

The CRISPR system provides adaptive immunity against mobile genetic elements in bacteria and archaea. Type III CRISPR systems detect viral RNA, resulting in activation of a HD nuclease domain for DNA degradation 1,2 and a Cyclase domain that synthesises cyclic oligoadenylate (cOA) from ATP 3–5. cOA activates defence enzymes with a CARF (CRISPR Associated Rossmann Fold) domain 6, sculpting a powerful antiviral response 7–10 that can drive viruses to extinction 7,8. Cyclic nucleotides are increasingly implicated as playing an important role in host-pathogen interactions 11–13. Here, we identify a new family of viral anti-CRISPR (Acr) enzymes that rapidly degrade cyclic tetra-adenylate (cA4). The viral ring nuclease (AcrIII-1) is widely distributed in archaeal and bacterial viruses, and proviruses. The enzyme uses a novel fold to bind cA4 specifically and utilizes a conserved active site to rapidly cleave the signalling molecule, allowing viruses to neutralise the type III CRISPR defence system. The AcrIII-1 family has a broad host range as it targets cA4 signalling molecules rather than specific CRISPR effector proteins. This study highlights the crucial role of cyclic nucleotide signalling in the conflict between viruses and their hosts.
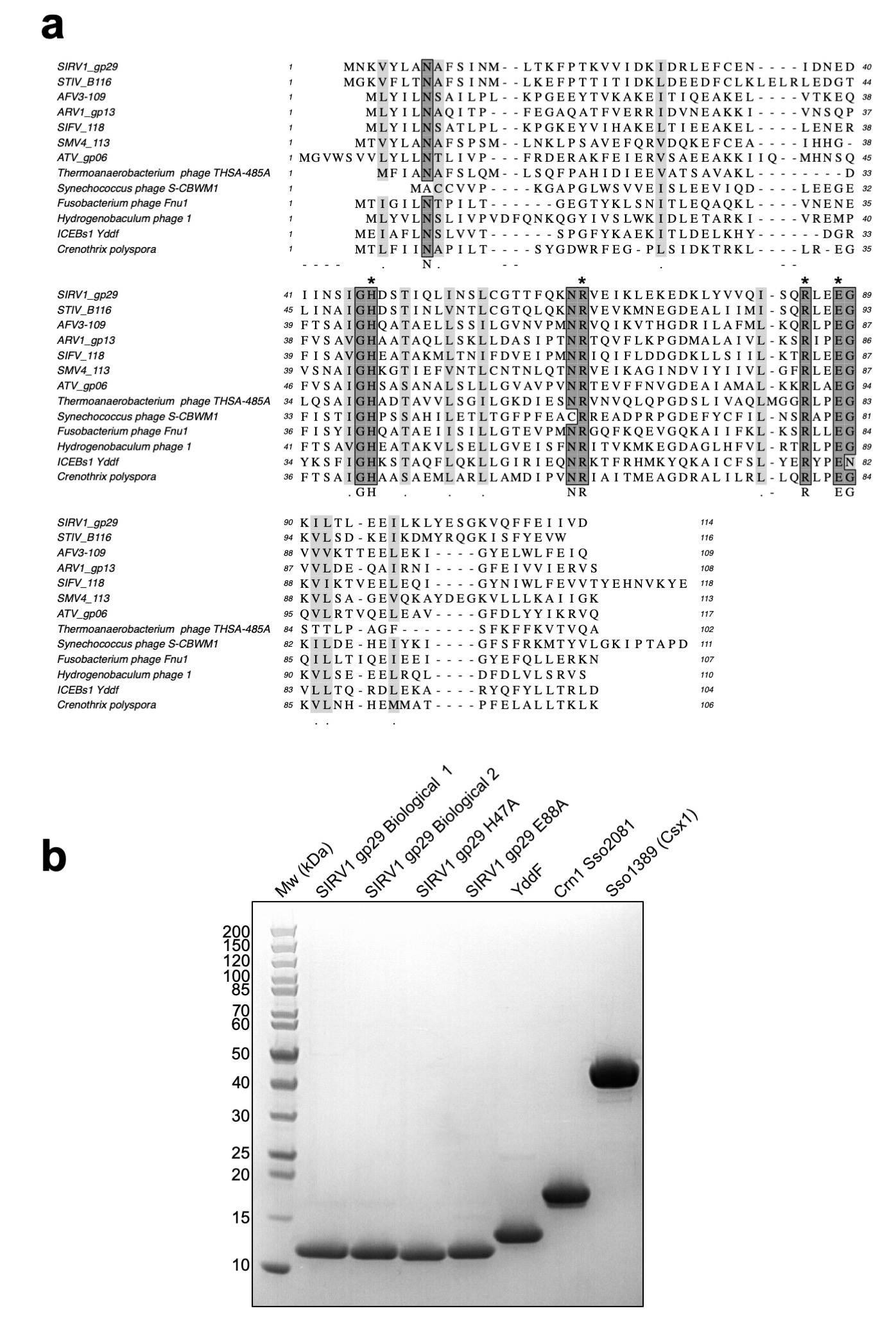
Recently, we identified a cellular enzyme in Sulfolobus solfataricus, hereafter referred to as the Crn1 family (for “CRISPR associated ring nuclease 1”), that degrades cyclic tetra-adenylate (cA4) molecules and deactivates the Csx1 ribonuclease 14. These enzymes are thought to act by mopping up cA4 molecules in the cell without compromising the immunity provided by the type III CRISPR system. In the absence of a mechanism to remove cOA following clearance of viral infection, cells could be pushed towards dormancy or cell death under inappropriate circumstances 7,14. Unsurprisingly, viruses have responded to the threat of the CRISPR system by evolving a range of anti-CRISPR (Acr) proteins, which are used to inhibit and overcome the cell’s CRISPR defences (reviewed in 15). Acrs have been identified for many of the CRISPR effector subtypes, and number over 40 families 16. Here, we investigated the DUF1874 protein, which is conserved and widespread in a variety of archaeal viruses and plasmids, bacteriophages and prophages (Extended Data Fig.1), for an Acr function. Structures are available for several family members, including gp29 of Sulfolobus islandicus rod-shaped virus 1 (SIRV1) 17 and B116 of Sulfolobus turreted icosahedral virus (STIV) 18. The structures revealed an intriguing dimeric structure with a large central pocket flanked by conserved residues. B116 is important for normal virus replication kinetics, as deletion of the gene results in a marked “small plaque” phenotype 19, consistent with an Acr function.
DUF1874 is a type III anti-CRISPR, AcrIII-1
To investigate this, we deleted the genes for the type I-A system in Sulfolobus islandicus M.16.4 so that it had only a type III-B CRISPR system for defence 20 (Extended Data Fig. 2). We challenged this strain with the archaeal virus SSeV (Fig. 1a), a lytic virus isolated from Kamchatka Russia with an exact CRISPR-spacer match of 100% in M.16.4 and several other potentially active CRISPR-spacers. SSeV lacks a duf1874 gene and failed to form plaques on a lawn of S. islandicus M.16.4 with type III-B CRISPR defence unless the csx1 effector gene was deleted (Fig. 1a and Extended Data Fig. 2). However, the same cells expressing the SIRV1 gp29 gene from a plasmid were readily infected, giving rise to plaque formation. These data are consistent with the hypothesis that SIRV1 gp29 is functioning as an Acr specific for type III CRISPR defence.
Figure 1. DUF1874 is an anti-CRISPR specific for cA4 signalling.

(a) SSeV infection assay showing the DUF1874 gene SIRV1 gp29 can neutralise the type III-B system in S. islandicus. S. islandicus RJW007∆type I-A or RJW007∆type I-A∆csx1 mutant strains were challenged with SSeV, in the presence or absence of duf1874 (SIRV1 gp29) expressed on a replicative plasmid. Plaques were observed when csx1 was deleted, or when the resistant strain expressed duf1874 (n= 3 biological replicates) (Extended data Fig. 2d). (b) Schematic showing the recombinant M. tuberculosis type III-A CRISPR interference system established in E. coli. By swapping the native Csm6 ancillary nuclease for a Csx1 protein, the system can be converted from cA6 to cA4-mediated immunity. (c) Plasmid transformation assay (1 day’s growth) using a plasmid with a match to a spacer in the CRISPR array. If the plasmid was successfully targeted by the CRISPR system, fewer transformants were expected. Plasmids with or without the duf1874 gene were targeted successfully when cA6 (Csm6) mediated antiviral signalling was active. In contrast, cells using a cA4-based (Csx1) system only reduced transformation when the DUF1874 protein was not present, suggesting that DUF1874 was effective in neutralising cA4-based CRISPR interference. The control strain lacked cOA-dependent ribonucleases. These results are representative from 2 biological replicates with 4 technical replicates each (n= 8). (d) Colony counts for transformants visible after 1- and 4-days growth in the presence or absence of DUF1874 and the indicated effector proteins. DUF1874 antagonises Csx1 but not Csm6-mediated immunity. Values and error bars reflect the mean ± standard deviation from 2 biological replicates with 4 technical replicates each (n= 8).
To explore this possibility further, we utilised a recently developed recombinant type III CRISPR system from Mycobacterium tuberculosis, which allows the cOA effector protein to be swapped to provide effective immunity based on either cA6 or cA4 signalling 21 (Fig. 1). Strains capable of cA4 or cA6-based immunity were transformed with a plasmid targeted for interference due to a match in the tetracycline resistance gene to a spacer in the CRISPR array. Efficient interference (lack of plasmid transformation) was observed after 1 day for either strain in the absence of the duf1874 gene from bacteriophage THSA-485A (Fig, 1c, d). However, the presence of the duf1874 gene on the plasmid reduced immunity for cA4, but not for cA6-mediated CRISPR defence. This observation supports the hypothesis that DUF1874 acts as an Acr against cA4-mediated type III CRISPR defence. Hereafter we propose the collective name AcrIII-1 for this family. The “-“ in place of a subtype reflects the fact that AcrIII-1 will inhibit any type III CRISPR subtype that utilizes cA4 for defence 22. After 4 days of growth, Csm6-mediated immunity was lost, regardless of the presence of DUF1874. This could indicate that alternative mechanisms exist to remove cA6 (Fig. 1d and Extended Data Fig. 3).
AcrIII-1 degrades cA4 rapidly
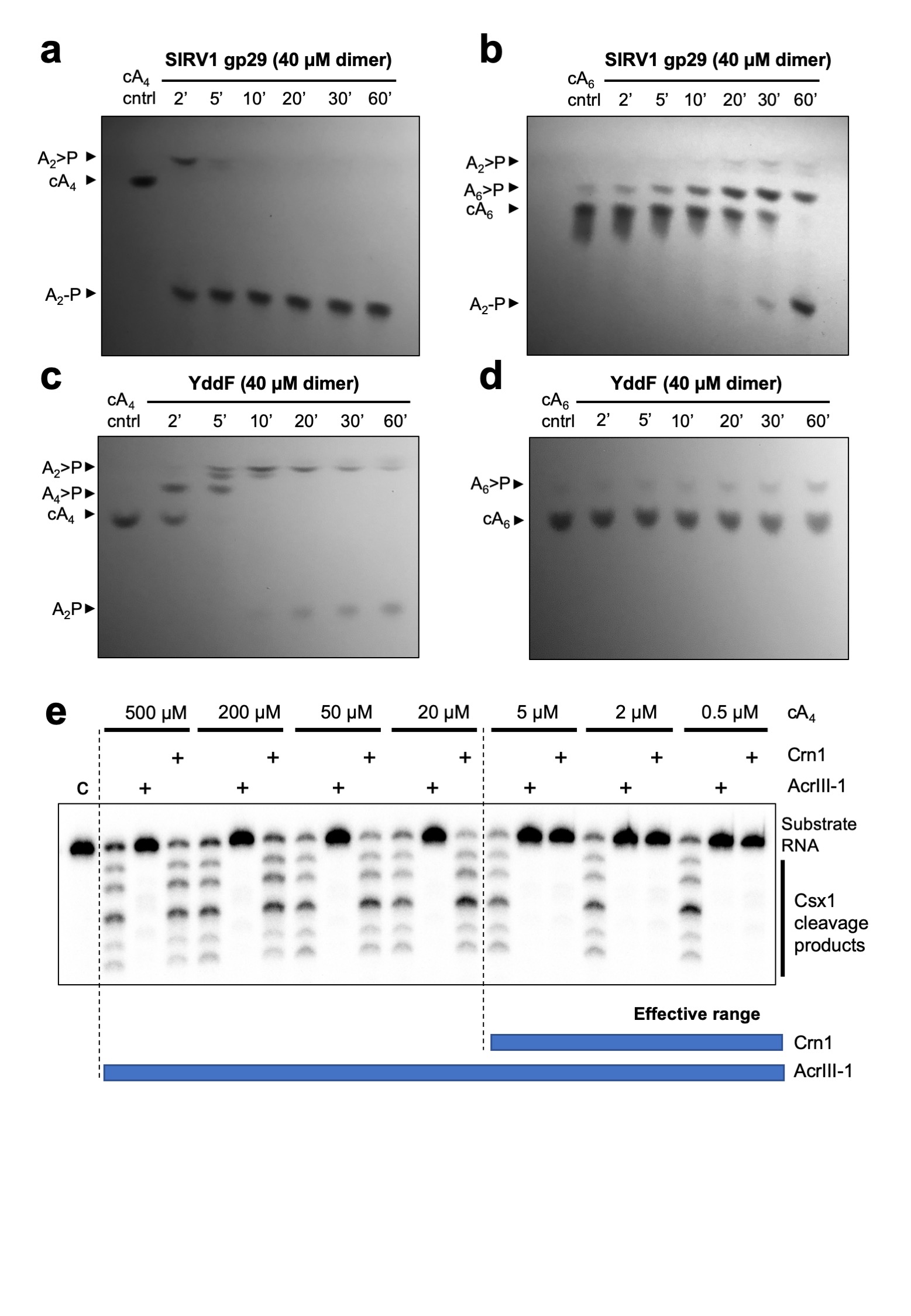
To explore the mechanism of the AcrIII-1 family, we cloned and expressed two family members in E. coli: the SIRV1 gp29 protein and the YddF protein encoded by an integrative and conjugative element ICEBs1 from B. subtilis 23 (Extended Data Fig.1b). Both proteins possess a potent ring nuclease activity, rapidly degrading cA4 to generate linear di-adenylate (ApA>P) with a cyclic 2’,3’ phosphate (Fig. 2 and Extended Data Fig. 4). With a catalytic rate exceeding 5 min-1, the Acr enzyme is at least 60-fold more active than the cellular ring nuclease Crn1 from S. solfataricus. Both SIRV1 gp29 and YddF enzymes show a strong preference for cA4 over cA6, with the latter degraded very slowly by comparison (Extended Data Fig. 4). We showed previously that the type III-D CRISPR effector of S. solfataricus generates cA4 in proportion to the amount of cognate target RNA present 14. By varying target RNA input and following cA4 levels and Csx1 activity, we compared the abilities of Crn1 and AcrIII-1 to destroy the signalling molecule and deactivate the ancillary defence nuclease Csx1. In keeping with its low turnover number, the Crn1 enzyme was effective at degrading cA4 and thus deactivating Csx1 only at the lowest levels of target RNA (Fig. 2c). In contrast, AcrIII-1 degraded cA4 completely at the highest target RNA concentration examined, preventing Csx1 activation. We investigated the ability of each enzyme to prevent Csx1 activation over a range of cA4 concentrations spanning four orders of magnitude (Extended Data Fig. 4e). Crn1 (2 µM) provided protection only up to 5 µM cA4, but in contrast 2 µM AcrIII-1 provided complete protection at the highest level of cA4 tested (500 µM). Thus, AcrIII-1 has the potential to destroy large concentrations of the cA4 second messenger rapidly, preventing activation of Csx1.
Figure 2. AcrIII-1 rapidly degrades cA4 to linear products.

(a) Liquid chromatography-high resolution mass spectrometry analysis confirms that AcrIII-1 SIRV1 gp29 converts cA4 to A2>P and A2-P. The experiment was repeated twice with similar results. (b) Kinetic comparison of cA4 degradation by the AcrIII-1 enzymes SIRV1 gp29 and YddF with the cellular ring nuclease Crn1. Values and error bars plotted reflect the mean ± standard deviation (n= 3 technical replicates). (c) Upper panel shows activation of Csx1 in a coupled assay containing type III Csm complex when activated with indicated amounts of (unlabeled) target RNA to initiate cA4 synthesis. Each set of three lanes after the control (C) reaction with Csx1 and substrate RNA alone, is first in the absence and then with AcrIII-1 (SIRV1 gp29) or Crn1 (Sso2081). Whereas AcrIII-1 degraded all cA4 generated with up to 50 nM RNA target, the Crn1 enzyme deactivated Csx1 only when less than 5 nM RNA target was used to initiate cA4 synthesis. The lower panel shows thin layer chromatography (TLC) of the same reactions to visualize cA4 production and degradation. Csx1 deactivation correlated with complete cA4 degradation (n= 3 technical replicates). For gel source data, see Supplementary Figure 1.
Structure and mechanism of AcrIII-1
The structure of AcrIII-1 is unrelated to the CARF domain, which is the only protein family thus far known to bind cOA 6. To elucidate the mechanism of cA4 binding and cleavage by AcrIII-1, we co-crystallised an inactive variant (H47A) of SIRV1 gp29 with cA4 and solved the structure to 1.55 Å resolution (Fig. 3; Extended Data Table 1). The complex reveals a molecule of cA4 bound at the dimer interface. Comparison of the cA4-bound and apo structures reveals a significant movement of a loop, comprising residues 82-94, and subsequent α-helix, to bury cA4 within the dimer. These loops adopt variable or unstructured conformations in the various apo protein structures. Once bound, the ligand is completely enclosed by the protein – a considerable accomplishment when one considers the relative sizes of protein and ligand (Fig. 3b). Superimposition of the cA4 ligand on the apo-protein structure reveals that the binding site is largely pre-formed, with the exception of the mobile loops that form the lid (Fig. 3c). The overall change is like two cupped hands catching a ball, with the loops (fingers) subsequently closing around it.
Figure 3. Structure of AcrIII-1 bound to cA4.

(a) Superimposition of the apo SIRV1 gp29 structure (salmon) and in complex with cA4 (blue), highlighting the movement of the loop and α-helix upon cA4 binding. cA4 is shown coloured by element. (b) Surface representation of the structure of SIRV1 gp29 (blue) in complex with cA4, emphasising the complete burial of the ligand. (c) Surface representation of the apo structure of SIRV1 gp29 (salmon) with cA4 in the position observed in the complex structure, indicating that the binding site is pre-formed. (d) Structure of cA4 bound to SIRV1 gp29. The two active site histidine residues (modelled based on the position of the alanine side chain in the H47A variant crystallised with cA4; coloured to represent residues from different monomers) are in suitable positions to act as the general acid, protonating the oxyanion leaving group. The corresponding ribose sugars have 2’-hydroxyl groups suitably positioned for in-line nucleophilic attack on the phosphodiester bond.
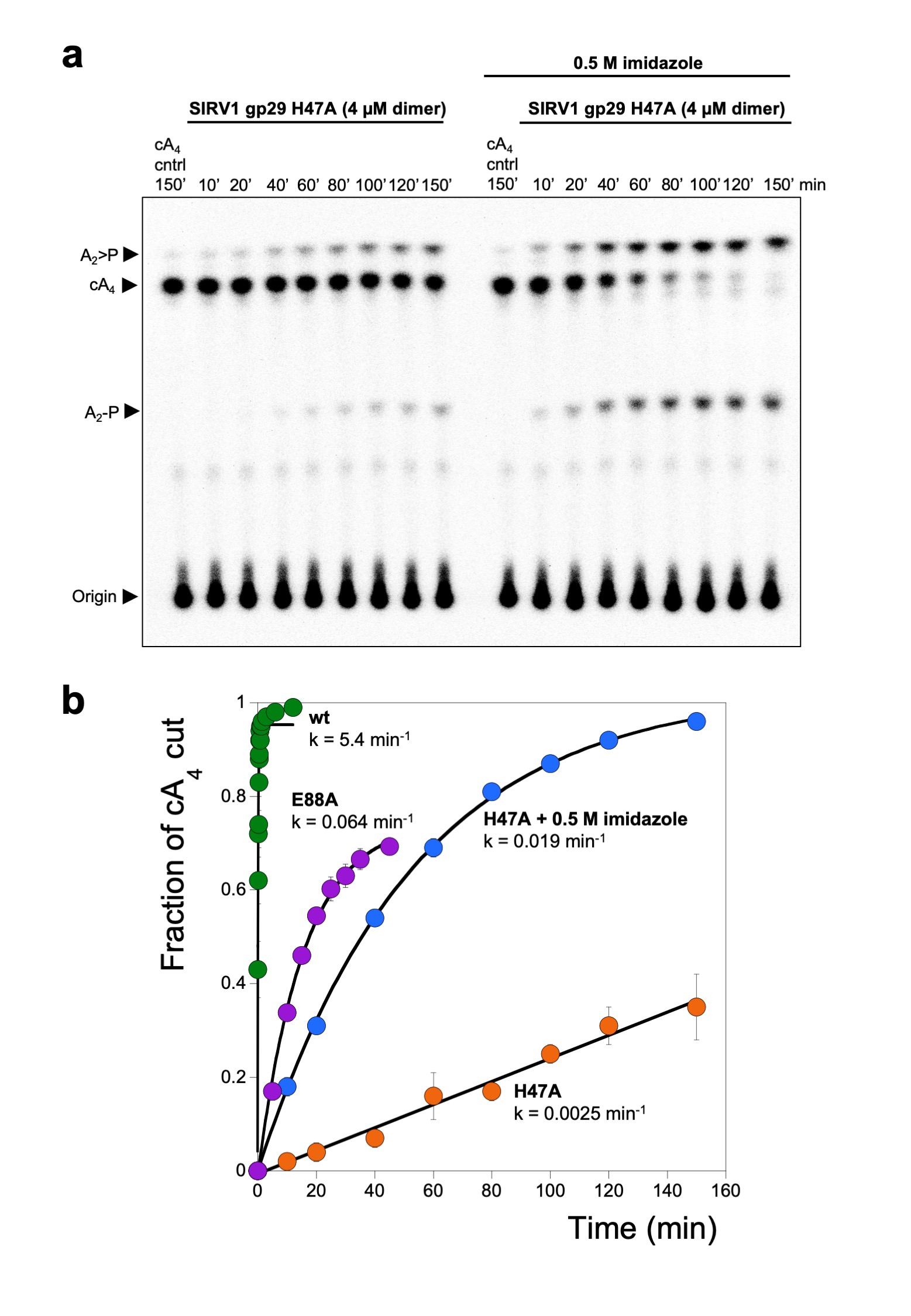
The cA4 molecule makes symmetrical interactions with each monomer of AcrIII-1 (Extended Data Fig. 5). Arginine R85 on the loop from one monomer interacts with the distant half of the cA4 molecule and appears to ‘lock’ the closed dimer. Other important interactions are made with main chain L92, I69, and N8, and side chains R66, N8, Q81, S11, T50, S49, and N13, most of which are semi or fully conserved (Extended Data Fig. 1 & 5), suggesting they have important roles in cA4 binding and/or catalysis in this whole family of enzymes. At two positions, on opposite sides of the ring, the 2’-hydroxyl of the ribose is positioned correctly for in-line attack on the phosphodiester bond, consistent with the observed bilateral cleavage (Fig. 3d). The catalytic power of the AcrIII-1 family likely derives from active site residues that position the 2’-hydroxyl group for in-line nucleophilic attack, stabilise the transition state and protonate the oxyanion leaving group 24. For the AcrIII-1 family, the absolutely conserved residue H47 is suitably positioned to act as a general acid and fulfil the latter role (Fig. 3d). To test this hypothesis, we assayed variant H47A of AcrIII-1. The variant enzyme suffered a >2500-fold decrease in catalytic power, which could be partially reversed by chemical rescue with 500 mM imidazole in the reaction buffer (Extended Data Fig. 6). We also noted that conserved glutamate E88, situated on the tip of the loop that covers the binding site, is positioned close to the H47 residue of the opposite subunit. When mutated to alanine, the catalytic rate was reduced by 84-fold to 0.064 min-1 (Extended Data Fig 6b), consistent with a role for E88 in positioning H47 and/or increasing the pKa of the catalytic histidine residue to enhance catalysis 25.
By targeting a key signalling molecule, a single AcrIII-1 enzyme should have broad utility in the inhibition of endogenous cA4-specific type III CRISPR systems in any species. Of the CRISPR ancillary nucleases studied to date, most are activated by cA4; activation by cA6 appears to be limited to certain bacterial phyla including the Firmicutes and Actinobacteria 21. Recently, a type III Acr (AcrIIIB1) has been reported that appears to function by binding and inhibiting the type III-B effector complex 26. Two other Acr proteins with enzymatic functions have been described: AcrVA1 which catalyses crRNA cleavage of Cas12a 27 and AcrVA5, which acetylates the PAM-sensing site of Cas12a 28. These and other Acrs target a protein (or protein:nucleic acid complex), implying a requirement for specific interactions that could be evaded by sequence variation. This is not a limitation of AcrIII-1.
Phylogenetic analysis of AcrIII-1
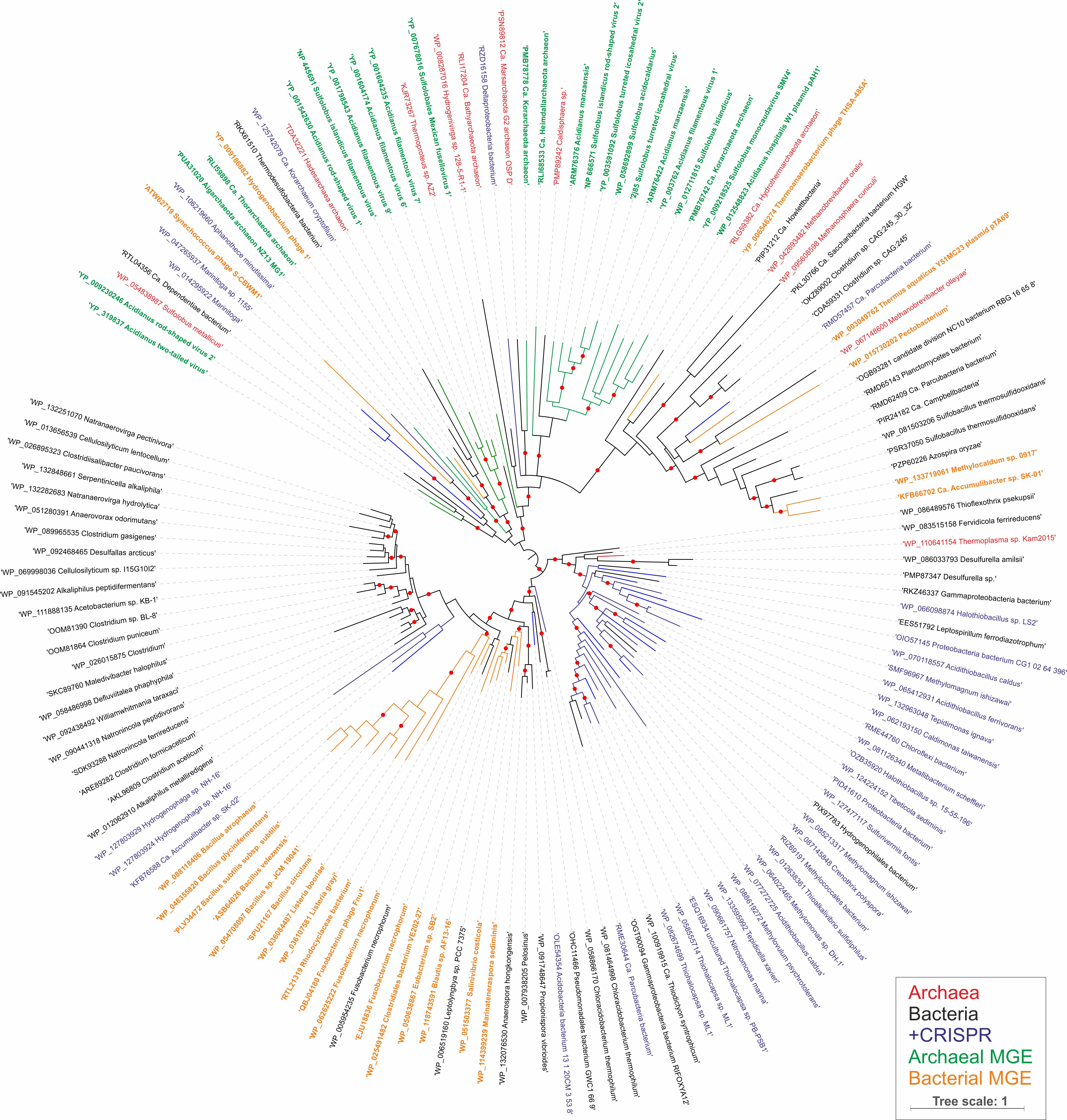
The gene encoding AcrIII-1 is found in representatives of at least five distinct viral families, making it one of the most widely conserved of all archaeal virus proteins 29 (Extended Data Fig. 1; Dataset S1). The distribution of AcrIII-1 in archaea is sporadic but covers most of the main lineages (Dataset S1) and is typically adjacent to ORFs from mobile genetic elements rather than CRISPR loci. A good example is the STIV virus integrated into S. acidocaldarius genomes 30. AcrIII-1 is also present in several bacteriophages of the order Caudovirales, and there are many instances of acrIII-1 genes in sequenced bacterial genomes, with homologues found in the Firmicutes, Cyanobacteria, Proteobacteria, Actinobacteria and other phyla (Dataset S1). Maximum likelihood phylogenetic analysis of the AcrIII-1 proteins suggests multiple horizontal gene transfers between unrelated viruses as well as between bacteria and archaea (Extended Data Fig. 7-9). Sometimes the acrIII-1 gene is clearly part of an integrated mobile genetic element, like the yddF gene in B. subtilis 29. However, in other species (n=49) the gene is associated with cellular type III CRISPR systems. In Marinitoga piezophilia, AcrIII-1 is fused to a cOA-activated HEPN ribonuclease of the Csx1 family. Since both active sites are conserved, this fusion protein may have cA4 activated ribonuclease activity coupled with a cA4 degradative ring nuclease, thus providing an explicit linkage between the AcrIII-1 family and the type III CRISPR system. In this context the enzyme is likely acting as a host-encoded ring nuclease, like Crn1, rather than an Acr. We therefore propose the family name of Crn2 (CRISPR associated ring nuclease 2) to cover DUF1874 family members that are associated with type III CRISPR systems (Extended Data Fig. 8).
Cyclic nucleotides in defence systems
AcrIII-1 is the first Acr predicted to have functional roles in both “offense and defence”. It remains to be determined whether the acrIII-1 gene arose in viruses and was appropriated by cellular type III systems or vice versa. However, the extremely broad distribution of acrIII-1 and limited distribution of crn2 suggests the former. Adoption of an anti-CRISPR protein as a component of a cellular CRISPR defence system seems counter-intuitive. However, the enzyme could be harnessed for a role in defence by putting it under tight transcriptional control so that it is expressed at appropriate times or levels. The unprecedented wide occurrence of this Acr across many archaeal and bacterial virus families reflects the fact that this enzyme degrades a key signalling molecule to subvert cellular immunity. This makes it very hard for cells to evolve counter-resistance, other than by switching to a different signalling molecule. Recent discoveries have highlighted the existence of diverse cyclic nucleotide signalling cellular defence systems in bacteria 11–13. It is possible that cOA, and the enzymes that metabolize it, have functions that extend beyond type III CRISPR systems. The identification here of a new class of cA4 binding proteins highlights the potential for further discoveries in this area.
Methods
Construction of S. islandicus strains
The type I-A CRISPR defence module including seven genes i.e. cas3b, csa5, cas7, cas5, cas3’, cas3”, and casX 31 was in-frame deleted from the genetic host S. islandicus RJW007, derived from wild-type strain S. islandicus M.16.4 carrying a double pyrEF and argD deletion 32, by employing a modified Plasmid Integration and Segregation knockout strategy 33, in line with the methodology developed by the She laboratory 34. The resultant type I-A deletion mutant (RJW007∆type I-A) was then used as a parental strain to further delete csx1 gene, generating the mutant strain RJW007∆type I-A∆csx1. Mutant strains were confirmed by PCR analysis using primers that bind outside of the homologous flanking arms of genes to be deleted.
Synthesised SIRV1 gp29 gene was purchased from IDT as a g-block and was cloned into a Sulfolobus-E.coli shuttle vector pSeSd-SsoargD 32 (referred to as pOE hereafter) at the NdeI and NotI sites, generating the gp29 expression plasmid pOE-gp29 in which the gp29 gene was placed under the control of the arabinose promoter. The pOE-gp29 and pOE plasmids were then transformed into competent cells of the ∆type I-A mutant and ∆type I-A∆csx1 mutant via electroporation as described previously 32, generating strains expressing and not expressing SIRV1 gp29, respectively.
Viral quantification
The genome sequence of SSeV is available in Genbank (accession code MN53972). To calculate the titer of SSeV, 100 µL diluted virus (10-5, 10-6, and 10-7) was co-incubated with 500 µL S. islandicus Y08.82.36 host 20 (10-fold concentrated) without shaking at 76-78°C for 30 min. Afterwards, the virus-infected cells were transferred into a glass test tube containing 5 mL of pre-warmed Sucrose-Yeast extract (SY) and 0.8% gelrite mixture, and plated onto SY plates. The plates were put into a plastic bag, and incubated for two days at 76-78°C. Plaques were counted in plates with proper virus dilutions, and the titer of SSeV was determined as 4.96×108 plaque forming units (PFU) per ml.
Infection by virus SSeV of S. islandicus M.16.4 with or without a type III CRISPR defence
The SSeV infection assay was carried out according to the procedure described previously 20 with minor modifications. In brief, approximately 6 × 108 cells of S. islandicus cells taken from the exponential stage were spun down at 4000 rpm × 12 min, and resuspended in 1mL of Arabinose-Tryptone (AT) medium. The resuspensions were then co-incubated with 20 mL of fresh AT medium or SSeV supernatant at different dilutions (100, 10-1, 10-2, 10-3, 10-4, 10-5, and 10-6) in a Falcon tube at 76-78°C for 1 h without shaking. The SSeV-infected cells were washed twice with 10 mL of AT medium and resuspended into 500 µL of AT medium. Afterwards, the concentrated SSeV-infected cells were mixed with 5 mL of top layer (2.5 mL of 2×Arabinose-Yeast extract (AY) medium+2.5 mL of 0.8% gelrite), and then plated onto the AY plates. PFU were counted after 4 days of incubation at 76-78°C. Three independent experiments were performed.
Cloning and purification
For cloning, synthetic genes (g-blocks) were purchased from Integrated DNA Technologies (IDT), Coralville, USA, and cloned into the vector pEhisV5spacerTev between the NcoI and BamHI sites 35. Competent DH5α (Escherichia coli) cells were transformed with the construct and sequence integrity confirmed by sequencing (Eurofins Genomics). The plasmid was transformed into Escherichia coli C43 (DE3) cells for protein expression. Cloning of AcrIII-1 SIRV1 gp29, Crn1 Sso2081 and SsoCsx1 has been described previously 14,17. For expression of SIRV1 gp29 and Bacillus subtilis YddF, 2 L of Luria-Broth culture was grown at 37°C to an OD600 of 0.8 with shaking at 180 rpm. Protein expression was induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside and cells were grown at 25°C overnight before harvesting by centrifugation at 4000 rpm (Beckman Coulter Avanti JXN-26; JLA8.1 rotor) at 4°C for 15 min.
For protein purification the cell pellet was resuspended in four volumes equivalent of lysis buffer containing 50 mM Tris-HCl 7.5, 0.5 M NaCl, 10 mM imidazole and 10% glycerol supplemented with EDTA-free protease inhibitor tablets (Roche; 1 tablet per 100 ml buffer) and lysozyme (1 mg/ml). Cells were lysed by sonicating six times for 1 min with 1 min rest intervals on ice at 4°C, and the lysate was ultracentrifuged at 40,000 rpm (70 Ti rotor) at 4°C for 35 min. The lysate was then loaded onto a 5 ml HisTrap FF Crude column (GE Healthcare) equilibrated with wash buffer containing 50 mM Tris-HCl pH 7.5, 0.5 M NaCl, 30 mM imidazole and 10% glycerol. Unbound protein was washed away with 20 column volumes (CV) of wash buffer prior to elution of his-tagged protein using a linear gradient (holding at 10% for 3 CV, and 50% for 3 CV) of elution buffer containing 50 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.5 M imidazole and 10% glycerol. SDS-PAGE was carried out to identify fractions containing the protein of interest, and relevant fractions were pooled and concentrated using a 10 kDa molecular weight cut-off centrifugal concentrator (Merck). The his-tag was removed by incubating concentrated protein overnight with Tobacco Etch Virus (TEV) protease (1 mg per 10 mg protein) while dialysing in buffer containing 50 mM Tris-HCl pH 7.5, 0.5 M NaCl, 30 mM imidazole and 10% glycerol at room temperature. The protein with his-tag removed was isolated using a 5 ml HisTrapFF column, eluting the protein using 4 CV wash buffer. His-tag removed protein was further purified by size-exclusion chromatography (S200 16/60; GE Healthcare) in buffer containing 20 mM Tris-HCl pH 7.5, 0.125 M NaCl using an isocratic gradient. After SDS-PAGE, fractions containing protein of interest were concentrated and protein was aliquoted and stored at -80°C. Variant enzymes were generated using the QuickChange Site-Directed Mutagenesis kit as per manufacturer’s instructions (Agilent technologies) and purified as for the wild-type proteins.
Radiolabelled cA4 cleavage assays
Cyclic oligoadenylate (cOA) was generated by incubating 120 μg Sulfolobus solfataricus (Sso) type III-D (Csm) complex with 5 nM α-32P-ATP, 1 mM ATP, 120 nM A26 RNA target and 2 mM MgCl2 in Csx1 buffer containing 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 5.5, 100 mM K-glutamate, 1 mM DTT and 3 units SUPERase•In™ Inhibitor for 2 h at 70°C in a 100 μl reaction volume. cOA was extracted by phenol-chloroform (Ambion) extraction followed by chloroform extraction (Sigma-Aldrich), and stored at -20°C.
For single turnover kinetics experiments, AcrIII-1 SIRV1 gp29 and variants (4 μM protein dimer) were assayed for radiolabelled cA4 degradation by incubating with 1/400 diluted 32P-labelled SsoCsm cOA (~200 nM cA4; when generated in a 100 μl cOA synthesis reaction as described above) in Csx1 buffer supplemented with 1 mM EDTA at 50°C. AcrIII-1 YddF (8 μM dimer) was incubated with cOA in buffer containing 20 mM MES pH 6.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA and 3 units SUPERase•In™ Inhibitor at 37°C. Crn1 Sso2081 (4 μM dimer) was incubated with cOA in buffer containing 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM DTT and 3 units SUPERase•In™ Inhibitor at 50°C. For SIRV1 gp29 H47A chemical rescue, reactions were supplemented with 0.5 M imidazole. Two experimenters were involved in kinetic experiments involving 5 s time points. At desired time points, a 10 μl aliquot of the reaction was removed and quenched by adding to phenol-chloroform and vortexing. Subsequently, 5 μl of deproteinised reaction product was extracted into 5 μl 100% formamide xylene-cyanol loading dye if intended for denaturing polyacrylamide gel electrophoresis (PAGE), or products were further isolated by chloroform extraction if intended for thin-layer chromatography (TLC). A reaction incubating cOA in buffer without protein to the endpoint of each experiment was included as a negative control. All experiments were carried out in triplicate. For SIRV1 gp29 two biological samples were assayed in triplicate. cA4 degradation was visualised by phosphor imaging following denaturing PAGE (7M urea, 20% acrylamide, 1x TBE) or TLC.
For TLC, 1 μl of radiolabelled product was spotted 1 cm from the bottom of a 20 x 20 cm silica gel TLC plate with fluorescence indicator 254 nm (Supelco Sigma-Aldrich). The TLC plate was placed in a sealed glass chamber pre-warmed and humidified at 37°C and containing 0.5 cm of a running buffer composed of 30% H2O, 70% ethanol and 0.2 M ammonium bicarbonate, pH 9.2. The temperature was lowered to 35°C and the buffer was allowed to rise along the plate through capillary action until the migration front reached 17 cm. The plate was air dried and sample migration was visualised by phosphor imaging.
To examine degradation of cA4 and cA6 by AcrIII-1 proteins, unlabelled cA4 or cA6 (450 μM, BIOLOG Life Science Institute, Bremen, Germany) was incubated with SIRV1 gp29 or YddF (40 μM dimer), in reaction buffers described above, at 70°C and 37°C, respectively. Reactions were quenched at the indicated timepoints and prepared for TLC as described above. Reaction substrate and products, which block fluorescence of the indicator on the plate, were visualised under short wave UV light (254 nm) and the plates were photographed using a 12-megapixel ƒ/1.8 aperture camera.
For kinetic analysis, cA4 cleavage was quantified using the Bio-Formats plugin 36 of ImageJ as distributed in the Fiji package 37 and fitted to a single exponential curve (y = m1 + m2*(1 - exp(-m3*x));m1=0.1;m2=1;m3=1;) using Kaleidagraph (Synergy Software), as described previously 38. The cA4 cleavage rate by the H47A variant in the absence of imidazole was obtained by linear fit. Raw data for kinetic analyses are available in Dataset S2.
Deactivation of HEPN nucleases by ring nucleases in coupled assays
In the absence and presence of Crn1 Sso2081 (2 μM dimer) or AcrIII-1 SIRV1 gp29 (2 μM dimer) 4 μg S. solfataricus Csm complex (~140 nM Csm carrying crRNA targeting A26 RNA target) was incubated with A26 RNA target (50, 20, 5, 2, or 0.5 nM) in buffer containing 20 mM MES pH 6.0, 100 mM NaCl, 1 mM DTT and 3 units SUPERase•In™ Inhibitor supplemented with 2 mM MgCl2 and 0.5 mM ATP at 70°C for 60 min. 5’-end 32P-labelled A1 RNA (AGGGUAUUAUUUGUUUGUUUCUUCUAAACUAUAAGCUAGUUCUGGAGA) and 0.5 μM dimer SsoCsx1 was added to the reaction at 60 min and the reaction was allowed to proceed for a further 60 min before quenching by adding phenol-chloroform. A1 RNA cleavage was visualised by phosphor imaging after denaturing PAGE. A control reaction incubating SsoCsx1 with A1 RNA in the absence of cOA was carried out to determine SsoCsx1 background activity. cA4 synthesis by Csm in response to A26 RNA target and subsequent cA4 degradation in the presence of Crn1 Sso2081 or AcrIII-1 SIRV1 gp29, was visualised by adding 5 nM α-32P-ATP with 0.5 mM ATP at the start of the reaction. Reactions were quenched at 60 min with phenol-chloroform and cA4 degradation products were visualised by phosphor imaging following TLC. A control reaction incubating Csm with ATP and α-32P-ATP in the absence of A26 RNA target was also carried out, quenching the reaction after 60 min.
cA4 degradation capacity of AcrIII-1 SIRV1 gp29 versus the Crn1 enzyme (Sso2081) was determined by incubating 2 μM dimer of each enzyme with 500-0.5 μM unlabelled cA4 (BIOLOG Life Science Institute, Bremen, Germany) in Csx1 buffer at 70°C for 20 min before introducing SsoCsx1 (0.5 μM dimer) and 32P-labelled A1 RNA (50 nM). The reaction was left to proceed for a further 60 min at 70°C before quenching by adding phenol-chloroform. Deproteinised products were separated by denaturing PAGE to visualise RNA degradation.
Tsac 2833 mediated plasmid immunity from a reprogrammed type III system in E. coli
Plasmids pCsm1-5_∆Csm6 (containing the type III Csm interference genes cas10, csm3, csm4, csm5 from M. tuberculosis and csm2 from M. canettii), pCRISPR_TetR (containing M. tuberculosis cas6 and tetracycline resistance gene-targeting CRISPR array), pRAT-Target (tetracycline-resistance, target plasmid) and M. tuberculosis (Mtb)Csm6/ Thioalkalivibrio sulfidiphilus (Tsu)Csx1 expression constructs have been described previously 21. pRAT-Duet was constructed by replacing the pUC19 lacZα gene of pRAT-Target with the MCSs of pACYCDuet-1 by restriction digest (5’-NcoI, 3’-XhoI). The viral ring nuclease (duf1874) gene from Thermoanaerobacterium phage THSA_485A, tsac_2833, was PCR-amplified from its pEHisTEV expression construct and cloned into the 5’-NdeI, 3’-XhoI sites of MCS-2. The cOA-dependent nucleases (mtb csm6, tsu csx1) were cloned into the 5’-NcoI, 3’-SalI sites of MCS-1 by restriction digest from their respective expression constructs. Each nuclease was cloned with and without the viral ring nuclease; pRAT-Duet without insert and pRAT-Duet containing only the viral ring nuclease were used as controls. The plasmid transformation assay was carried out essentially as described in 21. E. coli C43 containing pCsm1-5_∆Csm6 and pCRISPR_TetR were transformed by heat shock with 100 ng of pRAT-Duet target plasmid containing different combinations of cOA-dependent nuclease and viral ring nuclease. After outgrowth at 37°C for 2 h, cells were collected and resuspended in 200 µl LB. A series of 10-fold dilutions was applied onto LB agar containing 100 µg ml-1 ampicillin and 50 µg ml-1 spectinomycin to determine the cell density of the recipient cells and onto LB agar additionally containing 25 µg ml-1 tetracycline, 0.2% (w/v) d-lactose and 0.2% (w/v) l-arabinose to determine the cell density of viable transformants. Plates were incubated at 37°C for 16 – 18 h; further incubation was carried out at room temperature. Colonies were counted manually and corrected for dilution and volume to obtain colony-forming units (cfu) ml-1. Raw data for plasmid counts are available in Dataset S3.
Liquid chromatography high-resolution mass spectrometry
AcrIII-1 SIRV1 gp29 (40 μM dimer) was incubated with 400 μM cA4 in Csx1 buffer for 2 min at 70°C and deproteinised by phenol-chloroform extraction followed by chloroform extraction. Liquid chromatography-high resolution mass spectrometry (LC-HRMS) analysis was performed on a Thermo Scientific™ Velos Pro instrument equipped with HESI source and Dionex UltiMate 3000 chromatography system. Compounds were separated on a Kinetex® EVO C18 column (2.6 µm, 2.1 × 50 mm, Phenomenex) using the following gradient of acetonitrile (B) against 20 mM ammonium bicarbonate (A): 0 – 2 min 2% B, 2 – 10 min 2 – 8% B, 10 – 11 min 8 – 95% B, 11 – 14 min 95% B, 14 – 15 min 95 – 2% B, 15 – 20 min 2% B at a flow rate of 300 µl min-1 and column temperature of 40°C. UV data were recorded at 254 nm. Mass data were acquired on the FT mass analyzer in negative ion mode with scan range m/z 150 – 1500 at a resolution of 30,000. Source voltage was set to 3.5 kV, capillary temperature was 350°C, and source heater temperature was 250°C. Data were analysed using Xcalibur™ (Thermo Scientific).
Phylogenetic analysis
AcrIII-1 homologs were collected by using gp29 (NP_666617) of SIRV1 as a query and running two iterations (E=1e-05) of PSI-BLAST 39 against the non-redundant protein database at NCBI. The sequences were aligned using PROMALS3D 40. Redundant sequences (95% identity threshold) and sequences with mutated H47 active site residue were removed from the alignment. Poorly aligned (low information content) positions were removed using the gt 0.2 function of Trimal 41. The final alignment contained 124 positions. The maximum likelihood phylogenetic tree was constructed using the PhyML program 42 with the automatic selection of the best-fit substitution model for a given alignment. The best model identified by PhyML was LG +G+I. The branch support was assessed using aBayes implemented in PhyML. The tree was visualized using iTOL 43.
Crystallisation
AcrIII-1 H47A variant was concentrated to 10 mg ml-1, incubated at 293 K for 1 hour with a 1.2 M excess of cA4, and centrifuged at 13,000 rpm for 10 minutes prior to crystallization. Sitting drop vapor diffusion experiments were set up at the nanoliter scale using commercially available and in-house crystallization screens and incubated at 293 K. Crystals appeared in various conditions, but those used for data collection grew from 40% 2-methyl-2,4-pentanediol, 5% polyethylene glycol 8000 and 0.1 M sodium cacodylate, pH 6.5. Crystals were harvested and transferred briefly into cryoprotectant containing mother liquor with 20% glycerol immediately before cryo-cooling in liquid nitrogen. The H47A variant was used to avoid cleavage of the cA4 substrate during co-crystallisation. The position of the active site histidine was inferred from the structure of the apo-protein.
Data collection and processing
X-ray data were collected from two crystals at 100 K, at a wavelength 0.9686 Å, on beamline I24 at Diamond Light Source, to 1.49 and 1.60 Å resolution. Both data sets were automatically processed using Xia2 44, utilizing XDS and XSCALE 45. The data were merged in Aimless 46 and the overall resolution truncated to 1.55 Å. The data were phased by molecular replacement using Phaser 47 with a monomer from PDB file 2X4I stripped of water molecules as the search model. Model refinement of AcrIII-1 was achieved by iterative cycles of REFMAC5 48 in the CCP4 suite 49 and manual manipulation in COOT 50. Electron density for cA4 was clearly visible in the maximum likelihood/σA weighted Fobs - Fcalc electron density map at 3σ. The coordinates for cA4 were generated in ChemDraw (Perkin Elmer) and the library was generated using acedrg 51, before fitting of the molecule in COOT. Model quality was monitored throughout using Molprobity 52 (score 1.13; centile 99). Ramachandran statistics were 98.5% favoured, 0% disallowed. Data and refinement statistics are shown in Extended Data Table 1.
Data availability statement
The structural coordinates and data have been deposited in the Protein Data Bank with deposition code 6SCF. The genome sequence of the SSeV virus has been submitted to Genbank with accession code MN53972.
Raw data is available for the plasmid immunity analysis presented in figure 1/ ED figure 3 and the kinetic analysis presented in figure 2 / ED figure 5 & 6 as supplementary files.
Extended Data
Extended Data Figure 1. Multiple sequence alignment of DUF1874 family members and purity of DUF1874 and CRISPR ancillary enzymes used in biochemical assays.

(a) Multiple sequence alignment includes the AcrIII-1 proteins from the archaeal viruses SIRV1, STIV, AFV3, ARV1, SIFV, SMV4 and ATV, the integrated conjugative element ICEBs1 protein YddF from B. subtilis, the bacteriophage proteins from Thermoanaerobacterium phageTHSA-485A, Synechococcus phage S-CBWM1, Fusobacterium phage Fnu1, Hydrogenobaculum phage 1 and the Crn2 protein from Crenothrix polyspora. Conserved residues H47, R66, R85 and E88 are indicated by asterisks. (b) SDS-PAGE of AcrIII-1 SIRV1 gp29 and YddF, CRISPR associated ring nuclease 1 (Crn1) Sso2081 and CRISPR ancillary nuclease Csx1 (Sso1389). The gel is representative for 2 or more biological replicates.
Extended Data Figure 2. Construction of RJW007Δtype I-A and RJW007Δtype I-AΔcsx1mutant strains.

(a) Genomic context of CRISPR system in the genetic host and the mutant strains. A1 and A2 denote two different CRISPR arrays, the orientation of which are indicated with arrows. (b) PCR verification of Δtype I-A mutants. A representative Sulfolobus transformant with type I-A knockout plasmid integrated was grown in Dextrin-Tryptone liquid medium, and the cell cultures were subsequently plated on Dextrin-Tryptone plates containing 5-FOA (5-fluoroorotic acid, 50 µg/mg), uracil (20 µg/ml), and agamatine (1 mg/ml). Seven randomly selected 5-FOA resistant (5-FOAR) colonies were screened using the primers that bind outside of the flanking homologous regions to confirm the type I-A deletion. A representative Δtype I-A mutant was further colony-purified for following experiment. The expected size of PCR product amplified from the genomic DNA of parental strain (referred to wt) and Δtype I-A mutant is 8830 bp and 3001 bp, respectively. -, negative control (water as DNA template for PCR). L, 2-log DNA ladder (NEB, USA). Seven biological replicates were screened (c) PCR analysis of the RJW007Δtype I-AΔcsx1 mutant and its parental strain RJW007Δtype I-A using the primers that anneal to the outside of the flanking homologous regions of csx1, which generated amplicons with 2312 bp and 3650 bp, respectively. -, negative control (water as DNA template for PCR). L, Gene Ruler Express DNA ladder (Thermo Scientific, USA). Experiment carried out once. (d) Plaque counts for the three strains tested (n = 3 biological replicates).
Extended Data Figure 3. Effect of DUF1874 on plasmid immunity provided by a heterologously expressed M. tuberculosis type III-A CRISPR system providing cA4 or cA6 mediated immunity.

Unprocessed images of sample plates for all replicates (2 biological replicates with 4 technical replicates each, n=8). Dilutions are indicated above the plates.
Extended Data Figure 4. Substrate preference of AcrIII-1 SIRV1 gp29 and YddF and effective range of cA4 degradation compared to Crn1.

Thin-layer chromatography (TLC) visualising (under 254 nm UV light) cA4 and cA6 (450 μM) degradation by (a-b) SIRV1 gp29 and (c-d) YddF over time (min). Both AcrIII-1 enzymes display a clear preference for cA4 over cA6. All TLC images are representative of 3 technical replicates (e) Denaturing PAGE showing activation of Csx1 (0.5 μM dimer) by indicated amounts (500-0.5 μM) of HPLC-purified cA4 and its subsequent deactivation when either AcrIII-1 or Crn1 (2 μM dimer) was present to degrade cA4. The AcrIII-1 enzyme degraded 100-fold more cA4 than Crn1. Control reaction (c) shows RNA incubated with Csx1 in the absence of cA4 (n= 3 technical replicates). For gel source data, see Supplementary Figure 1.
Extended Data Figure 5. Structure of SIRV1 gp29 bound to cA4.

a and b, Orthogonal views of SIRV1 gp29 dimer in complex with cA4. The protein monomers are coloured blue and orange, with catalytic residue H47 from the apo structure shown in salmon. cA4 is shown in spacefill with green, blue, red and orange representing carbon, nitrogen, oxygen and phosphorus atoms, respectively. Conserved residues (Extended data figure 1) in the AcrIII-1 family are indicated and discussed in the text. (c) Interactions between each monomer of the SIRV1 dimer (orange and blue), with cA4 shown in green. (d) Schematic showing the interaction. Dotted lines represent hydrogen bonds, with the distance annotated. Spheres represent water molecules.
Extended Data Figure 6. Single turnover cA4 cleavage by SIRV1 gp29 and variants and chemical rescue with imidazole.

(a) Phosphorimage of TLC visualising cA4 cleavage by SIRV1 gp29 H47A (4 μM dimer, 50°C) in the presence or absence of 500 mM imidazole, over time. The rate of cA4 cleavage to generate A2>P and A2-P was calculated by quantifying densiometric signals from the phosphorimage (n= 3 technical replicates). (b) Plot comparing the single turnover rates of cA4 by SIRV1 gp29, its E88A variant, and its H47A variant in the presence or absence of imidazole. cA4 cleavage by the H47A variant can be partially restored when the reaction is supplemented with 500 mM imidazole. Values and error bars plotted reflect the mean ± standard deviation (n= 3 technical replicates). For gel source data, see Supplementary Figure 1.
Extended Data Figure 7. Maximum likelihood phylogeny of AcrIII-1 homologs.

The maximum likelihood phylogenetic tree was constructed with the automatic selection of the best-fit substitution model for a given alignment (LG +G+I). Red circles indicate 95-100% branch support, as assessed using aBayes implemented in PhyML. The scale bar represents the number of substitutions per site. Branches and labels are colour coded: archaea, red; bacteria, black; bacteria and archaea in which AcrIII-1 homologs are associated with CRISPR loci, blue; archaeal viruses and plasmids, green; bacteriophages, orange.
Extended Data Figure 8. Genomic context of crn2 genes in selected bacteria.

Type III CRISPR loci in the bacterial species Crenothrix polyspora, Methylovulum psychrotolerans, Methylomagnum ishizawai, Thioalkalivibrio sufidiphilus and Marinitoga piezophilia are shown in cartoon form, with genes labelled and color-coded. The crn2 gene is shown in pale yellow with a bold outline; CRISPRs are indicated by small black arrows and unrelated/hypothetical genes shown as small white arrows. The size and orientation of genes is not reflected in the cartoon. Key to gene labels: RT, Reverse Transcriptase; Ago, Argonaute; CARF, CRISPR associated Rossman fold; DUF1887, predicted CARF nuclease; CARF-RelE, CARF domain fused to the RelE toxin.
Extended Data Figure 9. CRISPR-associated AcrIII-1 homologs.

Genomic neighbourhoods were analysed using the Enzyme Function Initiative-Genome Neighbourhood Tool (EFI-GNT) against the Pfam profile database 53. Gene annotations are colour coded and the key is provided on the right of the figure.
Extended Data Table 1. Data collection and refinement statistics for AcrIII-1 in complex with cA4.
| AcrIII-1 with cA4 | |
|---|---|
| Data collection | |
| Space group | Pl |
| Cell dimensions | |
| a, b, c (Å) | 49.8, 51.7, 85.6 |
| α, β, γ (°) | 80.2, 89.7, 83.4 |
| Resolution (Å) | 50.63-1.55 (1.58-1.55) * |
| Rsym or Rmerge | 0.12 (0.36) |
| I / σI | 12.3 (1.7) |
| Completeness (%) | 98.6 (92.4) |
| Redundancy | 2.9 (1.8) |
| Refinement | |
| Resolution (Å) | 84.26-1.55 |
| No. reflections | 113882 |
| Rwork / Rfree | 0.20 / 0.25 |
| No. atoms | |
| Protein | 7,365 |
| Ligand/ion | 352 |
| Water | 595 |
| B-factors | |
| Protein | 20.2 |
| Ligand/ion | 13.3 |
| Water | 30.7 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.64 |
Values in parentheses are for highest-resolution shell.
Supplementary Material
Supplementary Information is available for this paper.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (REF: BB/S000313/1 to MFW and REF: BB/R008035/1 to TMG) and by a NASA Exobiology and Evolutionary Biology grant (NNX14AK23G to RJW). We thank Jesse Black and Maria Alejandra-Bautista for isolating and characterizing the SSeV virus, and Rebecca Wipfler and Wenlong Zhu for technical assistance.
Footnotes
Author contributions J.S.A. designed experiments and carried out the enzyme assays and analysis; S.M. carried out the structural biology; C.Z. constructed the S. islandicus strains and performed the virus infection assays; S.Grü. carried out the plasmid transformation assays and mass spectrometry; S.Gra. generated expression plasmids and purified proteins; M.K. contributed to the conception of the project and performed phylogenetic analysis; T.M.G., R.J.W. and M.F.W. oversaw the work, analysed the data and wrote the manuscript. All authors contributed to data analysis and writing.
Competing interests The University of St Andrews has filed a patent application (UK Patent Application No. 1902256.5: Novel Enzyme for phage therapy. Filed 19/2/2019) for which J.S.A. and M.F.W. are inventors. The other authors declare no competing interests.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Samai P, et al. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamulaitis G, et al. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell. 2014;56:506–517. doi: 10.1016/j.molcel.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 3.Kazlauskiene M, Kostiuk G, Venclovas C, Tamulaitis G, Siksnys V. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017;357:605–609. doi: 10.1126/science.aao0100. [DOI] [PubMed] [Google Scholar]
- 4.Niewoehner O, et al. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017;548:543–548. doi: 10.1038/nature23467. [DOI] [PubMed] [Google Scholar]
- 5.Rouillon C, Athukoralage JS, Graham S, Grüschow S, White MF. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. eLife. 2018;7:e36734. doi: 10.7554/eLife.36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarova KS, Anantharaman V, Grishin NV, Koonin EV, Aravind L. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front Genet. 2014;5:102. doi: 10.3389/fgene.2014.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rostol JT, Marraffini LA. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR-Cas immunity. Nat Microbiol. 2019;4:656–662. doi: 10.1038/s41564-018-0353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyenson NC, Gayvert K, Varble A, Elemento O, Marraffini LA. Broad Targeting Specificity during Bacterial Type III CRISPR-Cas Immunity Constrains Viral Escape. Cell Host Microbe. 2017;22:343–353 e343. doi: 10.1016/j.chom.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng L, Garrett RA, Shah SA, Peng X, She Q. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol. 2013;87:1088–1099. doi: 10.1111/mmi.12152. [DOI] [PubMed] [Google Scholar]
- 10.Jiang W, Samai P, Marraffini LA. Degradation of Phage Transcripts by CRISPR-Associated RNases Enables Type III CRISPR-Cas Immunity. Cell. 2016;164:710–721. doi: 10.1016/j.cell.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whiteley AT, et al. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature. 2019;567:194–199. doi: 10.1038/s41586-019-0953-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maelfait J, Rehwinkel J. RECONsidering Sensing of Cyclic Dinucleotides. Immunity. 2017;46:337–339. doi: 10.1016/j.immuni.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Cohen D, et al. Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature. 2019 doi: 10.1038/s41586-019-1605-5. [DOI] [PubMed] [Google Scholar]
- 14.Athukoralage JS, Rouillon C, Graham S, Grüschow S, White MF. Ring nucleases deactivate Type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature. 2018;562:277–280. doi: 10.1038/s41586-018-0557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borges AL, Davidson AR, Bondy-Denomy J. The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu Rev Virol. 2017;4:37–59. doi: 10.1146/annurev-virology-101416-041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang S, Maxwell KL. Meet the Anti-CRISPRs: Widespread Protein Inhibitors of CRISPR-Cas Systems. CRISPR J. 2019;2:23–30. doi: 10.1089/crispr.2018.0052. [DOI] [PubMed] [Google Scholar]
- 17.Oke M, et al. The Scottish Structural Proteomics Facility: targets, methods and outputs. J Struct Funct Genom. 2010;11:167–180. doi: 10.1007/s10969-010-9090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson ET, et al. A new DNA binding protein highly conserved in diverse crenarchaeal viruses. Virology. 2007;363:387–396. doi: 10.1016/j.virol.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 19.Wirth JF, et al. Development of a genetic system for the archaeal virus Sulfolobus turreted icosahedral virus (STIV) Virology. 2011;415:6–11. doi: 10.1016/j.virol.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Bautista MA, Zhang C, Whitaker RJ. Virus-induced dormancy in the archaeon Sulfolobus islandicus. mBio. 2015;6:e02565–02514. doi: 10.1128/mBio.02565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grüschow S, Athukoralage JS, Graham S, Hoogeboom T, White MF. Cyclic oligoadenylate signalling mediates Mycobacterium tuberculosis CRISPR defence. Nucl Acids Res. 2019;47:9259–9270. doi: 10.1093/nar/gkz676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bondy-Denomy J, et al. A Unified Resource for Tracking Anti-CRISPR Names. CRISPR J. 2018;1:304–305. doi: 10.1089/crispr.2018.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auchtung JM, Aleksanyan N, Bulku A, Berkmen MB. Biology of ICEBs1, an integrative and conjugative element in Bacillus subtilis. Plasmid. 2016;86:14–25. doi: 10.1016/j.plasmid.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broo KS, Brive L, Sott RS, Baltzer L. Site-selective control of the reactivity of surface-exposed histidine residues in designed four-helix-bundle catalysts. Fold Des. 1998;3:303–312. doi: 10.1016/S1359-0278(98)00041-8. [DOI] [PubMed] [Google Scholar]
- 26.Bhoobalan-Chitty Y, Johansen TB, Di Cianni N, Peng X. Inhibition of Type III CRISPR-Cas Immunity by an Archaeal Virus-Encoded Anti-CRISPR Protein. Cell. 2019 doi: 10.1016/j.cell.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Knott GJ, et al. Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat Struct Mol Biol. 2019;26:315–321. doi: 10.1038/s41594-019-0208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong L, et al. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat Struct Mol Biol. 2019;26:308–314. doi: 10.1038/s41594-019-0206-1. [DOI] [PubMed] [Google Scholar]
- 29.Keller J, et al. Crystal structure of AFV3-109, a highly conserved protein from crenarchaeal viruses. Virol J. 2007;4:12. doi: 10.1186/1743-422X-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson RE, Kouris A, Seward CH, Campbell KM, Whitaker RJ. Structured Populations of Sulfolobus acidocaldarius with Susceptibility to Mobile Genetic Elements. Genome Biol Evol. 2017;9:1699–1710. doi: 10.1093/gbe/evx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Held NL, Herrera A, Whitaker RJ. Reassortment of CRISPR repeat-spacer loci in Sulfolobus islandicus. Environ Microbiol. 2013;15:3065–3076. doi: 10.1111/1462-2920.12146. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C, Whitaker RJ. Microhomology-Mediated High-Throughput Gene Inactivation Strategy for the Hyperthermophilic Crenarchaeon Sulfolobus islandicus. Appl Environ Microbiol. 2018;84 doi: 10.1128/AEM.02167-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Cooper TE, Krause DJ, Whitaker RJ. Augmenting the genetic toolbox for Sulfolobus islandicus with a stringent positive selectable marker for agmatine prototrophy. Appl Environ Microbiol. 2013;79:5539–5549. doi: 10.1128/AEM.01608-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng L, Zhu H, Chen Z, Liang YX, She Q. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles. 2009;13:735–746. doi: 10.1007/s00792-009-0254-2. [DOI] [PubMed] [Google Scholar]
- 35.Rouillon C, Athukoralage JS, Graham S, Grüschow S, White MF. Investigation of the cyclic oligoadenylate signalling pathway of type III CRISPR systems. Methods Enzymol. 2019;616:191–218. doi: 10.1016/bs.mie.2018.10.020. [DOI] [PubMed] [Google Scholar]
- 36.Linkert M, et al. Metadata matters: access to image data in the real world. J Cell Biol. 2010;189:777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sternberg SH, Haurwitz RE, Doudna JA. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA. 2012;18:661–672. doi: 10.1261/rna.030882.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pei J, Grishin NV. PROMALS3D: multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol Biol. 2014;1079:263–271. doi: 10.1007/978-1-62703-646-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 43.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucl Acids Res. 2019;47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallog. 2010;43:186–190. [Google Scholar]
- 45.Kabsch W. Xds. Acta Crystallog D Biol Crystallog. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallog D Biol Crystallog. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallog. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog D Biol Crystallog. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallog D Biol Crystallog. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallog D Biol Crystallog. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Long F, et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallog D Struct Biol. 2017;73:112–122. doi: 10.1107/S2059798317000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallog D Biol Crystallog. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerlt JA. Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence-Function Space and Genome Context to Discover Novel Functions. Biochemistry. 2017;56:4293–4308. doi: 10.1021/acs.biochem.7b00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structural coordinates and data have been deposited in the Protein Data Bank with deposition code 6SCF. The genome sequence of the SSeV virus has been submitted to Genbank with accession code MN53972.
Raw data is available for the plasmid immunity analysis presented in figure 1/ ED figure 3 and the kinetic analysis presented in figure 2 / ED figure 5 & 6 as supplementary files.