

ABSTRACT
Apart from its well-documented role in long-term promoter silencing, the genome-wide distribution patterns of ~ 28 million methylated or unmethylated CpG dinucleotides, e. g. in the human genome, is in search of genetic functions. We have set out to study changes in the cellular CpG methylation profile upon introducing foreign DNA into mammalian cells. As stress factors served the genomic integration of foreign (viral or bacterial plasmid) DNA, virus infections or the immortalization of cells with Epstein Barr Virus (EBV). In all instances investigated, alterations in cellular CpG methylation and transcription profiles were observed to different degrees. In the case of adenovirus DNA integration in adenovirus type 12 (Ad12)-transformed hamster cells, the extensive changes in cellular CpG methylation persisted even after the complete loss of all transgenomic Ad12 DNA. Hence, stress-induced alterations in CpG methylation can be inherited independent of the continued presence of the transgenome. Upon virus infections, changes in cellular CpG methylation appear early after infection. In EBV immortalized as compared to control cells, CpG hypermethylation in the far-upstream region of the human FMR1 promoter decreased four-fold. We conclude that in the wake of cellular stress due to foreign DNA entry, preexisting CpG methylation patterns were altered, possibly at specific CpG dinucleotides. Frequently, transcription patterns were also affected. As a working concept, we view CpG methylation profiles in mammalian genomes as a guarding sensor for genomic stability under epigenetic control. As a caveat towards manipulations of cells with foreign DNA, such cells can no longer be considered identical to their un-manipulated counterparts.
KEYWORDS: DNA methylation, foreign DNA, transgenomic cells, alterations of CpG methylation profiles, inheritable epigenetic response, genomic stability
Our concept
The human genome sequence, contains about 28 million CpG pairs which are potential targets for the modification of cytidine- to 5-mC (5-methyl-deoxycytidine)-residues by DNA methyltransferases. The distribution of 5-mC’s across the human genome most likely varies with cell type. Depending on environmental conditions, CpG methylation patterns are subject to alterations. Lacking a complete map of 5-mC distributions in the human genome, how might one visualize these patterns with high functional significance for genomic stability and activity? Definite information is not available; tentative models have been proposed [1,2]. The real challenge raised by CpG methylation landscapes emerges from the large number of CpG’s, and from deciphering their functional meaning. By selecting two examples of the complex problem for more detailed inspection, we are fully aware that there will be many additional ones worth consideration.
The presence of 5-mC residues in specific, functionally decisive positions of the genome is undoubtedly related to genetic activity [3,4].
Perhaps as importantly, the genome-inherent 5-mCpG versus CpG algorithm might be an important guardian of genomic stability, and capable of recognizing any threat against it. Much like innate and acquired immunity respond to the intrusion of foreign, often pathogenic molecules or cells, the CpG arrays are thought to be highly sensitive to the invasion of foreign DNA into the cell. The CpG guard might already be alerted by the contact of foreign nucleic acids with the cell surface. This CpG alarm clock has probably developed early in evolution and, like other ancient biological defenses, has progressed and evolved over evolutionary times. This system is flexible and permits alterations, not always under strictly controlled conditions. Altered methylation patterns can be transmitted over cell generations, i. e. are inheritable.
The notion of a guardian for genome stability is caught between two contradicting, equally essential options. (i) Maintaining the inherited genome is the precondition for survival in the real world that abounds with a gamut of competing molecules and organisms. However, will defense suffice as the major principle for survival? (ii) More realistically, the system requires the genetic and epigenetic potential to exploit competing organisms and their intruding foreign genetic information. Novel genetic and epigenetic information from foreign sources would be convenient to have around and could be constantly scanned for internal usefulness. Ubiquitous non-homologous recombination mechanisms enable the cell to incorporate newly-acquired foreign DNA into its own genome. Subsequently, this acquisition could be screened for internal advantage or, if necessary, might be eliminated from the cell’s indigenous nucleotide sequence. Selection in a competitive environment would then determine survival of propitious acquisitions of foreign DNA sequences.
This hypothetical concept is founded on the results of studies on the consequences of foreign DNA entry into mammalian cells and/or the integration of this DNA into established mammalian genomes. The description of the experimental results adduced over more than two decades, which helped formulate these thoughts, will be the matter of this invited review article.
Integration of foreign DNA into mammalian genomes: inverse correlations between DNA segment methylation and transcription
Starting in the late 1960s, this laboratory has been studying the integration of human adenovirus type 12 (Ad12) DNA into cellular genomes in Ad12-transformed cells or in Ad12-induced hamster tumor cells. At that time, the total intracellular DNA was cleaved with the restriction endonuclease HpaII which generated small DNA fragments linking cellular and Ad12 DNA. Ad12 virion DNA from purified virions was properly cleaved by HpaII, but integrated Ad12 DNA proved refractory to cleavage by methylation-sensitive HpaII [5]. The methylation-insensitive MspI, an isoschizomer of HpaII, did cut the integrated Ad12 DNA. Similarly, HhaI, another methylation-sensitive enzyme, cut virion, but not the integrated Ad12 DNA. The diagnostic restriction enzyme pair HpaII/MspI had just been discovered at that time [6]. We had shown previously that Ad12 DNA from purified virions was not methylated [7]. Hence, we concluded that the integrated Ad12 genomes had become de novo methylated [5,8,9]. Later, this conclusion was confirmed by applying the bisulfite sequencing technique to the analyses of integrated Ad12 DNA in revertant TR12 [10] of the Ad12-transformed cell line T637. As foreign integrate the revertant carried only one Ad12 DNA copy and a flipped right-terminal fragment of a second one. All 1.634 CpG’s in the integrated Ad12 DNA sequences were interrogated for their methylation status by bisulfite sequencing [11]. This method had been developed by Frommer et al. [12,13]. Furthermore, when DNA methylation levels were compared between the transcribed and the majority of the non-transcribed segments of the integrated Ad12 genomes in Ad12-transformed cells, an inverse correlation was discovered between the levels of methylation in the integrated Ad12 DNA segments and their transcriptional activity [8,9]. These reports were arguably the first to establish an inverse correlation and led us and others to document the notion that promoter methylation correlated with the long-term silencing of mammalian (eukaryotic) genes [for review 3,4].
Integration of foreign DNA into mammalian genomes can alter profiles of genome-wide DNA methylation
Continuing studies on the biological function of DNA methylation, we have investigated the consequences of DNA methylation in mammalian systems in several projects. In one of them, the analyses of methylation levels in cellular DNA segments from the Ad12-transformed hamster cell line T637 led to an interesting observation [14]. This cell line, which had been derived from the parent hamster cell line BHK21 [15], carries about 12 copies of chromosomally integrated Ad12 DNA at a single genomic site [14,16] (Figure 1(c,d)). When the total intracellular DNA from T637 cells was cleaved with HpaII, MspI or HhaI and screened for the cleavage patterns of integrated Ad12 DNA, the methylation-sensitive enzymes (HpaII, HhaI) failed to cleave the integrated viral genomes correctly, whereas MspI cut integrated Ad12 DNA. The control virion-derived Ad12 DNA was correctly cleaved by all enzymes as expected, since virion DNA had been shown to be unmethylated [7] (Figure 1(a), right panel). In contrast, when the cellular IAPI (intracellular A particle I) DNA segment from a 900 copy endogenous retroviral sequence [17,18] was used as hybridization probe to analyze the cleavage pattern on Southern blots [19], the following results emerged: The cleavage patterns of BHK21 DNA with HpaII and HhaI revealed a distinct methylation pattern for the IAPI sequences in this cell line (Figure 1a, left panel). However, DNA from the Ad12-transformed T637 cells unexpectedly showed a marked increase in IAPI methylation as documented by the complete failure of HpaII or HhaI to cleave the IAPI sequences, because these sequences had become hypermethylated (Figure 1a, left panel). In T637 cells, methylation levels of several additional cellular DNA sequences were also increased (data not shown here [14]). Methylation analyses of the IAPI sequences in another Ad12-transformed hamster cell line, A2497-3, yielded results very similar to those for T637 DNA. We concluded that the methylation levels in an endogenous retrotransposon cellular DNA sequence, IAPI, were strongly increased in DNA from the Ad12-transformed cell lines in comparison to the parent cell line BHK21 whose genome did not carry integrated Ad12 DNA sequences. In DNA from Ad12-infected BHK21 cells increases in IAPI cellular sequences were not observed. As a corollary, cell line TR3, a morphological revertant of T637 cells [10] with all Ad12 sequences deleted (Figure 1(b)), also showed marked hypermethylation of IAPI sequences (Figure 1(a), left panel). This finding documented that the continued presence of the transgenomic Ad12 DNA was not required to maintain hypermethylation levels in IAPI cellular DNA sequences after the loss of all Ad12 DNA sequences. These results suggested a hit-and-run mechanism for the foreign DNA integrate leading to genome-wide alterations in the levels of DNA methylation in the recipient cells. The guard for stability had been called upon, remained alerted, and was stably transmitted to the next generations of cells. Hence, altered DNA methylation profiles proved inheritable in this instance.
Figure 1.
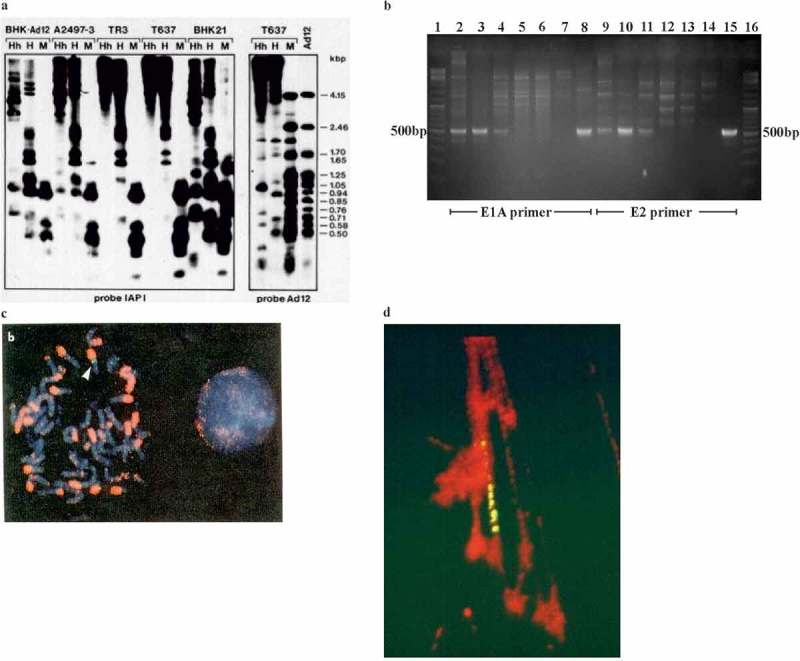
Genome-wide increases in DNA methylation in intracisternal A particle (IAPI) endogenous retrotransposon DNA in Ad12-transformed hamster cells.
a. DNA samples from hamster cell lines as designated in the top line of graph were cleaved with methylation-sensitive restriction endonuclease (re) HpaII (H) or HhaI (Hh) or the methylation-insensitive re MspI (M) (second line from top). Subsequently, DNA fragments were separated by electrophoresis on an agarose gel, DNA fragments were transferred to a Nylon membrane by Southern blotting [19], and hybridized to 32P-labelled DNA probes IAPI [17,18] or Ad12 (bottom line). This figure was taken from Heller et al. [14].
b. The DNA from cell line TR3 (a revertant [10] of Ad12-transformed hamster cell line T637) has lost all Ad12 DNA sequences. DNAs from cell lines TR3 (devoid of Ad12 DNA), TR12 (carries 1 copy and a fragment of Ad12 [11]) or T637 [15] with about 12 copies of integrated Ad12 DNA, see (d) [16] were screened for the presence of Ad12 DNA by PCR using primers from the E1A region of Ad12 DNA (tracks 2–8) or from the E2 promoter region (tracks 9–15), as indicated. Expected Ad12 DNA amplicon lengths were 524 bp and 540 bp, respectively; compare to gene ruler, tracks 1, 16. The following DNAs were used for PCR amplifications: Tracks 2, 9 – TR12 DNA; tracks 3, 10 – T637 DNA; tracks 4, 11 – Ad12 control; tracks 5–7 and 12–14 – TR3 DNA; tracks 8, 15 – Ad12 virion DNA. Tracks containing TR3 DNA (5–7 and 12–14) are devoid of Ad12-specific PCR amplicons, i. e. the TR3 revertant has lost all Ad12 DNA sequences. S. Weber and W. Doerfler, unpublished results.
c. Karyogram of the Ad12-transformed hamster cell line T637. The chromosomes were stained in the following ways: DAPI (4′, 6-diamidin-2-phenylindol)-stain (blue); fluorescent in situ hybridization (FISH) with biotin-16-dUTP-labeled Ad12 DNA (green, see arrow) or with digoxigenin-dUTP labeled IAPI DNA (pink). IAPI is widely distributed on many chromosomes, frequently on their short arms. The 12 copies of Ad12 DNA reside in a single chromosomal spot (arrow). This figure was taken from Heller et al. [14].
d. The Ad12-transformed cell line T637 carries about 12 copies of integrated Ad12 DNA, as demonstrated on this stretched chromosome preparation by a FISH experiment using biotin-16-dUTP-labeled Ad12 DNA as probe (yellow) and ethidium bromide staining of the stretched hamster chromosome. This figure was taken from Schröer et al. [16].
Genome-wide changes in DNA methylation and transcription in cells transgenomic for bacteriophage lambda DNA
We have performed a comparable study with the DNA of bacteriophage lambda as the transgenome in BHK21 hamster cells. Similar, though less pronounced changes in DNA methylation in the hamster cells’ IAP1 segments were documented by using the bisulfite sequencing method [20]. Moreover, when the transcriptional profiles in cellular genes were investigated both in Ad12 and in lambda DNA transgenomic BHK21 hamster cells, alterations in the transcription of a number of cellular genes could be detected [21].
Epigenetic consequences of the insertion of a 5.6 kbp bacterial plasmid as transgenome in human cells
Altered transcription of cellular genes in transgenomic versus non-transgenomic human cells
How general are alterations in the recipient cells’ methylation and transcription profiles upon entry of foreign DNA and/or the integration of foreign DNA into mammalian genomes? We designed a reductionist, proof-of-principle experiment [22]. Human colon tumor (HCT116) cells were transfected with bacterial plasmid pC1–5.6 DNA by using a nucleofection protocol (Lonza Cologne GmbH, Cologne, Germany). The 5.6 kbp plasmid contained as the sole transcribed element the CMV promoter-controlled kanamycin resistance gene for clonal selection. Five single transgenomic clones were isolated as were five non-transgenomic control clones devoid of an added transgenome. From five clones of non-transgenomic HCT116 cells total RNA was isolated. By using an Affymetrix Expression Array system with fluorescent hybridization probes, transcriptional activities were measured in 28,869 genomic DNA segments covered by 764,885 distinct probes. Expression profiles of all five independently isolated and propagated non-transgenomic cell clones were analyzed in double determinations and had a pair-wise Pearson’s correlation coefficient above 0.98 for all transcripts measured. This finding was the critical precondition for meaningful comparisons of transcriptional activities between non-transgenomic and transgenomic cell clones, the latter with the chromosomally integrated p-5.6 plasmid DNA as transgenome. All analytical work on transcriptional profiles was performed by a commercial laboratory (KFB – Center of Excellence for Fluorescent Bioanalytics, Regensburg, Germany) which had not been informed about the derivation of the samples nor the purpose of the experiment.
Subsequently, differential gene segment expression was assessed between mean values of two biological replicates from five non-transgenomic control clones against seven plasmid-transgenomic cell clones. Differences in transcription were called significant when fold changes were > plus/minus 2 and p-values < 0.05. All p-values were adjusted for false discovery rates (FDR) (23). Among the 28,869 human DNA segments analyzed, 1343 were differentially expressed, 907 were upregulated and 436 were downregulated in transgenomic cell clones (Figure 2(a)). These gene segments could be assigned to 43 canonical pathways which included EIF2 signaling, regulation of eIF4 and p70S6K signaling, glutathione-mediated detoxification, FAK signaling, insulin receptor signaling and ErbB4 signaling, and most predominantly to snoRNA (small nucleolar RNA) genes. SnoRNAs affect the biochemistry of many RNA populations, ribosomal, transfer and small nuclear RNA’s. We conclude that the integration of a small bacterial plasmid into the human genome has altered transcriptional activities in 4.7% of the analyzed gene segments in this experiment [22].
Figure 2.
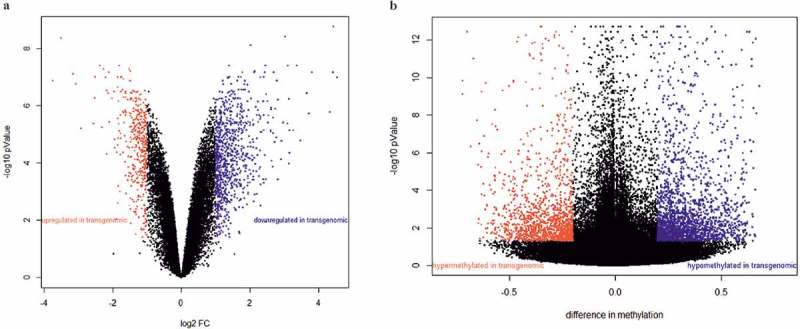
Alterations in patterns of transcription (a) and methylation (b) in pC1-5.6 transgenomic HCT116 cell clones as compared to non-transgenomic cells. (a) Volcano plot displays non-standardized signal (log2 fold-change) on the x-axis against standardized signal (-log10 FDR-adjusted p-value), on the y-axis for the comparison of five non-transgenomic against seven transgenomic cell clones of all 28,869 gene segments analyzed. Up-regulated genes in transgenomic cell clones were displayed in red and down-regulated genes in blue (FC ± 2, adjusted p-values < 0.05; n = 1343 genes). (b) Volcano plot displays differences in methylation on the x-axis against standardized methylation (-log10 FDR-adjusted p-value), on the y-axis for the comparison of four non-transgenomic against five pC1-5.6 transgenomic cell clones of all 361,983 CpG’s interrogated. Hypermethylated CpG’s in transgenomic cell clones were displayed in red and hypomethylated CpG’s in blue (Δβ value ≥ 0.2, adjusted p-value < 0.05; n = 3,791 CpG’s). This figure and its legends were taken from Weber et al., 2015 [22].
Alterations of DNA methylation profiles in transgenomic gene segments
Genome-wide methylation profiles were assessed in over 480,000 CpG sites by the bisulfite method in four non-transgenomic clones and five pC1–5.6 DNA transgenomic clones using Illumina 450K Infinium arrays. After correction for possibly cross-reacting CpG’s in the transgenomic plasmid, 361,983 CpG’s were left for analyses. Samples were processed on the Human Methylation 450 K Bead Chips from Illumina (San Diego, CA, USA). Similarly to transcription profiling, the mean correlation across all non-transgenomic human cell clones was calculated as 0.97 ± 0.02 and thus facilitated comparisons between non-transgenomic and transgenomic clones. By applying the rigorous statistical Benjamini – Hochberg correction [23], 3.791 CpG’s proved differentially methylated in DNA samples from p-5.6 DNA transgenomic as compared to the non-transgenomic cell clones [22]: 1504 were hyper- and 2287 were hypo-methylated (Figure 2(b)). The 3.791 CpG’s could be assigned to 2.622 genes and 109 pathways, including those associated with neuron signaling. Thus, in the p-5.6 plasmid-transgenomic human cells, CpG methylation was significantly altered when compared to non-transgenomic clones. These alterations were maintained over many cell generations.
Epicritical considerations
It appeared that the insertion of foreign DNA, like Ad12 viral DNA, bacteriophage lambda DNA or 5.6 kbp bacterial plasmid DNA, in some way led to genome-wide alterations of CpG methylation and transcription profiles in mammalian cells.
Genome-wide effect. – In the case of cell line T637 with 12 copies of viral DNA integrated at one chromosomal location [14,16], several specific points deserve emphasizing. The increase in CpG methylation had obviously affected the majority of the about 900 copies of intracisternal A particle (IAPI) endogenous retro-transposon DNA [17,18] (see Figure 1a, lane T637), and these IAPI sequences were distributed across most of the hamster chromosomes (Figure 1(c)). Therefore, the hypermethylation upon Ad12 integration had affected the IAPI sequences on many of the hamster chromosomes, hence constituted a genome-wide event. Insertion of foreign DNA and the subsequent alterations of CpG methylation profiles must have been a distinct stress signal for the cells and their genomes.
Inheritability. – We had isolated morphological revertants of cell line T637 [10]. One of these revertants, TR3, had lost all of the twelve Ad12 DNA copies originally present in cell line T637 (Figure 1(b)). The hypermethylation of IAPI sequences discovered in T637 cells was maintained in the revertant cell line TR3 after the loss of all transgenomic Ad12 DNA [14]. We conclude that the foreign DNA-induced alterations of genome-wide methylation profiles in the cell are independent of the persistence of the stress signal, which has been elicited by the integration of foreign DNA. These alterations have been transmitted to daughter generations of the revertant cell line. This stability raises the generally important possibility that induced epigenetic alterations can be inheritable, at least at the level of many sub-sequent cell generations.
General significance of observation? – The genome-wide patterns of methylation in 28 million CpG’s (figure for the human genome) seem to represent a highly sensible stress sensor for genome stability. We have exploited and further interrogated the sensitivity of this sensor and searched for similar effects by using the following foreign DNA molecules: The p-5.6 kbp bacterial plasmid [22] or the telomerase gene as transgenomes as well as Epstein Barr Virus as a non-integrating episomal challenge (see below) [24]. In the small plasmid p-5.6, the only transcriptionally active element was the kanamycin resistance gene which had to be used for the selection of transgenomic cell clones. Again, alterations in CpG methylation patterns could be documented, although not at the level as described for Ad12 transgenomic cells.
It is conceivable that the activity of Ad12 gene products from the viral genome’s E1 and E4 regions [25] in cell line T637 might have played a modulating or even enhancing role in altering CpG methylation in the Ad12-transformed cell line T637. The transactivation functions of the adenovirus E1A proteins on viral and cellular gene activities have long been recognized [26–28]. More recently, it has been shown that Ad5 E1A proteins directly associate in vitro and in vivo with DNA methyltransferase Dnmt1 [29]. This finding demonstrates how adenoviral gene products might mediate the activity of the DNA methyltransferase system. In addition, the E1 functions of Ad2 can counteract promoter inactivation by CpG methylation [30]. Furthermore, E1A proteins can lead to the redistribution of histone H3K18ac and thus exert epigenetic activities in adenovirus infected cells [31,32]. There may be additional, hitherto unknown, ways in which viral gene products influence the epigenetic profile of a cell.
HERVs. – In the work with integrated Ad12 or lambda DNA integrates, the retroviral IAPI sequences had responded to foreign DNA integration by alterations in their CpG methylation patterns. Would the human endogenous retroviral (HERV) sequences (recent reviews [33,34]) also respond in the p-5.6 plasmid transgenomic human cell clones? Detailed analyses with the bisulfite sequencing technique failed to reveal alterations in the complex HERV DNA methylation profiles in the same transgenomic human cell clones which had shown such alterations in other parts of their genomes [35]. We are still left with very limited understanding of the forces at work in changing cellular methylation patterns in response to foreign DNA integration, and hence have to be cautious in the interpretation of the limited data available.
Role of transfection method. – In the experiments with the p-5.6 plasmid, the non-transgenomic cell clones had been selected and grown under identical conditions as the plasmid-transgenomic ones, except that we had not exposed them to the electric shock treatment applied for the transfection of plasmid DNA. Hence, the alterations in CpG methylation and transcription patterns described [22] could have in part been influenced by the transfection method. In the section ‘Research on the stability of CpG methylation profiles: Targets for future projects’ (see below), we recommend a systematic study on the mode of administration and on the fate of foreign DNA in mammalian cells, an essential premise for further work on epigenetic effects of foreign DNA on mammalian genomes.
Altered CpG methylation in the human genome upon Ad12 infection?
As will be outlined in the following section, in human cells immortalized by transformation with the herpesvirus EBV, a four-fold decrease of DNA methylation in the far-upstream region of the human FMR1 (fragile X mental retardation 1) gene has been observed [24]. The discovery of methylation changes in the far-upstream region had been a serendipitous observation, since the immortalized human cells had been generated for a different project. In a proactively designed approach, we then chose to investigate the consequences of virus infections on the stability of the methylation profiles of the about 28 × 106 CpG’s in the human genome, and chose Ad12 as the challenging virus. Profiles of DNA methylation were determined both in the newly synthesized Ad12 DNA and in about 400.000 CpG’s out of a total of about 28 million CpG’s in the human genome by using bisulfite sequencing. During the Ad12 infection cycle up to 48 h p. i., the viral genome escaped de novo methylation by the host’s de novo methylation machinery [36]. The analyses of changes in CpG methylation in DNA from Ad12-infected cells at different times after infection have not yet been completed. Preliminary findings suggest that there are changes in some of the analyzed CpG’s which appear early after Ad12 infection.
Alterations of CpG methylation in the far-upstream region of the human FMR1 promoter
The fragile X syndrome (FXS) is one of the most frequent heritable forms of mental impairment and a frequent cause of comorbid autism in humans. The molecular analyses of the fragile X mental retardation 1 (FMR1) gene on human chromosome Xq27.3 revealed a naturally occurring unstable CGG-repeat in the 5′-untranslated region of the first exon and a CpG cluster in the promoter of the FMR1 gene. In non-FXS individuals, the CGG-repeat length is < 50. In pre-mutation females, the trinucleotide CGG repeat expands to 55–200, in FXS individuals this value exceeds > 200. The CGG-repeats become expanded mainly upon passage of FXS chromosomes through the female germ line. In most, but not all, cases of FXS, the CpG’s in the promoter and upstream regions of the FMR1 gene are hypermethylated. FXS and its molecular characteristics have been investigated in great detail, but the actual cause of this genetic disorder have not been revealed (for recent reviews see [37,38]). The mechanisms leading to CGG-repeat expansions and to promoter hyper-methylation remain unknown, although several interesting hypotheses have been proposed [39,40].
The human genome segment upstream of the FMR1 gene contains several genetic signals: The FMR1 promoter and the initiation site for FMR1 gene transcription, an origin of DNA replication, three CCTC-factor binding sites, and > 100 CpG dinucleotides. We have discovered a distinct DNA-methylation boundary at a site between 650 and 800 nucleotides upstream of the CGG repeat (Figure 3(a)). This boundary has been identified by bisulfite sequencing and is present in all human cell lines and cell types, irrespective of age, gender, and developmental stage, type of organ or tumor phenotype [41]. The same boundary exists also in different mouse tissues, although sequence homology in this region between humans and mice is only 46.7%. The boundary sequence, in both the unmethylated and the CpG-methylated modes, binds specifically to nuclear proteins from human cells. We interpret this boundary to exhibit a specific chromatin structure which demarcates a hypermethylated upstream sequence from the unmethylated downstream FMR1 promoter. The boundary sequence protects the promoter from the spreading of DNA methylation and helps maintain its activity [41]. In contrast, in most FXS individuals the methylation boundary is lost, CpG methylation penetrates into the FMR1 promoter and inactivates the gene. Loss of the FMR1 gene product during fetal development causes the syndrome. It has still not been resolved how loss of FMR1 promoter function and CGG-repeat expansion are mutually contributing to the fragile X syndrome. Figure 3 depicts the methylation profiles in the FMR1 promoter region in unaffected (Figure 3(a)) and FXS individuals (Figure 3(b)) [41]. A genetic map of the region is shown in Figure 3(c). During the analyses of females with unstable, expansion-prone CGG-repeats and their families, rare unaffected males were identified [42–44] who carried a > 200 CGG-repeat expansion but an unmethylated FMR1 promoter [24,44,45]. These men were not affected by the syndrome and were designated high functioning males (HFM). The methylation boundary in the FMR1 promoter region in the genomes of two HF males, was found to be stable [24,45], although their CGG-repeats had expanded to ~ 330 and ~ 400 expansions, respectively, i. e. to values characteristic for FXS individuals (data not shown). Hence, CpG hypermethylation in the FMR1 promoter region can be considered a major causative factor in promoter inactivation in FXS. The result leaves unexplained why the CGG repeat was expanded in the two exceptional cases described in our report.
Figure 3.
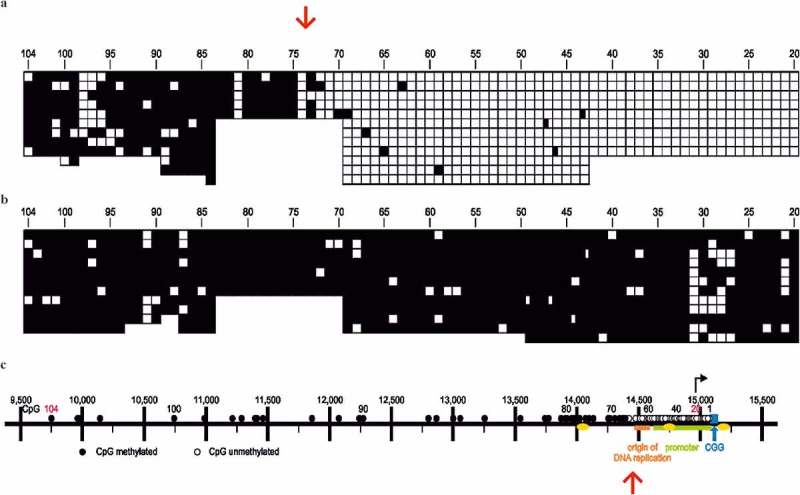
The CpG DNA methylation boundary (red arrows) is located in the upstream region of the FMR1 promoter in the human genome [41]. Methylated CpG`s were identified by the bisulfite sequencing method [12,13]. (a) DNA from a non-FXS male with an intact methylation boundary. Unmethylated CpG’s □, methylated CpG’s ■. CpG’s have been arranged next to each other and not according to their actual map positions which has been used in the map panel (c) of this figure. (b) DNA from an FXS male. The methylation boundary has been lost, and most of the CpG’s in the upstream region of the promoter are methylated: ●/■. (c) Genomic map of the FMR1 promoter region on human chromosome Xq27.3. Numbers 1–104 designate CpG’s in the region: ○/□, unmethylated, ●/■, methylated. Blue – CGG repeats; yellow – CTCF binding sites; green – FMR1 promoter; orange – origin of DNA replication; red arrows – location of methylation boundary. This figure was taken from Naumann et al. [24].
As a corollary to these findings, an unexpected observation was made. In some of the FXS genomes and in genomes from cells, which had been immortalized by transformation with EBV or by transfection with the telomerase gene, as well as in a cell line from fibroblasts (possibly immortalized) of an FXS fetus, the high methylation level in a genomic region far-upstream from the methylation boundary had been four-fold decreased (Figure 4(a-d) [24]). We pursue the possibility that this marked loss of 5-mC residues is related to the introduction of foreign DNA (EBV DNA or telomerase gene) into the human cells. Destabilization of DNA methylation has been described by Grafodatskaya et al. in EBV-transformed human lymphoblastoid cell lines [46]. It is also conceivable that the expansion of CGG repeats in FXS had been registered as a stress factor akin to foreign DNA intrusion. Of course, there could be additional and more complex reasons for this ‘induced’ hypomethylation. We intend to further investigate this finding by exposing human cells to different environmental stress conditions and study CpG methylation in this region of the human genome by bisulfite sequencing. If successful, this system would provide a tool for more extended and controlled studies on the effect of diverse environmental factors on CpG methylation profiles.
Figure 4.
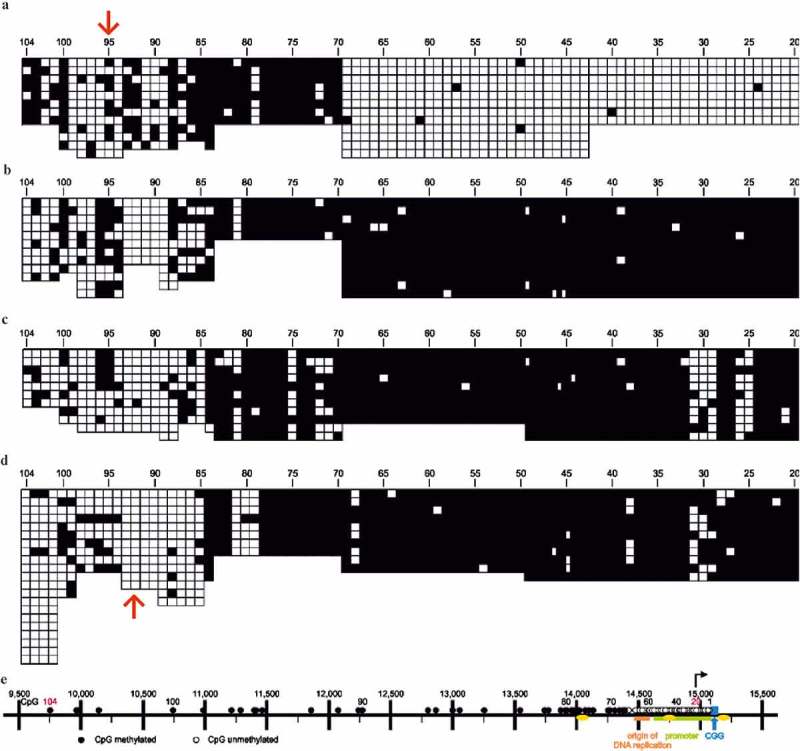
Loss of methylation profile in the far-upstream region of the FMR1 promoter boundary (red arrows). In human cells which have been immortalized by EBV infection or by transfection with the human telomerase gene, methylation in the far-upstream region (approximately between CpG’s 80 and 104) from the methylation boundary has been four-fold decreased. Figure design is similar to the one in Figure 3. The analyzed DNA preparations have been taken from: (a) EBV-immortalized PBMC’s (peripheral blood mononuclear cells) of a non-FXS individual with an intact methylation boundary. (b) EBV-immortalized PBMC’s of an FXS male. (c) Similar to (b), except that the PBMC’s had been immortalized by transformation with the human telomerase gene. (d) Cultured fibroblasts from a 22-week-old male FXS fetus (ATCC number GMO7072). (e) This figure was taken from Naumann et al. [24].
What mechanisms could be at work in altering cellular CpG methylation patterns upon foreign DNA entry?
Clearly, we cannot provide a satisfactory answer at this point. However, it will be helpful for future research to consider plausible options, although predictions can fall far from reality in basic research. In the section ‘Our concept’ of this review, we have postulated that the patterns of CpG methylation in the entire genome are not only of importance for the stability of the genome, but are constantly screened for maintenance or alterations, e.g., due to the entry of foreign DNA which jeopardizes the uniqueness of a given genome. In trying to understand the processes related to this surveillance system, there are at least three fundamental issues: (i) How is the intrusion of foreign DNA monitored; (ii) what mechanisms effect alterations of the CpG methylation landscape; lastly, (iii) could the functional consequences of these changes not be potentially enormous?
By proceeding in a step-wise manner, we are aware that we will now be entering hypothetical territory.
Foreign DNA (perhaps also RNA) could be recognized by cells already after its interaction with the cell membrane which can respond with a multitude of known (and/or hitherto unknown) receptor and signal transduction portfolios. Inside the cell, there must be recipients for these signals and pathways of functions which start working on changing DNA methylation patterns.
What are the crucial number and specific locations of CpG’s to be methylated or de-methylated? In effecting such changes, again a multitude of factors will participate. It can neither be exclusively the activity of DNA methyltransferases or the chromatin structure at a given location in the genome, but rather a complex combination of structural, enzymatic and guidance mechanisms. Existing or co-evolving patterns of histone modifications will play a further role. The potential of small RNAs could rank high during these reactions and their targeting.
At this stage, we have no information on the sequence or site specificities of the alterations in CpG methylation. The functional consequences for genome stability and/or transcriptional profile in the cell might well depend on the location of these changes in methylation landscapes. In instances of foreign DNA integration into an established genome, we have previously raised the question of in what way site of insertion and location of functional alterations in the genome might be interdependent [22]. The outcome of these events could range from moderately harmful, to catastrophic, to possibly advantageous for the cell and its survival. Of course, a cell’s phenotype, particularly a malignant one, is co-determined by its CpG methylation and transcription profiles.
How to proceed in investigating these problems? We intend to choose a manageable system out of the selection we have so far been working with. With current technology, it will be unrealistic to alter experimental parameters systematically and after each step screen genome-wide alterations in CpG methylation. Compromises in this respect carry the risk of ending up with incomplete answers. Nevertheless, we might start out with a genome segment of limited size, vary parameters and analyze the effect on CpG methylation in this more realistically manageable environment. More far-reaching decisions will depend on the results from this low scale investigation.
Research on the stability of CpG methylation profiles: targets for future projects
The modification of C-residues to 5-mC’s, mainly, but not exclusively, in CpG dinucleotides, can affect the binding of specific proteins at functionally essential sites in mammalian genomes. On that basis, the structure, function, and stability of the genome could be altered by changes in CpG methylation profiles. A well-documented case in point is the long-term silencing of genes through promoter CpG methylation. With that recognition, we have barely scratched the surface of a major problem in epigenetic research – the meaning of genome-wide CpG methylation patterns involving about 28 million CpG’s, e. g. in the human genome. So far, methods and data that would provide a complete map of CpG methylation patterns in the human genome are not available. In the projects described in this review, we have challenged the stability of these patterns by using foreign DNA or viruses that entered mammalian cells as biologically relevant stress factors. In this way, we tried to evoke responses from these patterns, since cells and organisms encounter foreign DNA in significant amounts nearly constantly.
What are the mechanisms recognizing foreign DNA in different compartments of the cell? Important signals might be elicited depending on the site of impact or the origin of foreign DNA. The following items have to be considered:
Contact with the cell surface,
any of the frequently used methods to introduce foreign DNA into intact cells,
foreign DNA immediately after its addition to the cell and in a time course after entry into the cell,
foreign DNA in the cell’s nucleus,
persisting foreign DNA in an episomal,
or a chromosomally integrated configuration.
- Derivation, amount, nucleotide sequence, size, and configuration of foreign DNA.
- (iii) Foreign DNA enclosed in virus particles represents a very different challenge. Are the effects elicited by virus infections discernible from those caused by the invasion of foreign DNA molecules?
- (iv) Does the location of foreign DNA integration into the recipient genome play a role in determining extent and sites of alterations of CpG methylation and transcription programs?
- (v) Could RNA molecules lead to similar consequences for the stability of the cell’s CpG methylation profiles?
- (vi) Foreign DNA from the gastrointestinal tract and its fate in the organism have been studied earlier in our laboratory [47,48]. This topic deserves more detailed investigations. Food-ingested DNA in fragmented form might have major con-sequences in mammalian organisms. Hence, projects directed at these questions deserve active support, since large amounts of foreign DNA are regularly taken up with the food supply.
- (vii)Of course, there are many additional, non-DNA related stress factors in the environment to which CpG methylation profiles respond.
- (viii) Caveats for work with manipulated cells and genomes – a perspective [49]. The genetic and epigenetic sequelae of exposing mammalian or plant cells and organisms to foreign DNA, let alone manipulations on a cell’s genome, are poorly understood; they have been barely investigated. This apparent lack of interest is all the more surprising since almost all fields of genetic research, particularly in medicine, have employed techniques involving massive manipulations on the cellular genomes of many different organisms. With a long-standing commitment to research on the fate of foreign DNA in cells, organisms and the environment, the authors wish to recommend launching a major project in this area of molecular genetics. There are many fundamental questions begging to be answered. The gain of insights with far-reaching relevance will prove rewarding.
Funding Statement
This work was supported by the Fritz Thyssen Stiftung [Az. 10.07.2.138];Staedtler Stiftung [WW/eh 01/15].
Acknowledgments
At different times, W.D.’s research has been supported by the Deutsche Forschungsgemeinschaft in Bonn-Bad Godesberg (SFB 74 and SFB 274), by the Center for Molecular Medicine Cologne (CMMC, TP13), by the Thyssen Foundation in Cologne (Az. 10.07.2.138 plus a stipend to A. Naumann Az. 40.12.0.029.), by the Staedtler Stiftung in Nürnberg (WW/eh 01/15), and by the continued support of W.D.’s research group at the Institute for Clinical and Molecular Virology, Friedrich-Alexander University, Erlangen-Nürnberg.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Saxonov S, Berg P, Brutlag DL.. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lövkvist C, Dodd IB, Sneppen K, et al. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016;44:5123–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doerfler W. DNA methylation - a regulatory signal in eukaryotic gene expression. J Gen Virol. 1981;57:1–20. [DOI] [PubMed] [Google Scholar]
- 4.Doerfler W. DNA methylation and gene activity. Ann Rev Biochem. 1983;52:93–124. [DOI] [PubMed] [Google Scholar]
- 5.Sutter D, Westphal M, Doerfler W. Patterns of integration of viral DNA sequences in the genomes of adenovirus type 12-transformed hamster cells. Cell. 1978;14:569–585. [DOI] [PubMed] [Google Scholar]
- 6.Waalwijk C, Flavell RA. MspI, an isoschizomer of hpaII which cleaves both unmethylated and methylated hpaII sites. Nucleic Acids Res. 1978;5:3231–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Günthert U, Schweiger M, Stupp M, et al. DNA methylation in adenovirus, adenovirus-transformed cells, and host cells. Proc Natl Acad Sci USA. 1976;73:3923–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutter D, Doerfler W. Methylation of integrated viral DNA sequences in hamster cells transformed by adenovirus 12. Cold Spring Harbor Symp Quant Biol. 1979;44:565–568. [DOI] [PubMed] [Google Scholar]
- 9.Sutter D, Doerfler W. Methylation of integrated adenovirus type 12 DNA sequences in transformed cells is inversely correlated with viral gene expression. Proc Natl Acad Sci USA. 1980;77:253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groneberg J, Sutter D, Soboll H, et al. Morphological revertants of adenovirus type 12-transformed hamster cells. J Gen Virol. 1978;40:635–645. [DOI] [PubMed] [Google Scholar]
- 11.Hochstein N, Muiznieks I, Mangel L, et al. The epigenetic status of an adenovirus transgenome upon long-term cultivation in hamster cells. J Virol. 2007;81:5349–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark SJ, Harrison J, Paul CL, et al. High sensitivity mapping of methylated cytosines. Nucl Acids Res. 1994;22:2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heller H, Kämmer C, Wilgenbus P, et al. Chromosomal insertion of foreign (adenovirus type 12, plasmid, or bacteriophage lambda) DNA is associated with enhanced methylation of cellular DNA segments. Proc Natl Acad Sci USA. 1995;92:5515–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strohl WA, Rabson AS, Rouse H. Adenovirus tumorigenesis: role of the viral genome in determining tumor morphology. Science. 1967;156:1631–1633. [DOI] [PubMed] [Google Scholar]
- 16.Schröer J, Hölker I, Doerfler W. Adenovirus type 12 DNA firmly associates with mammalian chromosomes early after virus infection or after DNA transfer by the addition of DNA to the cell culture medium. J Virol. 1997;71:7923–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lueders KK, Kuff EL. Sequences homologous to retrovirus-like genes of the mouse are present in multiple copies in the Syrian hamster genome. Nucleic Acids Res. 1981;9:5917–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ono M, Ohishi H. Long terminal repeat sequences of intracisternal A particle genes in the Syrian hamster genome: identification of tRNAPhe as a putative primer tRNA. Nucleic Acids Res. 1983;11:7169–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. [DOI] [PubMed] [Google Scholar]
- 20.Remus R, Kämmer C, Heller H, et al. Insertion of foreign DNA into an established mammalian genome can alter the methylation of cellular DNA sequences. J Virol. 1999;73:1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Müller K, Heller H, Doerfler W. Foreign DNA integration. Genome-wide perturbations of methylation and transcription in the recipient genomes. J Biol Chem. 2001;276:14271–14278. [DOI] [PubMed] [Google Scholar]
- 22.Weber S, Hofmann A, Herms S, et al. Destabilization of the human epigenome: consequences of foreign DNA insertions. Epigenomics. 2015;7:745–755. [DOI] [PubMed] [Google Scholar]
- 23.Benjamini Y, Hochberg Y. Controlling of false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;289:289–300. [Google Scholar]
- 24.Naumann A, Kraus C, Hoogeveen A, et al. Stable DNA methylation boundaries and expanded trinucleotide repeats: role of DNA insertions. J Mol Biol. 2014;426:2554–2566. [DOI] [PubMed] [Google Scholar]
- 25.Ortin J, Scheidtmann KH, Greenberg R, et al. Transcription of the genome of adenovirus type 12. III. Maps of stable RNA from productively infected human cells and abortively infected and transformed hamster cells. J Virol. 1976;20:355–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berk AJ. Adenovirus promoters and E1Â transactivation. Ann Rev Genet. 1986;20:45–79. [DOI] [PubMed] [Google Scholar]
- 27.Flint J, Shenk T. Adenovirus E1A protein paradigm viral transactivator. Annu Rev Genet. 1989;23:141–161. [DOI] [PubMed] [Google Scholar]
- 28.Nevins JR. Control of cellular and viral transcription during adenovirus infection. CRC Crit Rev Biochem. 1986;19:307–322. [DOI] [PubMed] [Google Scholar]
- 29.Burgers WA, Blanchon L, Pradhan S, et al. Viral oncoproteins target the DNA methyltransferases. Oncogene. 2007;26:1650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langner KD, Weyer U, Doerfler W. Trans-effect of the E1 region of adenoviruses on the expression of a prokaryotic gene in mammalian cells: resistance to 5′-CCGG-3′ methylation. Proc Natl Acad Sci USA. 1986;83:1598–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari R, Su T, Li B, et al. Reorganization of the host epigenome by a viral oncogene. Genome Res. 2012;22:1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–7685. [DOI] [PubMed] [Google Scholar]
- 33.Kim HS. Genomic impact, chromosomal distribution and transcriptional regulation of HERV elements. Mol Cell. 2012;33:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss RA. On the concept and elucidation of endogenous retroviruses. Philos Trans R Soc London B Biol Sci. 2013;368:201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber S, Jung S, Doerfler W. DNA methylation and transcription in HERV (K, W, E) and LINE sequences remain unchanged upon foreign DNA insertions. Epigenomics. 2016;8:157–165. [DOI] [PubMed] [Google Scholar]
- 36.Weber S, Doerfler W. Altered CpG methylation in the human genome upon Ad12 infection? [Manuscript in preparation]. 2018. [Google Scholar]
- 37.Dockendorff TC, Labrador M. The fragile X protein and genome function. Mol Neurobiol. 2018. DOI: 10.1007/s12035-018-1122-9 [DOI] [PubMed] [Google Scholar]
- 38.Kraan CM, Godler DE, Amor DJ. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev Med Child Neurol. 2018. DOI: 10.1111/dmcn.13985 [DOI] [PubMed] [Google Scholar]
- 39.Cleary JD, Nichol K, Wang YH, et al. Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat Genet. 2002;31:37–46. [DOI] [PubMed] [Google Scholar]
- 40.Nichol Edamura K, Leonard MR, Pearson CE. Role of replication and CpG methylation in fragile X syndrome CGG deletions in primate cells. Am J Hum Genet. 2005;76:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naumann A, Hochstein N, Weber S, et al. A distinct DNA methylation boundary in the 5′-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am J Hum Genet. 2009;85:606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smeets HJ, Smits AP, Verheij CE, et al. Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet. 1995;4:2103–2108. [DOI] [PubMed] [Google Scholar]
- 43.Wöhrle D, Salat U, Gläser D, et al. Unusual mutations in high functioning fragile X males: apparent instability of expanded unmethylated CGG repeats. J Med Genet. 1998;35:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabolacci E, Moscato U, Zalfa F, et al. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur J Hum Genet. 2008;16:1487–1498. [DOI] [PubMed] [Google Scholar]
- 45.Naumann A, Weber S, Hochstein N, et al. Safeguarding the human FMR1 promoter: the methylation barrier in its 5′-upstream region. Abstract, Annual Meeting of the American Society of Human Genetics in Washington, DC; 2010.
- 46.Grafodatskaya D, Choufani S, Ferreira JC, et al. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics. 2010;95:73–83. [DOI] [PubMed] [Google Scholar]
- 47.Schubbert R, Renz D, Schmitz B, et al. Foreign (M13) DNA ingested by mice reaches peripheral leukocytes, spleen and liver via the intestinal wall mucosa and can be covalently linked to mouse DNA. Proc Natl Acad Sci USA. 1997;94:961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hohlweg U, Doerfler W. On the fate of plant or other foreign genes upon the uptake in food or after intramuscular injection in mice. Mol Genet Genomics. 2001;265:225–233. [DOI] [PubMed] [Google Scholar]
- 49.Doerfler W. Beware of manipulations on the genome: epigenetic destabilization through (foreign) DNA insertions. Epigenomics. 2016;8:587–591. [DOI] [PubMed] [Google Scholar]