, PR/SET Domain 16 (PRDM16) (Figure 2(b), bottom). Pyrosequencing was also performed for genes showing higher magnitude (27%–41%) and lower magnitude (6%–8%) methylation differences. Figure 2(c) shows very good agreement between methods for five significant sites detected for Collagen Type XVIII Alpha 1 chain (COL18A1) (R2 > 0.74; p < 0.0001) and that pyrosequencing results for all eight CpGs analyzed (five of which were significant by RRBS) distinguish users from non-users (p = 0.0012). For Protein Tyrosine Phosphatase, Receptor Type 2 (PTPRN2) with lower magnitude differences in methylation, there was also significant agreement between methods for the five CpG sites showing significance by RRBS (R2 > 0.20; p ≤ 0.03; Figure 2(d)). Pyrosequencing results for all seven PTPRN2 CpGs also distinguished users from non-users (p = 0.02). Together these data support the validity of the results obtained using RRBS.
Linear regression of data from the cannabis using men (n = 10; one statistical outlier, one without creatinine data) showed there were 183 individual CpG sites representing 177 named genes for which the level of methylation was significantly correlated with their measured THC levels. For example, for PTG1R, which encodes the Prostaglandin I2 Receptor (a powerful vasodilator), there was an inverse correlation between THC levels and DNA methylation with R2 = 0.839 (p = 1.97e-4; Figure 2(e)). PTGIR was hypomethylated in the sperm from the cannabis users relative to the non-users, and prior studies have shown it is also hypomethylated in human sperm with reduced fecundity [16]. In contrast, there was a positive correlation between THC levels and methylation at the gene encoding Casein Kinase 1 Epsilon (CSNK1E) (R2 = 0.686; p = 0.003; Figure 2(e); also see Figure. S2). CSNK1E phosphorylates circadian clock protein PER2 [17] and is implicated in sensitivity to opioids [18]. In addition, of the 3,979 differentially methylated CpG sites, linear regression analyses showed that methylation levels of 409 (10.3%) CpGs were significantly correlated with sperm count (representative data in Figure. S3).
To identify any functionally related groups of genes coordinately affected by methylation changes in sperm associated with cannabis use, we entered the 2,077 unique gene names for the 3,979 identified differentially methylated CpG sites into the DAVID Bioinformatics Database (29). The top three pathways significantly enriched in genes whose methylation was altered in cannabis users included hsa00053: ‘Ascorbate and aldarate metabolism’ (4.8-fold enrichment; 11 genes, Bonferroni p = 0.010), hsa04390: ‘Hippo signaling pathway’ (2.3-fold enrichment; 29 genes, Bonferroni p = 0.013) and hsa:05200: ‘Pathways in cancer’ (1.7-fold enrichment; 55 genes, Bonferroni p = 0.04) (Table 2). Sensitivity analyses showed that removing CpGs in upstream regions did not alter the findings (not shown).
Table 2.
KEGG pathways enriched with differentially methylated genes in humans and rats*.
| Term | Genes | Fold-enrichment | FDR |
|---|---|---|---|
| Human | |||
| hsa00053:Ascorbate and aldarate metabolism | UGT1A7, UGT1A10, UGT1A6, UGT1A9, UGT1A8, UGT1A3, UGT1A5, UGT1A4, UGT2A2, UGT2A1, UGT1A1 | 4.7235 | 0.0006 |
| hsa04390:Hippo signaling pathway | WNT5A, PRKCZ, MOB1A, APC2, GDF6, WNT3A, GDF5, GLI2, LLGL1, TCF7L1, LOC400927-CSNK1E, FRMD6, PPP2R2B, PPP2R2C, DLG2, AXIN1, WNT10A, DVL3, TEAD3, PATJ, WWTR1, YWHAE, CTNNA2, DVL1, WNT9A, PARD6G, BMP7, PPP2R2A, BMP6 | 2.2267 | 0.0010 |
| hsa05200:Pathways in cancer | FGF6, FGF18, GNA11, WNT3A, ADCY6, LPAR3, LPAR2, FGF12, GLI2, SHH, FLT3LG, AKT1, CUL2, RARA, PRKACA, GNG7, WNT10A, CTBP1, RET, CTBP2, BCR, BRAF, FGF22, FGF21, DAPK1, CTNNA2, CCDC6, GNB2, JUN, PDGFRA, PIAS2, GNAS, WNT9A, ITGA2B, DCC, WNT5A, GNAI2, APC2, GNAI1, BCL2L1, TCF7L1, RAC3, PLEKHG5, LAMB1, TRAF4, AXIN1, FH, PIK3R2, DVL3, EPAS1, VHL, DVL1, LAMA1, ADCY9, NTRK1, ABL1, CRK | 1.6816 | 0.0012 |
| hsa00040:Pentose and glucuronate interconversions | UGT1A7, UGT1A10, UGT1A6, UGT1A9, CRYL1, UGT1A8, UGT1A3, UGT1A5, UGT1A4, UGT2A2, UGT2A1, UGT1A1 | 3.8647 | 0.0019 |
| hsa04611:Platelet activation | PRKCZ, TBXAS1, GNAI2, GNAI1, ADCY6, ARHGAP35, PRKG1, ITPR2, AKT1, VWF, PTGIR, GP6, ADCY9, P2RX1, MAPK12, COL27A1, TBXA2R, PRKACA, GNAS, FCGR2A, MYLK, PIK3R2, ITGA2B | 2.0512 | 0.0198 |
| hsa04010:MAPK signaling pathway | FGF6, FGF18, IL1R1, MKNK2, FGF12, AKT1, MAP3K6, MAP3K5, HSPA2, RAC3, PRKACA, RAPGEF2, CACNA2D1, BRAF, TAOK1, CACNG8, CACNG6, TAOK3, CACNA1I, FGF22, CACNG3, FGF21, CDC25B, RPS6KA5, RASGRF2, MAPK12, RPS6KA2, JUN, NTRK1, PDGFRA, CACNA1H, MAPK8IP3, CACNA1C, MAP3K13, CRK, CACNA1A, CACNA1B | 1.6823 | 0.0254 |
| hsa00860:Porphyrin and chlorophyll metabolism | UGT1A7, UGT1A10, UGT1A6, UGT1A9, UGT1A8, UGT1A3, UGT1A5, UGT1A4, UGT2A2, UGT2A1, UGT1A1 | 3.0365 | 0.0315 |
| hsa04713:Circadian entrainment | GNAI2, GNAI1, ADCY6, GRIN1, CACNA1I, GRIN2A, GRIA3, PRKG1, RPS6KA5, ADCY9, GNB2, RYR1, CACNA1H, PER1, GNAS, PRKACA, CACNA1C, GNG7 | 2.1968 | 0.0358 |
| hsa04530:Tight junction | SHROOM1, PRKCZ, OCLN, EPB41, GNAI2, GNAI1, PATJ, AMOTL1, LLGL1, CLDN14, CTNNA2, AKT1, PRKCQ, EXOC3, MYH14, PARD6G, TJP3, PPP2R2B, YES1, PPP2R2C, JAM3, MYH10, PPP2R2A | 1.9464 | 0.0389 |
| hsa00830:Retinol metabolism | GNAI2, GNAI1, ADCY6, GRIK4, GRIN1, GRIN2A, GRIA3, GRIN3A, SHANK2, ITPR2, GRM4, GLUL, ADCY9, GNB2, SLC1A7, GNAS, PRKACA, CACNA1C, CACNA1A, GNG7 | 2.7725 | 0.0404 |
| hsa04724:Glutamatergic synapse | UGT1A7, UGT1A10, UGT1A6, UGT1A9, UGT1A8, UGT1A3, UGT1A5, UGT1A4, UGT2A2, UGT2A1, UGT1A1 | 4.0579 | 0.0476 |
| Rat | |||
| rno04390:Hippo signaling pathway | Fzd9, Apc2, Gdf6, Tgfbr2, Tgfb3, Ppp1cc, Tcf7l1, Llgl1, Wnt7b, Crb1, Csnk1e, Dlg4, Wnt11, Bmp7, Axin2, Myc, Bmp6 | 3.5721 | 0.0002 |
| rno05200:Pathways in cancer | Fgfr2, Ppard, Apc2, Fgf14, Tgfb3, Fgf12, Tcf7l1, Tpm3, Gli1, Rasgrp1, Pax8, Prkaca, Axin2, Myc, Csf2ra, Traf3, Gng7, Fzd9, Map2k1, Tgfbr2, Hgf, Mecom, Dapk3, Vegfb, Wnt7b, Gnb2, Wnt11, Ptch1, Lamc1 | 2.3425 | 0.0005 |
| rno04010:MAPK signaling pathway | Fgfr2, Fgfr4, Cacna2d1, Map2k1, Fgf14, Cacna1i, Tgfbr2, Tgfb3, Fgf12, Mecom, Cacna2d2, Arrb2, Rasgrp1, Mapt, Ntrk2, Cacna1g, Prkaca, Myc, Cacna1a, Map2k6 | 2.4825 | 0.0053 |
| rno04810:Regulation of actin cytoskeleton | Fgfr2, Fgfr4, Apc2, Limk2, Map2k1, Fgf14, Wasf2, Nckap1l, Actn1, Pip5k1c, Fgf12, Itgb3, Ppp1cc, Src, Arpc1a, Ezr, Scin | 2.5070 | 0.0149 |
| rno04550:Signaling pathways regulating pluripotency of stem cells | Fgfr2, Fzd9, Fgfr4, Hnf1a, Tbx3, Apc2, Map2k1, Wnt7b, Pou5f1, Wnt11, Jak3, Axin2, Myc | 2.9852 | 0.0165 |
| rno05217:Basal cell carcinoma | Fzd9, Wnt7b, Apc2, Ptch1, Wnt11, Axin2, Tcf7l1, Gli1 | 4.6762 | 0.0183 |
*Bold text indicates differentially methylated genes in common between humans and rats in the same pathways
To determine if THC exposure induced similar effects in rodents, we dosed sexually mature male rats via oral gavage with 2 mg/kg THC or vehicle control daily for 12 days. Sperm DNA methylation was measured by RRBS and underwent the same analytical steps, resulting in identification of 627 genes whose methylation status was altered in association with THC exposure. The topmost pathways enriched with these altered genes were ‘rno:04390: Hippo signaling pathway’ (3.6-fold enrichment; 17 genes, Bonferroni p = 0.004) and ‘rno:05200: Pathways in cancer’ (2.3-fold enrichment; 29 genes, Bonferroni p = 0.009) (Table 2). There were six overlapping genes among those altered by cannabis/THC in the ‘Hippo signaling’ pathway between humans and rats (indicated in bold text, Table 2). For ‘Pathways in cancer’, there were also six overlapping genes.
We examined each differentially methylated CpG site for all 99 genes in common between human and rat by taking the human CpGs identified for those genes and determining whether they were conserved in rat, and vice-versa, using the UCSC Genome Browser (http://genome.ucsc.edu/) [19], human assembly GRCh37/hg19 and rat assembly RGSC 6.0/rn6. There was conservation of 39/123 (31.7%) CpG sites when comparing rat gene CpG sites to human genomic sequence and 51/276 (18.5%) when comparing human gene CpG sites to rat genomic sequence (Table S2). Conservation of CpG sites could indicate the presence of a functionally relevant transcription factor binding site or enhancer element. We queried the UCSC Genome Browser at the position of each differentially methylated human CpG site in the human-rat overlapping gene set and found there were 23 genes with differentially methylated CpGs contained within the recognition sequence of one or more transcription factor binding sites. Between human and rat, CpG conservation at these regions was present for GATA binding protein 2 (GATA2), plectin (PLEC) and outer dense fiber of sperm tails 2 (ODF2) (Table S2). There were no enhancer elements detected at these sequences.
A prior study by Watson and colleagues [20] reported that rat offspring born to parents who were both exposed for 21 days to THC during their adolescence had an increased propensity to self-administer heroin as adults. Analysis of DNA methylation in the offspring’s brains showed a large number of changes in DNA methylation relative to controls. These results strongly suggested that the effects of parental THC exposures were transmitted to the offspring, likely via DNA methylation changes in the gametes that were able to resist post-fertilization reprogramming, as has been demonstrated for methylation at thousands of non-imprinted genes [21]. These reprogramming-resistant, THC-induced methylation changes could then theoretically be carried forward and maintained in all of the somatic cells of the offspring, including the brain. Watson et al. showed that the altered methylation in the brains of the offspring was associated with increased drug-seeking behavior. We therefore compared our list of 627 genes exhibiting differential methylation between THC-exposed males versus controls to the list of 473 differentially methylated genes identified from the brains of the rats in the aforementioned study [20]. There were 55 overlapping genes between these two datasets, a markedly significant enrichment (p < 2e-7).
Discussion
This study is the first to report that cannabis use among males of child-bearing age compared to non-using males results in substantial disruption in the DNA methylome of their sperm. The results indicate that there are at least 6,640 CpGs whose methylation status is altered in conjunction with cannabis use. We found enrichment of altered CpG sites associated with genes involved in the Hippo signaling pathway and in Pathways in Cancer, findings that were replicated in THC-exposed rodents. These results indicate that the major epigenetic effect of cannabis exposure may be due to the THC component. Furthermore, only six gene members each in the Hippo signaling and Pathways in cancer groups were in common between the human and rodent results in spite of a larger number of affected genes (29 and 55 for humans and 17 and 29 for rats, respectively) amid the total gene membership in each defined pathway (151 and 393, and 153 and 398, respectively). This suggests that these two pathways, rather than the genes per se, are specific targets of exposure, although the reasons for this and mechanisms are presently unknown.
Consistent with previous epidemiological studies [22–24], we replicated the finding that cannabis users have lower sperm concentrations than non-users and, for the first time, demonstrated this in a sample with biologically verified use. These findings are significant, especially in light of the changing landscape of cannabis use in the US and other parts of the world. With the legal status of cannabis use shifting towards a more permissive stance, this may increase the prevalence of use among males of child-bearing potential. Risks of exposure may be exacerbated by two other recent trends in cannabis use. First, perceptions of risk associated with cannabis use are decreasing. In 2013 among US 12th graders, only 39.5% reported that regular cannabis use was potentially harmful, a significant decrease of 4.6% from 2012 and the lowest reported rate since the late 1970s [25]. Second, the potency of cannabis has been increasing substantially over the past 20 years with mean THC levels increasing from ~4% to ~12% between 1995 and 2014. The ratio of THC to cannabidiol has increased from ~14:1 to ~80:1 over the same time frame [26].
It is presently unclear whether DNA methylation changes identified in sperm as a result of environmental exposures are capable of being passed on to the next generation. Recent reports indicate that a great deal more sperm DNA methylation is retained and not erased, as previously thought, during post-fertilization epigenetic reprogramming [21,27]. As such, there is growing interest in the effects of preconceptional paternal exposures via epigenetic alterations [28,29], though there are only a few empirical studies in humans. For example, epigenetic changes in sperm of obese men have been reported that are similar to DNA alterations in cord blood from children born to obese men, suggestive of potential intergenerational heritability [8,30]. Epigenetic alterations were also reported in sperm from adult men who had undergone cancer-related chemotherapy during adolescence [31]. While no other studies have been published showing the influence of cannabis use on sperm DNA methylation, several studies have examined the impact of cigarette smoking on DNA methylation in sperm. Some of the identified CpG sites and/or affected genes for cigarette smokers are in common with those identified here for cannabis use. Laggan et al [32]. used the Illumina HumanMethylation450 beadchip and identified differentially methylated sites in the MAPK8IP3 and TKR genes. We also identified one differentially methylated CpG site with cannabis use in MAPK8IP3 (11% higher in user group) but none in TKR. The methylation difference in MAPK8IP3 for cannabis users was in the opposite direction of that reported for men who smoke cigarettes. It was also in a different region of the gene (chr16:1813414 versus chr16:1797050). Alkhaled et al [33]. identified differential methylation of PGAM5, PTPRN2 and TYRO3 comparing sperm DNA methylation of smokers to non-smokers. The cannabis users in our study showed both increased and decreased methylation for 12 CpG sites, depending on location, within PTPRN2. A number of these sites were in the same region as at least one differentially methylated CpG (cg23841288) identified for the cigarette smokers. Another study [34] examined DNA methylation in sperm from 78 smokers and 78 never-smokers on the HumanMethylation450 beadchip. They identified 141 CpGs with tobacco smoke-associated differential methylation. Comparing their associated gene list with our list of genes for 3,979 differentially methylated CpG sites showed 19 genes present in both datasets, including ABR, APC2, BCR, BEST2, BPNT1, DIPC2, FAM19A5, FCER2, GUCY2D, HGS, MMP16, PDCD1, PLEKHG5, PRIM2, PRSS16, PTGIR, RFPL2. RHOV and TBXA2R. Together, these results are suggestive of certain areas of the genome that may be more generally vulnerable to the effects of exposure to cannabis and tobacco smoke, but also support that the majority of methylation changes in sperm are uniquely responsive to the different types of compounds in the combusted smoke or to the compound mixtures.
If the methylation alterations detected here by RRBS in humans and rats at the Hippo signaling and Pathways in cancer pathway genes are retained in the zygote, the changes in methylation could lead to disrupted expression of important growth regulatory genes, resulting in nonviability, or if viable embryos are formed, shifted growth trajectories during development. If sustained, the changes in DNA methylation could increase risk of later cancer development, which is often characterized by these types of methylation alterations in one or both pathways identified. However, these possibilities remain speculative. Further research is essential to determine the potential for the methylation changes to be transmitted inter- and trans-generationally.
Results of the present study should be considered alongside several important limitations. First, our sample size was small, limiting generalizability. The RRBS method generated data for approximately one million CpG sites, and with the small sample size, none of the comparisons for single CpG sites between groups were significant after accounting for multiple comparisons (p < 10−8). Despite this, we found changes at multiple CpG sites associated with the same gene, agreement regarding the affected pathways between humans and rats, even with differences in the exposures (THC versus combusted cannabis) and routes of exposures (oral versus inhalation), a correlative relationship between methylation and THC levels, and significant overlap in targeted genes in an independent dataset from rat offspring of parents exposed to THC during adolescence. Nevertheless, it will be important to replicate these findings in larger samples. Sample size also limited the ability to definitively assess a dose-effect relationship between exposure and changes in methylation, although we found evidence supporting dose-response relationships for 177 genes. Given trends in cannabis potency noted above, analysis with a wider range of exposure levels will be important to determine the extent of dosage effects. We also note that the THC concentration used for the rat studies was higher than that typically consumed by humans. Comparatively effective THC doses in rats are higher than in humans as they are for many drugs because of higher rates of metabolism [35,36] with chronic doses up to 50 mg/kg/day causing moderate neurotoxic effects[37].
Another limitation of our study is that there are likely to be a wide range of potential confounders (e.g., life style habits, physical condition, diet/nutrition, sleep, alcohol use, etc.) that might alter sperm DNA methylation but that were not accounted for in the present study. The influence of such factors on sperm DNA methylation is for the most part unknown, but more comprehensive characterization of a larger number of samples will help elucidate the possible role of these variables in sperm epialterations.
The route and ingredient composition of the exposure differed between the human and rat studies. However, the marked increase in THC concentration of current cannabis strains may have helped minimize some of this difference. The duration of THC exposure in the rats was 12 days, limiting the detectable effects of the exposure to the latter stages of spermatogenesis. Our results suggest that there are substantial changes in DNA methylation even with this relatively brief exposure window. Supporting this, DNA methyltransferase enzymes are expressed in maturing sperm cells at every stage of spermatogenesis, including during final maturation in the epididymis and in ejaculated sperm [38]. Furthermore, a recent study in mice showed that sperm DNA methylation modifications continue to occur in the epididymis [9].
Since the present study was cross-sectional in design, we do not know how exposure over time might influence epigenetic processes – or if the observed changes are reversible. Though not explicitly a limitation, the design does not allow for inferences to be made regarding the heritability of epigenetic changes and potential implications for offspring of male cannabis users since we did not assess functional consequences of the observed DNA methylation changes. Future studies should evaluate the functional significance of the present findings, especially in light of evidence supporting intergenerational effects. If there is evidence that the epigenetic changes observed in this study are maintained post-fertilization, such findings should be considered with regard to cannabis use policy decisions in the US and worldwide.
Materials and methods
Participants
Twenty-four males participated based on inclusion criteria of age 18–40 years, no significant medical/psychiatric conditions, 0.0 breath alcohol level at Screening, and willingness to comply with all study requirements. Sample size was chosen based on power calculations for targeted analysis that showed we would have 90% power to detect a 5% methylation difference between groups at an alpha of 0.05. Exclusion criteria were predefined and included a positive result for drugs of abuse on a rapid urine test (except for cannabis in the User group, described below), currently prescribed any psychoactive medication, self-reported nicotine/tobacco use within the past 12 weeks and urine cotinine level >30 ng/mL, and an estimated IQ <80. Urine measurements for THC and other cannabinoids were performed by Dominion Diagnostics using Ultra Performance Liquid Chromatography/Tandem Mass Spectrometry (UPLC-MS/MS) and by enzyme immunoassay (EIA). Their EIA Cannabinoids assay has a Lower Reference Limit (LRL) of 25 ng/mL. Typically, this is sufficient to detect cannabinoids up to five days after occasional, or up to three days of more chronic use. The more definitive LC-MS/MS assay has a LRL of 5 ng/mL. At Screening, users (n = 12) reported ≥weekly cannabis use for the past six months, were positive for cannabis use on a rapid urinary test and had unadjusted urinary 11-nor-9-carboxy-ΔTHC (THCCOOH; the primary metabolite of cannabis) concentrations of ≥50 ng/mL, verified by enzyme immunoassay. At Screening, non-users (n = 12) self-reported no cannabis use in the past six months and fewer than ten lifetime uses, had a negative result for cannabis on a rapid urinary test, and a verified urinary THCCOOH level of 0 ng/mL.
Procedures
This study was reviewed and approved by the Duke Institutional Review Board, the Duke Institutional Animal Care and Use Committee, and was conducted in accordance to the Declaration of Helsinki of 2013. Following phone screening, eligible participants came to the clinic for a Screening visit. All participants provided written informed consent and were then evaluated by a clinician. Psychiatric functioning was assessed using the Mini International Neuropsychiatric Interview, version 7.0 [39,40]. Medical and medication history were also reviewed and vital signs (blood pressure, heart rate) as well as height and weight data were collected. A breath alcohol test was required to be 0.0 to continue. An expired breath carbon monoxide sample was taken to evaluate recent tobacco smoking, and a urine sample was collected to 1) measure cotinine to assess nicotine/tobacco use; 2) to conduct rapid screening of cannabis and other illicit drug use; and 3) to conduct EIA analyses for quantitative THCCOOH levels (Dominion Diagnostics). Both raw and creatinine adjusted values were assessed. Participants who met all inclusion criteria and no exclusion criteria were subsequently scheduled for a visit to provide a semen specimen at the University-affiliated fertility clinic using procedures as previously described [8]. A total of 24 participants (12 in each group) met all inclusion and no exclusion criteria for the study and were enrolled (Figure 1).
Semen analyses
Following liquefaction at room temperature and within 60 minutes of collection, semen was analyzed for appearance, volume, viscosity, pH, WBC concentration, sperm concentration, total motility, forward progression and total motile sperm count (TMC) using standard analytic methods. Normal appearance was considered off white to grayish opalescent fluid. Volume was determined by aspirating the sample into a 5 or 10 mL Falcon pipette (VWR; cat nos. 53,300–421 or 53,300–523) with normal considered ≥1.5 mL. Viscosity was determined by dispersing drops from a 5 mL pipette, with observation of >2 cm threads considered abnormal. Analyses for pH (normal: ≥ 7.2) and WBC (normal: < 1 x 106/mL) were performed with QwikCheckTM Test Strips (Medical Electronic Systems; Product No. 0700). All samples were within normal limits for WBCs. Concentration (normal: ≥15 x 106/mL) [41] and motility (normal: >40%) were determined with 5 μL samples using a Makler Counting Chamber (Sefi-Medical Instruments) examined at 20X with an inverted phase-contrast microscope (NIKON DIAPHOT inverted phase). Duplicate analysis with a minimum of 100 sperm was performed; >10% difference resulted in repeat analysis. Forward progression was characterized by the largest proportion of motile sperm and classified as: definite (normal), weak/sluggish or absent. Data collection was completed over approximately 3.5 months.
Rat exposure to tetrahydrocannabinol (THC)
Nine-week-old, sexually mature male Sprague Dawley rats were purchased from Charles River Laboratories and were housed 2–3 per cage and were randomized to two groups that were dosed daily for 12 days via oral gavage with 4 mls of vehicle only (n = 9; 10% ethanol, 1% Triton X-100 in saline) or 2 mg/kg THC (Sigma; T-2386), 10% ethanol, 1% Triton X-100 in saline (n = 8), equivalent to human moderate daily cannabis use [42–44]. Sample size was determined by power calculations which showed that with n = 8 per group, we would have 90% power to detect a 5% difference in DNA methylation at an alpha of 0.05. Rats were sacrificed two days following the conclusion of the exposure. The epididymis was placed in sterile PBS and the ‘swim out’ method enriched the solution for mature (motile) sperm. Sperm were washed with PBS and frozen. Animal studies were not blinded, were conducted in accordance with compliance regulations and were approved by the Duke University Institutional Animal Care and Use Committee. Duke University maintains an animal program that is registered with the USDA, assured through the NIH/PHS, and accredited with AAALAC International.
DNA purification
Samples were stored at – 80°C prior to use. DNA from up to ~5 million sperm was extracted using Qiagen’s Puregene DNA Extraction kit. The average A260:A280 ratio for the human samples was 1.75 (SD = 0.07) and the average yield was 3.4 µg (SD = 2.7). There was no significant difference in DNA yield between groups (p = 0.90).
Reduced Representation Bisulfite Sequencing (RRBS)
Between 200–500 ng of genomic DNA from each sperm sample was digested with 30U MspI (New England Biolabs; Cat. No. R0106S) to fragment the DNA and then was extracted with Zymo Research DNA Clean and Concentrator Kit (Zymo Research; Cat. No. D4003). Fragments were ligated to pre-annealed adapters containing 5ʹ-methylcytosine instead of cytosine, per Illumina’s specified guidelines (www.illumina.com). Adapter-ligated fragments of >50 bp in size were recovered using the DNA Clean and Concentrator Kit (Zymo Research; Cat. No. D4003). The fragments were bisulfite modified using the EZ DNA Methylation-Lightning Kit (Zymo Research; Cat. No. D5020). Preparative-scale PCR was performed and the resulting products were purified (DNA Clean and Concentrator Kit, Zymo Research; Cat. No. D4003) for 50-bp paired end sequencing on an Illumina HiSeq 1500/2500 instrument with no more than 12 samples per lane. Library construction and sequencing were performed by Zymo Research. Sequence reads were identified using Illumina’s base calling software and analyzed using a Zymo Research proprietary analysis pipeline, written in Python. Bismark was used to perform the alignment [45]. The methylation level for each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T.
Pyrosequencing
Verification of RRBS results was performed by bisulfite pyrosequencing on Pyromark Q96 MD Pyrosequencing Instruments (Qiagen) for select regions using 800 ng of genomic DNA isolated from sperm. PCR amplification and pyrosequencing were performed as previously described in detail [46,47]. Briefly, PCR primer design utilized the Pyromark CpG Assay Design Software (Qiagen). PCR amplification was optimized to produce a single, robust band by ethidium bromide staining of amplicons resolved on agarose gels. Pyrosequencing performance for each assay was tested in triplicate using mixtures of commercially available fully methylated and unmethylated control DNAs mixed in proportions to reflect 0%, 25%, 50%, 75% and 100% methylation. Once assay performance was deemed valid, samples were analyzed using the optimized assay conditions. Primer sequences and PCR conditions are available from the authors on request. Linear regression was performed to assess relationships between RRBS and pyrosequencing.
Data analyses
The human dataset included 1,861,760 CpG sites. We removed 625,262 CpG sites with missing data or less than 5X coverage. A two-tailed, unpaired student’s t-test with α = 0.05 was performed on the remaining 1,236,498 CpG sites. Sites were rejected if they had a calculated p-value <0.05 but a methylation difference <10%; 6,640 CpG sites met the criteria. These data were cross-listed with the UC Santa Cruz 2009 hg19 gene location list using a MATLAB script. A 5,000 bp window, upstream of the TSS, was included to account for promoter regulatory regions. This yielded 3,979 CpG sites and 2,077 unique gene names.
The chromosomal locations of the 3,979 CpG sites were entered into Kaviar, a SNP identification tool [48]. The hg19 gene set with zero-based input coordinates was used to match the UCSC 2009 hg19 data set. The resulting SNP data was cross-listed with the 3,979 CpG sites. CpG sites containing known SNPs with cumulative allele frequencies >0.05 were excluded from further analysis.
RRBS data for the rats covered 931,161 CpG sites. We removed 143,618 CpG sites with incomplete data or less than 5X coverage. We retained 1,822 CpG sites with a p < 0.05 and that had a > 10% average difference between groups. Data was cross-listed with the UCSC 2014 Rat Build to determine the locations of these CpG sites relative to annotated genes, including the 5,000 bp upstream of TSSs. This yielded 782 CpG sites with 627 unique gene names.
To determine potential intergenerational relevance, the list of differentially methylated rat genes from the cannabis experiment (n = 627 genes) was compared to differentially methylated rat genes from a dataset derived from the nucleus accumbens of rats whose parents were exposed to THC as adolescents (n = 473 genes) [20]. A probability simulation was used to assess whether the 55-gene overlap was significant. Each of the 17,299 genes documented in the UCSC Genome Browser was assigned a number from 1 to 17,299. A random number generator selected permutations of length 627 and 473 to represent each dataset. Overlapping genes between each set was recorded for five million simulations. The likelihood of 55 randomly-selected genes overlapping between two sets is p < 2e-7.
Bioinformatics analyses
The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 [49] was used to identify pathway annotations associated with differentially methylated CpG sites between cannabis users and controls.
Funding Statement
This work was supported by the John Templeton Foundation under Grant 60564.
Author contributions
SKM contributed to study conception, study design, acquisition, analysis and interpretation of data, and drafting and revising of the manuscript.
NIR was involved with study design, acquisition of data and revising the manuscript.
ZV contributed to data analysis and drafting and revising the manuscript.
ZH was involved with study design, acquisition and interpretation of data, and revising the manuscript.
CG acquired data and revised the manuscript.
RS acquired and analyzed data and revised the manuscript.
KA acquired data and revised the manuscript.
MHB acquired data and revised the manuscript.
TMP contributed to study design, acquisition and interpretation of data, and drafting and revising the manuscript.
DR contributed to data analysis and revising the manuscript.
DLC annotated the RRBS data results and revised the manuscript.
JEL contributed to study design, interpretation of data and revising the manuscript.
JTM contributed to study design, acquisition of data and revised and edited the manuscript.
FJM contributed to study design and revised and edited the manuscript.
MC acquired data and revised the manuscript.
BJH acquired data and revised the manuscript.
EDL contributed to study conception and design, acquisition, analysis and interpretation of data, and drafting and revising of the manuscript.
SHK contributed to study conception, study design, acquisition, analysis and interpretation of data, and drafting and revising of the manuscript.
All authors gave final approval of the version to be published.
Acknowledgments
This research was supported a grant from the John Templeton Foundation. The opinions expressed in this publication are those of the authors and do not necessarily reflect the views of the John Templeton Foundation. We thank Drs. Andrew Berchuck and Matthew Barber for critical review of the manuscript. We thank the study participants and gratefully acknowledge the expert assistance of Sabrina Simpson and Jamie Wylie.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- 1.Gunn JK, Rosales CB, Center KE, et al. Prenatal exposure to cannabis and maternal and child health outcomes: a systematic review and meta-analysis. BMJ Open. 2016;6:e009986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Funkhouser M. Governing the states and localities: state Marijuana laws in 2017 map. Governing News. Available from: http://www.governing.com: Dr. Mark Funkhouser, 2017.
- 3.Center for Behavioral Health Statistics and Quality (CBHSQ) SAaMHSAS U.S. Department of Health and Human Services (HHS), and RTI international. results from the 2015 national survey on drug use and health: detailed tables In: Types of illicit drug use in lifetime PY, and past month among persons aged 18 to 25: percentages, 2014 and 2015. https://www.samhsa.gov/data/ 2017. [Google Scholar]
- 4.Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73:292–297. [DOI] [PubMed] [Google Scholar]
- 5.Szutorisz H, Hurd YL.. High times for cannabis: epigenetic imprint and its legacy on brain and behavior. Neurosci Biobehav Rev. 2018;85:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pitsilis G, Spyridakos D, Nomikos GG, et al. Adolescent female cannabinoid exposure diminishes the reward-facilitating effects of Delta9-tetrahydrocannabinol and d-amphetamine in the adult male offspring. Front Pharmacol. 2017;8:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrnes JJ, Johnson NL, Schenk ME, et al. Cannabinoid exposure in adolescent female rats induces transgenerational effects on morphine conditioned place preference in male offspring. J Psychopharmacol. 2012;26:1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soubry A, Guo L, Huang Z, et al. Obesity-related DNA methylation at imprinted genes in human sperm: results from the TIEGER study. Clin Epigenetics. 2016;8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariel M, Cedar H, McCarrey J.. Developmental changes in methylation of spermatogenesis-specific genes include reprogramming in the epididymis. Nat Genet. 1994;7:59–63. [DOI] [PubMed] [Google Scholar]
- 10.Donkin I, Versteyhe S, Ingerslev LR, et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016;23:369–378. [DOI] [PubMed] [Google Scholar]
- 11.Meissner A, Gnirke A, Bell GW, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu AT, Hannon E, Levine ME, et al. Genetic architecture of epigenetic and neuronal ageing rates in human brain regions. Nat Commun. 2017;8:15353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huen K, Solomon O, Kogut K, et al. PON1 DNA methylation and neurobehavior in Mexican-American children with prenatal organophosphate exposure. Environ Int. 2018;121:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Non AL, Binder AM, Kubzansky LD, et al. Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics. 2014;9:964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joubert BR, Haberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120:1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laqqan M, Tierling S, Alkhaled Y, et al. Spermatozoa from males with reduced fecundity exhibit differential DNA methylation patterns. Andrology. 2017;5:971–978. [DOI] [PubMed] [Google Scholar]
- 17.Yang WS, Stockwell BR. Inhibition of casein kinase 1-epsilon induces cancer-cell-selective, PERIOD2-dependent growth arrest. Genome Biol. 2008;9:R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryant CD, Parker CC, Zhou L, et al. Csnk1e is a genetic regulator of sensitivity to psychostimulants and opioids. Neuropsychopharmacology. 2012;37:1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson CT, Szutorisz H, Garg P, et al. Genome-wide DNA methylation profiling reveals epigenetic changes in the rat nucleus accumbens associated with cross-generational effects of adolescent THC exposure. Neuropsychopharmacology. 2015;40:2993–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang WW, Dietmann S, Irie N, et al. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161:1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gundersen TD, Jorgensen N, Andersson AM, et al. Association between use of Marijuana and male reproductive hormones and semen quality: A study among 1,215 healthy young men. Am J Epidemiol. 2015;182:473–481. [DOI] [PubMed] [Google Scholar]
- 23.Hembree WC 3rd, Nahas GG, Zeidenberg P, et al. Changes in human spermatozoa associated with high dose marihuana smoking. Adv Biosci. 1978;22–23:429–439. [DOI] [PubMed] [Google Scholar]
- 24.Pacey AA, Povey AC, Clyma JA, et al. Participating centres of chaps UK. Modifiable and non-modifiable risk factors for poor sperm morphology. Hum Reprod. 2014;29:1629–1636. [DOI] [PubMed] [Google Scholar]
- 25.Johnston L, O’Malley P, Miech R, et al. Monitoring the future national results on drug use: 1975–2013: Overview, key findings on adolescent drug use.: Ann Arbor: Institute for Social Research. University of Michigan; 2014. [Google Scholar]
- 26.ElSohly MA, Mehmedic Z, Foster S, et al. Changes in Cannabis potency over the last 2 decades (1995–2014): analysis of current data in the United States. Biol Psychiatry. 2016;79:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hackett JA, Sengupta R, Zylicz JJ, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abbasi J. The paternal epigenome makes its mark. JAMA. 2017;317:2049–2051. [DOI] [PubMed] [Google Scholar]
- 29.Day J, Savani S, Krempley BD, et al. Influence of paternal preconception exposures on their offspring: through epigenetics to phenotype. Am J Stem Cells. 2016;5:11–18. [PMC free article] [PubMed] [Google Scholar]
- 30.Soubry A, Murphy SK, Wang F, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond). 2015;39:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shnorhavorian M, Schwartz SM, Stansfeld B, et al. Differential DNA methylation regions in adult human sperm following adolescent chemotherapy: potential for epigenetic inheritance. PLoS One. 2017;12:e0170085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laqqan M, Tierling S, Alkhaled Y, et al. Aberrant DNA methylation patterns of human spermatozoa in current smoker males. Reprod Toxicol. 2017;71:126–133. [DOI] [PubMed] [Google Scholar]
- 33.Alkhaled Y, Laqqan M, Tierling S, et al. Impact of cigarette-smoking on sperm DNA methylation and its effect on sperm parameters. Andrologia. 2018;50:e12950. [DOI] [PubMed] [Google Scholar]
- 34.Jenkins TG, James ER, Alonso DF, et al. Cigarette smoking significantly alters sperm DNA methylation patterns. Andrology. 2017;5:1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams IB, Martin BR. Cannabis: pharmacology and toxicology in animals and humans. Addiction. 1996;91:1585–1614. [PubMed] [Google Scholar]
- 36.Grotenhermen F, Russo E, eds. Cannabis and Cannabinoids: pharmacology, toxicology and therapeutic potential. New York: The Hawthorne Press, Inc; 2002. [Google Scholar]
- 37.Scallet AC. Neurotoxicology of cannabis and THC: a review of chronic exposure studies in animals. Pharmacol Biochem Behav. 1991;40:671–676. [DOI] [PubMed] [Google Scholar]
- 38.Marques CJ, Joao Pinho M, Carvalho F, et al. DNA methylation imprinting marks and DNA methyltransferase expression in human spermatogenic cell stages. Epigenetics. 2011;6:1354–1361. [DOI] [PubMed] [Google Scholar]
- 39.Sheehan DV, Lecrubier Y, Harnett-Sheehan K, et al. Reliability and validity of the M.I.N.I. international neuropsychiatric interview (M.I.N.I.): according to the SCID-P. Eur Psychiatry. 1997;12:232–241. [Google Scholar]
- 40.Lecrubier Y, Sheehan D, Weiller E, et al. The M.I.N.I. International Neuropsychiatric Interview (M.I.N.I.) A short diagnostic structured interview: reliability and validity according to the CIDI. Eur Psychiatry. 1997;12:224–231. [Google Scholar]
- 41.Cooper TG, Noonan E, von Eckardstein S, et al. World Health Organization reference values for human semen characteristics. Hum Reprod Update. 2010;16:231–245. [DOI] [PubMed] [Google Scholar]
- 42.Irimia C, Polis IY, Stouffer D, et al. Persistent effects of chronic Delta9-THC exposure on motor impulsivity in rats. Psychopharmacology (Berl). 2015;232:3033–3043. [DOI] [PubMed] [Google Scholar]
- 43.Harte LC, Dow-Edwards D. Sexually dimorphic alterations in locomotion and reversal learning after adolescent tetrahydrocannabinol exposure in the rat. Neurotoxicol Teratol. 2010;32:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubino T, Realini N, Braida D, et al. Changes in hippocampal morphology and neuroplasticity induced by adolescent THC treatment are associated with cognitive impairment in adulthood. Hippocampus. 2009;19:763–772. [DOI] [PubMed] [Google Scholar]
- 45.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy SK, Huang Z, Hoyo C. Differentially methylated regions of imprinted genes in prenatal, perinatal and postnatal human tissues. PLoS One. 2012;7:e40924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bassil CF, Huang Z, Murphy SK. Bisulfite pyrosequencing. Methods Mol Biol. 2013;1049:95–107. [DOI] [PubMed] [Google Scholar]
- 48.Glusman G, Caballero J, Mauldin DE, et al. Kaviar: an accessible system for testing SNV novelty. Bioinformatics. 2011;27:3216–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang Da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.