

Abstract
Innate immune complement activation may contribute to sickle cell disease (SCD) pathogenesis. Ischemia-reperfusion physiology is a key component of the inflammatory and vaso-occlusive milieu in SCD and is associated with complement activation. C5a is an anaphylatoxin, a potent pro-inflammatory mediator that can activate leukocytes, platelets, and endothelial cells, all of which play a role in vaso-occlusion. We hypothesize that hypoxia-reoxygenation (H/R) in SCD mice activates complement, promoting inflammation and vaso-occlusion. At baseline and after H/R, Townes-SS mice had increased C3 fragments and C5b-9 deposition in kidneys, livers and lungs and alternative pathway fragment Bb in plasma compared to control AA-mice. Activated complement promoted vaso-occlusion (microvascular stasis) in sickle (SS)-mice; infusion of zymosan-activated, but not heat-inactivated serum, induced substantial vaso-occlusion in the skin venules of SS-mice. Infusion of recombinant C5a induced stasis in SS, but not AA-mice that was blocked by anti-C5a receptor (C5aR) IgG. C5a-mediated stasis was accompanied by inflammatory responses in SS-mice including NF-κB activation and increased expression of TLR4 and adhesion molecules VCAM-1, ICAM-1, and E-selectin in the liver. Anti-C5aR IgG blocked these inflammatory responses. Also, C5a rapidly up-regulated Weibel-Palade body P-selectin and von Willebrand factor on the surface of human umbilical vein endothelial cells in vitro and on vascular endothelium in vivo. In SS-mice, a blocking antibody to P-selectin inhibited C5a-induced stasis. Similarly, an antibody to C5 that blocks murine C5 cleavage or an antibody that blocks C5aR inhibited H/R-induced stasis in SS-mice. These results suggest that inhibition of C5a may be beneficial in SCD.
1. INTRODUCTION
The pathophysiology of sickle cell disease (SCD) includes anemia, oxidative stress, hemolysis, inflammation and vaso-occlusion.1 Inflammatory vascular episodes are a hallmark of SCD. Red cell, platelet and leukocyte activation and aggregation, together with activation of endothelial cell adhesion molecules such as P-selectin mediate vascular occlusion and the consequent organ damage that is characteristic of SCD.2 Ischemia-reperfusion physiology underpins SCD biology when vessels are transiently occluded in organs throughout the body and subsequently reopened.3,4 In this ischemia-reperfusion paradigm, the innate immune system plays a key role in promoting the inflammatory response, which includes TLR4 signaling and complement activation.5–14
The first report that complement activation may be involved in SCD was published in 1967 by Francis and Womack.15 Since then, many studies have reported increased levels of complement-derived fragments in the blood of SCD patients, demonstrating that complement is activated in SCD and suggesting that complement may play an important role in the pathophysiology of the disease.9–13,16 Several studies suggested that complement activation takes place through the alternative pathway, which would be triggered by the membrane phospholipid changes that occur in SS red blood cells (RBCs).10,11 Moreover, recent investigations have suggested that alternative pathway activation may be increased by a high blood concentration of phosphatidylserine-rich microparticles as may occur in SCD; alternative pathway activation can also be demonstrated in stored RBCs and this pathway is activated due to heme released by lysed RBCs.11,14,17–21 Therefore, these studies of SCD patients showing evidence of complement activation suggest that complement, another major component of innate immunity, may play a fundamental role in the pathophysiology of SCD.
Despite these many investigations demonstrating that complement activation occurs in SCD patients, few studies have addressed the question as to whether complement plays any role in the severe clinical manifestations of SCD, especially vaso-occlusion and tissue injury that lead to substantial patient suffering and early mortality. Several complement-derived fragments, most prominently C5a, are known to activate the cells involved in SCD that mediate inflammation, pain, and vascular occlusion, including leukocytes, platelets, mast cells and endothelial cells.2,22–24
In this study we used Townes-SS mice, a well-defined model of SCD25 to investigate whether hypoxia/re-oxygenation (H/R), a mimic of ischemia-reperfusion physiology, activates complement and whether C5a induces P-selectin mediated vaso-occlusion. Our study demonstrates that C5a plays a fundamental role in inflammation, cell activation, and vaso-occlusion.
2. METHODS
2.1. Mice
All animal experiments were approved by the University of Minnesota’s Institutional Animal Care and Use Committee. These studies utilized male and female Townes-SS mice on a 129/B6 mixed genetic background.25 The SS mice were created by knocking in human α and AɣβS globins into the deletion sites for murine α- and β-globins. SS mice have anemia and an SS-RBC half-life of 2.5 days (d). Townes-AA control mice express normal human α and AɣβA globins with a 16 d RBC half-life. Townes-AS mice are heterozygous for βA and βS globins with a 11.5 d RBC half-life.26 All animals were housed in specific pathogen-free rooms to limit infections and kept on a 12 hour (h) light/dark cycle at 21°C. All animals were monitored daily including weekends and holidays for health problems, food and water levels and cage conditions. Littermates were randomly assigned to different treatment groups. All animals were included in each endpoint analysis and there were no unexpected adverse events that required modification of the protocol. Mice were aged 8 −14 weeks.
2.2. Immunofluorescence staining of tissues
Townes-SS and AA mice were exposed to H/R with 1 h in hypoxia (7% O2/93% N2) followed by 1 h in room air before tissue collection. Control SS and AA mice remained in room air before tissue collection. Alternatively, SS and AA mice were infused with vehicle (saline) or recombinant mouse complement fragment C5a (R&D Systems#2150-C5) before tissue collection. Kidneys, livers, lungs and dorsal skin were collected 30 min after infusion and placed in optimal cutting temperature (OCT) compound, snap-frozen in liquid N2 and stored at −850C prior to frozen sectioning in a microtome-cryostat into 6 μm (kidneys, livers, lungs) or 60 μm (skin) sections. Tissues collected from mice exposed to H/R and their room air controls were stained with primary antibodies to C3 activation fragments C3b/iC3b/C3c (Hycult #HM1065HycultHM1065), C5b-9 (Abcam #ab55811) and endothelial cell marker CD31 (Abcam #ab119341). Tissues collected from mice infused with C5a or vehicle were stained with primary antibodies to P-selectin (R&D Systems #AF737), von Willebrand factor (vWF, Cedarlane #CL20176A-R) and endothelial cell marker CD31 (Cedarlane #CL-8930AP) or a platelet marker CD41 (BD Biosciences #562957). Primary antibodies in tissues were identified with the appropriate fluorescent-labeled secondary antibodies (Jackson Immunoresearch). Slides were mounted using DPX mounting medium (Electron Microscope Sciences #13514),) visualized, and images acquired using a FluoView FV1000 BX2 upright confocal microscope (Olympus, Center Valley, PA) with UPlanSApo 20X/0.80 and UPlanApo N 60X/1.42 objectives with zoom (Z) 2 as indicated. Images were processed with FluoView (Olympus) and Adobe Photoshop software (San Jose, CA). C3 activation fragments, C5b-9, P-selectin and vWF were quantitated by counting the number of positive pixels and dividing that by the total number of tissue pixels in the image (Adobe Photoshop) to generate percent positive pixels for each protein of interest. Results were counted from 3 independent image fields collected from each mouse organ (n=3) for a total of 9 independent fields used for each condition. Results are expressed as mean percent ± SD.
2.3. Measurement of vaso-occlusion (microvascular stasis)
Townes-SS and AA mice were anesthetized with a mixture of ketamine (106 mg/kg) and xylazine (7.2 mg/kg) and implanted with dorsal skin-fold chambers. After implantation, mice were placed on an intravital microscopy stage and 20–24 flowing subcutaneous venules in the chamber window were selected and mapped as previously described.27 After baseline selection of flowing venules, mice were infused with zymosan-treated mouse serum, zymosan-treated heat-inactivated serum, recombinant murine C5a (R&D Systems#2150-C5) or vehicle (saline). Alternatively, mice were exposed to H/R (7% O2/93% N2 for 1 h followed by room air). Each of the same venules selected and mapped at baseline were visually re-examined for stasis (no flow) at 1 and 4 hours after infusion or H/R. The static venules in each mouse were counted and percent stasis at 1 and 4 h was calculated by dividing the number of static venules by the total (static + flowing) number of venules. In some experiments, SS mice were pre-treated 30 min prior to C5a infusion or H/R with either a low endotoxin/azide-free blocking monoclonal IgG (30 μg/mouse) to murine C5aR (Biolegend #135804) or to P-selectin (BD Biosciences #553741). Alternatively, a monoclonal IgG that blocks cleavage of murine C5 (BB5.1, a kind gift to the Taylor lab from Dr. B. Stockinger), or an appropriate isotype control IgG was used.
2.4. Immunoblots
Microsomes and nuclear extracts were isolated from tissues of mice as previously described.28 Immunoblots of cellular subfractions or EDTA plasma (5 μL) were immunostained with primary antibodies to complement activation fragment Bb (GeneTex #GTX86947), NF-ĸB phospho-p65 (Ser536, Cell Signaling #3031), total p65 (Cell Signaling #3034), TLR4 (Antibodies Online #361724), VCAM-1 (Abcam #174279), ICAM-1 (Abcam #ab124759) and loading controls GAPDH (Sigma-Aldrich #G9545) or IgG (Bio-Rad #170–6518). Primary antibodies were detected with appropriate secondary antibodies conjugated to alkaline phosphatase and visualized with ECF™ substrate (GE Healthcare) and a Typhoon FLA 9500 imager (GE Healthcare).
2.5. Statistics
Analyses were performed with SigmaStat 3.5 for Windows (Systat Software, San Jose, CA). Comparisons of two treatment groups were made using an unpaired Student’s t-test. Comparisons of three or more treatment groups were made using One Way Analysis of Variance using the Holm-Sidak method.
3. RESULTS
3.1. H/R induces complement activation
Ischemia-reperfusion physiology occurs in SCD with cycles of vaso-occlusion and reperfusion that can activate complement. We first investigated whether H/R induced complement activation and deposition in organs. We found that kidneys, livers and lungs of SS mice had significantly increased levels of C3 activation fragments and C5b-9 following H/R (Figures 1A-C, Supplemental Figures S1–3 and Supplemental Tables S1–3). In kidneys, control untreated SS mice had significantly more C3 activation fragments and C5b-9 deposition than control AA mice (Figure 1A [120X], Supplemental Figure S1 [20X] and Supplemental Tables S1–2 [Quantification]). Deposition of C3 fragments and C5b-9 increased significantly in AA and SS mice after H/R with SS+H/R being significantly higher than AA+H/R. In the kidney, C3 fragments were localized primarily on glomeruli and C5b-9 complexes were localized primarily around tubules.
Figure 1. Complement is activated after hypoxia-reoxygenation.




AA and SS mice (n=3/group) were exposed to 1 h of hypoxia (7% O2/93% N2) followed by 1 h of reoxygenation in room air (H/R). Control AA and SS mice were in room air. Organs were harvested, frozen in OCT compound and immunostained for C3 activation fragments (C3b/iC3b/C3c) (red), C5b-9 (green) and CD31 (blue). Representative images of organs from control and H/R-treated AA and SS mice are shown in (A) kidneys (60XZ2), (B) livers (60X) and (C) lungs (60X). Scale bars = 10 μm (kidneys) or 30 μm (livers and lungs). Gray insets in the merged images are magnified in the bottom row of each panel. (D) Plasma Bb activation fragments in AA and SS mice (n=4/group) were examined by immunoblot with and without exposure of mice to H/R. Each lane was loaded with 5 μL of EDTA plasma from untreated mice. Plasma IgG heavy chain was used as a loading control.
In livers, control untreated SS mice appeared to have slightly higher levels of C3 activation fragments and C5b-9 deposition (Figure 1B [60X2Z], Supplemental Figure S2 [20X]), but these differences were not significant (Supplemental Tables S3–4 [Quantification]). Deposition of C3 fragments and C5b-9 increased significantly in the livers of SS mice after H/R with SS+H/R being significantly higher than AA+H/R. In the livers of AA mice, C5b-9 complexes, but not C3 fragments, were significantly higher after H/R. In the liver, C3 fragments and C5b-9 complexes were localized around hepatic sinusoids and blood vessels.
In lungs, C3 activation fragments were similar in control untreated AA and SS mice. But C5b-9 deposition was greater in control SS mice compared to control AA mice (Figure 1C [60X], Supplemental Figure S3 [20X] and Supplemental Tables S5–6 [Quantification]). Deposition of C3 fragments and C5b-9 increased significantly in the lungs of AA and SS mice after H/R with SS+H/R being significantly higher than AA+H/R. In the lungs, C3 fragments and C5b-9 complexes were localized primarily around blood vessels. These results in kidneys, livers and lungs demonstrate that untreated SS mice have a low but significant level of ongoing complement activation and tissue deposition and this activation and deposition is greatly increased by H/R.
We confirmed that 1 h of hypoxia in 7% O2 induces RBC sickling in an SS mouse. When mice were normoxic, RBCs collected from venous blood of an SS mouse had 21% sickling. In contrast, after 1 h of hypoxia, RBCs collected from the same SS mouse had 69% sickling (Supplemental Figure S4). RBCs collected from an AA mouse had 0% sickling in normoxia and 5% after hypoxia.
Complement factor B, is a component of the alternative pathway that circulates in the blood as a single chain polypeptide. Upon activation of the alternative pathway, it is cleaved by factor D yielding the noncatalytic fragment Ba and the catalytic fragment Bb.29 We asked whether Bb was demonstrable in plasma. Immunoblots of plasma Bb showed more Bb in SS mice compared to AA mice and the Bb level was greatly increased in SS mice post-H/R (Figure 1D). However, plasma C5a levels were similar in AA and SS mice in normoxia and after 5, 15 or 30 minutes of H/R (Supplemental Figure S5A-B). We hypothesize that C5a might be generated locally on cell membranes by H/R and cleared rapidly by C5a receptors on cells without increasing C5a levels in the plasma.
3.2. C5a causes microvascular stasis (vaso-occlusion) in SS mice
Since H/R activates complement, we investigated whether complement activation products such as C5a would promote vaso-occlusion. We have previously shown that H/R triggers vaso-occlusion in sickle mice but not in normal control mice.27 We first tested the effect of mouse serum containing complement that was activated with zymosan, a strong, complement activator.30 We found that zymosan-activated mouse serum induced vaso-occlusion in SS mice, but not in SS mice infused with complement-inactive, zymosan-treated serum (heat-inactivated) (Figure 2A). This experiment suggests that a complement factor derived from complement activation may promote vaso-occlusion in SS mice.
Figure 2. C5a induces microvascular stasis (vaso-occlusion) in SS mice.



Dorsal skin-fold chambers were implanted onto SS mice (n=3/group) and AA mice (n=3/group) and 20 – 24 flowing venules were selected in each mouse at baseline (time 0). (A) SS mice were infused with 100 μl of zymosan-activated C57 mouse serum or zymosan-treated heat-inactivated C57 mouse serum at baseline after selection of flowing venules. Microvascular stasis (% non-flowing venules) was measured in the same venules at 1 and 4 hours after infusion. Bars represent means ± SD. *P<.05 and ***P<.001 versus heat-inactivated serum. (B) AA and SS-mice (n=3/group) were infused with 10 ng of C5a. Microvascular stasis was measured 1, 2, 3 and 4 h after infusion. Values are means ± SD. *P≤.05 and **P≤.01 versus AA mice. (C) SS mice (n=3/group) were infused with 200 ng of C5a. One group of SS mice was pre-treated with 30 μg of anti-C5a receptor (C5aR) IgG 30 minutes prior to C5a infusion. Microvascular stasis was measured 1, 2, 3 and 4 hours after C5a infusion. Values are means ± SD. **P≤0.01 and ***P≤0.001 versus C5a. Comparison of treatment groups at each time point was made using an unpaired t-test.
Since C5a is the most active complement-derived fragment that stimulates multiple vascular responses, including leukocyte, platelet and endothelial cell activation, we investigated whether C5a infused in SS mice would promote vaso- occlusion. As shown in Figure 2B, C5a induced microvascular stasis in SS mice but not in AA mice, an effect of C5a that was blocked by pre-treatment of the SS mice with an antibody against C5aR (Figure 2C).
3.3. C5a induces hepatic pro-inflammatory responses in SS mice that are blocked by anti-C5aR
In previous studies we have shown that H/R induces hepatic pro-inflammatory, pro-adhesive responses that are required for induction of microvascular stasis in SS mice.27,31–33 We investigated whether C5a would induce those pro-inflammatory responses in a specific manner. Supplemental FigureS6A shows that administration of C5a elicits activation of NF-κB as seen in the immunoblots of phospho-p65 in hepatic nuclear extracts; in addition, increased expression of TLR4 in hepatic microsomes is also evident. Moreover, administration of anti-C5aR antibody blocked the effect of C5a. Supplemental FigureS6B shows induced expression of adhesion molecules VCAM-1, ICAM-1 and E-selectin in hepatic microsomes, responses that were also blocked by administration of anti-C5aR antibody before giving C5a to these mice.
3.4. C5a induces expression of P-selectin and vWF on HUVEC in culture and on vascular endothelium in SS and AA mice
The P-selectin adhesion molecule has been shown to be very important in the mechanism of vaso-occlusion in SCD.34–37 Previous studies demonstrated that C5a induces P-selectin and vWF on endothelial cells in culture.38 As already noted above, an antibody to C5aR blocked stasis, therefore we tested whether an antibody against C5aR was able to block P-selectin and vWF expression on HUVEC. In HUVEC treated with hemin, histamine and C5a there was substantial induction of P-selectin and vWF expression on the cell surface within 30 minutes compared to untreated HUVEC (Figure 3A). The C5a response was abrogated by pre-treatment of the HUVECs with an anti-C5aR antibody.
Figure 3. C5a induces P-selectin and von Willebrand factor (vWF) expression on vascular endothelium in AA and SS mice through the C5a receptor (C5aR).


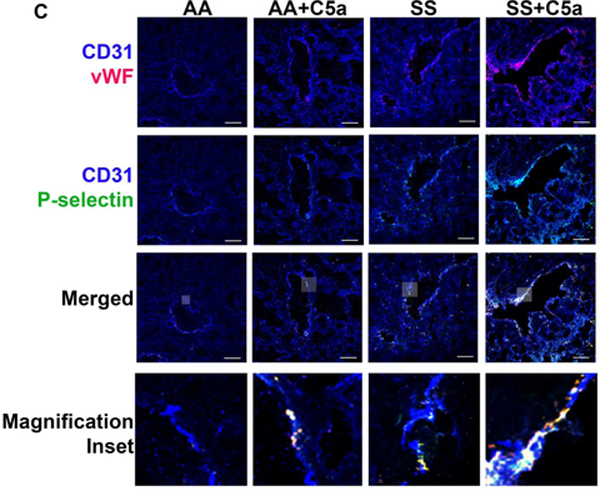


(A) Human umbilical vein endothelial cells (HUVEC) were untreated or incubated with 10 μM hemin, 100 μM histamine, 1 μg/ml C5a, or 5 μg/ml C5aR IgG + 1 μg/ml C5a for 30 minutes. At the end of incubation cells were washed and fixed in 4% paraformaldehyde and the cells were stained for cell surface P-selectin (green) and vWF (red), and nuclei were stained with DAPI (blue). Scale bars = 10 μm. (B-D) Vehicle (saline) or C5a (200 ng) was infused into AA and SS mice. Organs were collected 30 minutes after C5a infusion and frozen in OCT compound. Frozen tissues were cut into 6 μm (kidneys, livers and lungs) or 60 μm (skin) sections and stained for P-selectin (green), vWF (red) and CD31 (endothelial cell marker, blue). Representative images of organs from control and C5a-treated AA and SS mice are shown for (B) kidneys (60XZ2), (C) livers (60X), (D) lungs (60X) and (E) dorsal skin (60X). Scale bars = 10 μm (kidneys) or 30 μm (livers, lungs and skin). Gray insets in the merged images are magnified in the bottom row of each panel.
We then proceeded to investigate the effect of C5a in vivo on P-selectin and vWF in various organs of AA and SS mice. We found marked induction of P-selectin and vWF on the vessel walls of kidneys, liver, lungs and skin in response to C5a, especially in SS mice. In kidneys, control untreated SS mice had significantly more P-selectin and vWF expression on the vessel wall than control AA mice (Figure 3B [120X], Supplemental Figure S7 [20X] and Supplemental Tables S7–8 [Quantification]). P-selectin and vWF expression on the vessel wall increased significantly in the kidneys of AA and SS mice after C5a infusion with SS+C5a being significantly higher than AA+C5a. In the kidney, P-selectin and vWF were seen primarily on blood vessels in the glomeruli and to a lesser extent near extra-glomerular blood vessels and tubules.
In livers, control untreated SS mice had significantly more P-selectin and vWF expression on the vessel wall than control AA mice (Figure 3C [120X], Supplemental Figure S8 [20X] and Supplemental Tables S9–10 [Quantification]). P-selectin and vWF expression on liver vessel walls increased significantly in AA and SS mice after C5a infusion with SS+C5a being significantly higher than AA+C5a. In the liver, P-selectin and vWF were seen co-localized primarily on the vessel walls of hepatic veins.
In lungs, control untreated SS mice had significantly more P-selectin and vWF expression on the vessel wall than control AA mice (Figure 3D [120X], Supplemental Figure S9 [20X] and Supplemental Tables S11–12 [Quantification]). P-selectin and vWF expression on lung vessel walls increased significantly in AA and SS mice after C5a infusion with SS+C5a being significantly higher than AA+C5a. In the lungs, P-selectin and vWF were seen primarily co-localized on blood vessels.
In dorsal skin, control untreated SS mice had significantly more P-selectin and vWF expression in the vessels than control AA mice (Figure 3E [120X], Supplemental Figure S10 [20X] and Supplemental Tables S13–14 [Quantification]). P-selectin and vWF expression in skin vessels increased significantly in SS mice, but not AA mice, after C5a infusion with SS+C5a being significantly higher than AA+C5a. P-selectin and vWF were co-localized primarily with endothelial cell CD31 on the vessel wall.
We found little or no platelet CD41 staining in kidneys, livers or lungs (Supplemental Figure S11A). However, some platelet CD41 staining could be seen in the skin blood vessels (Supplemental Figure S11B). We saw no co-localization of P-selectin or vWF with platelet CD41 in any of the tissues. P-selectin and vWF were primarily co-localized with CD31. This is consistent with C5a activating P-selectin and vWF expression primarily on endothelial cells of the vessel wall.
3.5. P-selectin blockade inhibits microvascular stasis induced by C5a in SS mice
Since C5a promotes vaso-occlusion and the expression of endothelial P-selectin, we asked whether blocking P-selectin would interfere with C5a-induced vaso-occlusion. As shown in Figure 4A, a blocking antibody against P-selectin, but not an IgG control, given before infusion of C5a abolished the development of microvascular stasis. This experiment demonstrates that P-selectin is a key mediator of C5a-induced vaso-occlusion.
Figure 4.


(A) P-selectin blockade inhibits microvascular stasis in SS mice induced by C5a. Dorsal skin-fold chambers were implanted onto SS mice (n=3/group) and 20 – 24 flowing venules were selected in each mouse at baseline (time 0). (A) Anti-P-selectin IgG or isotype control IgG (30 μg) was infused 30 minutes before infusion of C5a (200 ng) at time 0. Percent stasis was measured in the same venules at 1, 2, 3 and 4 h after C5a infusion. (B) Anti-C5 or Anti-C5aR IgG inhibit stasis induced by H/R. Dorsal skin-fold chambers were implanted onto SS mice (n=3/group) and 20 – 24 flowing venules were selected in each mouse at baseline. Anti-C5 IgG (monoclonal antibody BB5.1; the murine equivalent of eculizumab), anti-C5aR IgG or isotype control IgG (30 μg) was infused 30 minutes before the mice were placed in hypoxia (7% O2/93% N2) for 1 h. After 1 h of hypoxia the mice were returned to room air (reoxygenation) and stasis (no flow) was measured in the same venules after 1 and 4 h of reoxygenation. Percent stasis was calculated and the data expressed as means ± SD. *P<.05, **P<.01 and ***P<.001.
3.6. Anti-C5 or Anti-C5aR IgG inhibit vaso-occlusion induced by H/R
Since both H/R and C5a induce microvascular stasis and H/R activates complement, we tested whether inhibition of C5 cleavage or blockade of C5aR would reduce H/R-induced stasis. As shown in Figure 4B, IgG antibodies against C5 or C5aR significantly inhibited the development of microvascular stasis at 1 and 4 h after H/R compared to IgG controls (p<0.01). Thus, these results clearly demonstrate the critical role of C5a, through P-selectin induction, in H/R-induced vaso-occlusion in the Townes-SS mouse model of SCD.
4. DISCUSSION
In these studies, we have demonstrated that complement plays a major role in the tissue injury of SCD focusing on ischemia-reperfusion, which is at the center of the pathophysiology of this condition. We elicited ischemia-reperfusion through the well-established, clinically relevant H/R model. First, we found that H/R causes complement activation and tissue deposition of C3 fragments and the membrane attack complex (MAC or C5b-9). As in other forms of ischemia-reperfusion, complement activation by H/R may begin through recognition molecules of the lectin pathway such as mannose binding protein, followed by activation of MASP1/MASP2, C4/C2, or C2 alone, and the alternative pathway.29,39–42 Then complement activation proceeds, yielding the main mediator of ischemia-reperfusion, C5a and also C5b-9 (MAC).
Without inducing H/R, we found increased plasma levels of Bb, a fragment of complement factor B, in SS mice relative to AA mice, indicating alternative pathway involvement.11 This suggests intermittent vaso-occlusion and ongoing ischemia-reperfusion pathophysiology in SS mice, since we found that exposure to H/R further increased Bb in the plasma of SS mice but not AA mice. Another contribution to increased Bb in SS mice that have not been subjected to H/R may be related to direct, ongoing, alternative pathway activation by RBCs, as discussed below. It is possible that the initiation of complement activation in H/R is through the lectin pathway.39,40,42 This requires additional studies because of its potential clinical relevance to define a possible therapeutic site to interfere with the effects of ischemia-reperfusion in SCD patients.
Given the prominent role of C5a in inflammation and its well-known action as the effector molecule of ischemia-reperfusion,40 our current studies focused on C5a. We demonstrated that C5a in zymosan-activated serum or C5a itself given to SS mice induced venous occlusion, an effect that is C5aR-dependent. We also found that Townes-SS mice demonstrate a C5a-dependent promotion of inflammation through activation of NF-κB and increased expression of TLR4. Our finding that C5a activates TLR4 expression in the context of H/R suggests that cross-talk between complement and TLR4 may be a convergence point of native immunity in SCD since we and others have previously demonstrated a key role for TLR4 in SCD pathophysiology.5–7,43–45 Like TLR4, the complement system also contributes to SCD pathogenesis, in part due to ischemia-reperfusion pathophysiology.
Importantly, we also found that C5a increased the expression of the adhesion molecules VCAM-1, ICAM-1 and E-selectin in SS mice. This observation directed us to investigate the possible role of C5a in expression of P-selectin and its role in mediating vaso-occlusion in SCD. A recent report demonstrated that a monoclonal antibody against P-selectin reduces vaso-occlusive crises in SCD patients.37 Therefore, we first confirmed the ability of C5a to activate P-selectin expression in vitro38 and then demonstrated in vivo that C5a also induces P-selectin-mediated vaso-occlusion in SS mice. As shown in Figure 4A, anti-P-selectin IgG, but not isotype control IgG, markedly inhibits C5a-induced vascular stasis in SS mice. Finally, the anti-mouse C5 antibody BB5.1 as well as an anti-C5aR antibody could block H/R-induced stasis as shown in Figure 4B. Therefore, our current study demonstrates a central role of complement activation triggered by H/R in SS mice and its pathogenic role in vaso-occlusion through C5a in activation of inflammatory mechanisms. We propose that this form of complement activation plays a major role in the pathogenesis of patients with SCD over their lifespan, since they undergo multiple severe crises triggered by episodes of ischemia-reperfusion, often of an H/R nature.
With regard to the clinical import of our study, it is essential to consider that complement may also be activated in SCD through a different mechanism from the one we report here. This other mechanism could be initiated by direct activation of the alternative pathway by membrane biochemical abnormalities of SS-RBCs consisting of increased phosphatidylserine and reduced sialic acid on the outer leaflet of the RBCs and microparticles.10,46 These membrane features promote the formation and binding of the alternative pathway convertase. Recently Merle et al. found C3b/iC3b and C5b-9 deposits in the kidneys of SCD patients and two mouse models of SCD, including Townes mice as confirmed here. They induced intravascular hemolysis or injection of heme or hemoglobin in WT mice to provide evidence that these complement deposits could be triggered by cell-free heme.14 Heme and heme-loaded microvesicles induced activation of the alternative complement pathway and heme triggered endothelial expression of P-selectin, C3aR and C5aR and down regulation of CD46. These effects were reduced by the heme-scavenger hemopexin. Details on the action of heme on complement and other plasma systems, relevant for SCD, can be found in a recent review.21
Another potential mechanism of complement activation in SCD is that phosphatidylserine molecules on the surface of SS-RBCs and activated platelets are able to induce the assembly of prothrombin in complexes that lead to thrombin generation, which could generate biologically active C3 and C5 fragments, including C3a and C5a.20,47 In addition, SS-RBCs might be more susceptible to complement activation through reduced CD55 and CD59 expression.48
In conclusion, our studies demonstrate a critical role for C5a in vaso-occlusion and inflammation in a murine model of SCD. C5a, by activating endothelial cell P-selectin, can mediate multicellular responses leading to vascular occlusion. Binding of C5a to C5aR on neutrophils, monocytes, platelets and the endothelium can promote inflammation, oxidative stress and vaso-occlusion. Activation of C5aR on mast cells could potentially release pain mediators provoking a pain crisis. The pro-inflammatory responses to C5a in organ blood vessels support this model and inhibition by C5aR antibody demonstrates its importance. The organ deposition of complement in response to H/R in SS mice, especially in the vasculature with consequent P-selectin and vWF up-regulation, provides insights as to what might happen to a sickle patient undergoing a crisis with attendant vaso-occlusion.
From a clinical perspective, complement activation in SCD patients can occur in response to inciting factors which can trigger an acute crisis, for example infection or surgery. A comprehensive analysis of ischemia-reperfusion physiology is likely to be fundamental for our understanding SCD and its ability to activate complement and the cells involved in vaso-occlusion. Complement is therefore an attractive target for inhibition to prevent vaso-occlusive crisis. The human anti-C5 antibody eculizumab has been beneficial in SCD patients with delayed transfusion reactions and hyperhemolysis.49 These data suggest that an anti-C5 antibody may be beneficial in certain other patients with SCD. In recommending this approach we temper our enthusiasm due to the potential attendant compromise of host defense, and we suggest that further studies are indicated to test this hypothesis in SCD patients.
Supplementary Material
ACKNOWLEDGEMENT
We acknowledge the University of Minnesota’s ‘University Imaging Center’ for imaging equipment used to capture pictures of immunoblots, cells and organs. This work was funded by National Heart, Lung and Blood Institute
Grant/Award Number: R01 HL114567 (Vercellotti).
Funding information
National Heart, Lung and Blood Institute
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Hebbel R, Vercellotti GM. Pathobiology of Sickle Cell Disease In: Hoffman R, Benz EJ Jr, Silberstein LE, et al. , eds. Hematology: Principles and Practice. 7th ed Philadelphia, PA: Elsevier; 2018:571–583. [Google Scholar]
- 2.Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. 2018;68(2–3):263–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106(3):411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96(1):314–320. [PubMed] [Google Scholar]
- 5.Ghosh S, Adisa OA, Chappa P, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. 2013;123(11):4809–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014;123(3):377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H, Wandersee NJ, Guo Y, et al. Sickle cell disease increases high mobility group box 1: a novel mechanism of inflammation. 2014;124(26):3978–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Beers EJ, Yang Y, Raghavachari N, et al. Iron, inflammation, and early death in adults with sickle cell disease. Circ Res. 2015;116(2):298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson WA, Thomas EJ, Sissons JG. Complement activation in asymptomatic patients with sickle cell anaemia. Clin Exp Immunol. 1979;36(1):130–139. [PMC free article] [PubMed] [Google Scholar]
- 10.Wang RH, Phillips G, Jr., Medof ME, Mold C. Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. J Clin Invest. 1993;92(3):1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chudwin DS, Papierniak C, Lint TF, Korenblit AD. Activation of the alternative complement pathway by red blood cells from patients with sickle cell disease. Clin Immunol Immunopathol. 1994;71(2):199–202. [DOI] [PubMed] [Google Scholar]
- 12.Mold C, Tamerius JD, Phillips G, Jr. Complement activation during painful crisis in sickle cell anemia. Clin Immunol Immunopathol. 1995;76(3 Pt 1):314–320. [DOI] [PubMed] [Google Scholar]
- 13.Gavriilaki E, Mainou M, Christodoulou I, et al. In vitro evidence of complement activation in patients with sickle cell disease. Haematologica. 2017;102(12):e481–e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merle NS, Grunenwald A, Rajaratnam H, et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI insight. 2018;3(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francis WG, Womack CR. Serum complement activity in normal individuals and patients with sickle cell hemoglobin abnormalities. Am J Med Technol. 1967;33(2):77–86. [PubMed] [Google Scholar]
- 16.Merle N, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pawluczkowycz AW, Lindorfer MA, Waitumbi JN, Taylor RP. Hematin promotes complement alternative pathway-mediated deposition of C3 activation fragments on human erythrocytes: potential implications for the pathogenesis of anemia in malaria. J Immunol. 2007;179(8):5543–5552. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology. 2012;217(11):1106–1110. [DOI] [PubMed] [Google Scholar]
- 19.Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282–292. [DOI] [PubMed] [Google Scholar]
- 20.Zecher D, Cumpelik A, Schifferli JA. Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler Thromb Vasc Biol. 2014;34(2):313–320. [DOI] [PubMed] [Google Scholar]
- 21.Roumenina LT, Rayes J, Lacroix-Desmazes S, Dimitrov JD. Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol Med. 2016;22(3):200–213. [DOI] [PubMed] [Google Scholar]
- 22.Gaudenzio N, Sibilano R, Marichal T, et al. Different activation signals induce distinct mast cell degranulation strategies. J Clin Invest. 2016;126(10):3981–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quadros AU, Cunha TM. C5a and pain development: An old molecule, a new target. Pharmacol Res. 2016;112:58–67. [DOI] [PubMed] [Google Scholar]
- 24.Ward PA, Gao H. Sepsis, complement and the dysregulated inflammatory response. J Cell Mol Med. 2009;13(10):4154–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108(4):1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belcher JD, Nath KA, Vercellotti GM. Vasculotoxic and pro-inflammatory effects of plasma heme: Cell signaling and cytoprotective responses. ISRN Oxidative Medicine. 2013;2013:Article ID 831596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalambur VS, Mahaseth H, Bischof JC, et al. Microvascular blood flow and stasis in transgenic sickle mice: utility of a dorsal skin fold chamber for intravital microscopy. Am J Hematol. 2004;77(2):117–125. [DOI] [PubMed] [Google Scholar]
- 28.Belcher JD, Vineyard JV, Bruzzone CM, et al. Heme oxygenase-1 gene delivery by Sleeping Beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med (Berl). 2010;88(7):665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pangburn MK, Muller-Eberhard HJ. The alternative pathway of complement. Springer Semin Immunopathol. 1984;7(2–3):163–192. [DOI] [PubMed] [Google Scholar]
- 30.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. C3b deposition during activation of the alternative complement pathway and the effect of deposition on the activating surface. J Immunol. 1983;131(4):1930–1935. [PubMed] [Google Scholar]
- 31.Belcher JD, Chen C, Nguyen J, et al. The fucosylation inhibitor, 2-fluorofucose, inhibits vaso-occlusion, leukocyte-endothelium interactions and NF-kB activation in transgenic sickle mice. PLoS One. 2015;10(2):e0117772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. 2006;116(3):808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belcher JD, Young M, Chen C, et al. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood. 2013;122(15):2757–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99(5):3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Embury SH, Matsui NM, Ramanujam S, et al. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood. 2004;104(10):3378–3385. [DOI] [PubMed] [Google Scholar]
- 36.Wood KC, Hebbel RP, Granger DN. Endothelial cell P-selectin mediates a proinflammatory and prothrombogenic phenotype in cerebral venules of sickle cell transgenic mice. Am J Physiol Heart Circ Physiol. 2004;286(5):H1608–1614. [DOI] [PubMed] [Google Scholar]
- 37.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376(5):429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foreman KE, Vaporciyan AA, Bonish BK, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. 1994;94(3):1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrar CA, Asgari E, Schwaeble WJ, Sacks SH. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front Immunol. 2012;3:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology. 2012;217(11):1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asgari E, Farrar CA, Lynch N, et al. Mannan-binding lectin-associated serine protease 2 is critical for the development of renal ischemia reperfusion injury and mediates tissue injury in the absence of complement C4. FASEB J. 2014;28(9):3996–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark JE, Dudler T, Marber MS, Schwaeble W. Cardioprotection by an anti-MASP-2 antibody in a murine model of myocardial infarction. Open heart. 2018;5(1):e000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vinchi F, Costa da Silva M, Ingoglia G, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127(4):473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Godefroy E, Liu Y, Shi P, et al. Altered heme-mediated modulation of dendritic cell function in sickle cell alloimmunization. Haematologica. 2016;101(9):1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res. 2010;44(11):1267–1288. [DOI] [PubMed] [Google Scholar]
- 46.Onyemelukwe GC, Esievo KA, Kwanashie CN, Kulkarni AG, Obinechie EN. Erythrocyte sialic acid in human sickle-cell disease. J Comp Pathol. 1987;97(2):143–147. [DOI] [PubMed] [Google Scholar]
- 47.Krisinger MJ, Goebeler V, Lu Z, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120(8):1717–1725. [DOI] [PubMed] [Google Scholar]
- 48.Al-Faris L, Al-Rukhayes M, Al-Humood S. Expression pattern of CD55 and CD59 on red blood cells in sickle cell disease. Hematology. 2017;22(2):105–113. [DOI] [PubMed] [Google Scholar]
- 49.Chonat S, Quarmyne MO, Bennett CM, et al. Contribution of alternative complement pathway to delayed hemolytic transfusion reaction in sickle cell disease. Haematologica. 2018;103(10):e483–e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.