

ABSTRACT
During the recording of whole cell currents from stably transfected HEK-293 cells, the decline of currents carried by the recombinant human Cav2.3+β3 channel subunits is related to adenosine triphosphate (ATP) depletion after rupture of the cells. It reduces the number of functional channels and leads to a progressive shift of voltage-dependent gating to more negative potentials (Neumaier F., et al., 2018). Both effects can be counteracted by hydrolysable ATP, whose protective action is almost completely prevented by inhibition of serine/threonine but not tyrosine or lipid kinases. These findings indicate that ATP promotes phosphorylation of either the channel or an associated protein, whereas dephosphorylation during cell dialysis results in run-down. Protein phosphorylation is required for Cav2.3 channel function and could directly influence the normal features of current carried by these channels. Therefore, results from in vitro and in vivo phosphorylation of Cav2.3 are summarized to come closer to a functional analysis of structural variations in Cav2.3 splice variants.
KEYWORDS: Covalent modification, exon skipping, facilitation, protein kinase A, protein kinase C, splice variants
Introduction
Covalent modification of ion channels and other proteins is a common event during cellular growth, metabolic regulation and neuronal signalling. Many cell signalling pathways act on voltage-gated calcium channels to fine tune their activities and to control their cell surface expression. They are often modulated via G protein-coupled receptors, which control downstream events by posttranslational modification including phosphorylation of the pore-forming Cavα1-subunit or interacting auxiliary proteins [1].
The exact molecular mechanisms of these biochemical changes are only partially known for individual calcium channel subtypes. Best investigated are certain members of the L-type (Cav1) subfamily, which comprises the skeletal muscle Cav1.1 subunit and three additional homologous subunits (Cav1.2, Cav1.3 and Cav1.4). They are expressed in various tissues including cardiac, smooth muscle and neuronal regions (for details see [2,3]). It was the sympathetic regulation of cardiac activity [4], which was investigated first and most intensive to understand how Cav1.2 calcium channels may modulate heart beat frequency and power during rest and working activity (for details [5]).
Members of the second calcium channel subfamily (Cav2) are mainly expressed in the neuronal system [6] and were early analyzed as in vitro phosphorylation targets of protein kinase A [7]. Cav2.3 containing voltage-gated calcium channels are known since 1993, when the first clone was found in Discopyge ommata [8]. During the same time a mammalian counterpart was identified by electrophysiological measurements [9,10]. Initially, it was proposed that the Cav2.3-gene (cacna1e) may encode a structurally unknown T-type calcium channel [11]. However, depending on the recording conditions in recombinant systems, the human counterpart proved to be a mid-voltage gated calcium channel [12,13].
Members of the third calcium channel subfamily (Cav3) show unique gating properties, which make them important for neuronal spontaneous firing, for pacemaker activities, for rebound burst firing and additional oscillating processes [14]. They contribute to hyperexcitability disorders [15,16] such as epilepsy [17] and their blockade prevents tonic-clonic seizures [18]. At physiological temperatures (30 – 37°C), activation of PKA as well as PKC (but not PKG) result in a potent increase of currents mediated by the members of this subfamily (Cav3.1, Cav3.2 and Cav3.3) [18]. So far, the Cav3.2 T-type Ca2+ channel is the only voltage-gated Ca2+-channel, which was immunopurified from rat brain to identify in vivo phosphorylation sites by high-resolution mass spectroscopy [19]. Interestingly, a hot spot locus of phosphorylation sites in the I-II loop was found to be critical for the channels regulation. Even more important, within the 34 sites found in vivo, a total of 26 sites was never described among in vivo phosphosites and 4 sites were not even predicted to be phosphorylated by widely used computer algorithms, underscoring the limitations of algorithm-based predictions [19].
Physiological and pathophysiological roles of Cav2.3/R-type voltage-gated Ca2+ channels
Using subtype-specific antibodies, the regional and subcellular localization of Cav2.3 was characterized in mice and rats at both light and electron microscopic levels [20]. Cav2.3 immunogold particles were found to be predominantly presynaptic in the interpeduncular nucleus, but postsynaptic in other brain regions. Serial section analysis of electron microscopic images from the hippocampal CA1 region revealed a higher density of immunogold particles in the dendritic shaft plasma membrane compared with the pyramidal cell somata [20].
Cav2.3 channel expression appears to be required for long term potentiation (LTP) induced by short brief tetani [21], as presynaptic LTP and posttetanic potentiation (PTP) are impaired in Cav2.3-deficient mice as well as in control mice which were pharmacologically antagonized [21,22].
Additional evidence that Cav2.3 channels contribute to neurotransmission arises from expression studies in neurons. Exogenous Cav2.3 channels expressed by cDNA injection in cultured superior cervical ganglion neurons mediate cholinergic synaptic transmission [23]. This supports earlier results from Wu et al. (1998) who demonstrated that calcium influx through Cav2.3 channels is able to evoke transmitter release in the calyx of Held from young mice large enough to initiate an action potential in the postsynaptic neuron. However, transmitter release mediated by Cav2.3 channels is less efficient than transmitter release by Cav2.1 (P/Q-) and Cav2.2 (N-type) Ca2+ currents. The exact mechanisms underlying these differences are not fully clear [24].
The coupling to synaptic transmission varies within the subfamily of Cav2 channels over different frequency ranges with consequences for the frequency tuning of both synaptic dynamics and presynaptic neuromodulation [25]. Relative to Cav2.1, Cav2.2 had a disproportionately reduced contribution to synaptic transmission at frequencies >20 Hz, while Cav2.3 had a disproportionately increased contribution to synaptic transmission at frequencies >1 Hz. These activity-dependent effects of different Cav2 family members shape the filtering characteristics of GABAB receptor-mediated presynaptic inhibition [25].
Mice lacking the Cav2.3 channel showed altered pain responses. Obviously, the Cav2.3 channel controls pain behavior by both spinal and supraspinal mechanisms [26]. In HEK-293 cells expressing members of the Cav2 subfamily, the mechanism of Ca2+ channel modulation by opioid receptor (OR) activation was analyzed and the different types of ORs were cotransfected with Cav2.3. Selective agonists of µ-, δ-, and κ-ORs inhibited IBa through Cav2.3 channels by 35%. Cav2.2 channels were inhibited more potently [27].
Within several unexpected tissues the ablation of Cav2.3 channels disturbed the regularity of rhythmic processes like heart beat [28–30], peptide hormone secretion [31–33] and sensory signal transduction [34,35]. Depending on the mouse model used for ablation of Cav2.3, opposite results have been reported with regard to the role of Cav2.3 channels during sleep [36,37].
The general ablation of Cav2.3 channels alters seizure susceptibility after treatment with kainate in mice, so that hippocampal seizure resistance is increased [38] and neuronal excitotoxicity reduced [39]. Thus, the molecular understanding of the Cav2.3 channel gating and its modulation is needed to adopt future therapies for cardiac, endocrinal and neuronal diseases.
Pharmacological properties of Cav2.3
When the primary sequence for the ray Cav2.3 was deduced and its cRNA was functionally expressed in Xenopus oocytes, it was finally described as a so called “pharmacoresistant” (R-type) voltage-gated Ca2+ channel. In cerebellar granule cells the counterpart for the ray version was identified and its kinetic and pharmacological properties differed from the known Ca2+ subtypes [9,10]. In 1998 the peptide toxin SNX-482 was identified in the tarantula Hysterogrates gigas, which blocked in rat neurophypophyseal nerve terminals at low nanomolar concentrations the R-type Ca2+ currents, but not in several types of rat central neurons [40]. Later on, it was found that SNX-482 dramatically reduces the A-type K+ current in acutely dissociated dopamine neurons from the mouse substantia nigra pars compacta with an IC50 of less than 3 nM [41]. Therefore, a highly selective antagonist of Cav2.3/R-type currents is not available today but needed urgently.
Cav2.3 channels are very sensitive towards divalent metal cations, similar as some T-type channels [42,43]. Further, testing the convulsive drug kainate, which is routinely used for experimentally induced epilepsy, led us to the conclusion that the sensitivity of Cav2.3 channels towards divalent bioavailable metal cations must be investigated more in detail to understand physiological imbalances of Zn2+ and Cu2+ concentrations [44] in neuronal and other tissues, which may explain changes in excitability causing neurological and other diseases.
As an important side remark, recombinant Cav2.3 Ca2+ channels represent low affinity dihydropyridine receptors for nicardipine in COS-7 cells [45] and for racemic isradipine in HEK-293 cells [28]. Therefore, it may not be excluded that the therapeutical use of high local dihydropyridine concentrations after subarachnoidal hemorhage (SAH) do target the Cav2.3/R-type channel, which increases its expression after SAH [46–48].
Early investigations of Cav2.3 phosphorylation
Early after the detection of the pharmacoresistant Ca2+ channel types and based on the published deduced primary sequences, channel-specific antibodies were raised and used for in vivo characterization of Cav2.3 expression in the rat brain [49]. The authors determined in partially purified preparations the phosphorylation of the anti-Cav2.3 immunopositive protein(s) by 4 different kinases, comprising cAMP-dependent protein kinase (PKA), cGMP-dependent protein kinase (PKG), protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). Immunoblotting identified a polypeptide of 245 to 255 kDa, which was not a high affinity receptor for classical Ca2+ channel blockers [49].
Prediction of putative phosphorylation sites increases over time
As soon as the deduced primary sequences of newly cloned voltage-gated Ca2+ channels became identified, software packages (Genetics Computer Group, 1991) could be used to predict putative phosphorylation sites within the deduced sequences. For example, more than a dozen different sites for PKA are predicted to be present in the human Cav2.3d variant, 3 of them located in the I-II loop (close to a putative EF-hand), 4 of them in the II-III loop, one of them in the distal and a cluster of 6 in the proximal C-terminus [13]. Meanwhile, about 10 different online predictors are available as software and the number of potential sites for phosphorylation by various protein kinases has increased dramatically. According to the information on their website (http://gps.biocuckoo.org/index.php), the newly released Group-based Prediction System (GPS 3.0) covers a novel peptide selection and more than 6000 phosphorylation sites for training, allowing for the prediction of kinase-specific phosphorylation sites for 464 human protein kinases in hierarchy.
Using the human primary Cav2.3 sequence of the fetal brain (Cav2.3d, L27745.2) at the highest prediction threshold, the software predicts a total of 95 consensus sites for the cAMP-dependent protein kinase. Thus, the number of predicted sites is large and cannot be tested easily. But in vivo analysis as done recently for the Cav3.2/T-type channel [19] could be performed as a more reliable approach.
Functional differences of recombinant Cav2.3 splice variants modulated by phorbol ester stimulation and protein kinase c inhibition
The subfamily Cav2 of voltage-gated calcium channels had been investigated more systematically for its function as targets for PKC-dependent modulation in combination with a molecular crosstalk to other second messenger pathways [50]. For the tested recombinant channel types in Xenopus oocytes, only Cav2.2 and Cav2.3 but not Cav2.1 were sensitive towards PKC activation by PMA (phorbol 12-myristate 13 acetate) [51]. During these investigations, the cytosolic loop between domain I and II was identified as an important integration center between PKC-mediated activation and G protein dependent modulation of the Cav2.2/N-type Ca2+ channels [52].
Structure – function relation of the PKC-mediated Cav2.3 activation was investigated in more detail in HEK-293 cells when comparing the transient facilitation of different Cav2.3 splice variants by Ca2+-influx. With Ba2+ as charge carrier, the tested biophysical properties were similar. In Ca2+, the inactivation time course was slower and the recovery from short-term inactivation was faster only in splice variants containing a 19-amino-acid-long arginine-rich insertion, which is typical for subset of neuronal Cav2.3 calcium channel subunits [53–55].
The mechanism underlying the Ca2+ dependent increase of channel activity was investigated by introduction of the II-III loop of Cav2.1 channels, which abolished the transient Ca2+ mediated stimulation as well as the phorbol ester (PMA or PDBu) sensitivity of the resulting construct [56]. Also the 19 amino acids long arginine-rich insertion (encoded by exon 19) was needed in the recombinant system for PKC-mediated stimulation of Cav2.3, leading to the conclusion that only the splice variants Cav2.3c and Cav2.3d (both containing exon 19 encoded sequences) are subject to Ca2+ and phorbolester mediated stimulation. Interestingly, the arginine-rich segment contains no serine or threonine residues, as would be expected if PKC-mediated stimulation involves phosphorylation of this region. Instead, it could harbor a PKC binding site or other determinants for the association of Cav2.3 channels with PKC, which may be required for phosphorylation of other sites in the Cav2.3 channel protein. This interpretation is partly supported by the observation that the FLAG-tagged II-III loop of Cav2.3 augments the autophosphorylation of exogenously added PKCα [57].
During the years several serine/threonine sites in Cav2.3 were identified, which mediate stimulatory and inhibitory effects of protein kinase C isozymes (for an overview see [58]). Some sites reside in the I-II linker, where G-protein binding was identified close to the predicted phosphorylation sites for the other two members of the Cav2 subfamily [59–61]. Interestingly, the effects on Cav2.3 are dependent on the PKC-isozyme used for activation, and PKC mediates stimulatory and inhibitory effects on Cav2.3-mediated currents [58,62].
Quantitative phospho-proteomic studies get closer to the in vivo situation
Compared to other posttranslational modifications, protein phosphorylation causes one of the most widespread covalent structural changes, which involves transfer of a negatively charged phosphate group to certain amino acid side chains of newly synthetized proteins [63]. Apart from the formation of acid stable phosphomonoesters of serine, threonine and tyrosine, other less stable covalent modifications have been described, such as low stability phosphohistidine residues, which are problematic to analyze by conventional proteomic methods [64,65]. A quantitative phosphoproteomic analysis is highly useful for identifying and characterizing changes in protein function and for mapping associated regulatory pathways. Mass spectrometric analysis of stable isotope labelled samples has progressed and meanwhile the enrichment of phosphopeptides has improved to a level that allows for identification of less stable and low abundant phospopeptides. The new techniques pay attention to the fact that in vivo phosphorylation of a target protein is often a transient event [66–68] and typically occurs at low stoichiometry [65]. Also, phosphoproteomic analyzes initially restricted to single organs of the mouse [69] have been extended to large scale human interactome analyzes [70], which provide insights into hundreds of poorly characterized proteins and interconnecting signal transduction pathways.
Interestingly, patterns of Ca2+ channel phosphorylation observed in phosphoproteomic analyzes reveal a clustering of phosphorylation sites at two cytoplasmic regions of the Cavα1-subunits, which comprise the II-III loop and the carboxy terminus [71]. For the Cav2.3 α1-subunit, most identified phosphorylation sites are located within the II-III loop (Table 1).
Table 1.
Phosphorylation sites identified in the mouse brain Cav2.3 α1-subunit (Q61290) [71]. The GenBank entry Q61290 mentions only part of the sites, which were originally listed as in vivo identified phosphopeptides (labelled in bold). In addition, this so called large scale analysis identified residue Ser-816 in the II-III loop as an additional phosphorylation site.
| Location within the Cav2.3 α1-subunit: | ||||
|---|---|---|---|---|
| N-terminus | I-II loop | II-III loop | III-IV loop | C-terminus |
|
Ser-15 Ser-20 Thr-29 |
Ser-428 Thr-441 |
Ser-737 Ser-740 Ser-746 Ser-793 Ser-794 Ser-856 Thr-865 Ser-866 Ser-873 Ser-876 Ser-1051 Ser-1056 Thr-1094 |
– none - |
Ser-2054 Thr-2067 Ser-2073 |
For the vertebrate Cav2.3 at least 3 major splice variants (Cav2.3c, Cav2.3d, and Cav2.3e) have been identified by cloning and RT-PCR. They have been characterized electrophysiologically showing significant differences for the fast component of the time constants for the recovery from short-term inactivation, when lacking the arginine-rich insert within the II-III loop [53].
One of the in vivo identified phosphopeptides of the II-III loop from the mouse brain (Ser-746) is located in close neighbourhood to the arginine-rich insert of the II-III loop, which is encoded by the alternative exon 19 (Figure 1(b) and 1(e)).
Figure 1.
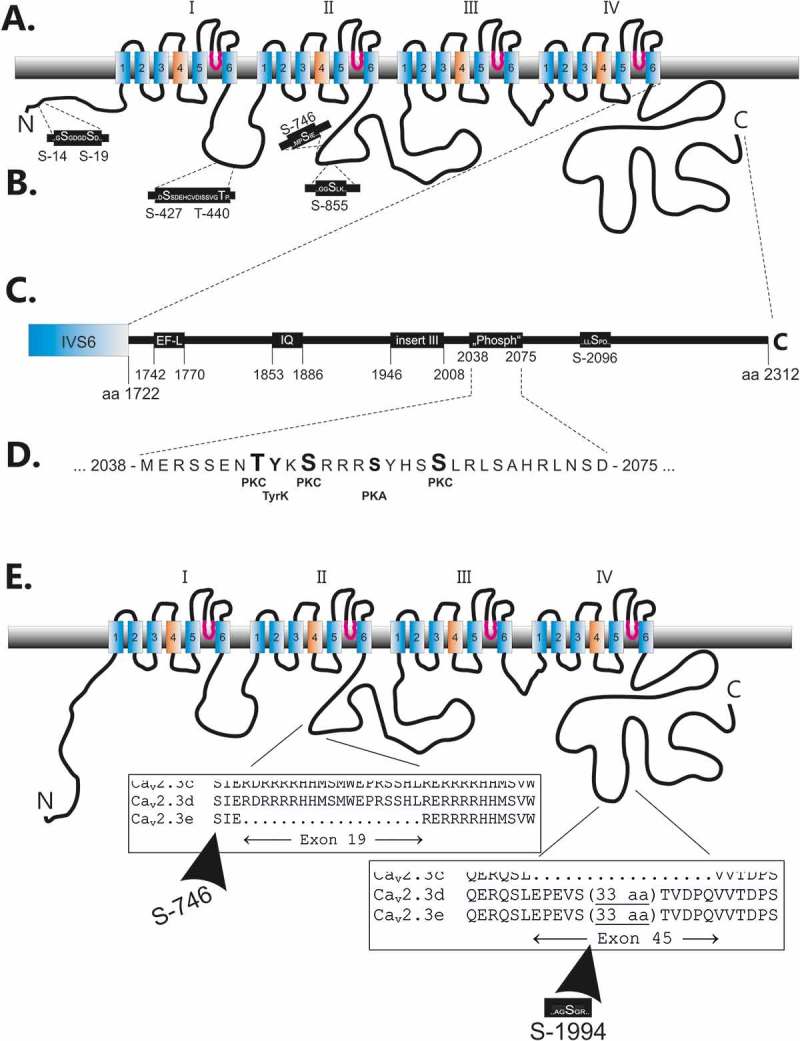
Location of predicted phosphorylation sites within human Cav2.3d. (a). Cartoon of the deduced transmembrane organization of the human Cav2.3d subunit. (b). Predicted phosphorylation sites within the amino terminus, the I-II loop and the II-III loop. (c). and (d). Predicted phosphorylation sites within the carboxy terminus including a cluster of putative phosphorylation sites for PKC, PKA and tyrosine kinase downstream of important functional domains (EF-like and IQ site as well as the inserted exon 45 of the splice variant d of Cav2.3). (E). A detailed view of the alternate exons 19 and 45, which are identified in different Cav2.3 splice variants (for details see 53) is shown. Note within exon 45 one additional PKC site, which was predicted.
Another remarkable but only putative phosphorylation site, predicted to be phosphorylated by protein kinase C, is located within the alternative exon 45 (Figure 1(e)). So far, no homologous phosphopeptide was identified in mouse brain studies.
Cloned human Cav2.3d voltage gated calcium channels [13], co-expressed with human β3 [72], are regulated by protein kinases and phosphatases in HEK-293 cells [73]. This is reflected in a spontaneous decline of current responses (run-down) and changes in channel gating in dialysed cells, which can be prevented by provision of hydrolysable ATP. Importantly, the protective action of ATP is prevented by inhibition of serine/threonine but not tyrosine or lipid kinases, suggesting that protein phosphorylation is required to prevent rapid de-phosphorylation by one or more associated protein phosphatases and thus maintain the normal function of Cav2.3 channels.
It will be a future challenge to correlate the identified putative and real phosphorylation sites with observed molecular movements in the channel protein structure, in order to understand the macroscopic changes in function of the body.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [SCHN 387/21-1].
Acknowledgments
We thank Renate Clemens for their excellent technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Huang J, Zamponi GW.. Regulation of voltage gated calcium channels by GPCRs and post-translational modification. Curr Opin Pharmacol. 2017;32:1–8. [DOI] [PubMed] [Google Scholar]
- [2].Hofmann F, Lacinova L, Klugbauer N.. Voltage-dependent calcium channels: from structure to function. Rev Physiol Biochem Pharmacol. 1999;139:33–87. [DOI] [PubMed] [Google Scholar]
- [3].Catterall WA. Voltage-Gated Calcium Channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reuter H. Discovery of Cardiac Calcium Channels. Curr Mol Pharmacol. 2015;8:4–7. [DOI] [PubMed] [Google Scholar]
- [5].Hofmann F, Flockerzi V, Kahl S, et al. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev. 2014;94:303–326. [DOI] [PubMed] [Google Scholar]
- [6].Mochida S. Presynaptic calcium channels. Neurosci Res (N Y). 2018;127:33–44. [DOI] [PubMed] [Google Scholar]
- [7].Hell JW, Yokoyama CT, Breeze LJ, et al. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Horne WA, Ellinor PT, Inman I, et al. Molecular diversity of Ca2+ channel alpha 1 subunits from the marine ray Discopyge ommata. Proc Natl Acad Sci USA. 1993;90:3787–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ellinor PT, Zhang JF, Randall AD, et al. Functional expression of a rapidly inactivating neuronal calcium channel. Nature. 1993;363:455–458. [DOI] [PubMed] [Google Scholar]
- [10].Zhang J-F, Randall AD, Ellinor PT, et al. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. [DOI] [PubMed] [Google Scholar]
- [11].Soong TW, Stea A, Hodson CD, et al. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. [DOI] [PubMed] [Google Scholar]
- [12].Williams ME, Marubio LM, Deal CR, et al. Structure and functional characterization of neuronal alpha 1E calcium channel subtypes. J Biol Chem. 1994;269:22347–22357. [PubMed] [Google Scholar]
- [13].Schneider T, Wei X, Olcese R, et al Molecular analysis and functional expression of the human type Eneuronal Ca2+ channel alpha-1 subunit. Receptors Channels. 1994;2:255–270. [PubMed] [Google Scholar]
- [14].Cheong E, Shin HS. T-type Ca2+ channels in normal and abnormal brain functions. Physiol Rev. 2013;93:961–992. [DOI] [PubMed] [Google Scholar]
- [15].Kim D, Song I, Keum S, et al. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron. 2001;31:35–45. [DOI] [PubMed] [Google Scholar]
- [16].Song I, Kim D, Choi S, et al. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004;24:5249–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Becker AJ, Pitsch J, Sochivko D, et al. Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J Neurosci. 2008;28:13341–13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sakkaki S, Gangarossa G, Lerat B, et al. Blockade of T-type calcium channels prevents tonic-clonic seizures in a maximal electroshock seizure model. Neuropharmacology. 2016;101:320–329. [DOI] [PubMed] [Google Scholar]
- [19].Blesneac I, Chemin J, Bidaud I, et al. Phosphorylation of the Cav3.2 T-type calcium channel directly regulates its gating properties. Proc Natl Acad Sci USA. 2015;112:13705–13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Parajuli LK, Nakajima C, Kulik A, et al. Quantitative regional and ultrastructural localization of the Ca(v)2.3 subunit of R-type calcium channel in mouse brain. J Neurosci. 2012;32:13555–13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dietrich D, Kirschstein T, Kukley M, et al. Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron. 2003;39:483–496. [DOI] [PubMed] [Google Scholar]
- [22].Breustedt J, Vogt KE, Miller RJ, et al. 1E-Containing Ca2+ channels are involved in synaptic plasticity. Proc Natl Acad Sci USA. 2003;100:12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mochida S, Westenbroek RE, Yokoyama CT, et al. Subtype-selective reconstitution of synaptic transmission in sympathetic ganglion neurons by expression of exogenous calcium channels. Proc Natl Acad Sci USA. 2003;100:2813–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kamp MA, Krieger A, Henry M, et al. Presynaptic ‘Ca2.3-containing’ E-type Ca channels share dual roles during neurotransmitter release. Eur J Neurosci. 2005;21:1617–1625. [DOI] [PubMed] [Google Scholar]
- [25].Ricoy UM, Frerking ME. Distinct roles for Cav2.1-2.3 in activity-dependent synaptic dynamics. J Neurophysiol. 2014;111:2404–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saegusa H, Kurihara T, Zong SQ, et al. Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proc Natl Acad Sci USA. 2000;97:6132–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Berecki G, Motin L, Adams DJ. Voltage-gated R-type calcium channel inhibition via human μ-, δ-, and κ-opioid receptors is voltage-independently mediated by Gβγ protein subunits. Mol Pharmacol. 2016;89:187–196. [DOI] [PubMed] [Google Scholar]
- [28].Lu Z-L, Pereverzev A, Liu H-L, et al Arrhythmia in isolated prenatal hearts after ablation of the Cav2.3(alpha 1E) subunit of voltage-gated Ca2+ channels. Cell Physiol Biochem. 2004;14:11–22. [DOI] [PubMed] [Google Scholar]
- [29].Galetin T, Tevoufouet EE, Sandmeyer J, et al. Pharmacoresistant Cav2·3 (E-type/R-type) voltage-gated calcium channels influence heart rate dynamics and may contribute to cardiac impulse conduction. Cell Biochemistry Funct. 2013;31:434–449. [DOI] [PubMed] [Google Scholar]
- [30].Tevoufouet EE, Nembo EN, Distler F, et al Multiple nickel-sensitive targets elicit cardiac arrhythmia inisolated mouse hearts after pituitary adenylate cyclase-activatingpolypeptide-mediated chronotropy. Pharmacol Res. 2017;117:140–147. [DOI] [PubMed] [Google Scholar]
- [31].Pereverzev A, Mikhna M, Vajna R, et al. Disturbances in glucose-tolerance, insulin-release, and stress-induced hyperglycemia upon disruption of the Ca(v)2.3 (alpha 1E) subunit of voltage-gated Ca(2+) channels. Mol Endocrinol. 2002;16:884–895. [DOI] [PubMed] [Google Scholar]
- [32].Jing X, Li DQ, Olofsson CS, et al. CaV2.3 calcium channels control second-phase insulin release. J Clin Invest. 2005;115:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang Q, Bengtsson M, Partridge C, et al. R-type Ca(2+)-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat Cell Biol. 2007;9:453–460. [DOI] [PubMed] [Google Scholar]
- [34].Weiergräber M, Henry M, Ho MSP, et al. Altered thalamocortical rhythmicity in Ca(v)2.3-deficient mice. Mol Cell Neurosci. 2008;39:605–618. [DOI] [PubMed] [Google Scholar]
- [35].Zaman T, Lee K, Park C, et al. Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron. 2011;70:95–108. [DOI] [PubMed] [Google Scholar]
- [36].Münch A, Dibué M, Hescheler J, et al. Modulation des Schlafs durch spannungsgesteuerte Cav2.3-E- und Cav2.3-R-Typ-Kalziumkanäle in Mäusen. Somnologie. 2013;17:185–192. [Google Scholar]
- [37].Siwek ME, Muller R, Henseler C, et al. The CaV2.3 R-type voltage-gated Ca2+ channel in mouse sleep architecture. Sleep. 2014;37:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Weiergräber M, Henry M, Radhakrishnan K, et al. Hippocampal seizure resistance and reduced neuronal excitotoxicity in mice lacking the Cav2.3 E/R-type voltage-gated calcium channel. J Neurophysiol. 2007;97:3660–3669. [DOI] [PubMed] [Google Scholar]
- [39].Dibue-Adjei M, Kamp MA, Alpdogan S, et al. Cav2.3 (R-Type) Calcium Channels are Critical for Mediating Anticonvulsive and Neuroprotective Properties of Lamotrigine In Vivo. Cell Physiol Biochem. 2017;44:935–947. [DOI] [PubMed] [Google Scholar]
- [40].Newcomb R, Szoke B, Palma A, et al. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. [DOI] [PubMed] [Google Scholar]
- [41].Kimm T, Bean BP. Inhibition of A-type potassium current by the peptide toxin SNX-482. J Neurosci. 2014;34:9182–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shcheglovitov A, Vitko I, Lazarenko RM, et al. Molecular and biophysical basis of glutamate and trace metal modulation of voltage-gated Ca(v)2.3 calcium channels. J Gen Physiol. 2012;139:219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Neumaier F, Dibué-Adjei M, Hescheler J, et al. Voltage-gated calcium channels: determinants of channel function and modulation by inorganic cations. Prog Neurobiol. 2015;129:1–36. [DOI] [PubMed] [Google Scholar]
- [44].Neumaier F, Akhtar-Schafer I, Luke JN, et al. J Neurochem. 2018. DOI: 10.1111/jnc.14546 [DOI] [PubMed] [Google Scholar]
- [45].Stephens GJ, Page KM, Burley JR, et al. Functional expression of rat brain cloned alpha1E calcium channels in COS-7 cells. Pflugers Arch. 1997;433:523–532. [DOI] [PubMed] [Google Scholar]
- [46].Ishiguro M, Wellman TL, Honda A, et al. Emergence of a R-type Ca2+ channel (CaV 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ Res. 2005;96:419–426. [DOI] [PubMed] [Google Scholar]
- [47].Van Lieshout JH, Dibue-Adjei M, Cornelius JF, et al. Neurosurg Rev. 2017. DOI: 10.1007/s10143-017-0827-y [DOI] [Google Scholar]
- [48].Albanna W, Neumaier F, Luke JN, et al. CNS Neurosci Ther. 2017. DOI: 10.1111/cns.12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yokoyama CT, Westenbroek RE, Hell JW, et al. Biochemical properties and subcellular distribution of the neuronal class E calcium channel alpha 1 subunit. J Neurosci. 1995;15:6419–6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zamponi GW, Bourinet E, Nelson D, et al. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. [DOI] [PubMed] [Google Scholar]
- [51].Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. [DOI] [PubMed] [Google Scholar]
- [52].Hamid J, Nelson D, Spaetgens R, et al. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem. 1999;274:6195–6202. [DOI] [PubMed] [Google Scholar]
- [53].Pereverzev A, Leroy J, Krieger A, et al. Alternate splicing in the cytosolic II-III loop and the carboxy terminus of human E-type voltage-gated Ca(2+) channels: electrophysiological characterization of isoforms.. Mol Cell Neurosci. 2002;21:352–365. [DOI] [PubMed] [Google Scholar]
- [54].Leroy J, Pereverzev A, Vajna R, et al. Ca2+-sensitive regulation of E-type Ca2+ channel activity depends on an arginine-rich region in the cytosolic II-III loop. Eur J Neurosci. 2003;18:841–855. [DOI] [PubMed] [Google Scholar]
- [55].Weiergräber M, Kamp MA, Radhakrishnan K, et al. The Ca(v)2.3 voltage-gated calcium channel in epileptogenesis--shedding new light on an enigmatic channel. Neurosci Biobehav Rev. 2006;30:1122–1144. [DOI] [PubMed] [Google Scholar]
- [56].Klöckner U, Pereverzev A, Leroy J, et al. The cytosolic II-III loop of Cav2.3 provides an essential determinant for the phorbol ester-mediated stimulation of E-type Ca2+ channel activity. Eur J Neurosci. 2004;19:2659–2668. [DOI] [PubMed] [Google Scholar]
- [57].Krieger A, Radhakrishnan K, Pereverzev A, et al. The molecular chaperone hsp70 interacts with the cytosolic II-III loop of the Cav2.3 E-type voltage-gated Ca2+ channel. Cell Physiol Biochem. 2006;17:97–110. [DOI] [PubMed] [Google Scholar]
- [58].Rajagopal S, Burton BK, Fields BL, et al. Stimulatory and inhibitory effects of PKC isozymes are mediated by serine/threonine PKC sites of the Cav2.3α1 subunits. Arch Biochem Biophys. 2017;621:24–30. [DOI] [PubMed] [Google Scholar]
- [59].Zhang JF, Ellinor PT, Aldrich RW, et al. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. [DOI] [PubMed] [Google Scholar]
- [60].Page KM, Stephens GJ, Berrow NS, et al. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Herlitze S, Hockerman GH, Scheuer T, et al. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel 1A subunit. Proc Natl Acad Sci USA. 1997;94:1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rajagopal S, Fang H, Patanavanich S, et al. Protein kinase C isozyme-specific potentiation of expressed Cav 2.3 currents by acetyl-beta-methylcholine and phorbol-12-myristate, 13-acetate. Brain Res. 2008;1210:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. [DOI] [PubMed] [Google Scholar]
- [64].Ciesla J, Fraczyk T, Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol. 2011;58:137–148. [PubMed] [Google Scholar]
- [65].Kweon HK, Andrews PC. Quantitative analysis of global phosphorylation changes with high-resolution tandem mass spectrometry and stable isotopic labeling. Methods. 2013;61:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Stadtman ER, Chock PB. Superiority of interconvertible enzyme cascades in metabolic regulation: analysis of monocyclic systems. Proc Natl Acad Sci USA. 1977;74:2761–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shacter E, Chock PB, Stadtman ER. Regulation through phosphorylation/dephosphorylation cascade systems. J Biol Chem. 1984;259:12252–12259. [PubMed] [Google Scholar]
- [68].Karin M, Hunter T. Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr Biol. 1995;5:747–757. [DOI] [PubMed] [Google Scholar]
- [69].Huttlin EL, Jedrychowski MP, Elias JE, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Huttlin EL, Bruckner RJ, Paulo JA, et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cerda O, Baek JH, Trimmer JS. Mining recent brain proteomic databases for ion channel phosphosite nuggets. J Gen Physiol. 2011;137:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Murakami M, Wissenbach U, Flockerzi V. Gene structure of the murine calcium channel beta3 subunit, cDNA and characterization of alternative splicing and transcription products. Eur J Biochem. 1996;236:138–143. [DOI] [PubMed] [Google Scholar]
- [73].Neumaier F, Alpdogan S, Hescheler J, et al Protein phosphorylation maintains the normal function of clonedhuman Cav2.3 channels. J Gen Physiol. 2018. doi: 10.1085/jgp.201711880. [Epub aheadof print] PMID: 29453293. [DOI] [PMC free article] [PubMed] [Google Scholar]