

Abstract
In-cell NMR spectroscopy is a powerful tool to study protein structures and interactions under near physiological conditions in both prokaryotic and eukaryotic living cells. The low sensitivity and resolution of in-cell NMR spectra and limited lifetime of cells over the course of an in-cell experiment have presented major hurdles to wide acceptance of the technique, limiting it to a few select systems. These issues are addressed by introducing the use of the CRINEPT pulse sequence to increase the sensitivity and resolution of in-cell NMR spectra and the use of a bioreactor to maintain cell viability for up to 24 h. Application of advanced pulse sequences and bioreactor during in-cell NMR experiments will facilitate the exploration of a wide range of biological processes.
Keywords: Protein interactions, Protein structure, Protein-drug interactions, In-cell biochemistry, In vivo biochemistry, Atomic resolution structure, Thioredoxin, Antibiotics, Ribosome, Nucleic acids, RNA
1. INTRODUCTION
In-cell NMR spectroscopy utilizes NMR spectroscopy to study the structure, dynamics and interactions of proteins in living cells. The use of in-cell NMR spectroscopy to study proteins has expanded at a steady pace over the past two decades. Since the initial experiments of overexpressing target proteins in bacterial cells (Serber et al., 2001; Serber & Dotsch, 2001; Wieruszeski, Bohin, Bohin, & Lippens, 2001; Williams, Haggie, & Brindle, 1997), the field has developed a variety of methods for isotopic labeling (Hamatsu et al., 2013; Li et al., 2010; Serber et al., 2004), delivering labeled targets to prokaryotic and eukaryotic cells (Banci et al., 2013; Bertrand, Reverdatto, Burz, Zitomer, & Shekhtman, 2012; Bodart et al., 2008; Hamatsu et al., 2013; Inomata et al., 2009; Ogino et al., 2009; Sakai et al., 2006; Selenko, Serber, Gadea, Ruderman, & Wagner, 2006; Theillet et al., 2016), identifying high and low affinity specific and non-specific protein-protein interactions (Burz, Dutta, Cowburn, & Shekhtman, 2006a, 2006b), determining in-cell atomic resolution structures (Ikeya et al., 2016; Muntener, Haussinger, Selenko, & Theillet, 2016; Sakakibara et al., 2009), studying interactions of the target with the cytosol, mapping the interactions surfaces of target proteins (Danielsson et al., 2015; Kyne & Crowley, 2017; Luh et al., 2013; Majumder, DeMott, Burz, & Shekhtman, 2014; Smith, Zhou, Gorensek, Senske, & Pielak, 2016), detecting targets at physiological concentrations, high throughput drug screening, interaction proteomics, data collection and analysis (Cobbert et al., 2015; DeMott et al., 2018; Ikeya et al., 2010; Theillet et al., 2016; Xie, Thapa, Reverdatto, Burz, & Shekhtman, 2009). Despite these innovations, two major problems continue to plague in-cell NMR experiments: spectral peak broadening and cell viability. In this work we present protocols that help alleviate these difficulties by improving the resolution of in-cell NMR spectra.
1.1. In-cell NMR peak broadening
Multi-dimensional NMR spectroscopy such as heteronuclear single quantum coherence, HSQC, NMR spectroscopy has traditionally been used to investigate target proteins in-cell (Serber & Dotsch, 2001). In-cell spectra are compared to a well-resolved 1H–15N HSQC spectrum of purified isotope-labeled protein in vitro or in cell lysates to assign chemical shifts. However, in-cell, many of the NMR crosspeaks of folded proteins exhibit reduced intensity (broadening) due to a reduced rate of tumbling arising from the increased viscosity of the intracellular medium and interactions with macromolecular components of the cytosol (quinary interactions) that increase the apparent molecular weight of the complex (Crowley, Chow, & Papkovskaia, 2011; Majumder et al., 2015; Ye et al., 2013) (Fig. 1). The contribution from increased viscosity and molecular crowding contributes a comparatively small amount to the peak broadening; the dominant effect arises from quinary interactions (Majumder et al., 2015; Ye et al., 2013). Notable exceptions to this are intrinsically disordered proteins, IDPs, which lack persistent secondary or higher structure, and fail to interact with intracellular constituents; the in-cell spectra of IDPs are much sharper than those typically observed for folded proteins (Pielak et al., 2009). Modifications of traditional NMR pulse sequences (Felli, Gonnelli, & Pierattelli, 2014) have provided major advancements in the ability to resolve crosspeaks that are typically broadened during in-cell NMR experiments.
Fig. 1.
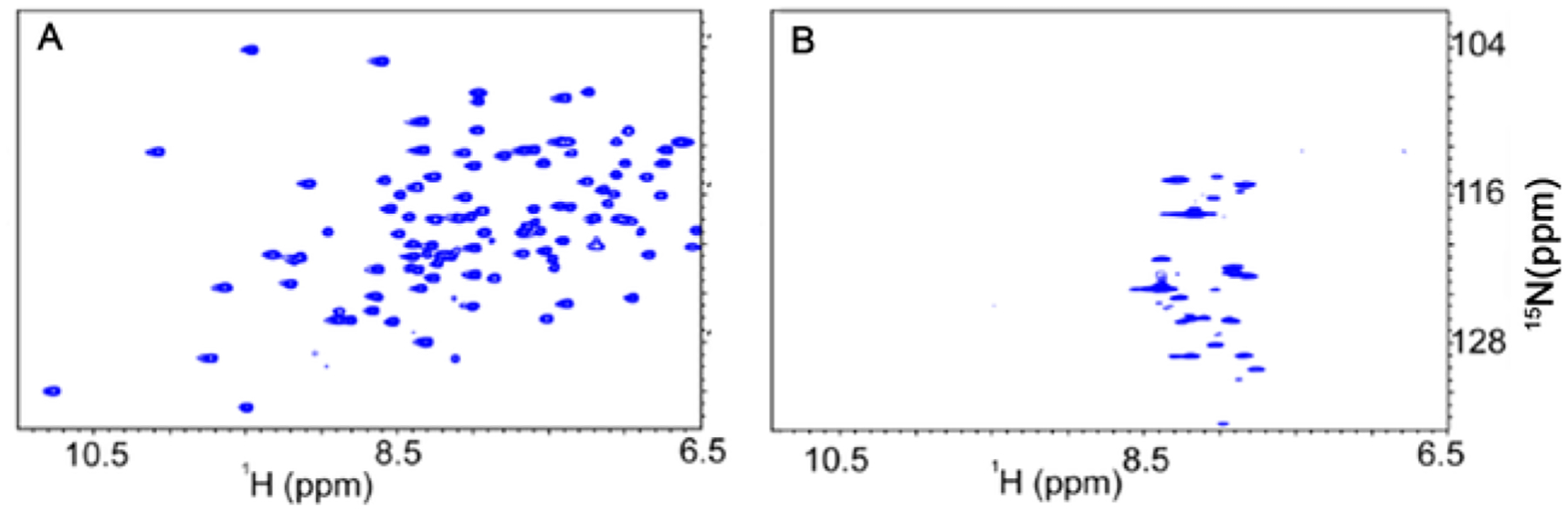
The in-cell spectra of most folded proteins are undetectable using HSQC NMR spectroscopy. (A). In vitro 1H{15N}-HSQC spectrum of Trx. B). 1H{15N}-HSQC spectrum of Trx in Escherichia coli. Crosspeaks are not resolved due to an increase in the apparent molecular weight of thioredoxin, only small metabolites are observed.
The increase in apparent molecular weight of a target protein in-cell resulting from quinary interactions leads to a reduced transverse relaxation, T2, which dictates the rate of free induction decay, and results in broader crosspeaks (Wuthrich, 1986). T2 is inversely related to the rotational correlation time, τc, of a molecule or molecular complex. τc is a time constant that describes the rotation diffusion of a molecule in solution. It is the time required for a molecule to rotate by 1 radian and depends on the Stokes radius. Increasing viscosity or molecular weight increases τc. The resulting NMR signal from larger molecules decays more rapidly and leads to line broadening. By using transverse relaxation-optimized spectroscopy, TROSY, which suppresses transverse nuclear spin relaxation in heteronuclear NMR experiments during evolution and acquisition cycles (Pervushin, Wider, & Wuthrich, 1998), TROSY provides increased sensitivity and resolution of in-cell NMR peaks for large, > 100 kDa, molecular weight species.
The HSQC and HMQC pulse sequences widely used for in-cell NMR spectroscopy use an insensitive nuclei enhanced by polarization transfer, INEPT, pulse sequence to transfer magnetization from protons to heteronuclei, but the efficiency of INEPT deteriorates with increasing τc (Riek, Pervushin, & Wuthrich, 2000). Further improvement in resolving the NMR spectra of large molecular species is achieved by using 15N-edited cross relaxation-enhanced polarization transfer, CRINEPT, NMR spectroscopy (Riek, Wider, Pervushin, & Wuthrich, 1999), which increases the efficiency of magnetization transfers between heteronuclei for molecules with τc between 50 and 300 ns. The [15N,1H]-HMQC pulse sequence contains two CRINEPT-like magnetization transfers delay times that can be optimized for molecular species with large rotational correlation times.
The combination of TROSY and CRINEPT creates transverse relaxation optimization during the entire pulse sequence. Importantly, since the efficiency of a CRINEPT polarization transfer increases with the strength of the external magnetic field, the sensitivity of the NMR experiment improves at higher magnetic fields (Riek et al., 2000). Relaxation optimized 15N-edited CRINEPT-heteronuclear multiple quantum coherence, HMQC, transverse relaxation optimized spectroscopy, TROSY, [1H–15N]-CRINEPT-HMQC-[1H]-TROSY experiments (Riek et al., 1999) in combination with REDuced PROton density (REDPRO) labeling (Shekhtman, Ghose, Goger, & Cowburn, 2002), which exchanges alpha and beta protons of amino acids for deuterons to minimize proton relaxation, have proven to be useful to detect high molecular weight species in-cell due to superior sensitivity to NMR signals and relative insensitivity to magnetic field inhomogeneity (Wider, 2005). Despite these innovative advances and other improvements such as shortening the data collection and analysis time, in-cell NMR spectroscopic experiments must still confront the dilemma associated with cell viability/stability.
1.2. Cell viability
A major consideration when performing in-cell NMR is verifying the integrity of the cells. Control experiments are routinely performed to verify that cell viability remains at or > 90–95% over the course of an experiment. Cells stained with trypan blue, which does not penetrate cells with intact membranes, or colony plated, are quantitatively compared before and after in-cell NMR experiments to assess viability (Burz & Shekhtman, 2010; Serber et al., 2006). In addition, samples are centrifuged following data acquisition and HSQC spectra collected on the supernatants. Target proteins in the supernatant will yield sharp signals indicative of leakage or lysis due to cell death over the course of the experiment.
In-cell NMR experiments often require 2–3 h or more to achieve at least a 3:1 signal to noise ratio.(Banci et al., 2013; Maldonado, Burz, & Shekhtman, 2011; Sakakibara et al., 2009; Serber, Corsini, Durst, & Dotsch, 2005; Theillet et al., 2016). Intracellular changes induced by stimuli, such as cellular adaptation to metabolic (Masters & Stacey, 2007; Wellen & Thompson, 2010) or antibiotic (Galhardo, Hastings, & Rosenberg, 2007) stress, can take hours to fully manifest into detectable signals. During this time the cells are deprived of nutrients and are in essence dying. The metabolic environment is therefore in a state of decay with an ever-changing background. As a result, it is important to ensure that the cells remain in a metabolically active state during in-cell NMR experiments.
To maintain a consistent cellular background during in-cell NMR experiments a bioreactor can be used to ensure that the experiments are conducted with stable, metabolically active cells. In these bioreactors fresh growth medium is continually flowed into the cells while metabolic byproducts are removed. Most bioreactors are complex and require pumps to circulate the medium in a closed system (Inomata, Kamoshida, Ikari, Ito, & Kigawa, 2017; Kubo et al., 2013; Sharaf, Barnes, Charlton, Young, & Pielak, 2010). Current bioreactors can maintain cell viability, as assessed by constant elevated levels of ATP, for up to 12 h. Real time, RT, in-cell NMR reactor (Breindel, DeMott, Burz, & Shekhtman, 2018) significantly simplifies previously described flow in-cell NMR methods (Kubo et al., 2013; Sharaf et al., 2010) and maintains cell viability for up to 24 h.
In RT-NMR flow is passively sustained by using a gravity siphon. Continuous exchange of medium minimizes cell leakage of labeled proteins. Cells are cast in low melting agarose to keep the sample at the bottom of the NMR tube. To lock on solvent and eliminate the need for deuterated solvent in the medium, an agarose plug in 100% D2O is located at the bottom of the NMR tube. A horizontal “drip irrigation” system maximizes exposure of the cells to fresh medium and minimizes the upward drag that often dislodges cells from their positions in the NMR tube, which can significantly reduce the quality of the in-cell NMR spectrum.
The metabolic state of the cells is assessed by monitoring the ATP levels. Reduction in the intracellular concentration of ATP is indicative of cell death, whereas constant levels are consistent with a viable metabolically active steady state condition.
1.3. Real time STINT-NMR spectroscopy
Broadening of in-cell spectral peaks arises primarily because of quinary interactions between the target protein and RNA (Majumder et al., 2015; Majumder, DeMott, Reverdatto, Burz, & Shekhtman, 2016). As a result, the in-cell spectrum must be analyzed relative to an in vitro or lysate spectrum to identify the interacting surfaces of the target molecule that define the quinary state. Changes in the in-cell target spectrum due to overexpression of an interactor protein or externally administered compounds are, in turn, analyzed relative to the quinary state. Structural interactions NMR spectroscopy, STINT-NMR, is used to identify the interacting surfaces (Burz et al., 2006a; Burz, DeMott, Aldousary, Dansereau, & Shekhtman, 2018; Majumder et al., 2014).
During a STINT-NMR experiment, a series of in-cell spectra are collected and a matrix is created that contains the changes in target protein peak intensities or chemical shifts versus time. Individual signals change at different rates as the concentration of the interactor protein increases. Time dependent degradation of the target protein inside the cell or differences in sample preparations also changes the in-cell spectrum. To distinguish spectral changes that are due to specific binding interactions as the concentration of the interactor or external stimulus increase a rigorous objective analysis, singular value decomposition, SVD, is used (Golub & Van Loan, 2012; Majumder et al., 2014).
SVD analysis discriminates between changes in in-cell spectra due to concentration-dependent and -independent binding interactions (Burz et al., 2018; Majumder et al., 2014). The analysis is simplified by maintaining cells in a steady state. Results of the analysis are presented as a Scree plot (Cattell, 1966) that shows the distribution of singular values that defines the relative contribution of each binding mode to the change in spectral peak intensities or chemical shifts. An abrupt drop in singular values following the first binding mode indicates a change due to specific interactions; a gradual decrease corresponds to random binding of the target protein to components of the cytosol (Majumder et al., 2014).
We describe two techniques for improving the resolution of in-cell NMR experiments. The first describes the application of 1H–15N CRINEPT-HMQC-TROSY spectroscopy to study large slowly tumbling molecular complexes in an effort to improve the resolution and sensitivities of traditional 2D experiments. The second describes the construction of a bioreactor that maintains cells in a steady state for periods of up to 24 h. A third protocol combines the use of 1H–15N CRINEPT-HMQC-TROSY spectroscopy with RT and STINT-NMR, and analysis of the data via SVD, to study the effect of the ribosome-binding antibiotic on the quinary structure of Escherichia coli thioredoxin.
2. MATERIALS
2.1. Equipment
Shaker incubator
Centrifuge
600 MHz or greater Bruker NMR spectrometer equipped with a cryoprobe
150 cm2 culture flasks
LB-kanamycin agar plates
2.5 Cm diameter × 20 cm cylinder
Polytetrafluoroethylene (PTFE) tubing, 0.5 mm I.D.
Stainless steel dissection needle with 10 μm tip (Roboz Surgical)
Microdissection needle holder (Roboz Surgical)
Low pressure tee (Idex Peek)
10 mL syringe
5 mm NMR tube
5 mm screw-cap NMR tube
Teflon membrane/PTFE/silicone septum (New Era)
Inlet reservoir flask
Outlet reservoir flask
Sonicator
Lyophilizer
Inversion microscope equipped with digital camera
Topspin data analysis software
CARA analysis software
MATLAB® software
EXCEL software
2.2. Chemicals
pRSF-Trx expression plasmid
E. coli strain BL21(DE3) codon +
HeLa cells
LB medium
Kanamycin
M9 medium
Deuterated M9 medium
15NH4Cl
Glucose
1 M IPTG
NMR buffer, 10 mM potassium phosphate buffer pH 6.5
D2O
LB-Kn agar plates
Dulbecco’s modified Eagle medium, DMEM (Gibco)
Fetal bovine serum, FBS (Gibco)
Trypsin/EDTA (Sigma-Aldrich)
Phosphate buffered saline, PBS
Cambrex Seaprep agarose
35% perchloric acid
2 M KOH
0.5 M EDTA, pH 8
Trypan blue (Hyclone)
Streptomycin
3. METHODS
3.1. In-cell [1H–15N]-CRINEPT-HMQC-[1H]-TROSY NMR spectroscopy
For this protocol E. coli thioredoxinA, trxA, was cloned into pRSF (No-vagen) (Majumder et al., 2015). pRSF-Trx confers kanamycin resistance, codes for lac repressor and expresses N-terminal His-tagged bacterial Trx from the T7 promoter/lac operon, which is induced by IPTG in E. coli strain BL21(DE3) codon +. The His-tag was not removed from the protein since it does interfere with the experiments (Majumder et al., 2015). NMR peak assignments for the target protein are required to complete the analysis.
3.1.1. Over-expression and labeling of Trx
Transform E. coli BL21(DE3) codon + with plasmid pRSF-Trx.
Inoculate 10 mL of LB containing 35 μg/mL of kanamycin, LB-Kn with a single colony of transformed cells.
Grow the culture overnight at 37 °C with shaking at 275 rpm.
Dilute the overnight culture to OD600 ~ 0.07 in 50 mL of LB-Kn and grow to an OD600 of ~ 0.9–1.0 at 37 °C, 275 rpm.
Centrifuge the cells at 200 × g for 15 min at room temperature.
Wash the cells twice with 50 mL of minimal, M9, medium.
Resuspend the cells in 200 mL of deuterated M9 supplemented with 1.0 g/L of 15NH4Cl and 0.2% glucose for uniform [U-2D,15N] labeling.
Incubate the culture for 20 min at 37 °C.
Induce protein overexpression by adding by adding 0.1% culture volume of 1 M isopropyl β-D-1-thiogalactopyranoside, IPTG.
Allow overexpression of REDPRO-labeled target protein to proceed for up to 36 h at 37 °C, 275 rpm.
3.1.2. Sample preparation for in-cell NMR spectroscopy
Remove a 100 mL aliquot from the overexpressing cell culture.
Centrifuge the cells at 200 × g for 15 min at room temperature.
Wash the cells twice with NMR buffer, and re-suspend in 450 μL of NMR buffer supplemented with 60 μL of D2O.
Remove 10 μL of the sample for a cell viability assay.
Prepare serial dilutions at 10−4, 10−5 and 10−6.
Plate 100 μL of each dilution onto LB-Kn plates in triplicate.
Incubate the plates overnight at 37 °C and count the resulting colonies.
Transfer the remaining sample to a standard 5 mm NMR tube.
3.1.3. In-cell NMR spectroscopy
NMR spectra were recorded at 298 K on a 700 MHz Avance II NMR spectrometer (Bruker) equipped with a TXI z-gradient cryoprobe.
Shim the sample manually using the z, z2 and z3 shims.
Select/create the [1H–15N]-CRINEPT-HMQC-[1H]-TROSY experiment as described in Riek et al. (1999), Fig. 3B.
Apply water suppression by using water selective pulses along the + z-axis for the entire sequence with a recycle delay of 300 ms.
Set the number of transients to 512.
Set the spectral widths to 12 ppm and 30 ppm in the 1H and 15N dimensions, respectively.
Collect 1024 and 128 points in the 1H and 15N dimensions, respectively.
Use the first block of [1H–15N]-CRINEPT-HMQC-[1H]-TROSY to optimize the CRINEPT transfer delay time by increasing the CRIPT transfer time by 0.2–0.3 ms starting from 1 ms.
Monitor the amide envelope and tryptophan imide peaks to determine the transfer time that produces maximum peak volume, i.e., Topt.
Set Topt to the optimal value determined in step 7 (Fig. 2).
Collect a [1H–15N]-CRINEPT-HMQC-[1H]-TROSY spectrum of the cells (Fig. 2).
When data collection is complete, remove a 10 μL aliquot for cell viability assays (Section 3.1.2). A final colony count within 90% of the original count is considered viable.
Centrifuge the remaining sample at 4500 × g for 10 min at RT.
Transfer the supernatant to a standard 5 mm NMR tube and test for cell leakage by collecting a [1H–15N]-HSQC spectrum.
Re-suspend the cell pellet in 0.3 mL of NMR buffer.
Freeze-thaw (− 80 °C-RT) five times to lyse the cells.
Centrifuge the sample at 18000 × g for 15 min at RT.
Collect a [1H–15N]-CRINEPT-HMQC-[1H]-TROSY spectrum on the clarified lysate using the optimized value for Topt.
Fig. 3.
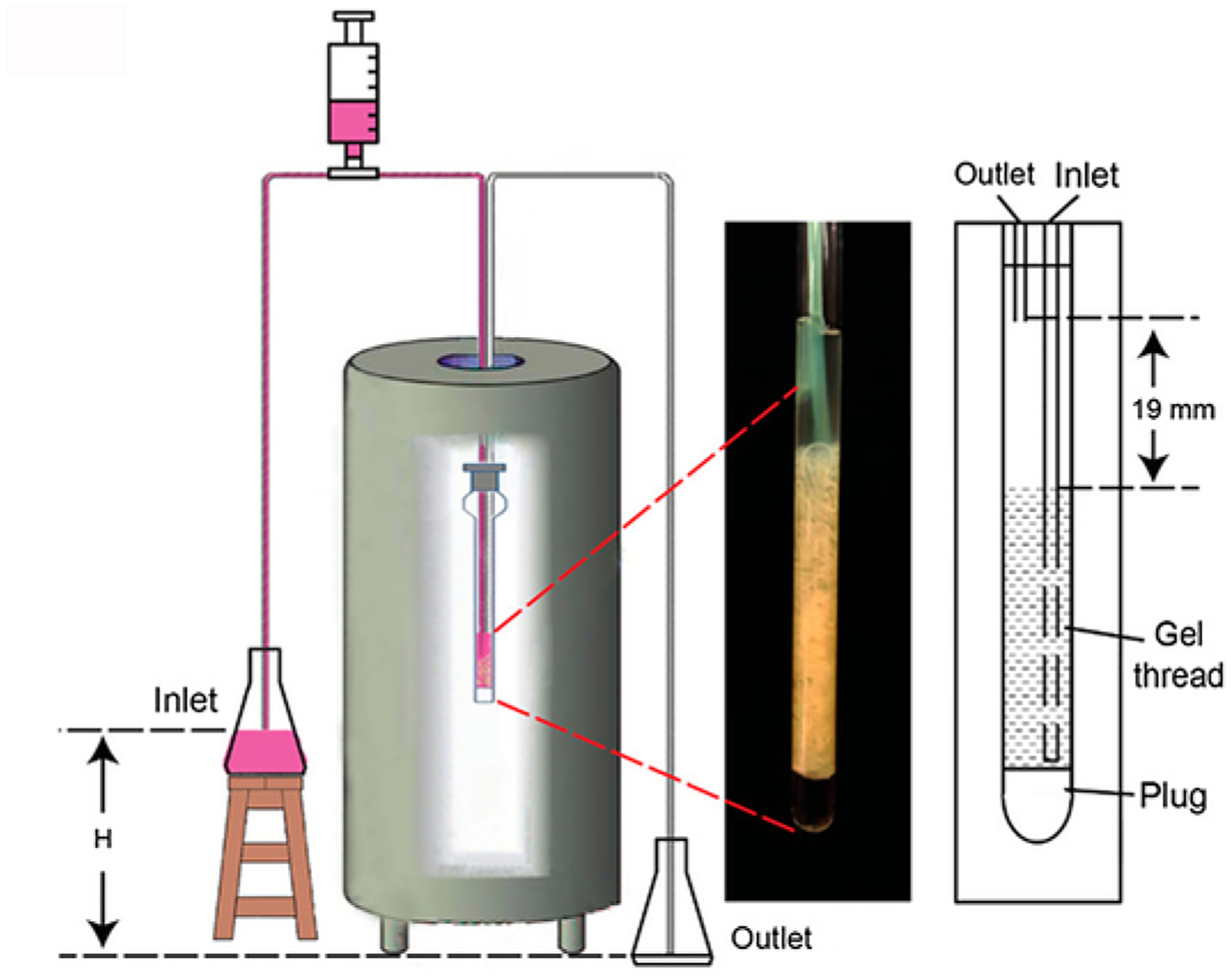
RT in-cell NMR. A gravity siphon drives the continuous flow of fresh medium through the cells.
Fig. 2.
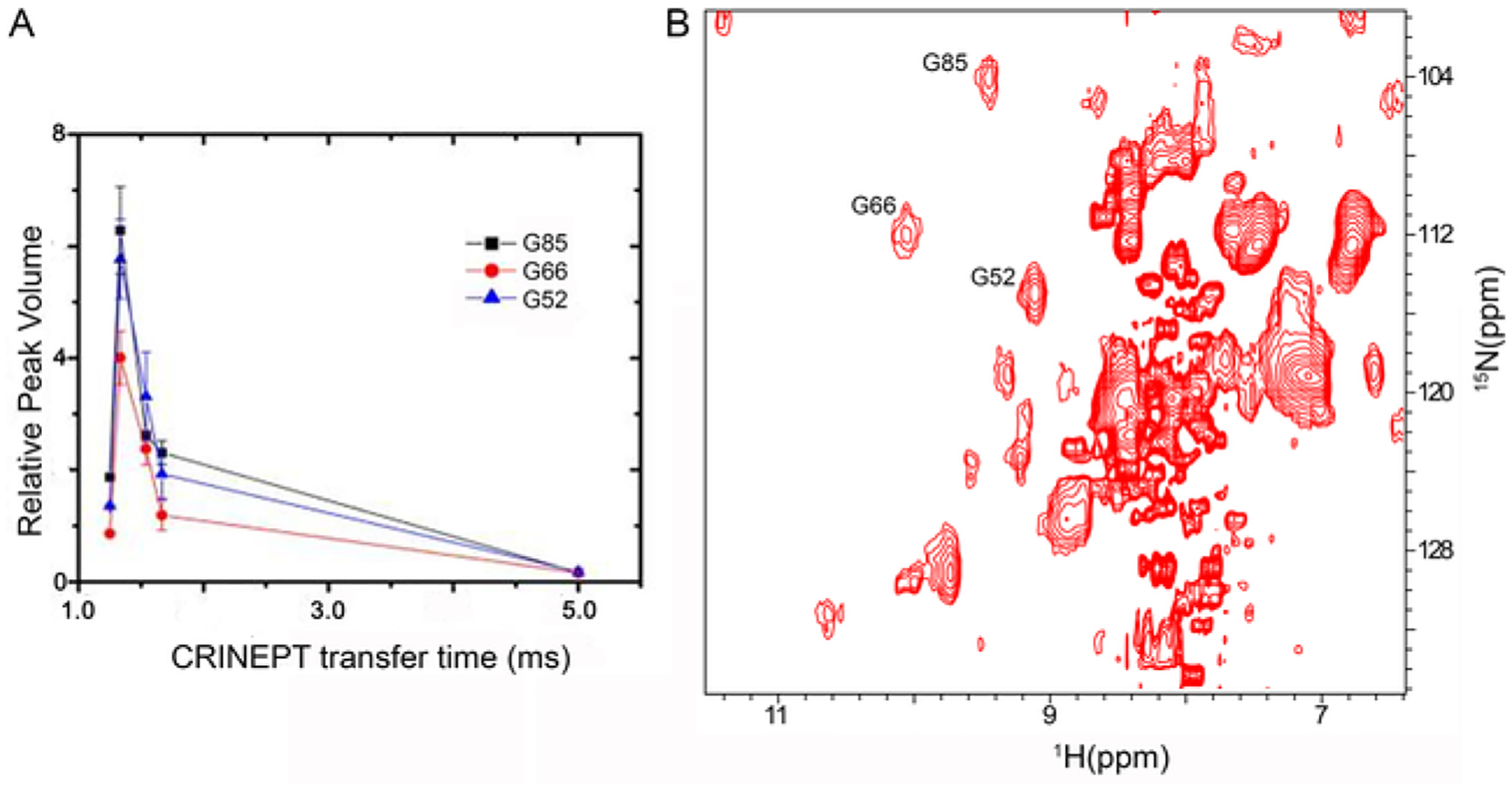
CRINEPT NMR spectroscopy resolves in-cell crosspeaks for proteins with high apparent molecular weights due to quinary interactions. (A) The relative volumes of G52, G66 and G85 crosspeaks are plotted against the CRINEPT transfer delay times. An endogenous tryptophan indole amide peak in the in-cell spectra is used as a reference. (B) 1H–15N CRINEPT–HMQC–TROSY spectrum of REDPRO-labeled Trx in E. coli. The G52, G66 and G85 crosspeaks used for optimizing the CRINEPT transfer delay time are indicated.
3.2. Bioreactor for cell viability
This protocol describes the preparation of E. coli and HeLa cells for viability testing. E. coli are transformed with expression vector to mimic the cellular background used in in-cell NMR experiments.
3.2.1. Preparation of E. coli cells
Transform E. coli BL21(DE3) codon + with plasmid pRSF-Trx.
Inoculate 10 mL of Luria broth (LB) medium supplemented with 35 μg/mL of kanamycin with a single colony of pRSF-Trx and grow overnight at 37 °C and 225 rpm in a shaker incubator.
Transfer the overnight culture to 200 mL of LB medium supplemented with 35 μg/mL of kanamycin and grow overnight at 37 °C and 225 rpm to an OD600 of 0.7–1.0.
Centrifuge the culture at 200 × g for 20 min and wash the cells twice with minimal, M9, medium.
3.2.2. Preparation of HeLa cells
To assess cell viability two sets of cast threads are prepared, one for NMR spectroscopy and one for trypan blue staining (Section 3.2.7).
Seed HeLa cells onto four 150 cm2 flasks in complete medium, low-glucose Dulbecco’s modified Eagle’s medium, DMEM, supplemented with 10% fetal bovine serum, FBS, and culture in a 5% CO2 incubator at 37 °C.
Culture the cells to 80% confluence (2 × 107 cells/flask).
Treat the plates with 0.25% trypsin/EDTA for 5 min at 37 °C to harvest the cells.
Neutralize the trypsin by diluting 5 × with complete medium.
Centrifuge the cells at 200 × g for 10 min at 25 °C and wash with 15 mL of pre-warmed (37 °C) phosphate-buffered saline, PBS.
3.2.3. Cell casting
The same procedure is used for E. coli and HeLa cells.
Centrifuge the cells at 200 × g for 20 min at 4 °C.
Re-suspend the pellet 1:1 (v/v) with 3.0% Cambrex Seaprep agarose in M9 medium (for E. coli) or DMEM-FSB medium (HeLa cells) at 37 °C so that the final concentration of the gel is 1.5%.
Inject the 37 °C mixture into 2 m of polytetrafluoroethylene, PTFE, tubing (I.D. 0.5 mm). Wrap the tubing around a 2.5 cm diameter cylinder and place in an ice bath for 15 min to allow the gel to solidify.
Insert a 135 μL plug of 3.0% agarose in D2O in the bottom of a tube 5 mm screw cap NMR tube. This is for the sample lock.
Extrude the immobilized cells into the NMR tube by injecting sterile M9 medium into the tubing.
3.2.4. The perfusion system
Thermally seal one end of a 2 m length of PTFE inlet line.
Make eight 50 μm orifices over a 3 cm length at the sealed end of the inlet line using a stainless steel dissection needle with 10 μm tip.
Prepare a 3 m length of PTFE outlet line.
Attach a low-pressure tee 4 in. from the unsealed end of the inlet line.
Insert the two lines through the septum in the screw cap of the NMR tube with the orifices of the inlet line inside the tube.
Attach a 10 mL syringe to the low-pressure.
Insert the inlet and outlet lines into an NMR tube that contains cast cell threads. Set the end of the inlet tubing just above the D2O agarose plug and the outlet tubing ~ 2 cm above the cell threads (Fig. 3).
Insert the free end of the inlet line into a reservoir of M9 medium.
Insert the free end of the outlet line into a waste reservoir.
Use the syringe to prime the inlet line with fresh M9 medium.
Initiate the flow of medium, using the syringe if necessary.
Adjust the height difference between the reservoirs to maintain a constant flow rate of 100 μL/min. In this experiment the height difference was 0.86 m.
Equilibrate the system to for 30 min to eliminate any air from the system.
3.2.5. Cell viability-I: Intracellular ATP levels
This experiment was performed using a Bruker 600 MHz Avance III NMR spectrometer equipped with a QCI-P cryoprobe,
Incubate the sample containing either E. coli or Hela cells for 30 min at RT before positioning it into the NMR spectrometer.
Collect a proton-decoupled 31P spectrum.
Continue collecting spectra at 3 h intervals over a 24 h period.
Repeat the experiment without the perfusion system (Fig. 4).
Integrate the 31P peak intensities at − 11.5 ppm using three different ranges to calculate a mean standard error for each spectrum.
Plot the average intensity versus time to assess the cell viability of cells in the perfusion system.
Fig. 4.
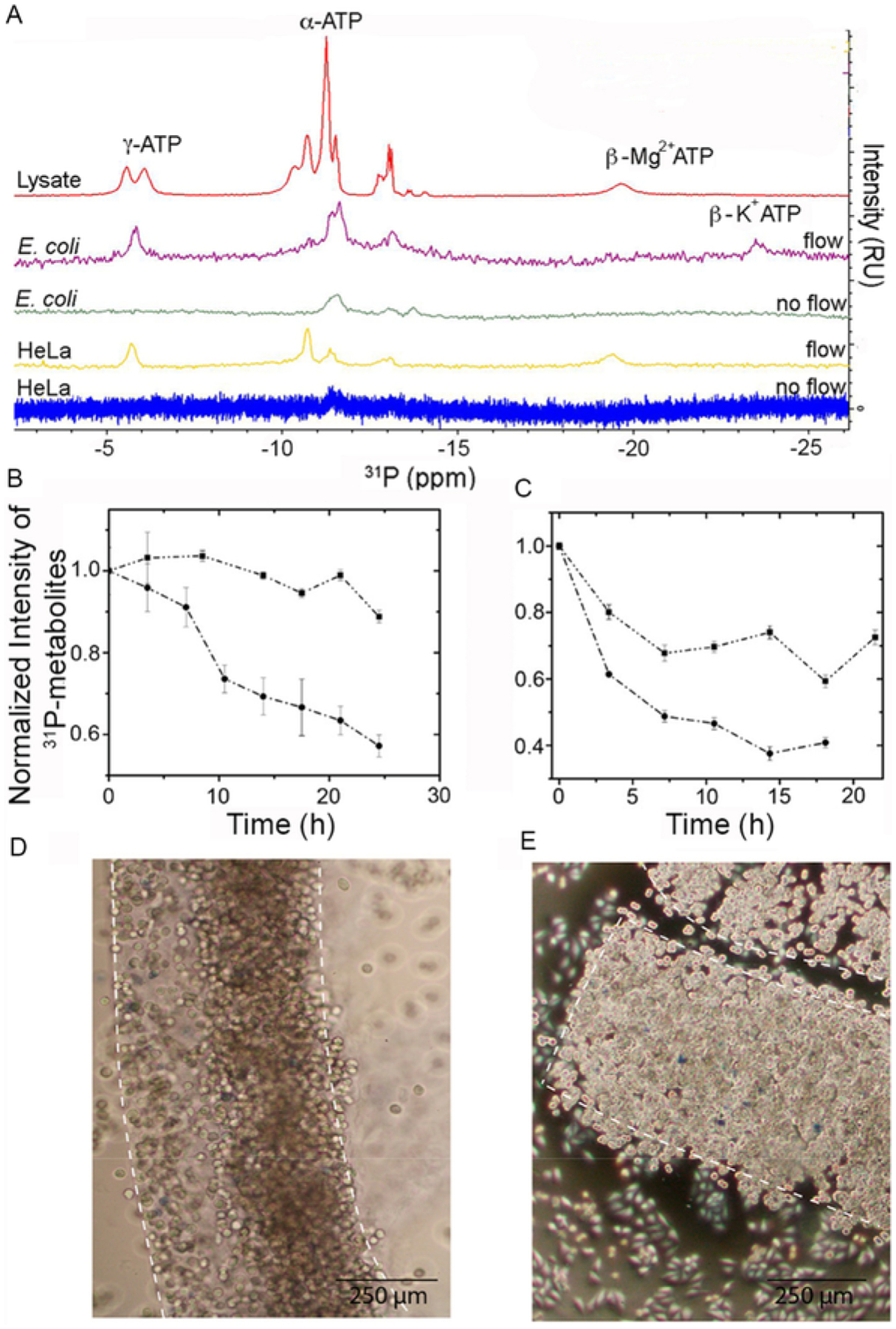
Cell viability is maintained for up to 24 h in a bioreactor. (A) 31P NMR spectra showing α-, β- and γ-ATP levels after 6 h for E. coli and HeLa cells in the bioreactor and in E. coli cell lysate. (B) Signal intensity of 31P-metabolites over time in E. coli with (squares) and without (circles) M9 flow. (C) Intensity of 31P-metabolites over time in HeLa cells with (squares) and without (circles) DMEM flow. (D) Trypan blue staining of cast HeLa cells before in-cell spectroscopy. (E) Trypan blue staining of cast HeLa cells 24 h after casting.
3.2.6. Cell viability-II: Prokaryotic cells
In this procedure cellular metabolites are extracted from E. coli as a baseline control and to identify in-cell peaks.
Suspend 3 g of E. coli cells (Section 3.2.1) in 5 mL of H2O and 0.5 mL of 35% (v/v) perchloric acid.
Sonicate the cells for 5 min with a 50% duty cycle.
Centrifuge at 13,300 × g for 1 h at 4 °C.
Neutralize the supernatant by using 2 M KOH. Monitor the process with pH indicator paper.
Centrifuge at 13,300 × g for 1 h at 4 °C.
Lyophilize the supernatant and re-dissolve in 1 mL of 90% H2O/10% D2O supplemented with 5 mM EDTA from a 0.5 M EDTA, pH 8 stock.
Transfer to an NMR tube and collect a proton-decoupled 31P spectrum.
3.2.7. Cell viability-III: Eukaryotic cells
Stained cells were imaged using a Nikon Eclipse TS100 inversion microscope equipped with a Canon EOS digital camera.
Divide the HeLa cells cast in agarose threads (Section 3.2.3) into two samples.
Incubate the first sample in a 5% CO2 incubator at 37 °C for 24 h.
Suspend the second sample in 200 μL of DMEM at 37 °C and add 200 μL of Trypan blue stock solution.
Allow the cells to stain for ~ 10 min and examine the cells under a microscope.
After 24 h, stain the second aliquot with trypan blue and examine under a microscope.
Compare the extent of cell staining to assess cell viability (Fig. 4). A final cell count within 90% of the original count is considered viable.
3.3. Real time in-cell STINT-NMR spectroscopy
For this procedure, Trx is overexpressed and labeled in E. coli, and the effects of externally administered ribosome-binding antibiotic streptomycin on the interaction surface of Trx is monitored in real time.
3.3.1. NMR spectroscopy
Overexpress and label Trx in E. coli (Section 3.1.1).
Cast the cells into agarose threads (Section 3.2.3).
Set up the perfusion system for the bioreactor (Section 3.2.4).
Set Topt to 1.3 ms and collect [1H–15N]-CRINEPT-HMQC-[1H]-TROSY in-cell basis spectrum (Section 3.1.3).
Add streptomycin to a final concentration of 50 μg/mL to the inlet reservoir.
Collect five consecutive [1H–15N]-CRINEPT-HMQC-[1H]-TROSY interaction spectra over the next 18 h (Fig. 5).
Fig. 5.
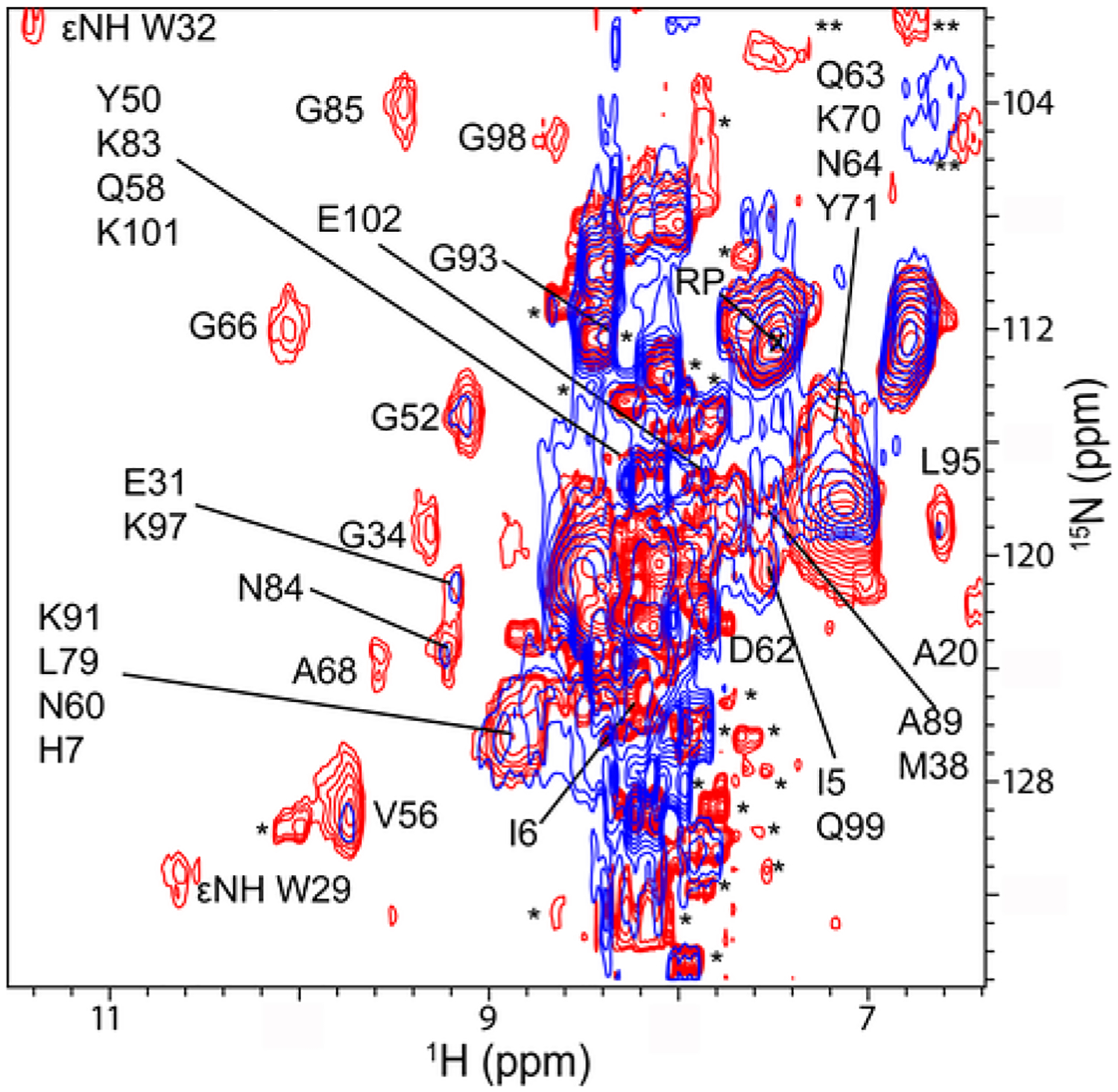
Binding of streptomycin to ribosomes changes the quinary structure of Trx in E. coli. Overlay of in-cell 1H −15N CRINEPT − HMQC − TROSY spectra of Trx without (red) and with (blue) streptomycin (final spectrum). Residues that broaden in the presence of streptomycin are indicated. Single and double asterisks indicate peaks from metabolites and unassigned side chain protons, respectively. The spectra are shown at the same contour levels. The reference peak used for intensity normalization is indicated by RP.
3.3.2. Data processing
Process the data with Topspin 3.0 (Bruker).
Upload in vitro assignments of the Trx from the BMRB databank and open the Trx [1H–15N] CRINEPT − HMQC − TROSY spectrum using CARA software.
Assign in-cell and lysate chemical shifts based on the proximity of each crosspeak to the in vitro assignments. Disregard residues with chemical shifts that differ by > 0.1 ppm between the lysate and purified protein.
Integrate crosspeak intensities by using base rectangle intensity summing.
Normalize intensities between the in-cell and lysate spectra using an amino proton peak from either glutamine or asparagine side chain that does not change chemical shift between the two spectra. For Trx a glutamine amide side chain at 7.49 and 112.4 ppm in the proton and nitrogen dimensions, respectively, was used.
Export the chemical shifts and peak integrations from CARA as a. TXT file to EXCEL.
- Calculate changes in peak intensities for each amino acid residue, ΔI, using
where Ilysate and Iin − cell are normalized, integrated peak intensities from lysate and in-cell spectra. - Calculate changes in chemical shifts for each amino acid residue, Δδ, using
where δH and δN are the changes in hydrogen and nitrogen chemical shifts, respectively, between the lysate and in-cell spectra. Assemble the data into matrices with residue numbers in columns and the corresponding changes in chemical shift or peak intensity values in rows.
Save the matrices as ASCII text files
3.3.3. Data processing of interaction spectra
Process the interaction spectra data with Topspin 3.0 (Bruker).
Upload the in-cell assignments of Trx from Section 3.3.2 using CARA software (Masse & Keller, 2005).
Assign in-cell chemical shifts for each spectrum based on the proximity of each crosspeak to the in-cell assignments.
Integrate crosspeak intensities by using base rectangle intensity summing.
Normalize intensities between the in-cell interaction spectra and in-cell basis spectrum using an amino proton peak from either glutamine or asparagine side chain that does not change chemical shift between the two spectra.
Export the chemical shifts and peak integrations from CARA as a. TXT file to EXCEL.
- Calculate changes in peak intensities for each amino acid residue, ΔI, using
where Ibasis and Iinteraction are normalized, integrated peak intensities from the in-cell Trx spectra in the absence and presence of streptomycin. - Calculate changes in chemical shifts for each amino acid residue, Δδ, using
where δH and δN are the changes in hydrogen and nitrogen chemical shifts, respectively, between the basis and interaction in-cell spectra. Assemble the data into matrices with residue numbers in columns and the corresponding changes in chemical shift or peak intensity values in rows.
Save the matrices as ASCII text files.
3.3.4. SVD analysis of interaction spectra
Start MATLAB® (Mathworks).
Load the chemical shift or intensity matrix text file into MATLAB®: e.g., LOAD TRXIntensitySVD.txt.
Define the uploaded matrix, M: M = TRXIntensitySVD.
Initiate SVD analysis of matrix M: [U,S,V] = svd(M).
Save the S matrix: save (‘S.txt’, ‘S’,’-ascii’).
Calculate the first binding mode: B1 = S(1,1)*U(:,1).
Save the first binding mode: save (‘B1.txt’, ‘B1’,’-ascii’).
Calculate the second binding mode: B2 = S(2,2)*U(:,2).
Save the second binding mode: save (‘B2.txt’, ‘B2’,’-ascii’).
Open MATLAB® results in EXCEL.
Create a Scree (bar) plot of singular values versus the binding mode.
Fit the singular values using linear regression to calculate the R2 value (Fig. 6).
Bar plot the first and the second binding modes for the protein residue sequence.
Set a threshold line at 1.5 times the maximum contribution of the second binding mode (Fig. 6).
Identify interacting residues as those whose first binding mode exceeds the threshold.
Map broadened residues onto the protein surface based on PDB file(s) using SWISS-PDB Viewer (Fig. 6).
Fig. 6.
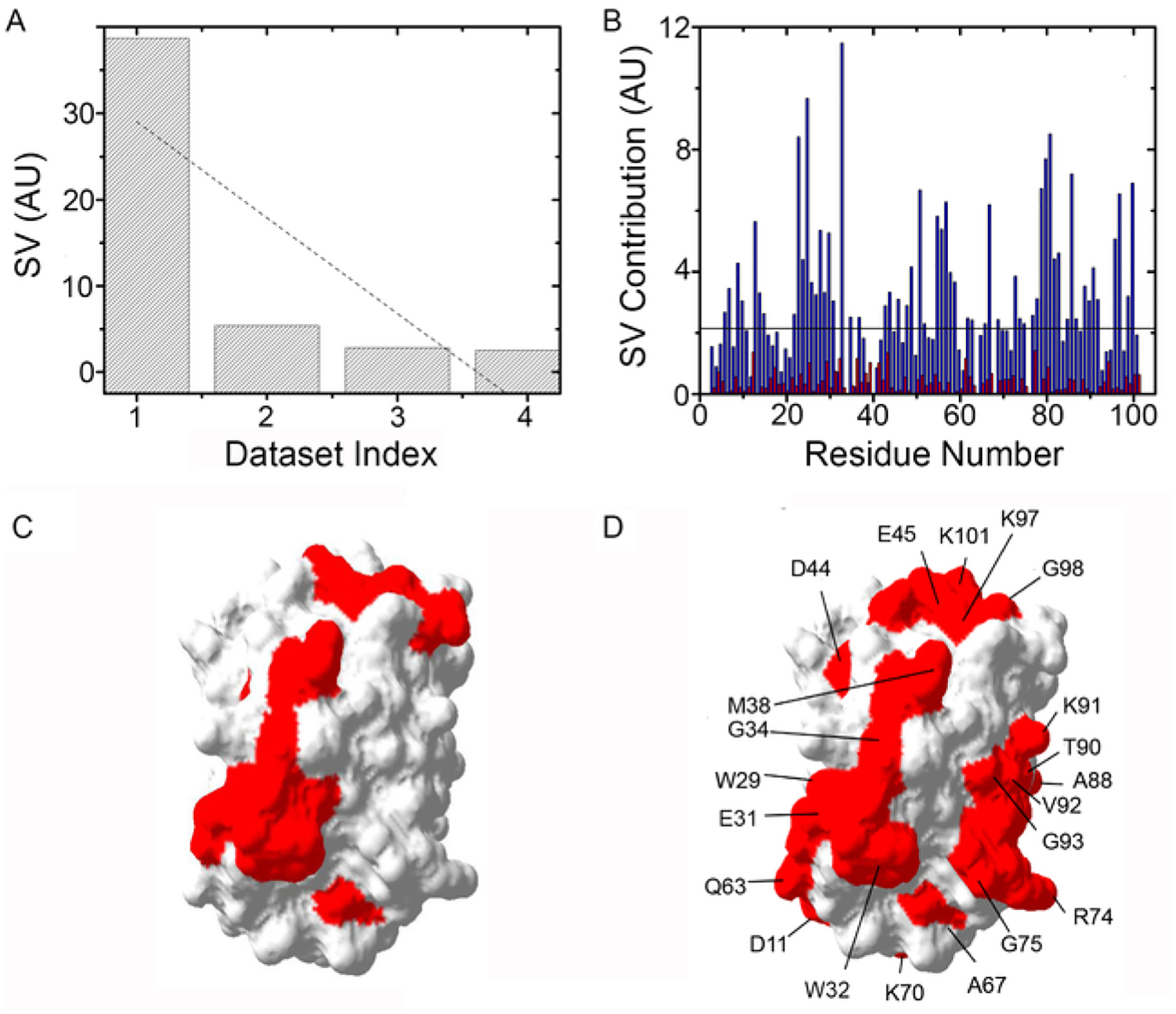
SVD analysis of Trx RT STINT NMR spectra. (A) Distribution of singular values, SV, of each data set index (binding mode) for Trx residues in the presence of streptomycin. (B) The contribution of each residue in response to adding streptomycin for the first (blue) and second (red) binding modes. (C) Surface map of Trx (Protein Data Bank entry 1X0B) showing the quinary interaction surface (red) in the absence of antibiotics. (D) Surface map of Trx showing the quinary interaction surface (red) in the presence of streptomycin. Residues that comprise the interaction surface are indicated.
4. RESULTS
Typical results for selected protocols are presented.
4.1. In-cell [1H-15N]-CRINEPT-HMQC-[1H]-TROSY NMR spectroscopy of Trx
Optimizing the CRINEPT transfer delay for in-cell 1H-15N CRINEPT-HMQC-TROSY spectra maximizes resolution. For Trx the optimum CRINEPT transfer delay was ~ 1.3 ms, which corresponds to an apparent molecular weight of ~ 1.1 MDa (Fig. 2A). The resulting spectrum shows crosspeaks that were not visible using HSQC NMR spectroscopy (Fig. 2B).
4.2. Bioreactor for cell viability
A schematic for RT-NMR is shown in Fig. 3. The height difference between the reservoirs provides the hydrodynamic force to drive the flow and allows external stimuli to be introduced into the cells from the inlet reservoir. The inlet line terminates at the bottom of the gel thread of cast cells. The outlet line terminates above the cast cells thread. An agarose plug in D2O at the base of the NMR tube is used to lock the sample.
4.3. Cell viability
Cell viability is assessed by measuring ATP levels during in-cell experiments using 31P NMR spectroscopy. In the presence of flow the phosphate levels are maintained. In the absence of flow, phosphate levels are rapidly depleted. In E. coli the intracellular level of Mg2 + is ~ 1 mM, which is lower than the concentration of ATP, 3–10 mM, the charge is mostly counterbalanced by K+ ions (Fig. 4A). Flow of medium maintains cellular levels of ATP, ADP, NAD+, and NAD(H), at ≥ 90% in E. coli and ≥ 75% in HeLa cells for up to 24 h (Fig. 4B and C). Staining HeLa cells with trypan blue shows that the cells remain 99 ± 1% viable over 24 h. An increase in cell density is consistent with ongoing metabolic activity (Fig. 4D and E).
4.4. Real time in-cell STINT-NMR spectroscopy
Changes in the in-cell NMR spectra resulting from the introduction of external stimulus reveal corresponding changes in quinary structure for Trx (Fig. 5).
4.5. SVD analysis of interaction spectra
Residues that form the quinary interacting surface are identified through SVD analysis of in-cell NMR spectra (Fig. 6A). The sharp drop and poor linear fit of the singular values (r2 = 0.66) are consistent with a specific change in quinary structure. Amino acids whose contribution to individual singular values exceed a threshold value make up the quinary interaction surface (Fig. 6B). The threshold line in Fig. 6B represents the average noise level of the NMR spectra. The amino acids identified in the SVD analysis are mapped onto the surface of the target molecule (Fig. 6C and D). Trx engages in quinary interactions in-cell that change when the antibiotic streptomycin, which binds to the 30S ribosome particle but not to Trx, is introduced into the cells during the course of an RT-STINT NMR experiment.
5. SUMMARY
The protocols presented addressed the major limitations facing most in-cell NMR experiments: spectral sensitivity and resolution, and cell viability. The introduction of CRINEPT-based NMR spectroscopy allowed for observation of target proteins that were previously invisible by conventional HSQC NMR. The gravity flow-based bioreactor provided a simple solution to maintain metabolically active cells over the time course of the experiment. Because of these modifications the field of in-cell NMR is now well positioned to study the complex interplay between cytosolic components, cellular processes and protein structural interactions.
ACKNOWLEDGMENT
This work was supported by NIH R01 GM085006.
REFERENCES
- Banci L, Barbieri L, Bertini I, Luchinat E, Secci E, Zhao Y, et al. , 2013. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nature Chemical Biology 9 (5), 297–299. 10.1038/nchembio.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand K, Reverdatto S, Burz DS, Zitomer R, Shekhtman A, 2012. Structure of proteins in eukaryotic compartments. Journal of the American Chemical Society 134 (30), 12798–12806. 10.1021/ja304809s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodart JF, Wieruszeski JM, Amniai L, Leroy A, Landrieu I, Rousseau-Lescuyer A, et al. , 2008. NMR observation of Tau in Xenopus oocytes. Journal of Magnetic Resonance 192 (2), 252–257, (doi:S1090–7807(08)00089-X [pii) 10.1016/j.jmr.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Breindel L, DeMott C, Burz DS, Shekhtman A, 2018. Real-time in-cell nuclear magnetic resonance: Ribosome-targeted antibiotics modulate Quinary protein interactions. Biochemistry 57, 540–546. 10.1021/acs.biochem.7b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burz DS, DeMott CM, Aldousary A, Dansereau S, Shekhtman A, 2018. Quantitative determination of interacting protein surfaces in prokaryotes and eukaryotes by using in-cell NMR spectroscopy. Methods in Molecular Biology 1688, 423–444. 10.1007/978-1-4939-7386-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burz DS, Dutta K, Cowburn D, Shekhtman A, 2006. In-cell NMR for protein-protein interactions (STINT-NMR). Nature Protocols 1 (1), 146–152. [DOI] [PubMed] [Google Scholar]
- Burz DS, Dutta K, Cowburn D, Shekhtman A, 2006. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR). Nature Methods 3 (2), 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burz DS, Shekhtman A, 2010. The STINT-NMR method for studying in-cell protein-protein interactions. Current Protocols in Protein Science 61, 17.11.1–17.11.15, Chapter 17, Unit 17 11 10.1002/0471140864.ps1711s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattell RB, 1966. The scree test for the number of factors. Multivariate Behavioral Research 1, 245–276. [DOI] [PubMed] [Google Scholar]
- Cobbert JD, DeMott C, Majumder S, Smith EA, Reverdatto S, Burz DS, et al. , 2015. Caught in action: Selecting peptide aptamers against intrinsically disordered proteins in live cells. Scientific Reports 5, 9402 10.1038/srep09402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley PB, Chow E, Papkovskaia T, 2011. Protein interactions in the Escherichia coli cytosol: An impediment to in-cell NMR spectroscopy. Chembiochem 12 (7), 1043–1048. 10.1002/cbic.201100063. [DOI] [PubMed] [Google Scholar]
- Danielsson J, Mu X, Lang L, Wang H, Binolfi A, Theillet FX, et al. , 2015. Thermodynamics of protein destabilization in live cells. Proceedings of the National Academy of Sciences of the United States of America 112 (40), 12402–12407. 10.1073/pnas.1511308112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMott CM, Girardin R, Cobbert J, Reverdatto S, Burz DS, McDonough K, et al. , 2018. Potent inhibitors of Mycobacterium tuberculosis growth identified by using in-cell NMR-based screening. ACS Chemical Biology 13, 733–741. 10.1021/acschembio.7b00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felli IC, Gonnelli L, Pierattelli R, 2014. In-cell (1)(3)C NMR spectroscopy for the study of intrinsically disordered proteins. Nature Protocols 9 (9), 2005–2016. 10.1038/nprot.2014.124. [DOI] [PubMed] [Google Scholar]
- Galhardo RS, Hastings PJ, Rosenberg SM, 2007. Mutation as a stress response and the regulation of evolvability. Critical Reviews in Biochemistry and Molecular Biology 42 (5), 399–435. 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub GH, Van Loan CF, 2012. Matrix computations, 4th ed The Johns Hopkins University Press, Baltimore, USA. [Google Scholar]
- Hamatsu J, O’Donovan D, Tanaka T, Shirai T, Hourai Y, Mikawa T, et al. , 2013. High-resolution heteronuclear multidimensional NMR of proteins in living insect cells using a baculovirus protein expression system. Journal of the American Chemical Society 135 (5), 1688–1691. 10.1021/ja310928u. [DOI] [PubMed] [Google Scholar]
- Ikeya T, Hanashima T, Hosoya S, Shimazaki M, Ikeda S, Mishima M, et al. , 2016. Improved in-cell structure determination of proteins at near-physiological concentration. Scientific Reports 6, 38312 10.1038/srep38312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeya T, Sasaki A, Sakakibara D, Shigemitsu Y, Hamatsu J, Hanashima T, et al. , 2010. NMR protein structure determination in living E. coli cells using nonlinear sampling. Nature Protocols 5 (6), 1051–1060. 10.1038/nprot.2010.69. [DOI] [PubMed] [Google Scholar]
- Inomata K, Kamoshida H, Ikari M, Ito Y, Kigawa T, 2017. Impact of cellular health conditions on the protein folding state in mammalian cells. Chemical Communications (Cambridge, England) 53 (81), 11245–11248. 10.1039/c7cc06004a. [DOI] [PubMed] [Google Scholar]
- Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, et al. , 2009. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 458 (7234), 106–109, nature07839 [pii] 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- Kubo S, Nishida N, Udagawa Y, Takarada O, Ogino S, Shimada I, 2013. A gel-encapsulated bioreactor system for NMR studies of protein-protein interactions in living mammalian cells. Angewandte Chemie (International Ed. in English) 52 (4), 1208–1211. 10.1002/anie.201207243. [DOI] [PubMed] [Google Scholar]
- Kyne C, Crowley PB, 2017. Short arginine motifs drive protein stickiness in the Escherichia coli cytoplasm. Biochemistry 56 (37), 5026–5032. 10.1021/acs.biochem.7b00731. [DOI] [PubMed] [Google Scholar]
- Li C, Wang GF, Wang Y, Creager-Allen R, Lutz EA, Scronce H, et al. , 2010. Protein (19)F NMR in Escherichia coli. Journal of the American Chemical Society 132(1), 321–327. 10.1021/ja907966n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luh LM, Hansel R, Lohr F, Kirchner DK, Krauskopf K, Pitzius S, et al. , 2013. Molecular crowding drives active pin1 into nonspecific complexes with endogenous proteins prior to substrate recognition. Journal of the American Chemical Society 135(37), 13796–13803. 10.1021/ja405244v. [DOI] [PubMed] [Google Scholar]
- Majumder S, DeMott CM, Burz DS, Shekhtman A, 2014. Using singular value decomposition to characterize protein-protein interactions by in-cell NMR spectroscopy. Chembiochem 15 (7), 929–933. 10.1002/cbic.201400030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, DeMott CM, Reverdatto S, Burz DS, Shekhtman A, 2016. Total cellular RNA modulates protein activity. Biochemistry 55 (32), 4568–4573. 10.1021/acs.biochem.6b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Xue J, DeMott CM, Reverdatto S, Burz DS, Shekhtman A, 2015. Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry 54 (17), 2727–2738. 10.1021/acs.biochem.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado AY, Burz DS, Shekhtman A, 2011. In-cell NMR spectroscopy. Progress in Nuclear Magnetic Resonance Spectroscopy 59 (3), 197–212, 10.1016/j.pnmrs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse JE, Keller R, 2005. AutoLink: Automated sequential resonance assignment of biopolymers from NMR data by relative-hypothesis-prioritization-based simulated logic. Journal of Magnetic Resonance 174 (1), 133–151. [DOI] [PubMed] [Google Scholar]
- Masters JR, Stacey GN, 2007. Changing medium and passaging cell lines. Nature Protocols 2 (9), 2276–2284. 10.1038/nprot.2007.319. [DOI] [PubMed] [Google Scholar]
- Muntener T, Haussinger D, Selenko P, Theillet FX, 2016. In-cell protein structures from 2D NMR experiments. Journal of Physical Chemistry Letters 7 (14), 2821–2825. 10.1021/acs.jpclett.6b01074. [DOI] [PubMed] [Google Scholar]
- Ogino S, Kubo S, Umemoto R, Huang S, Nishida N, Shimada I, 2009. Observation of NMR signals from proteins introduced into living mammalian cells by reversible membrane permeabilization using a pore-forming toxin, streptolysin O. Journal of the American Chemical Society 131 (31), 10834–10835. 10.1021/ja904407w. [DOI] [PubMed] [Google Scholar]
- Pervushin KV, Wider G, Wuthrich K, 1998. Single transition-to-single transition polarization transfer (ST2-PT) in [15N,1H]-TROSY. Journal of Biomolecular NMR 12(2), 345–348. 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- Pielak GJ, Li C, Miklos AC, Schlesinger AP, Slade KM, Wang GF, et al. , 2009. Protein nuclear magnetic resonance under physiological conditions. Biochemistry 48(2), 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R, Pervushin K, Wuthrich K, 2000. TROSY and CRINEPT: NMR with large molecular and supramolecular structures in solution. Trends in Biochemical Sciences 25 (10), 462–468. [DOI] [PubMed] [Google Scholar]
- Riek R, Wider G, Pervushin K, Wuthrich K, 1999. Polarization transfer by cross-correlated relaxation in solution NMR with very large molecules. Proceedings of the National Academy of Sciences of the United States of America 96 (9), 4918–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Tochio H, Tenno T, Ito Y, Kokubo T, Hiroaki H, et al. , 2006. In-cell NMR spectroscopy of proteins inside Xenopus laevis oocytes. Journal of Biomolecular NMR 36 (3), 179–188. 10.1007/s10858-006-9079-9. [DOI] [PubMed] [Google Scholar]
- Sakakibara D, Sasaki A, Ikeya T, Hamatsu J, Hanashima T, Mishima M, et al. , 2009. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 458 (7234), 102–105, nature07814 [pii] 10.1038/nature07814. [DOI] [PubMed] [Google Scholar]
- Selenko P, Serber Z, Gadea B, Ruderman J, Wagner G, 2006. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proceedings of the National Academy of Sciences of the United States of America 103(32), 11904–11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serber Z, Corsini L, Durst F, Dotsch V, 2005. In-cell NMR spectroscopy. Methods in Enzymology 394, 17–41. [DOI] [PubMed] [Google Scholar]
- Serber Z, Dotsch V, 2001. In-cell NMR spectroscopy. Biochemistry 40 (48),14317–14323. [DOI] [PubMed] [Google Scholar]
- Serber Z, Keatinge-Clay AT, Ledwidge R, Kelly AE, Miller SM, Dotsch V, 2001. High-resolution macromolecular NMR spectroscopy inside living cells. Journal of the American Chemical Society 123 (10), 2446–2447. [DOI] [PubMed] [Google Scholar]
- Serber Z, Selenko P, Hansel R, Reckel S, Lohr F, Ferrell JE Jr., et al. , 2006. Investigating macromolecules inside cultured and injected cells by in-cell NMR spectroscopy. Nature Protocols 1 (6), 2701–2709. [DOI] [PubMed] [Google Scholar]
- Serber Z, Straub W, Corsini L, Nomura AM, Shimba N, Craik CS, et al. , 2004. Methyl groups as probes for proteins and complexes in in-cell NMR experiments. Journal of the American Chemical Society 126 (22), 7119–7125. [DOI] [PubMed] [Google Scholar]
- Sharaf NG, Barnes CO, Charlton LM, Young GB, Pielak GJ, 2010. A bioreactor for in-cell protein NMR. Journal of Magnetic Resonance 202 (2), 140–146. 10.1016/j.jmr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhtman A, Ghose R, Goger M, Cowburn D, 2002. NMR structure determination and investigation using a reduced proton (REDPRO) labeling strategy for proteins. FEBS Letters 524 (1–3), 177–182. [DOI] [PubMed] [Google Scholar]
- Smith AE, Zhou LZ, Gorensek AH, Senske M, Pielak GJ, 2016. In-cell thermo-dynamics and a new role for protein surfaces. Proceedings of the National Academy of Sciences of the United States of America 113 (7), 1725–1730. 10.1073/pnas.1518620113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, et al. , 2016. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530 (7588), 45–50. 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB, 2010. Cellular metabolic stress: Considering how cells respond to nutrient excess. Molecular Cell 40 (2), 323–332. 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wider G, 2005. NMR techniques used with very large biological macromolecules in solution. Methods in Enzymology 394, 382–398. 10.1016/S0076-6879(05)94015-9. [DOI] [PubMed] [Google Scholar]
- Wieruszeski JM, Bohin A, Bohin JP, Lippens G, 2001. In vivo detection of the cyclic osmoregulated periplasmic glucan of Ralstonia solanacearum by high-resolution magic angle spinning NMR. Journal of Magnetic Resonance 151 (1), 118–123. 10.1006/jmre.2001.2348. [DOI] [PubMed] [Google Scholar]
- Williams SP, Haggie PM, Brindle KM, 1997. 19F NMR measurements of the rotational mobility of proteins in vivo. Biophysical Journal 72 (1), 490–498. 10.1016/S0006-3495(97)78690-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuthrich K, 1986. NMR of proteins and nucleic acids. John Wiley&Sons, New York. [Google Scholar]
- Xie J, Thapa R, Reverdatto S, Burz DS, Shekhtman A, 2009. Screening of small molecule interactor library by using in-cell NMR spectroscopy (SMILI-NMR). Journal of Medicinal Chemistry 52 (11), 3516–3522. 10.1021/jm9000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Liu X, Zhang Z, Wu Q, Jiang B, Jiang L, et al. , 2013. (19) F NMR spectroscopy as a probe of cytoplasmic viscosity and weak protein interactions in living cells. Chemistry 19 (38), 12705–12710. 10.1002/chem.201301657. [DOI] [PubMed] [Google Scholar]