

Abstract
The first edition of the ‘Standardized Pathology Report for Colorectal Cancer,’ which was developed by the Gastrointestinal Pathology Study Group (GIP) of the Korean Society of Pathologists, was published 13 years ago. Meanwhile, there have been many changes in the pathologic diagnosis of colorectal cancer (CRC), pathologic findings included in the pathology report, and immunohistochemical and molecular pathology required for the diagnosis and treatment of colorectal cancer. In order to reflect these changes, we (GIP) decided to make the second edition of the report. The purpose of this standardized pathology report is to provide a practical protocol for Korean pathologists, which could help diagnose and treat CRC patients. This report consists of “standard data elements” and “conditional data elements.” Basic pathologic findings and parts necessary for prognostication of CRC patients are classified as “standard data elements,” while other prognostic factors and factors related to adjuvant therapy are classified as “conditional data elements” so that each institution could select the contents according to the characteristics of the institution. The Korean version is also provided separately so that Korean pathologists can easily understand and use this report. We hope that this report will be helpful in the daily practice of CRC diagnosis.
Keywords: Colorectal neoplasms, Pathology report, Standardization, Protocol
The first edition of the ‘Standardized Pathology Report for Colorectal Cancer’ was developed by the Gastrointestinal Pathology Study Group of the Korean Society of Pathologists and published in the Korean Journal of Pathology (predecessor of the Journal of Pathology and Translational Medicine) in 2006 [1]. Colorectal cancer (CRC), which was the fourth most common cancer in Korea at the time, is now the second most common cancer in Korea. Meanwhile, there have been many changes in the pathologic diagnosis of CRC, such as the diagnostic criteria for carcinoma, and pathologic findings included in the pathology report [1,2]. Molecular pathology tests for CRC have also become necessary tests, as targeted therapy and immunotherapy were introduced into the treatment of CRC. The existing standardization report does not reflect the recent changes in colon cancer diagnosis.
There has been considerable demand for the revision of the standardized pathology report, which is used by many Korean pathologists. From September 2017 to October 2018, the committee for the revision of the report was organized, and after several discussions and meetings, the second edition of the ‘Standardized Pathology Report for Colorectal Cancer’ was completed. This new report is based on the previous report with reference to the College of American Pathologists (CAP) protocol, American Joint Committee on Cancer (AJCC) 8th edition, World Health Organization (WHO) classification of tumors of the digestive system 5th edition, and International Classification of Diseases for Oncology (ICD-O) [3-6].
The purpose of this standardized pathology report is to enable standardized diagnosis and treatment of CRC patients and, furthermore, to help select the same patient group and exchange information in multicenter clinical trials or international studies. It would be desirable to include all the latest findings up to now in this report, but this would lead to an increase in the workload of the pathologist. In the current medical system of Korea, increased workloads cannot be appropriately reflected in the medical fee, which may lead this report to be unpractical for Korean pathologists. Therefore, the basic pathologic findings and parts necessary for prognostication of CRC patients are classified as “standard data elements,” and other prognostic factors and factors related to adjuvant therapy are classified as “conditional data elements” so that each institution can select the contents according to the characteristics of the institution. As with the first edition, we have written this in English for the internationalization of pathology reports. We also have restricted the use of abbreviations or numerical taxonomy so that we can keep track of future data elements even if the diagnostic criteria and classification methods change.
As The Korean Journal of Pathology has become an English-language journal named The Journal of Pathology and Translational Medicine, this report was written in English, but the Korean version is also provided separately so that Korean pathologists can easily understand and use this report (Supplementary Material 1).
STANDARD DATA ELEMENTS FOR RESECTED COLORECTUM
All report forms mentioned in this document are shown in Table 1. If there are two or more tumors, the data elements should be listed for each tumor, starting with the tumor that has the deepest level of invasion. ‘Regional lymph node metastasis,’ ‘Associated findings,’ and ‘Separate lesions’ are noted only in the deepest tumor.
Table 1.
Report form of pathologic diagnosis for resected coloectal cancer
| Standard data elements | |||
|---|---|---|---|
| Specimen type | |||
| □ Right hemicolectomy | |||
| □ Transverse hemicolectomy | |||
| □ Left hemicolectomy | |||
| □ Anterior resection | |||
| □ Low anterior resection | |||
| □ Abdominoperineal resection | |||
| □ Subtotal/total colectomy | |||
| □ Total proctocolectomy | |||
| □ Transanal excision | |||
| □ Endoscopic mucosal resection | |||
| □ Other: (specify: ) | |||
| Histopathologic type of invasive carcinoma | |||
| □ Adenocarcinoma, NOS | |||
| □ Low-grade (well differentiated and moderately differentiated) | |||
| □ High-grade (poorly differentiated) | |||
| □ Mucinous adenocarcinoma | |||
| □ Signet ring cell carcinoma | |||
| □ Medullary carcinoma | |||
| □ Serrated adenocarcinoma | |||
| □ Micropapillary adenocarcinoma | |||
| □ Squamous cell (epidermoid) carcinoma (excluding upwardly spreading anal tumors) | |||
| □ Adenosquamous carcinoma | |||
| □ Small cell neuroendocrine carcinoma | |||
| □ Large cell neuroendocrine carcinoma | |||
| □ Mixed neuroendocrine-non-neuroendocrine neoplasm | |||
| □ Undifferentiated carcinoma | |||
| □ Other: (specify: ) | |||
| Location | |||
| □ Cecum | |||
| □ Ascending colon | |||
| □ Hepatic flexure | |||
| □ Transverse colon | |||
| □ Splenic flexure | |||
| □ Descending colon | |||
| □ Sigmoid colon | |||
| □ Rectosigmoid junction | |||
| □ Rectum | |||
| □ Other: (specify: ) | |||
| Gross type | |||
| □ Fungating/polypoid | |||
| □ Ulcerofungating | |||
| □ Ulceroinfiltrative | |||
| □ Infiltrative | |||
| □ Unclassifiable | |||
| Tumor size | |||
| × × cm | |||
| Depth of invasion | |||
| □ Intramucosal carcinoma (pTis) | |||
| □ Tumor invades the submucosa (pT1) | |||
| □ Tumor invades the muscularis propria (pT2) | |||
| □ Tumor invades through the muscularis propria into pericolorectal tissue (pT3) | |||
| □ Tumor invades through the visceral peritoneum (pT4a) | |||
| □ Tumor directly invades or adheres to adjacent organs or structures (pT4b) | |||
| - [Endoscopic excision (Endoscopic submucosal dissection/polypectomy) or transanal excision] | |||
| □ Tumor invades the lamina propria with no extension through muscularis mucosae (pTis) | |||
| □ Tumor invades the submucosa (pT1) | |||
| For sessile lesion: | |||
| Distance of tumor from muscularis mucosae: mm | |||
| For pedunculated lesion: | |||
| Haggitt level (head, neck, stalk, beyond stalk) | |||
| Distance of tumor invasion in stalk: mm | |||
| Resection margin | |||
| Proximal margin | |||
| □ Free from carcinoma | |||
| □ Involved by carcinoma | |||
| Distal margin | |||
| □ Free from carcinoma | |||
| □ Involved by carcinoma | |||
| Circumferential margin (rectum only) | |||
| □ Free from carcinoma | |||
| □ Involved by carcinoma | |||
| Safety margin: proximal cm, distal cm, circumferential cm | |||
| - [Endoscopic excision (Endoscopic mucosal resection/submucosal dissection/polypectomy) or transanal excision] | |||
| Deep margin | |||
| □ Free from carcinoma | |||
| □ Involved by carcinoma | |||
| □ Not applicable | |||
| Horizontal margin | |||
| □ Free from carcinoma | |||
| □ Involved by carcinoma | |||
| □ Involved by adenoma: (specify grade: ) | |||
| □ Not applicable | |||
| Safety margin: deep cm, horizontal cm | |||
| Regional lymph node metastasis | |||
| □ No metastasis in all regional lymph nodes (pN0) | |||
| □ Metastasis to out of regional lymph nodes pN | |||
| Lymphatic (small vessel) invasion | |||
| □ Not identified | |||
| □ Present | |||
| Venous invasion | |||
| □ Not identified | |||
| □ Present | |||
| □ Intramural | |||
| □ Extramural | |||
| Perineural invasion | |||
| □ Not identified | |||
| □ Present | |||
| Pre-existing adenoma | |||
| □ Absent | |||
| □ Tubular/Tubulovillous/Villous adenoma | |||
| Low grade dysplasia/High grade dysplasia | |||
| □ Sessile serrated lesion (sessile serrated adenoma/polyp) | |||
| □ Sessile serrated lesion (sessile serrated adenoma/polyp) with dysplasia | |||
| □ Traditional serrated adenoma | |||
| □ Other (specify: ) | |||
| Associated findings | |||
| □ Absent | |||
| □ Tumor perforation (pT4a) | |||
| □ Perforation (non-tumor perforation) | |||
| □ Metastasis to one site or organ without peritoneal metastasis (pM1a) | |||
| □ Metastasis to two or more sites or organs without peritoneal metastasis (pM1b) | |||
| □ Metastasis to the peritoneal surface with or without other site or organ metastasis (pM1c) | |||
| Specify metastatic sites or organs: | |||
| Separate lesions | |||
| □ Absent | |||
| □ Adenoma | |||
| □ Polyp | |||
| □ GIST | |||
| □ Ulcerative colitis/Crohn’s disease | |||
| □ Others | |||
| Specify: | |||
|
Conditional data elements | |||
| Tumor budding | |||
| □ Not identified | |||
| □ Present | |||
| □ ≤ 4 buds (low) | |||
| □ 5–9 buds (intermediate) | |||
| □ ≥ 10 buds (high) | |||
| □ Cannot be assessed (specify: ) | |||
| Completeness of total mesorectal excision | |||
| □ Complete | |||
| □ Nearly complete | |||
| □ Incomplete | |||
| □ Cannot be determined | |||
| Preoperative chemoradiotherapy | |||
| □ Yes □ No □ Not known | |||
| If yes) Tumor regression grade | |||
| □ Grade 0: No viable cancer cells (complete response) | |||
| □ Grade 1: Single cells or rare small groups of cancer cells (near-complete response) | |||
| □ Grade 2: Residual cancer with evident tumor regression, but more than single cells or rare small groups of cancer cells (partial response) | |||
| □ Grade 3: Extensive residual cancer with no evident tumor regression (poor or no response) | |||
| DNA mismatch repair immunohistochemistry | |||
| MLH1: | □ Positive (retained expression) | ||
| □ Negative (loss of expression) | |||
| MSH2: | □ Positive (retained expression) | ||
| □ Negative (loss of expression) | |||
| PMS2: | □ Positive (retained expression) | ||
| □ Negative (loss of expression) | |||
| MSH6: | □ Positive (retained expression) | ||
| □ Negative (loss of expression) | |||
| Summary: DNA mismatch repair deficiency (was/was not) observed | |||
| Microsatellite instability (MSI) | |||
| Summary: | □ MSI-stable (MSS) | ||
| □ MSI-low (MSI-L) | |||
| □ MSI-high (MSI-H) | |||
| KRAS mutation analysis | |||
| □ No mutation detected | |||
| □ Mutation detected (specify: example: c.35G > A, p.Gly12Asp ) | |||
| NRAS mutation analysis | |||
| □ No mutation detected | |||
| □ Mutation detected (specify: example: c.35G>A, p.Gly12Asp ) | |||
| BRAF mutation analysis | |||
| □ No mutation detected | |||
| □ BRAF V600E (c.1799T > A) mutation | |||
| □ Other BRAF mutation (specify: ) | |||
Comment: This report is intended to be applicable to endoscopic resection or transanal excision specimens as well as surgical resection of CRC.
NOS, not otherwise specified; CRC, colorectal cancer.
Histopathologic type of invasive carcinoma
Histologic classification of tumors is based on WHO classification (5th edition) [5]. Although most CRCs are “adenocarcinoma, not otherwise specified (NOS),” if there are other histologic variants, it is recommended to mention them separately. This is because some histologic variants may be associated with specific molecular alteration or patient prognosis [5,7]. Representative histologic types of CRC described in WHO classification and AJCC 8th edition are shown in Table 1 [4,5].
Mucinous adenocarcinoma and signet ring cell carcinoma can be diagnosed when extracellular mucins are over 50% of tumor areas and signet ring cells are over 50% of tumor component, respectively. At levels of 50% or less, it is recommended to describe the ratio of mucin or signet ring cells along with histologic type [5]. When patients received preoperative neo-adjuvant therapy, which may produce mucin, it is advisable to describe the diagnosis of the preoperative specimen [8]. Medullary carcinoma is a rare histologic type that requires differentiation from undifferentiated carcinoma and it is diagnosed when cancer cells appear as solid or sheet-like structures and lymphocytic infiltration is prominent with intraepithelial (tumor-infiltrating) lymphocytes and neutrophils [5,7]. Tumor cells of medullary carcinoma usually show abundant eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli [5]. Another rare histologic type, micropapillary adenocarcinoma, can be diagnosed when small tumor cell clusters are surrounded by empty spaces, resembling lymphatic or small vessel invasion [9-11]. Micropapillary adenocarcinoma has a high risk of lymph node metastasis and is frequently accompanied with poor prognostic factors such as lymphatic and vascular invasion [9-13]. However, CRC with pure micropapillary patterns are extremely rare and most micropapillary lesions coexist with another histologic type [5,14]. The WHO classification suggests that tumors consisting of 5% or more micropapillary component should be diagnosed as micropapillary adenocarcinoma [5]. However, the minimal proportion of micropapillary components required for the diagnosis of micropapillary adenocarcinoma is controversial, and our committee could not reach a consensus. Serrated adenocarcinoma is also a special subtype of CRC that is morphologically similar to serrated polyps and is characterized by neoplastic glands with prominent epithelial serrations, low nucleus-to-cytoplasm ratio, eosinophilic and abundant cytoplasm, and vesicular nuclei [5,15,16]. Although some studies have suggested diagnostic criteria for serrated adenocarcinoma [17], a consensus has not yet been reached. Adenosquamous carcinoma is diagnosed when an area of definite squamous differentiation is present in the tumor. Although the WHO classification suggests a greater than 20% and 25% adenocarcinoma component or squamous cell carcinoma component, respectively, for the diagnosis of adenosquamous carcinoma in esophageal and gastric cancer, respectively, there is no standardized diagnostic criteria for adenosquamous carcinoma in terms of the squamous cell carcinoma component [5]. We recommend diagnosing adenosquamous carcinoma when the squamous cell carcinoma component is clearly seen in “more than occasional small foci” as described in the previous report [1]. Undifferentiated carcinoma is diagnosed when the epithelial tumor lacks morphological, immunohistochemical, and molecular evidence of specific differentiation [5]. Adenoma-like adenocarcinoma, also called villous adenocarcinoma, was first introduced as a histological subtype of CRC in the WHO 5th edition [5]. Adenoma-like adenocarcinoma is composed of villous adenoma-like well differentiated tumors in the invasive portion, showing pushing border and minimal desmoplasia [5]. Whether adenoma-like adenocarcinoma should be classified as a specific subtype of CRC is still controversial and has not been added as a subtype in this report.
Differentiation of tumors is determined by the area ratio of gland or tubule formation by tumor cells [7]. The degree of differentiation of the tumor is applicable to adenocarcinoma, NOS. This is because other histologic types show their own prognosis [7]. Recently, tumor differentiation has been shown to affect the prognosis of patients with mucinous adenocarcinoma [5,18]. However, standardized tumor grading of mucinous adenocarcinoma has not been presented yet. Tumor grading is preferably performed using a two-tiered system with low-grade and high-grade [5]. In the 3-tiered grading system, tumor differentiation is graded as well differentiated (> 95% gland formation), moderately differentiated (50%–95% gland formation), or poorly differentiated (< 50% gland formation). The “well- and moderately differentiated” grades correspond to low-grade, while “poorly differentiated” corresponds to high-grade of the two-tiered grading system [5,19].
Location
The location of the tumor follows the ICD-O classification [6]. The length of the cecum is approximately 6 cm and the length of the sigmoid colon is approximately 40 cm. The rectum begins at 1–2 cm above the dentate line and is 12–15 cm long. The upper third of the rectum is covered by peritoneum on the anterior and lateral sides, the middle third is covered by peritoneum on the anterior side, and the lower third is not covered by peritoneum. In this revision, “overlapping lesion of the colon” was removed from the tumor location section because it is practically not used in most institutions. When tumors are involved in two locations of the colon, the tumor epicenter and more involved area should be considered in the determination of tumor location.
Gross type
Superficial type is not recommended for describing tumor gross morphology, because superficial type could be defined by microscopic examination. Fungating/polypoid type can substitute most gross morphology of the previously established superficial type. Nevertheless, if superficial type is used, it should be applied to tumors that are confined to mucosa or submucosa and with tumor thickness of no more than two-fold thickness of adjacent mucosa. Other criteria for tumor gross types are the same as in the previous version. Fungating/polypoid, ulcerofungating, ulceroinfiltrative, and infiltrative gross types correspond to the Borrmann classification of gastric cancer.
Tumor size
The tumor size is expressed as the product of the longest axis and the length perpendicular to it. The depth of the tumor is measured with a microscope at the thickest point.
Depth of invasion
According to the AJCC 8th edition, “intraepithelial carcinoma” that is confined to the crypt epithelium and lacks invasion beyond the basement membrane is considered synonymous to high-grade dysplasia and is excluded from the pTis category. Intramucosal carcinoma that shows invasion into lamina propria with no penetration through the muscularis mucosae into the submucosa is assigned to pTis considering the negligible risk for metastasis [4]. The histopathologic features of intramucosal carcinoma are defined as follows: (1) infiltration of the stroma by either single or small clusters of tumor cells with basement membrane disruption; (2) a stromal response such as desmoplasia or inflammatory cell infiltrates around the invasion front; (3) definite nuclear anaplasia and severe glandular structural abnormality including marked glandular crowding or excessive branching and budding that endows high suspicion of basement membrane destruction (Fig. 1A, B) [20]. In comparison, high-grade dysplasia is diagnosed when the tumor gland shows (1) nuclear pleomorphism with loss of polarity, (2) architectural complexity of intraluminal cribriforming or necrosis, and (3) back-to-back fusion of multiple glands without lamina propria invasion. Low-grade dysplasia is used when the pseudostratified nucleus is seen without complex architectural change of the glands, regardless of the nuclear ratio to the cell length (Fig. 1C) (refer to ‘Preexisting adenoma’). However, intraepithelial carcinoma of the colorectum had been classified as pTis in the AJCC 7th or earlier editions, while intraepithelial carcinoma of other gastrointestinal tracts except the colorectum is still diagnosed as pTis [4]. Therefore, we recommend minimizing the use of intraepithelial carcinoma or adenocarcinoma in situ in order to reduce confusion and suggest using high-grade dysplasia or intramucosal carcinoma for the diagnosis of mucosal high-grade lesions.
Fig. 1.
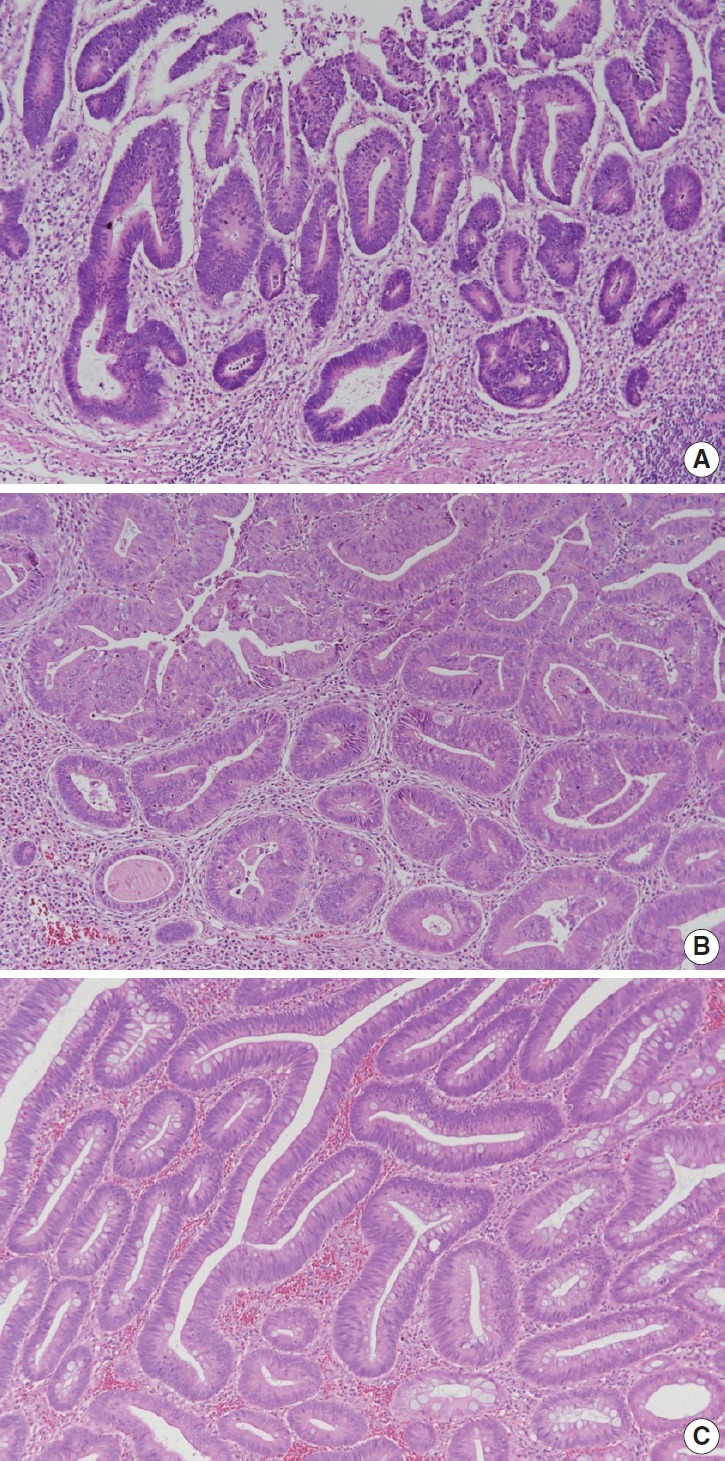
Histologic features of intramucosal carcinoma. (A) The intramucosal carcinoma shows irregular invasive glands accompanied by desmoplasia. (B) The glands show excessive budding and luminal serration, which is highly suspicious for disruption of the basement membrane. (C) The elongated nuclei are seen in low-grade dysplasia, regardless of the ratio to the cell length.
If there is a tumor at the site where the proper muscle has disappeared by ulceration, it is defined as subserosal infiltration of the tumor (pT3). If tumor cells approach the serosal surface by a gap of ≤ 1 mm with a fibroinflammatory reaction, scrupulous examinations using deeper sections and/or additional tissue blocks are needed to uncover serosal surface involvement [4,21]. In colorectal tumors, similar to other gastrointestinal organs, pT4 is subcategorized into pT4a and pT4b. Although pT4a is basically defined as direct involvement of the serosal surface (visceral peritoneum) by tumor invasion, pT4a encompasses the cases in which tumors with perforation display carcinoma cells running through inflammation to the serosal surface and are accompanied by mesothelial proliferation. The pT4a category is not applicable in nonperitonealized portions of the colorectum, including posterior aspects of the ascending and descending colon and lower portion of the rectum. The pT4b category is assigned when the tumor directly invades the adjacent organs or structures. For distal rectal tumors, tumors with involvement of the external sphincter are assigned to pT3, whereas those with the involvement of the levator ani muscle are assigned to pT4b.
The presence of tumor cells within the lymphatic or venous vessels is not considered when determining the depth of invasion. The presence of vascular invasion should be recorded in parentheses separately (e.g., invades proper muscle [involvement of subserosa by lymphatic emboli]). The skip metastasis of multiple lesions in mucosa or submucosa of the adjacent bowel is not classified as distant metastasis. The presence of a peritumoral abscess or acellular mucin pool in cancers with preoperative chemoradiotherapy has been known to be unrelated to patients’ prognosis in CRC, and also is not considered in determining the depth of invasion [22-24].
Endoscopic excision (endoscopic submucosal dissection/polypectomy) or transanal excision
Invasive adenocarcinomas arising in colorectal adenomas have been called “malignant polyps,” in which tumor cells penetrate through the muscularis mucosae into the submucosa. Independent prognostic factors that prompt further surgical treatment of endoscopically resected polyps are as follows: (1) poorly differentiated carcinoma, (2) tumors at or less than 1 mm from the resection margin, and (3) presence of lymphatic and/or venous vessel involvement. Submucosal invasion depth is also an important factor in determining subsequent surgical treatment, since a submucosal invasion depth of ≥ 1,000 μm in malignant polyps of sessile (non-pedunculated) morphology indicates a significantly increased risk for lymph node metastasis [25,26].
In this committee, measurement methods were discussed to reduce measurement error in submucosal invasion depth. Polyps are classified into pedunculated and nonpedunculated polyps. For pedunculated polyps, submucosal invasion is classified as ‘head,’ ‘neck,’ ‘stalk,’ and ‘beyond stalk’ according to the Haggitt classification, in which the neck of the polyp (level 2) is the reference point for measuring stalk invasion (Fig. 2A, B) [27,28]. In nonpedunculated polyps, when the muscularis mucosae is preserved, submucosal invasion depth is measured vertically from the lowest part of the muscularis mucosae. When the muscularis mucosae has been disrupted or has disappeared, the depth of submucosa invasion is measured from the imaginary line that is continuous from the residual muscularis mucosae of the neighboring mucosa (Fig. 2C, D) [29].
Fig. 2.

Measuring depth of invasion in tumors with submucosal invasion. (A) Haggitt level of invasion is composed of head, neck, stalk, and beyond stalk in pedunculated tumors. (B) The depth of invasion should be measured from the neck of the polyp (Haggitt level 2). (C) In cases with disrupted muscularis mucosae, the depth of submucosa invasion is measured from a continuous line of the residual muscularis mucosae. (D) To highlight indistinct muscularis mucosae, immunohistochemistry for desmin may be performed.
Resection margin
The distance from the proximal and distal resection margins is the length from the edge of the carcinoma to the nearest resection margin. In rectal cancer, the circumferential resection margin should be measured in the areas uncovered by peritoneum. After confirming the circumferential resection margin of the resected specimen, mark the margin with ink, and obtain sections including the site where the tumor is most deeply infiltrated. The involvement of carcinoma in the margin is finally assessed by microscopic examination. If the distance between circumferential resection margin and carcinoma (including lymph node, neural invasion, and intravascular tumor emboli) is 0.1 cm or less than 0.1 cm, the resection margin is regarded as positive [3,4]. Circumferential margins of colon cancers other than rectal cancers can be reported when necessary.
Endoscopic excision (endoscopic mucosal resection/submucosal dissection/polypectomy) or transanal excision
This form is applied to the endoscopic resection (polypectomy, mucosal resection, and submucosal dissection) and trans-anal resection. The deep and horizontal margins are examined microscopically. Margin involvement by adenoma is reported in the horizontal margin. When it is impossible to evaluate the resection margins, such as in specimen fragmentation, it shall be marked as ‘not applicable.’
Regional lymph node metastasis
Diagnosis of regional lymph node metastasis is made according to the AJCC 8th edition [4]. Although it is recommended to examine 12 or more lymph nodes for determination of accurate prognosis, pN0 can be used if fewer lymph nodes are available, but no lymph node metastasis is observed. Preoperative chemotherapy and radiotherapy can cause fewer lymph nodes to be harvested. Metastasis to lymph nodes other than regional lymph nodes should be diagnosed as distant metastasis and should not be included in lymph node metastasis numbers.
If the size of the metastatic tumor is less than 2 mm and more than 0.2 mm, it is classified as micrometastasis. If it is less than 0.2 mm, it is classified as isolated tumor cell [4]. A lymph node with micrometastasis is counted as a metastatic lymph node. Although isolated tumor cells are known to be associated with poor prognosis in some stages, pN0 is recommended in AJCC [4]. However, this is controversial and difficult to apply in routine pathologic diagnosis. Thus, we recommend that isolated tumor cells found on hematoxylin and eosin (H&E) slides are considered to be lymph node metastasis and included in the metastatic lymph node numbers, as in the first edition of this report [1].
Tumor deposits are tumor nodules observed separately from the primary tumor in the subserosa, mesentery, and perirectal tissues, regardless of size, shape, and border. Lymph node, vessel, and neural invasions are excluded from tumor deposits [4]. If a blood vessel or nerve structure is observed within the tumor nodule, it should be regarded as a blood vessel invasion or a neural invasion. Elastic stain or immunohistochemical stain (IHC), such as smooth muscle actin, may be helpful in differentiating tumor deposits. pN1c is used when there is no lymph node metastasis, regardless of the invasion depth of the primary tumor, and when there is a tumor deposit. If there are metastatic lymph nodes, tumor deposits are not included in the number of positive lymph nodes. Care should be taken if the patient received preoperative therapy, because regressed residual tumor in subserosa or perirectal soft tissue can be seen as a tumor deposit.
Lymphatic (small vessel) invasion and venous invasion
Identification of lymphatic invasion by the tumor has been recognized as a predictor of lymph node metastasis [26], and identification of venous invasion is a well-known independent prognostic indicator [30]. IHC for D2-40 may be performed additionally to identify endothelial cells because it can be difficult to differentiate the lymphatic vessel from retraction artifacts on H&E sections (Fig. 3A). It is commonly difficult to distinguish lymphatic vessels from blood vessels on H&E sections. Thus, it is considered as lymphatic (small vessel) invasion when the tumor cells involve small vessels, such as lymphatics, capillaries, and postcapillary venules (Fig. 3B), whereas venous invasion is when the tumor cells involve large vessels with an identifiable smooth muscle layer or elastic lamina (Fig. 3C) [31]. This should be considered lymphatic (small vessel) invasion if the size of the involved vessel corresponds to that of small vessel, even though there are red blood cells in the involved vessel. Additionally, special stains for elastic fiber or IHCs for CD31, D2-40, and smooth muscle actin can be used if necessary. It is recommended extramural venous invasion be reported separately from the intramural venous invasion, because the former is an adverse prognostic factor and an independent risk factor for liver metastasis [30].
Fig. 3.
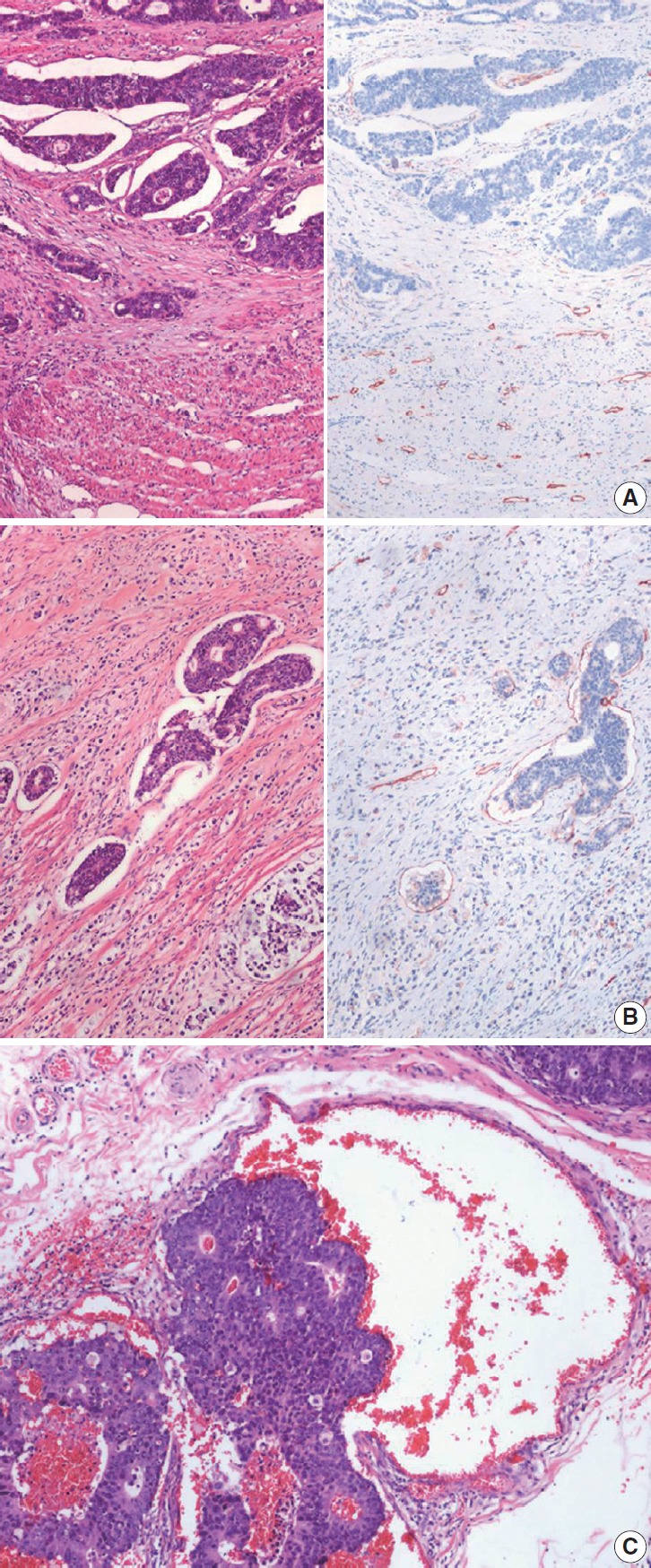
Histologic features of lymphatic invasion and venous invasion. (A) Tumor clusters with retraction artifacts can be misinterpreted as lymphatic invasion (H&E stain and D2-40 immunohistochemical stain). (B) Tumor invasion of small vessels is considered as lymphatic invasion (H&E stain and D2-40 immunohistochemical stain). (C) Tumors involving vessels with identifiable smooth muscle layer or elastic lamina are considered as venous invasion.
Perineural invasion
Perineural invasion has been known as an independent predictor of poor prognosis in CRC [32]. Perineural invasion is an essential element of the pathologic report for CRC and it can be assessed on routine H&E sections. Although there is no clear definition of perineural invasion, it is considered as perineural invasion when the tumor cells invade any of the three layers of nerve sheath, around the nerve, as well as into the nerve [33].
Pre-existing adenoma
Two major categories of CRC precursor lesions include conventional adenomas and serrated lesions [34]. According to the latest WHO classification of tumors of the digestive system, conventional adenomas are morphologically classified into tubular (villous component < 25%), tubulovillous (villous component 25%–75%), or villous (villous component > 75%) adenomas based on the proportion of tubular and villous architectures in an adenoma [5]. By definition, all conventional adenomas show dysplastic epithelium, and dysplasia in conventional adenomas can be graded as low-grade or high-grade [5]. High-grade dysplasia is characterized by architectural complexities (e.g., intraluminal cribriforming or necrosis, or back-to-back fusion of multiple glands), nuclear pleomorphisms, loss of nuclear polarity, and increased nuclear-to-cytoplasmic ratio (Fig. 4A). Features of low-grade dysplasia include non-complex architecture, pseudostratified/elongated nuclei with hyperchromasia, and preserved cellular polarization (Fig. 4B). The most important part of the distinction between high-grade and low-grade dysplasia is a complex glandular structure formation of tumor glands without invasion of lamina propria. Dysplastic glands only showing cytologic atypia without complex structural change are not diagnosed as high-grade dysplasia. In the previous edition, we recommended the diagnosis of high-grade dysplasia when there are three contiguous dysplastic gland structures showing high-grade features, which include cytologic atypia. According to the previous edition, there was a tendency to diagnose high-grade dysplasia with only focal cytologic atypia, but tumors with glandular structural abnormalities were sometimes missed as low-grade dysplasia. So, in this edition, complex structural abnormality is described as an essential part of the diagnosis of high-grade dysplasia. When high-grade and low-grade dysplasia components are mixed in a conventional adenoma, the adenoma should be diagnosed as high-grade dysplasia [1].
Fig. 4.

Histologic features of premalignant lesions of the colorectum. (A) Tubular adenoma with high-grade dysplasia. Note the architectural complexity including cribriform pattern or back-to-back fusion of dysplastic glands. (B) Tubulovillous adenoma with low-grade dysplasia. Note the retained cellular polarity with pseudostratified, elongated nuclei. (C) Sessile serrated adenoma without dysplasia. Note the dilated base of crypts. (D) Traditional serrated adenoma. Note the deep-invaginated pattern of crypt serration with hypereosinophilic cytoplasm and pencillated nuclei.
Serrated lesions include hyperplastic polyps (HPs), sessile serrated lesions (SSLs, formerly called sessile serrated adenoma/polyp), and traditional serrated adenomas (TSAs), according to the latest WHO classification [5]. However, the classification and diagnostic criteria of serrated lesions have continuously changed [35,36]. Based on a comprehensive review of the latest WHO classification (2019) [5], expert panel recommendations from the US (2012) [35], and UK guidance (2015) [36], we recommend the use of three diagnostic terms for serrated lesions as pre-existing adenomas in CRC: SSL, SSL with dysplasia (SSLD), and TSA. Because HP is regarded as a benign, non-precancerous lesion, it is excluded from serrated lesions as a pre-existing adenoma of CRC.
All SSLs are regarded as premalignant lesions, and morphologic dysplasia in SSLs can be present or absent. In comparison to SSL without dysplasia, SSLD is regarded to have a higher risk of progression into carcinoma. SSL is morphologically characterized by crypt luminal serrations extending to the crypt base, horizontal growth of the crypt, and asymmetrical proliferation with typical dilated/branching bases of crypts (Fig 4C). The diagnosis of SSL can be made when there is at least one typical architecturally distorted crypt [5,35]. Morphologic changes in SSLD can be diverse, and multiple morphologic patterns can be seen in a single lesion. Structural changes in SSLD include villous change, elongation of crypts, crowding of crypts, cribriform formation, and excessive decrease of luminal serration. Cytologically, dysplastic cells can show conventional adenoma-like dysplasia (so-called intestinal dysplasia) or round atypical nuclei with prominent nucleoli (serrated dysplasia) [5]. Loss of MLH1 expression is frequently found in SSLD and MLH1 immunohistochemical staining may help to define dysplasia [37]. However, the grading system for SSLD has not been suggested yet. Therefore, we simply recommend the presence or absence of dysplasia, regardless of dysplasia subtype or grade, for the description of SSL as a pre-existing adenoma of CRC.
All TSAs are regarded as precancerous dysplastic lesions, although classification or grading systems for dysplasia in TSAs have not been established. Thus, we recommend the term TSA without any description of dysplasia as a pre-existing adenoma of CRC. TSA is morphologically characterized by broad-topped, sharplyinvaginated luminal serration, tall columnar cells with abundant eosinophilic cytoplasm, pencillate nuclei, and ectopic crypt formation (Fig. 4D). TSAs can frequently be found as mixed lesions accompanying other serrated lesions or conventional adenomas [37,38].
In the case of a premalignant lesion consisting of two or more different histologic types of conventional adenomas and/or serrated lesions, the lesion can be referred to as a mixed adenoma.
Associated findings
Tumor perforation is associated with a poor outcome in CRC [39]. It is known that perforation of the uninvolved colon proximal to an obstructing tumor can cause peritonitis or sepsis, and is associated with poor prognosis. Distant metastasis is now divided into metastasis to one site or organ (pM1a), or metastasis to two or more sites or organs (pM1b), in the absence of peritoneal surface metastasis according to the AJCC 8th edition. Peritoneal surface metastasis is classified as pM1c [4].
Separate lesions
When there are lesions other than colorectal cancer, such as separate adenomas, polyps, gastrointestinal stromal tumors, or inflammatory bowel disease, they can be denoted in this section.
CONDITIONAL DATA ELEMENTS FOR RESECTED COLORECTUM
Tumor budding
Tumor budding is a well-known independent adverse prognostic factor of CRC [40]. Tumor budding components in the pathologic report can be potentially helpful in the decision-making process for the management of patients with CRC. First, in patients presenting with endoscopically resected submucosal invasive CRC, the presence of tumor budding is positively correlated with increased risk of lymph node metastases [26,41,42]. Therefore, patients with tumor budding can be potential surgical candidates. Second, stage II CRC patients with high-grade tumor budding show worse disease-free survival than those with no or low-grade tumor budding. Therefore, high-grade tumor budding may be useful for screening patients who need adjuvant therapy [43]. Finally, the presence of tumor budding in pre-operative biopsies can aid in the selection of high-risk rectal cancer patients for neoadjuvant therapy [44,45]. As mentioned above, in endoscopically resected pT1 CRC patients and stage II CRC patients, it is recommended to include tumor budding in the pathologic report.
The International Tumor Budding Consensus Conference (ITBCC) held in 2016 has recommended the following criteria for evaluation of tumor budding [46]. (1) Tumor budding is defined as a single tumor cell or a cell cluster containing ≤ 4 tumor cells. (2) Tumor budding should be assessed in the hotspot at the invasive tumor front (in a field measuring 0.785 mm2, which corresponds to a 20 × objective lens with an eyepiece having a field number that is 20 mm in diameter) chosen after a review of the available slides. (3) Tumor budding can be assessed on H&E slides. IHC for keratin can be helpful in evaluation of obscure cases showing a peritumoral inflammatory infiltrate on H&E-stained slides, which is difficult to distinguish from reactive stromal cells. In such cases, IHC enables better visualization of tumor budding and superior reproducibility and inter-observer agreement [47]. However, keratin also stains apoptotic bodies and cellular debris, which should not be misinterpreted as tumor budding [46,48].
We also recommend a 3-tier grading system for reporting of tumor budding. However, this grading system is not an absolute standard. Measuring and reporting the number of tumor buddings could be more beneficial to avoid any loss of information that may occur when applying a cut-off value in borderline cases. In rare histopathological subtypes, such as mucinous, signet-ring cells, micropapillary, and medullary carcinomas, the evaluation of tumor budding should be performed with caution [46]. Therefore, when tumor budding cannot be accurately assessed, it is recommended that the findings be reported as “cannot be assessed” with an explanatory note added to the report.
Tumor border configuration
Regarding tumor border configuration, there is currently no recommendation in the AJCC 8th edition, nor in the CAP protocol for CRC, and only a brief mention in the WHO classification [3-5]. Although the assessment of tumor border configuration can be easily carried out using H&E slides, inter-observer reproducibility is lacking due to ambiguous definitions [49]. For these reasons, the tumor border configuration was excluded from this standardization report.
Completeness of total mesorectal excision
Total mesorectal excision is a surgical technique that dissects within the areolar plane outside the visceral mesorectal fascia to remove rectum [50]. A large number of non-randomized studies have shown that if total mesorectal excision is appropriately performed, then adequate resection margin is secured, and local recurrence rate is reduced. The surface of the fresh rectal specimen is examined circumferentially, and the completeness of the mesorectal excision is scored according to the worst area, as below [3,4,7].
- Complete: Mesorectum is totally resected. The surface of the specimen is smooth and there is no coning towards the distal margin (there is no surface defect greater than 0.5 cm in depth).
- Nearly complete: There is irregularity of the mesorectal surface. The surface defect is greater than 0.5 cm, but proper muscle layer is not exposed (except for levator ani muscles).
- Incomplete: Severe mesorectal defects down to the muscularis propria. Muscularis propria is exposed.
Preoperative chemoradiotherapy
Preoperative chemoradiotherapy (CRT) followed by curative resection with TME has been established as a standard treatment for patients with locally advanced rectal cancer [51,52]. Tumor regression after preoperative CRT has an important role that can improve patient outcomes and the potential for achieving no residual tumor status [53-55]. Many studies have been performed to perceive a practical and applicable tumor regression grading (TRG) system [4,53-59]. The tumor regression after CRT was assessed only in the primary tumor and the nodal status was not included in the assessment of the TRG system [4]. The TRG system recommended in the previous edition of the “standardized pathology report for colorectal cancer” used a descriptive 5-tier system and can be easily translated into the Mandard and Dworak TRG scoring system [1]. However, even with a descriptive TRG system there is an inter-observer variation between pathologists, and this descriptive TRG system is not widely used and is often confused with other existing TRG systems. To overcome this, we suggest a new TRG system, which can be widely used in the pathologic practice, based on the AJCC 8th edition and CAP cancer protocol (Fig. 5) [3,4].
Fig. 5.

Recommended tumor regression grading system. (A) Grade 0, complete response. No residual tumor cells are identified. (B) Grade 1, near complete response. The tumor bed contained abundant fibrosis with only a few or scattered tumor cells. (C) Grade 2, partial response. Residual tumor glands are easily identified in tumor bed. (D) Grade 3, poor or no response. The tumor cells do not demonstrate any response to chemoradiotherapy because abundant residual adenocarcinoma is present.
The number of blocks taken is dependent on the size of the residual tumor. If the tumor is grossly visible, a minimum of 4 paraffin blocks should be taken from the tumor, including samples from its closest point to the nearest margin. However, if the tumor is ill-defined and impossible to distinguish from fibrous stromal tissue or the tumor bed size is less than 2.0 cm, the entire embedding of the macroscopically identifiable tumor bed or entire scar area is recommended, orientated from proximal to distal in 0.4-cm levels. Careful examination of the residual tumor cells is essential and is mandatory for assessment of complete response after CRT in CRC patients. Histologically, the tumor bed is characterized by abundant fibrotic stroma with a moderate number of mononuclear inflammatory cells or foamy macrophages. In these areas, there may be edema or mucinous or myxoid changes of the stroma or even areas of necrosis. The presence of mucin lakes without viable tumor cells (acellular mucin) should be defined as a pathologically complete response. If it is difficult to differentiate between giant cells or fibroblast and tumor cells, it may be helpful to perform serial H&E sections, mucin staining, or cytokeratin IHC in TRG grading.
EGFR immunohistochemistry
Expression of epidermal growth factor receptor (EGFR) by IHC has been reported in 25% to 90% of patients with CRC [60-64]. In almost all of the initial studies regarding the efficacy of targeted therapies with cetuximab or panitumumab in CRC patients, evidence of EGFR expression by IHC was essential for the application of therapeutic agents [65]. However, subsequent studies have shown no correlation between the degree of EGFR expression and the therapeutic responses to these drugs [66-68]. Thus, immunohistochemical expression of EGFR is not considered a predictor of response to EGFR-targeted drugs, and a number of recent studies do not include EGFR expression in selection criteria for anti-EGFR targeted therapy. However, in Korea, EGFR IHC has been routinely performed on CRC tissues of patients with stage IV CRC who are candidates for EGFR-targeted therapy according to the reimbursement criteria proposed by the Health Insurance Review and Evaluation Center, which mandates patient selection based on EGFR testing prior to application of EGFR-targeted therapy. In conclusion, whether immunohistochemical expression of EGFR accurately predicts the patients who would benefit from EGFR-targeted drugs has not been demonstrated until now.
DNA mismatch repair immunohistochemistry
IHC for the detection of DNA mismatch repair (MMR) proteins in CRC samples is a simple and useful tool to determine MMR deficiency, which is caused by germline mutation in one of the MMR genes (MLH1, MSH2, MSH6, or PMS2) or promoter CpG island hypermethylation of the MLH1 gene [69-71]. MMR deficiency results in a high level of microsatellite instability (MSI-H) that is characterized by genome-wide length alterations of DNA microsatellite repeat sequences [72]. Because MLH1 dimerizes with PMS2 in order to conduct DNA MMR function, when a mutation or promoter hypermethylation in MLH1 occurs in CRC, IHC expression of both MLH1 and PMS2 is negative in the tumor [69,70]. Similarly, as MSH2 dimerizes with MSH6, MSH2-mutated CRCs show negativity for both MSH2 and MSH6 [69,70]. However, when mutations in PMS2 or MSH6 occur, expression of only PMS2 or MSH6, respectively, is lost, with the expression of MLH1 and MSH2 remaining intact [69,70].
Nuclear staining should be observed for the IHC detection of MMR positivity. MMR deficiency is determined when the nuclear expression of at least one of the MMR proteins is not observed in tumor cells (Fig. 6).
Fig. 6.
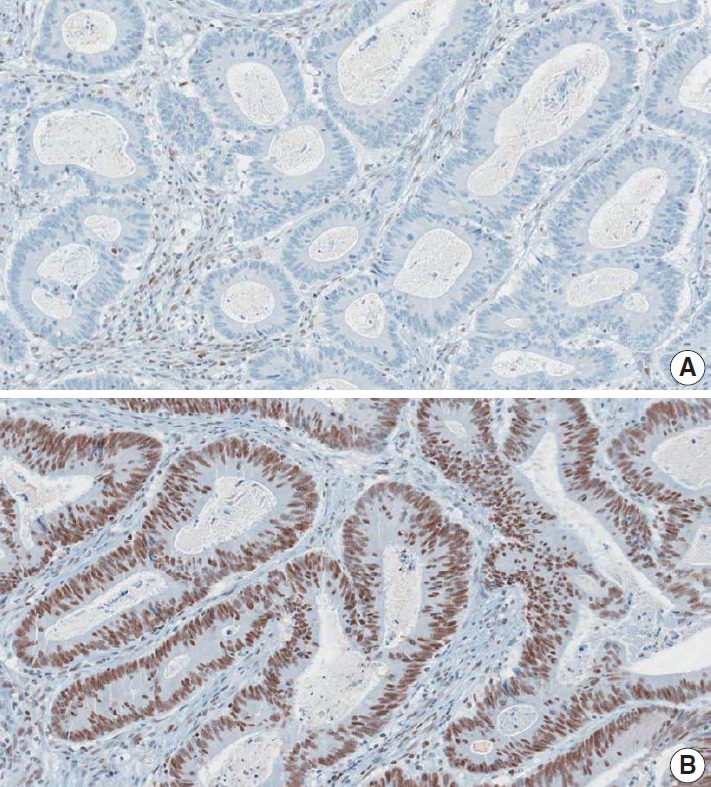
A representative case of colorectal cancer with MMR deficiency (MLH1 deficiency). (A) MLH1 immunohistochemical staining showed negativity of nuclear expression in tumor cells. Note the retained nuclear expression in adjacent inflammatory cells. (B) MSH2 immunohistochemical staining demonstrated positivity of nuclear expression in tumor cells.
MMR IHC is a well-established histopathological screening tool for Lynch syndrome or sporadic MSI-H molecular subtype in CRCs [69-71]. Previous data and meta-analyses have shown that MMR deficiency (or MSI-H) in CRCs is significantly associated with both a better prognosis as well as resistance to 5-fluorouracil-based adjuvant chemotherapy [72-74]. Moreover, recent evidence has indicated that MMR deficiency (or MSI-H) is a significant predictor of a positive response to immunotherapy using immune checkpoint blockade in solid tumors [75,76]. Therefore, the pathologic reporting of MMR or microsatellite instability (MSI) status is strongly recommended for all surgically resected CRC cases.
Immunoscore
Tumor-infiltrating lymphocytes (TILs) in tumor tissue has emerged as a strong prognostic factor in CRC. Many previous studies have consistently shown that TIL count is a strong prognostic factor with a high TIL count being significantly associated with a better prognosis in CRC [77]. Several investigations have reported that TIL count can be a strong predictor of patient prognosis in CRC regardless of both TNM staging and MSI [78,79]. The anti-tumor functions of TILs are conducted mainly by cytotoxic T cells. Thus, IHC for CD3+ and CD8+ T cells has been used as one of the major methods for the evaluation of TILs in CRC [78,80].
Based on the accumulating data, a TIL-based methodology named “immunoscore” has been under development and validation since 2012 by an international consortium led by Dr. Jerome Galon (http://www.immunoscore.org) [80,81]. The steps for determining the immunoscore of a CRC are as follows: (1) selection of a representative tumor section, (2) IHC for CD3 and CD8 on the tumor section, (3) evaluation of CD3+ and CD8+ cell densities at two tumor areas–invasive margin (IM) and tumor center (TC), (4) dichotomous categorization (high vs. low) of each of the four CD3/CD8 variables (CD3+ IM, CD3+ TC, CD8+ IM, and CD8+ TC), (5) determination of the final immunoscore based on the four CD3/CD8 variables (e.g., no “high” variable indicates I0; one “high” variable indicates I1; two “high” variables indicate I2; three “high” variables indicate I3; four “high” variables indicate I4).
Although there is strong evidence for the prognostic significance of the immunoscore in CRC, there is still a lack of global consensus regarding the application of immunoscore in terms of indication, evaluation, and classification. One of the critical limitations of the current immunoscoring system is the absence of a standard cut-off value for the high versus low categorization of CD3+ or CD8+ cell density. It is expected that a consensus established from digitalized image-based, automated analyses will aid the establishment of a standardized immunoscoring system.
Microsatellite instability
MSI is a condition of genome-wide alterations in the number of repeat nucleotide(s) caused by a defective mismatch repair (dMMR) system in the context of germline mutations in mismatch repair genes, including MLH1, MSH2, MSH6, and PMS2, or MLH1 promoter hypermethylation [72]. MSI testing is recommended for all CRC patients for the screening of Lynch syndrome in NCCN guidelines and guidelines from the American Society for Clinical Pathology (ASCP)/College of American Pathologists (CAP)/Association for Molecular Pathology (AMP)/American Society of Clinical Oncology (ASCO) [82]. Moreover, MSI-H is associated with better prognosis but poor therapeutic response to 5-fluorouracil-based cytotoxic chemotherapy [83,84]. Recently, MSI-H is considered as a predictive marker for PD-1 inhibitor-based cancer immunotherapy [75,85].
The gold standard method to evaluate MSI is the capillary gel electrophoresis, which compares the length in polymerase chain reaction (PCR) products of mono- or di-nucleotide repeats between cancer tissue and normal tissue. The Bethesda panel is composed of two mononucleotide repeat markers (BAT-25 and BAT-26) and three dinucleotide repeat markers (D5S346, D2S123, and D17S250), while the quasimonomorphic mononucleotides panel is composed of five mononucleotide repeat markers (NR-21, NR-24, NR-27 [or Mono-27], BAT-25, and BAT-26) [86,87]. Real-time PCR can be an alternative method for MSI evaluation in quasimonomorphic mononucleotides panel. Cancers classified as MSI-H should undergo immunohistochemistry of mismatch repair genes to screen for Lynch syndrome. If the BRAF V600E mutation is detected in MSI-H CRCs with MLH1 loss, Lynch syndrome can be ruled out [88]. Because MSI corresponds to dMMR, immunohistochemistry of four mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2) could be acceptable for an alternative method to capillary gel electrophoresis. The results of the MSI testing using the Bethesda panel are reported as MSI-H, MSI-low, and MSI-stable (MSS) if the tumor show instability in at least two, only one, and none of the 5 markers, respectively [87]. In determination of MSI using the quasimonomorphic mononucleotide panel, there is a controversy regarding the cut-off of MSI-H (≥ 3/5 or ≥ 2/5) [89,90].
KRAS and NRAS mutation analysis
The extended RAS mutation test is a molecular test for the detection of mutations in exons 2 to 4 of KRAS and NRAS genes [82,91]. It is widely known that EGFR blockers improve survival in patients with metastatic CRCs. However, mutations of exons 2 to 4 of KRAS and NRAS genes are associated with resistance to EGFR blockers. Thus, European Society for Medical Oncology (ESMO) guidelines for metastatic CRCs, NCCN guidelines, and ASCP/CAP/AMP/ASCO guidelines recommend extended RAS mutation tests in patients with metastatic CRCs [82,91-93]. Besides, a point mutation of KRAS exon 2 is associated with poor prognosis in CRCs [94-98]. Sanger sequencing, pyrosequencing, and real-time PCR-based methods can be used for extended RAS mutation testing. Next generation sequencing (NGS) panels for solid tumors should include KRAS and NRAS tests in South Korea. The presence or absence of nucleotide and amino acid changes in KRAS and NRAS exons 2–4 should be reported with appropriate nomenclature.
BRAF mutation analysis
BRAF V600E mutations are observed in approximately twothirds of MSI-H CRCs caused by MLH1 promoter hypermethylation, however, it never occurs in Lynch syndrome [99]. Thus, if the MSI-H CRCs harbor the BRAF V600E mutation, we can exclude Lynch syndrome. Genetic tests for germline mutation of mismatch repair genes are not indicated in MSI-high CRCs with the BRAF V600E mutation [88].
Of the molecular subtypes generated by MSI and BRAF/KRAS mutations, MSS CRCs with BRAF V600E mutations were associated with the worst prognosis. However, the association between BRAF V600E mutations and worse prognosis is weakened by the presence of MSI-H, because MSI-H CRCs with BRAF V600E mutations showed no difference in survival compared with MSS CRCs with wild-type KRAS/BRAF [95-98]. Hence, MSI tests and BRAF mutation tests should be performed in CRC patients. Two retrospective studies showed that the BRAF V600E mutation is associated with shorter relapse-free survival and overall survival in metastatic CRC patients that underwent liver resection [98,100].
Some studies reported that the BRAF V600E mutation is associated with poor therapeutic response to EGFR blockers. However, it is still controversial whether the BRAF V600E mutation is an independent predictive marker for EGFR blockers [101,102]. 2017 National Comprehensive Cancer Network (NCCN) guidelines recommend BRAF V600E mutation testing for metastatic CRC patients; however, ASCP/CAP/AMP/ASCO and ESMO guidelines for metastatic CRCs do not recommend BRAF V600E mutation testing for metastatic CRC [82,91].
Sanger sequencing, pyrosequencing, and real-time PCR-based methods can be used for BRAF mutation testing. NGS panels for solid tumors should include BRAF tests in South Korea. The presence or absence of nucleotide and amino acid changes in BRAF exon 15 should be reported. Appropriate nomenclature should be used.
Footnotes
Author contributions
Conceptualization: BHK, JMK, GHK, HJC, DWK, JHK, JMB, ANS, HSP, YKK, KHL, MYC, IGD, HSL, HKC, JHS, MSC, ESJ, SYJ, EY, HSH.
Project administration: BHK, JMK, GHK, HJC, DYP.
Supervision: JMK, GHK, HJC, SYJ, ESJ, YWK, MSC, JHS, EY.
Writing—original draft preparation: HSP, ANS, JHK, IGD, YKK, MYC, JMK, DWK, JMB, BHK, HSL, KHL.
Writing—review & editing: BHK, JMK, GHK, HJC, DWK, JHK, JMB, ANS, HSP, YKK, KHL, MYC, IGD, HSL, HKC, DYP, HJK, JHS, MSC, ESJ, SYJ, EY, HSH, YWK.
Conflicts of Interest
G.H.K., J.H.K., and H.S.L., contributing editors of the Journal of Pathology and Translational Medicine, were not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding
No funding to declare.
Supplementary Materials
The Data Supplement is available with this article at https://doi.org/10.4132/jptm.2019.09.28.
REFERENCES
- 1.Chang HJ, Park CK, Kim WH, et al. A standardized pathology report for colorectal cancer. Korean J Pathol. 2006;40:193–203. [Google Scholar]
- 2.Ministry of Health and Welfare . Annual report of cancer statistics in Korea in 2015. Sejong: Ministry of Health and Welfare; 2017. [Google Scholar]
- 3.Kakar S, Shi C, Berho ME, et al. Protocol for the examination of specimens from patients with primary carcinoma of the colon and rectum, version 4.0.0.0. Illinois: College of American Pathologists; 2017. [Google Scholar]
- 4.Amin MB, Edge S, Greene F, et al. AJCC cancer staging system. 8th ed. New York: Springer; 2017. [Google Scholar]
- 5.WHO Classification of Tumours Editorial Board . WHO classification of tumours: digestive system tumours. 5th ed. Geneva: World Health Organization; 2019. [Google Scholar]
- 6.Fritz AG, Percy C, Jack A, et al. International classification of diseases for oncology (ICD-O) 3rd ed. 1st rev. Lyon: International Agency for Research on Cancer; 2013. [Google Scholar]
- 7.Langman G, Loughrey M, Shepherd N, Quirke P. Association of Coloproctology of Great Britain & Ireland (ACPGBI): guidelines for the management of cancer of the colon, rectum and anus (2017)-pathology standards and datasets. Colorectal Dis. 2017;19 Suppl 1:74–81. doi: 10.1111/codi.13708. [DOI] [PubMed] [Google Scholar]
- 8.Nagtegaal I, Gaspar C, Marijnen C, Van De Velde C, Fodde R, Van Krieken H. Morphological changes in tumour type after radiotherapy are accompanied by changes in gene expression profile but not in clinical behaviour. J Pathol. 2004;204:183–92. doi: 10.1002/path.1621. [DOI] [PubMed] [Google Scholar]
- 9.Verdu M, Roman R, Calvo M, et al. Clinicopathological and molecular characterization of colorectal micropapillary carcinoma. Mod Pathol. 2011;24:729–38. doi: 10.1038/modpathol.2011.1. [DOI] [PubMed] [Google Scholar]
- 10.Lee HJ, Eom DW, Kang GH, et al. Colorectal micropapillary carcinomas are associated with poor prognosis and enriched in markers of stem cells. Mod Pathol. 2013;26:1123–31. doi: 10.1038/modpathol.2012.163. [DOI] [PubMed] [Google Scholar]
- 11.Nassar H. Carcinomas with micropapillary morphology: clinical significance and current concepts. Adv Anat Pathol. 2004;11:297–303. doi: 10.1097/01.pap.0000138142.26882.fe. [DOI] [PubMed] [Google Scholar]
- 12.Xu F, Xu J, Lou Z, et al. Micropapillary component in colorectal carcinoma is associated with lymph node metastasis in T1 and T2 Stages and decreased survival time in TNM stages I and II. Am J Surg Pathol. 2009;33:1287–92. doi: 10.1097/PAS.0b013e3181a5387b. [DOI] [PubMed] [Google Scholar]
- 13.Haupt B, Ro JY, Schwartz MR, Shen SS. Colorectal adenocarcinoma with micropapillary pattern and its association with lymph node metastasis. Mod Pathol. 2007;20:729–33. doi: 10.1038/modpathol.3800790. [DOI] [PubMed] [Google Scholar]
- 14.Jakubowska K, Guzinska-Ustymowicz K, Pryczynicz A. Invasive micropapillary component and its clinico-histopathological significance in patients with colorectal cancer. Oncol Lett. 2016;12:1154–8. doi: 10.3892/ol.2016.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuppurainen K, Makinen JM, Junttila O, et al. Morphology and microsatellite instability in sporadic serrated and non-serrated colorectal cancer. J Pathol. 2005;207:285–94. doi: 10.1002/path.1850. [DOI] [PubMed] [Google Scholar]
- 16.Patai AV, Molnár B, Tulassay Z, Sipos F. Serrated pathway: alternative route to colorectal cancer. World J Gastroenterol. 2013;19:607–15. doi: 10.3748/wjg.v19.i5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mäkinen MJ. Colorectal serrated adenocarcinoma. Histopathology. 2007;50:131–50. doi: 10.1111/j.1365-2559.2006.02548.x. [DOI] [PubMed] [Google Scholar]
- 18.Barresi V, Reggiani Bonetti L, Ieni A, Domati F, Tuccari G. Prognostic significance of grading based on the counting of poorly differentiated clusters in colorectal mucinous adenocarcinoma. Hum Pathol. 2015;46:1722–9. doi: 10.1016/j.humpath.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 19.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system. 4th ed. Lyon: International Agency for Research on Cancer; 2010. [Google Scholar]
- 20.Kojima M, Shimazaki H, Iwaya K, et al. Intramucosal colorectal carcinoma with invasion of the lamina propria: a study by the Japanese Society for Cancer of the Colon and Rectum. Hum Pathol. 2017;66:230–7. doi: 10.1016/j.humpath.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 21.Panarelli NC, Schreiner AM, Brandt SM, Shepherd NA, Yantiss RK. Histologic features and cytologic techniques that aid pathologic stage assessment of colonic adenocarcinoma. Am J Surg Pathol. 2013;37:1252–8. doi: 10.1097/PAS.0b013e3182960e7c. [DOI] [PubMed] [Google Scholar]
- 22.Uehara K, Nakanishi Y, Shimoda T, Taniguchi H, Akasu T, Moriya Y. Clinicopathological significance of microscopic abscess formation at the invasive margin of advanced low rectal cancer. Br J Surg. 2007;94:239–43. doi: 10.1002/bjs.5575. [DOI] [PubMed] [Google Scholar]
- 23.Smith KD, Tan D, Das P, et al. Clinical significance of acellular mucin in rectal adenocarcinoma patients with a pathologic complete response to preoperative chemoradiation. Ann Surg. 2010;251:261–4. doi: 10.1097/SLA.0b013e3181bdfc27. [DOI] [PubMed] [Google Scholar]
- 24.Kang CM, Lim SB, Hong SM, et al. Prevalence and clinical significance of cellular and acellular mucin in patients with locally advanced mucinous rectal cancer who underwent preoperative chemoradiotherapy followed by radical surgery. Colorectal Dis. 2016;18:O10–6. doi: 10.1111/codi.13169. [DOI] [PubMed] [Google Scholar]
- 25.Kitajima K, Fujimori T, Fujii S, et al. Correlations between lymph node metastasis and depth of submucosal invasion in submucosal invasive colorectal carcinoma: a Japanese collaborative study. J Gastroenterol. 2004;39:534–43. doi: 10.1007/s00535-004-1339-4. [DOI] [PubMed] [Google Scholar]
- 26.Bosch SL, Teerenstra S, de Wilt JH, Cunningham C, Nagtegaal ID. Predicting lymph node metastasis in pT1 colorectal cancer: a systematic review of risk factors providing rationale for therapy decisions. Endoscopy. 2013;45:827–34. doi: 10.1055/s-0033-1344238. [DOI] [PubMed] [Google Scholar]
- 27.Haggitt RC, Glotzbach RE, Soffer EE, Wruble LD. Prognostic factors in colorectal carcinomas arising in adenomas: implications for lesions removed by endoscopic polypectomy. Gastroenterology. 1985;89:328–36. doi: 10.1016/0016-5085(85)90333-6. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi R, Takano M, Takagi K, et al. Management of early invasive colorectal cancer: risk of recurrence and clinical guidelines. Dis Colon Rectum. 1995;38:1286–95. doi: 10.1007/BF02049154. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe T, Muro K, Ajioka Y, et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2016 for the treatment of colorectal cancer. Int J Clin Oncol. 2018;23:1–34. doi: 10.1007/s10147-017-1101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betge J, Pollheimer MJ, Lindtner RA, et al. Intramural and extramural vascular invasion in colorectal cancer: prognostic significance and quality of pathology reporting. Cancer. 2012;118:628–38. doi: 10.1002/cncr.26310. [DOI] [PubMed] [Google Scholar]
- 31.Kim WH, Park CK, Kim YB, et al. A standardized pathology report for gastric cancer. Korean J Pathol. 2005;39:106–13. [Google Scholar]
- 32.Liebig C, Ayala G, Wilks J, et al. Perineural invasion is an independent predictor of outcome in colorectal cancer. J Clin Oncol. 2009;27:5131–7. doi: 10.1200/JCO.2009.22.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer. 2009;115:3379–91. doi: 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- 34.Gibson JA, Odze RD. Pathology of premalignant colorectal neoplasia. Dig Endosc. 2016;28:312–23. doi: 10.1111/den.12633. [DOI] [PubMed] [Google Scholar]
- 35.Rex DK, Ahnen DJ, Baron JA, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012;107:1315–29. doi: 10.1038/ajg.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bateman AC, Shepherd NA. UK guidance for the pathological reporting of serrated lesions of the colorectum. J Clin Pathol. 2015;68:585–91. doi: 10.1136/jclinpath-2015-203016. [DOI] [PubMed] [Google Scholar]
- 37.Liu C, Walker NI, Leggett BA, Whitehall VL, Bettington ML, Rosty C. Sessile serrated adenomas with dysplasia: morphological patterns and correlations with MLH1 immunohistochemistry. Mod Pathol. 2017;30:1728–38. doi: 10.1038/modpathol.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chetty R, Hafezi-Bakhtiari S, Serra S, Colling R, Wang LM. Traditional serrated adenomas (TSAs) admixed with other serrated (socalled precursor) polyps and conventional adenomas: a frequent occurrence. J Clin Pathol. 2015;68:270–3. doi: 10.1136/jclinpath-2014-202827. [DOI] [PubMed] [Google Scholar]
- 39.Anwar MA, D'Souza F, Coulter R, Memon B, Khan IM, Memon MA. Outcome of acutely perforated colorectal cancers: experience of a single district general hospital. Surg Oncol. 2006;15:91–6. doi: 10.1016/j.suronc.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Mitrovic B, Schaeffer DF, Riddell RH, Kirsch R. Tumor budding in colorectal carcinoma: time to take notice. Mod Pathol. 2012;25:1315–25. doi: 10.1038/modpathol.2012.94. [DOI] [PubMed] [Google Scholar]
- 41.Choi DH, Sohn DK, Chang HJ, Lim SB, Choi HS, Jeong SY. Indications for subsequent surgery after endoscopic resection of submucosally invasive colorectal carcinomas: a prospective cohort study. Dis Colon Rectum. 2009;52:438–45. doi: 10.1007/DCR.0b013e318197e37f. [DOI] [PubMed] [Google Scholar]
- 42.Ueno H, Mochizuki H, Hashiguchi Y, et al. Risk factors for an adverse outcome in early invasive colorectal carcinoma. Gastroenterology. 2004;127:385–94. doi: 10.1053/j.gastro.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 43.Wang LM, Kevans D, Mulcahy H, et al. Tumor budding is a strong and reproducible prognostic marker in T3N0 colorectal cancer. Am J Surg Pathol. 2009;33:134–41. doi: 10.1097/PAS.0b013e318184cd55. [DOI] [PubMed] [Google Scholar]
- 44.Giger OT, Comtesse SC, Lugli A, Zlobec I, Kurrer MO. Intra-tumoral budding in preoperative biopsy specimens predicts lymph node and distant metastasis in patients with colorectal cancer. Mod Pathol. 2012;25:1048–53. doi: 10.1038/modpathol.2012.56. [DOI] [PubMed] [Google Scholar]
- 45.Rogers AC, Gibbons D, Hanly AM, et al. Prognostic significance of tumor budding in rectal cancer biopsies before neoadjuvant therapy. Mod Pathol. 2014;27:156–62. doi: 10.1038/modpathol.2013.124. [DOI] [PubMed] [Google Scholar]
- 46.Lugli A, Kirsch R, Ajioka Y, et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod Pathol. 2017;30:1299–311. doi: 10.1038/modpathol.2017.46. [DOI] [PubMed] [Google Scholar]
- 47.Koelzer VH, Zlobec I, Berger MD, et al. Tumor budding in colorectal cancer revisited: results of a multicenter interobserver study. Virchows Arch. 2015;466:485–93. doi: 10.1007/s00428-015-1740-9. [DOI] [PubMed] [Google Scholar]
- 48.Koelzer VH, Assarzadegan N, Dawson H, et al. Cytokeratinbased assessment of tumour budding in colorectal cancer: analysis in stage II patients and prospective diagnostic experience. J Pathol Clin Res. 2017;3:171–8. doi: 10.1002/cjp2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jass JR, Love SB, Northover JM. A new prognostic classification of rectal cancer. Lancet. 1987;1:1303–6. doi: 10.1016/s0140-6736(87)90552-6. [DOI] [PubMed] [Google Scholar]
- 50.Bosch SL, Nagtegaal ID. The importance of the pathologist’s role in assessment of the quality of the mesorectum. Curr Colorectal Cancer Rep. 2012;8:90–8. doi: 10.1007/s11888-012-0124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aklilu M, Eng C. The current landscape of locally advanced rectal cancer. Nat Rev Clin Oncol. 2011;8:649–59. doi: 10.1038/nrclinonc.2011.118. [DOI] [PubMed] [Google Scholar]
- 52.Bibeau F, Rullier A, Jourdan MF, et al. Locally advanced rectal cancer management: which role for the pathologist in 2011? Ann Pathol. 2011;31:433–41. doi: 10.1016/j.annpat.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 53.Dhadda AS, Dickinson P, Zaitoun AM, Gandhi N, Bessell EM. Prognostic importance of Mandard tumour regression grade following pre-operative chemo/radiotherapy for locally advanced rectal cancer. Eur J Cancer. 2011;47:1138–45. doi: 10.1016/j.ejca.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 54.Mihaylova I, Parvanova V, Velikova C, Kurteva G, Ivanova D. Degree of tumor regression after preoperative chemo-radiotherapy in locally advanced rectal cancer-Preliminary results. Rep Pract Oncol Radiother. 2011;16:237–42. doi: 10.1016/j.rpor.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vallböhmer D, Bollschweiler E, Brabender J, et al. Evaluation of histological regression grading systems in the neoadjuvant therapy of rectal cancer: do they have prognostic impact? Int J Colorectal Dis. 2012;27:1295–301. doi: 10.1007/s00384-012-1487-6. [DOI] [PubMed] [Google Scholar]
- 56.Kim SH, Chang HJ, Kim DY, et al. What is the ideal tumor regression grading system in rectal cancer patients after preoperative chemoradiotherapy? Cancer Res Treat. 2016;48:998–1009. doi: 10.4143/crt.2015.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mandard AM, Dalibard F, Mandard JC, et al. Pathologic assessment of tumor regression after preoperative chemoradiotherapy of esophageal carcinoma: clinicopathologic correlations. Cancer. 1994;73:2680–6. doi: 10.1002/1097-0142(19940601)73:11<2680::aid-cncr2820731105>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 58.Dworak O, Keilholz L, Hoffmann A. Pathological features of rectal cancer after preoperative radiochemotherapy. Int J Colorectal Dis. 1997;12:19–23. doi: 10.1007/s003840050072. [DOI] [PubMed] [Google Scholar]
- 59.Ryan R, Gibbons D, Hyland JM, et al. Pathological response following long-course neoadjuvant chemoradiotherapy for locally advanced rectal cancer. Histopathology. 2005;47:141–6. doi: 10.1111/j.1365-2559.2005.02176.x. [DOI] [PubMed] [Google Scholar]
- 60.Krasinskas AM. EGFR Signaling in Colorectal Carcinoma. Patholog Res Int. 2011;2011:932932. doi: 10.4061/2011/932932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldstein NS, Armin M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma: implications for a standardized scoring system. Cancer. 2001;92:1331–46. doi: 10.1002/1097-0142(20010901)92:5<1331::aid-cncr1455>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 62.McKay JA, Murray LJ, Curran S, et al. Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. Eur J Cancer. 2002;38:2258–64. doi: 10.1016/s0959-8049(02)00234-4. [DOI] [PubMed] [Google Scholar]
- 63.Spano JP, Fagard R, Soria JC, Rixe O, Khayat D, Milano G. Epidermal growth factor receptor signaling in colorectal cancer: preclinical data and therapeutic perspectives. Ann Oncol. 2005;16:189–94. doi: 10.1093/annonc/mdi057. [DOI] [PubMed] [Google Scholar]
- 64.Resnick MB, Routhier J, Konkin T, Sabo E, Pricolo VE. Epidermal growth factor receptor, c-MET, beta-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Cancer Res. 2004;10:3069–75. doi: 10.1158/1078-0432.ccr-03-0462. [DOI] [PubMed] [Google Scholar]
- 65.de Castro-Carpeno J, Belda-Iniesta C, Casado Saenz E, Hernandez Agudo E, Feliu Batlle J, Gonzalez Baron M. EGFR and colon cancer: a clinical view. Clin Transl Oncol. 2008;10:6–13. doi: 10.1007/s12094-008-0147-3. [DOI] [PubMed] [Google Scholar]
- 66.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 67.Vallböhmer D, Zhang W, Gordon M, et al. Molecular determinants of cetuximab efficacy. J Clin Oncol. 2005;23:3536–44. doi: 10.1200/JCO.2005.09.100. [DOI] [PubMed] [Google Scholar]
- 68.Ogino S, Meyerhardt JA, Cantor M, et al. Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin Cancer Res. 2005;11:6650–6. doi: 10.1158/1078-0432.CCR-05-0738. [DOI] [PubMed] [Google Scholar]
- 69.Geiersbach KB, Samowitz WS. Microsatellite instability and colorectal cancer. Arch Pathol Lab Med. 2011;135:1269–77. doi: 10.5858/arpa.2011-0035-RA. [DOI] [PubMed] [Google Scholar]
- 70.Pino MS, Chung DC. Microsatellite instability in the management of colorectal cancer. Expert Rev Gastroenterol Hepatol. 2011;5:385–99. doi: 10.1586/egh.11.25. [DOI] [PubMed] [Google Scholar]
- 71.Samowitz WS. Evaluation of colorectal cancers for Lynch syndrome: practical molecular diagnostics for surgical pathologists. Mod Pathol. 2015;28 Suppl 1:S109–13. doi: 10.1038/modpathol.2014.127. [DOI] [PubMed] [Google Scholar]
- 72.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guastadisegni C, Colafranceschi M, Ottini L, Dogliotti E. Microsatellite instability as a marker of prognosis and response to therapy: a meta-analysis of colorectal cancer survival data. Eur J Cancer. 2010;46:2788–98. doi: 10.1016/j.ejca.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 74.Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011;60:116–29. doi: 10.1136/gut.2009.206250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roxburgh CS, McMillan DC. The role of the in situ local inflammatory response in predicting recurrence and survival in patients with primary operable colorectal cancer. Cancer Treat Rev. 2012;38:451–66. doi: 10.1016/j.ctrv.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 78.Kirilovsky A, Marliot F, El Sissy C, Haicheur N, Galon J, Pages F. Rational bases for the use of the Immunoscore in routine clinical settings as a prognostic and predictive biomarker in cancer patients. Int Immunol. 2016;28:373–82. doi: 10.1093/intimm/dxw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 80.Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Galon J, Pagès F, Marincola FM, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med. 2012;10:205. doi: 10.1186/1479-5876-10-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sepulveda AR, Hamilton SR, Allegra CJ, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35:1453–86. doi: 10.1200/JCO.2016.71.9807. [DOI] [PubMed] [Google Scholar]
- 83.Zaanan A, Shi Q, Taieb J, et al. Role of deficient DNA mismatch repair status in patients with stage III colon cancer treated with FOLFOX adjuvant chemotherapy: a pooled analysis from 2 randomized clinical trials. JAMA Oncol. 2018;4:379–83. doi: 10.1001/jamaoncol.2017.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–26. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 88.Adar T, Rodgers LH, Shannon KM, et al. A tailored approach to BRAF and MLH1 methylation testing in a universal screening program for Lynch syndrome. Mod Pathol. 2017;30:440–7. doi: 10.1038/modpathol.2016.211. [DOI] [PubMed] [Google Scholar]
- 89.Buhard O, Cattaneo F, Wong YF, et al. Multipopulation analysis of polymorphisms in five mononucleotide repeats used to determine the microsatellite instability status of human tumors. J Clin Oncol. 2006;24:241–51. doi: 10.1200/JCO.2005.02.7227. [DOI] [PubMed] [Google Scholar]
- 90.Goel A, Nagasaka T, Hamelin R, Boland CR. An optimized pentaplex PCR for detecting DNA mismatch repair-deficient colorectal cancers. PLoS One. 2010;5:e9393. doi: 10.1371/journal.pone.0009393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 92.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 93.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 94.Rui Y, Wang C, Zhou Z, Zhong X, Yu Y. K-Ras mutation and prognosis of colorectal cancer: a meta-analysis. Hepatogastroenterology. 2015;62:19–24. [PubMed] [Google Scholar]
- 95.Phipps AI, Limburg PJ, Baron JA, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sinicrope FA, Shi Q, Smyrk TC, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst. 2017;109:djw272. doi: 10.1093/jnci/djw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schirripa M, Bergamo F, Cremolini C, et al. BRAF and RAS mutations as prognostic factors in metastatic colorectal cancer patients undergoing liver resection. Br J Cancer. 2015;112:1921–8. doi: 10.1038/bjc.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang L, Cunningham JM, Winters JL, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–12. [PubMed] [Google Scholar]
- 100.Loes IM, Immervoll H, Sorbye H, et al. Impact of KRAS, BRAF, PIK3CA, TP53 status and intraindividual mutation heterogeneity on outcome after liver resection for colorectal cancer metastases. Int J Cancer. 2016;139:647–56. doi: 10.1002/ijc.30089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 102.Pietrantonio F, Petrelli F, Coinu A, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–94. doi: 10.1016/j.ejca.2015.01.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.