

Abstract
Nasopharyngeal carcinoma has the highest rate of metastasis among head and neck cancers, and distant metastasis is the major reason for treatment failure. The underlying molecular mechanisms of nasopharyngeal carcinoma metastasis are not fully understood. Here, we report the identification of serine protease inhibitor Kazal-type 6 (SPINK6) as a functional regulator of nasopharyngeal carcinoma metastasis via EGFR signaling. SPINK6 mRNA was upregulated in tumor and highly metastatic nasopharyngeal carcinoma cells. Immunohistochemical staining of 534 nasopharyngeal carcinomas revealed elevated SPINK6 expression as an independent unfavorable prognostic factor for overall, disease-free, and distant metastasis–free survival. Ectopic SPINK6 expression promoted in vitro migration and invasion as well as in vivo lymph node metastasis and liver metastasis of nasopharyngeal carcinoma cells, whereas silencing SPINK6 exhibited opposing effects. SPINK6 enhanced epithelial–mesenchymal transition by activating EGFR and the downstream AKT pathway. Inhibition of EGFR with a neutralizing antibody or erlotinib reversed SPINK6-induced nasopharyngeal carcinoma cell migration and invasion. Erlotinib also inhibited SPINK6-induced metastasis in vivo. Notably, SPINK6 bound to the EGFR extracellular domain independent of serine protease–inhibitory activity. Overall, our results identified a novel EGFR-activating mechanism in which SPINK6 has a critical role in promoting nasopharyngeal carcinoma metastasis, with possible implications as a prognostic indicator in nasopharyngeal carcinoma patients.
Introduction
Nasopharyngeal carcinoma, the most common cancer originating in the nasopharynx, has a high incidence in Southern China and Southeast Asia (1, 2). The etiologic factors for nasopharyngeal carcinoma include Epstein–Barr virus infection, ethnics, genetic susceptibility, and environmental factors, including consumption of food with volatile nitrosamines (3). Upon diagnosis, most patients present with metastasis to the regional lymph nodes or even distant organs. More than 90% of nasopharyngeal carcinomas are undifferentiated carcinomas. Although nasopharyngeal carcinoma is sensitive to radiotherapy, distant metastasis is the primary cause of treatment failure (1, 4).
Nasopharyngeal carcinoma patients with advanced stages often have a poor prognosis because of limited knowledge on molecular pathogenesis, lack of reliable and robust biomarkers for early detection, and poor response to available therapies (5). Recently, aided by high-throughput technologies, including genomics, transcriptomics, proteomics, and metabolomics and bioinformatics together with integration and application of systems biology, many candidate biomarkers are identified, including BCL2, cyclin D1, EGFR, HER2, MET, MYC, and PI3KCA, etc.(6). Among them, omics data at multiple levels revealed that the Wnt, PI3K/AKT, and ERBB signaling pathways were often dysregulated in nasopharyngeal carcinoma (7–10). Secreted proteins were also involved in nasopharyngeal carcinoma progression, including TGFβ, Wnt5A, and CCL2, etc. (9, 11, 12). We also reported that SRGN, WNT5A, and IL8 can promote EMT and metastasis of nasopharyngeal carcinoma cells (13–15).
Using our previously established cellular and animal models of nasopharyngeal carcinoma metastasis (16), genomic expression profiling revealed that serine protease inhibitor Kazal-type 6 (SPINK6) was the most upregulated gene in the highly metastatic nasopharyngeal carcinoma cells (13). Human SPINK6 is encoded by the SPINK6 gene, which maps to chromosome 5q33.1 and is a selective inhibitor of kallikrein-related peptidases (KLK), including KLK4, KLK5, KLK6, KLK7, KLK12, KLK13, and KLK14 (17–20). SPINK6 is involved in maintaining skin homeostasis, and its expression decreases in atopic dermatitis but increases in psoriasis (17). Several KLKs have been reported to be useful tumor markers and involved in tumor pathogenesis, but their role is ambiguous and dependent on the tissue type and the tumor microenvironment (20–22). However, the exact role of SPINK6 in cancer progression is unknown.
Mature SPINK6 contains 57 amino acid residues and has three disulfide bonds in a Kazal-type serine protease inhibitor and follistatin-like domain (KAZAL-FS; ref. 23), which is similar to the EGF structure. STRING 9.1 analysis also revealed the protein–protein interaction of Kazal-type domain–containing proteins with EGFR and related tyrosine kinases (24, 25). We therefore hypothesize that SPINK6 could interact with EGFR. Recently, SPINK6 was reported to be crosslinked to fibronectin by transglutaminases and was shown to protect fibronectin from KLK5 cleavage (26). Because fibronectin is a putative EMT marker, SPINK6 may also play an important role in EMT regulation, which is a crucial process involved in development and differentiation, as well as motility of cancer cells (27).
In the current study, we found that SPINK6, as a secreted protein, could promote nasopharyngeal carcinoma cellular motility in vitro and metastasis in vivo via autocrine and paracrine mechanisms. In addition, SPINK6 induced EMT by binding to EGFR and activating EGFR and downstream AKT signaling pathway. Inhibition of EGFR by a neutralizing antibody or erlotinib could reverse the SPINK6-mediated induction of nasopharyngeal carcinoma cellular motility. Importantly, elevated SPINK6 expression in the primary nasopharyngeal carcinoma was an independent unfavorable prognostic factor for overall survival (OS), disease-free survival (DFS), and distant metastasis–free survival (DMFS) in nasopharyngeal carcinoma patients.
Materials and Methods
Cell lines and cell culture
Cell lines and cell culture were performed as described previously (28). Human nasopharyngeal carcinoma cell line CNE-2 and its clones (S18, S22, and S26) and SUNE-1 and its clone 5–8F, as well as CNE-1, HK1, HONE1, and HNE1, were maintained in DMEM (11995065, Thermo Fisher Scientific) supplemented with 10% FBS (10099141, Invitrogen) at 37°C in 5% CO2. As reported previously, the highly metastatic subclone S18 and 5–8F and poorly metastatic subclones (S22 and S26) were isolated using limiting dilution method and conformed for in vitro mobility and in vivo functional studies (16, 29). All the nasopharyngeal carcinoma cell lines used in the current study, including our isolated and established clones S18 and S26, have been maintained in our laboratory since 2000. The cell lines were authenticated using short tandem repeat profiling, and the cells were not cultured for more than 2 months.
Human tissue microarray
The human tissue samples were obtained from the Department of Pathology, Sun Yat-Sen University Cancer Center (SYSUCC; Guangzhou, China) with patients who had previously given consent and the approval of the Institutional Clinical Ethics Review Board at SYSUCC (approval number GZR2015–120). The tissue microarrays contained qualified primary nasopharyngeal carcinoma samples from 534 patients pathologically diagnosed at SYSUCC between December 30, 1997, and September 6, 2002, and the tissue microarray was constructed as described previously (13). The clinical characteristics of the 534 nasopharyngeal carcinoma patients were listed in the Supplementary Material in Supplementary Table S1.
Immunohistochemical staining
Immunohistochemical staining and scoring were performed as described previously (13). Rabbit anti-SPINK6 antibody (1:50 dilution, ab110830, Abcam), rabbit anti-EGFR-pY1068 (1:50 dilution, 3777, Cell Signaling Technology), and rabbit anti-vimentin (1:50 dilution, 5741, Cell Signaling Technology) were used. The intensity of immunohistochemical staining in the tumor cells was scored independently by two pathologists, and the average value from the two pathologists was used as the final score. The median score of SPINK6 (score = 3) was used as the cut-off value to divide the patients into the high (>median) and low (≤median) SPINK6 expression groups.
Immunoblotting assay
Cell lysates were prepared in RIPA lysis buffer (89900, Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (05056489001, Roche) and phosphatase inhibitor cocktail (04906837001, Roche). Thirty micrograms of each protein extract was examined after standard Western blotting protocols as described previously (13) and then detected using the ECL chemiluminescence system (NEL112001EA, PerkinElmer). Relative quantification of gradation was measured using Image-Pro Plus 6.0. The primary antibody and secondary antibody are listed in the Supplementary Material.
Lentiviral construction and transduction
The construction of the lentiviral vectors for stable SPINK6 overexpression or knockdown was carried out with a protocol from the Eric Campeau Lab (30). The lentiviral construction is described in the Supplementary Material, with the sequences for shRNAs listed in Supplementary Table S2. The lentiviral plasmids were cotransfected with pspAX2 and pMD2.G (kind gifts from Didier Trono, School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland, Addgene plasmid 12260 and 12259) in a 1:0.75:0.25 ratio in mass into 293T cells using X-tremeGENE HP DNA Transfection Reagent (06366236001, Roche). Two days after transfection, the viral supernatants were collected and filtered using a 0.45-μm filter (SLHU033RB, EMD Millipore) and were then used for transduction with 4 μg/mL polybrene (107689, Sigma-Aldrich). Twelve hours after transduction, the medium was exchanged for fresh medium, and 2 μg/mL puromycin (J593, GBCBIO Technologies) was used 1 day later to select the stably transfected cells for 3 days.
Coimmunoprecipitation and pull-down assays
For coimmunoprecipitation assays, cell-free–expressed 3× Flag-tagged EGFR or EGFR mutants were mixed with Myc-His–tagged SPINK6 or SPINK6 mutants. The mixtures were incubated at 4°C with inversion overnight and then subjected to incubation with an anti-FLAG M2 Affinity Gel (A2220, Sigma-Aldrich) for an additional 4 hours at 4°C. Then, the mixtures were centrifuged, and the precipitates were collected and subjected to Western blot detection, with the supernatants used as input. The cell lysates of wild-type cells and the condition medium of SPINK6-overexpres-sing or control cells (all in weak RIPA lysis) were also mixed for coimmunoprecipitation assay, followed by incubation with an anti-EGFR antibody (4267, Cell Signaling Technology) or an anti-His-tag antibody for 12 hours at 4°C and then coincubated with Protein A/G PLUS-Agarose (sc-2003, Santa Cruz Biotechnology) for an additional 4 houra at 4°C for precipitation. In pull-down assays, 2 μg/mL Fc-tagged human EGFR protein (10001-H02H, Sino Biological) was mixed with 2 μg/mL rSPINK6 or 2 μg/mL rhEGF (236-EG, R&D Systems) in the absence or presence of rSPINK6 (0.5, 2, 5, or 10 μg/mL) in 1 mL PBS buffer and subjected to coimmunoprecipitation using Protein A/G PLUS-Agarose.
Spontaneous lymph node metastasis model
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of SYSUCC (approval number L201501077). Female athymic mice ages 5 to 6 weeks were obtained from the Shanghai Institutes for Biological Sciences (Shanghai, China). The spontaneous lymph node metastasis model has been published previously (16, 31). Briefly, 1 × 105 cells in 20 μL serum-free DMEM were injected into the left hind footpad of the mouse to generate a primary tumor. Thirty days after inoculation, the mice were euthanized, and the popliteal lymph nodes from the left hind limbs were isolated and subjected to RNA extraction. Total RNA (1 μg) was used for cDNA synthesis with 10 ng cDNA used for the following real-time PCR detection of hHPRT1 with 40 cycles of amplification. The samples with Ct values lower than 38 were identified as metastasis positive. The primers for human HPRT1 are listed in the Supplementary Material in Supplementary Table S3.
Spontaneous spleen–liver metastasis model
The spontaneous spleen–liver metastasis model has been published previously (14). Male nude mice ages 5 to 6 weeks were anesthetized by continuous inhalation with isoflurane, and 2 × 105 cells in 30 μL DMEM with 33% Matrigel (354230, BD Biosciences) were injected into the spleen of laparotomized mouse using insulin syringes (328438, BD Biosciences). Twenty-eight days after inoculation, the mice were euthanized. The livers and spleens were weighed before the metastatic nodules in livers were counted, and the livers and spleens were also subjected to hematoxylin and eosin (H&E) staining. To assess the effects of erlotinib treatment, 5 × 105 cells were injected, and 15 days after cell inoculation, the mice were treated with erlotinib (S1023, Selleck Chemicals) in PBS with 6% Captisol (HY-17031, Med-ChemExpress) at 50 mg/kg (i.g.) daily for 14 days, and the mice were then euthanized for further assessment.
Statistical analysis
Data were analyzed using SPSS 21.0 software. Single comparisons were performed using unpaired Student t test or χ2 test (two tailed, P < 0.05 was considered significant). Spearman correlation analysis (two tailed) was used to calculate the correlation coefficient (r) and the P value between SPINK6 and pEGFR or vimentin staining scores. The censoring time distribution was estimated by the Kaplan–Meier method, and P values were calculated by log-rank analysis. Cox regression model was used for multivariate survival analysis. Predictors were judged to be significant at P < 0.05.
Results
SPINK6 expression as an independent, unfavorable prognostic indicator in nasopharyngeal carcinoma patients
To investigate the role of SPINK6 in nasopharyngeal carcinoma progression, SPINK6 expression in nasopharyngeal carcinoma cells and nasopharyngeal carcinoma tissues was determined. We found that the SPINK6 mRNA was upregulated in nasopharyngeal carcinoma tissues and even higher in lymph node metastases (Fig. 1A) and was highly expressed in poorly-differentiated nasopharyngeal carcinoma cells compared with well-differentiated nasopharyngeal carcinoma cells (CNE-1 and HK-1). Importantly, the highest SPINK6 mRNA levels were detected in the highly metastatic S18 and 5–8F cells (Fig. 1B). We found that SPINK6 protein was predominantly present in the condition medium, while some SPINK6 protein could be retained intracellularly following treatment with monensin (M5273, Sigma-Aldrich), an inhibitor of protein transmembrane transport (Supplementary Fig. S1). The highest levels of SPINK6 protein were detected in the condition medium of S18 and 5–8F cells (Fig. 1B).
Figure 1.
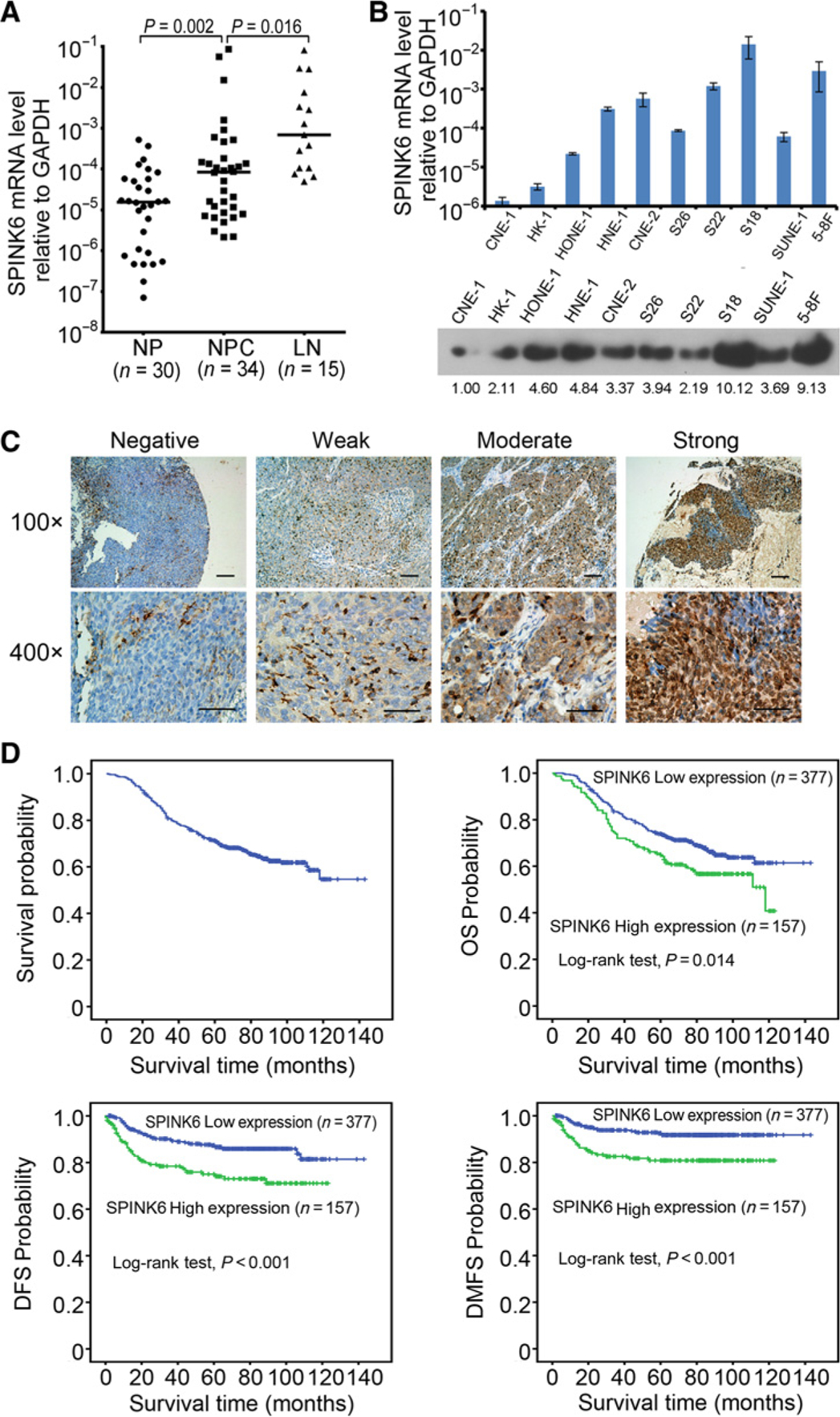
Elevated SPINK6 protein level in the primary nasopharyngeal carcinoma correlated with shorter OS, DFS, and DMFS in the patients. A, The mRNA levels of SPINK6 in nasopharyngeal carcinoma (NPC) tissues were higher than the noncancerous nasopharyngeal tissues (NP), and even higher in lymph node metastases (LN). P value, result of unpaired t test. B, Expression of SPINK6 in nasopharyngeal carcinoma cells is shown. Top, mRNA level of SPINK6; bottom, protein level of SPINK6 in condition medium with relative gradation shown below the band. C, Different levels of SPINK6 protein detected by IHC are shown under both low and high magnification of a light microscope. Scale bars, 100 μm. D, Tissue microarray analyses of a cohort of 534 nasopharyngeal carcinoma patients diagnosed at M0 stage were conducted. The median follow-up time for this cohort of patients was 81 months.
To evaluate the prognostic value of SPINK6 expression in primary nasopharyngeal carcinoma tissues, we performed immunohistochemical staining of SPINK6 in 534 nasopharyngeal carcinoma samples (Supplementary Table S1; Fig. 1C). Elevated SPINK6 expression was significantly correlated with mortality, disease progression, and distant metastasis (Supplementary Table S4). Univariate survival analyses showed that elevated SPINK6 expression was significantly correlated with shorter OS, DFS, and DMFS in patients (Fig. 1D). Multivariate analyses further indicated that elevated SPINK6 expression was an independent, unfavorable prognostic indicator in nasopharyngeal carcinoma patients (Table 1, P = 0.034 for OS, P = 0.001 for DFS, and P = 0.001 for DMFS, respectively). These analyses confirmed the critical role of SPINK6 in nasopharyngeal carcinoma progression.
Table 1.
Multivariate analysis of OS, DFS, and DMFS in nasopharyngeal carcinoma patients (Cox regression model)
| HR (95% CI) | P | |
|---|---|---|
| OS | ||
| Age (>46 vs. ≤46) | 1.548 (1.161–2.064) | 0.003 |
| Gender (female vs. male) | 0.584 (0.406–0.840) | 0.004 |
| Clinical staging (III + IV vs. I + II) | 3.543 (2.269–5.531) | <0.001 |
| SPINK6 (high vs. low level) | 1.379 (1.025–1.855) | 0.034 |
| DFS | ||
| T staging (T3–4 vs. T1–2) | 1.964 (1.234–3.125) | 0.004 |
| SPINK6 (high vs. low level) | 2.101 (1.378–3.206) | 0.001 |
| DMFS | ||
| Clinical staging (III + IV vs. I + II) | 4.265 (1.699–10.706) | 0.002 |
| SPINK6 (high vs. low level) | 2.555 (1.505–4.337) | 0.001 |
Abbreviation: CI, confidence interval.
SPINK6 as an autocrine factor that induces nasopharyngeal carcinoma cell migration, invasion, and metastasis
To examine the causal role of SPINK6 in nasopharyngeal carcinoma metastasis, we constructed stable SPINK6-overexpressing S26 and SUNE-1 cells (S26-SPINK6 and SUNE-1-SPINK6, respectively; Fig. 2A). Colony formation and cellular proliferation assays showed no significant difference in SPINK6-overexpressing cells and SPINK6 knockdown cells compared with control cells in whole medium, while SPINK6 overexpression moderately promoted the growth of nasopharyngeal carcinoma cells in DMEM with 1% FBS (Supplementary Fig. S2). However, SPINK6 overexpression dramatically increased the migration and invasion of S26 and SUNE-1 cells (Fig. 2B and C). Importantly, the condition medium collected from S26-SPINK6 and SUNE-1-SPINK6 cells could induce migration and invasion of wild-type S26 and SUNE-1 cells (Fig. 2D), suggesting that SPINK6 could promote nasopharyngeal carcinoma cellular motility via a paracrine manner. In murine lymph node metastasis model, SPINK6 overexpression significantly increased the metastatic rate of S26 cells (Fig. 2E). In addition, using murine liver metastasis model, we also found that SPINK6 overexpression dramatically enhanced S26 cell metastasis to liver with increased metastatic nodules in the liver, without changing primary tumor growth in the spleen (Fig. 2F and G). Taken together, these results showed that SPINK6 can promote nasopharyngeal carcinoma cellular migration, invasion, and metastasis.
Figure 2.
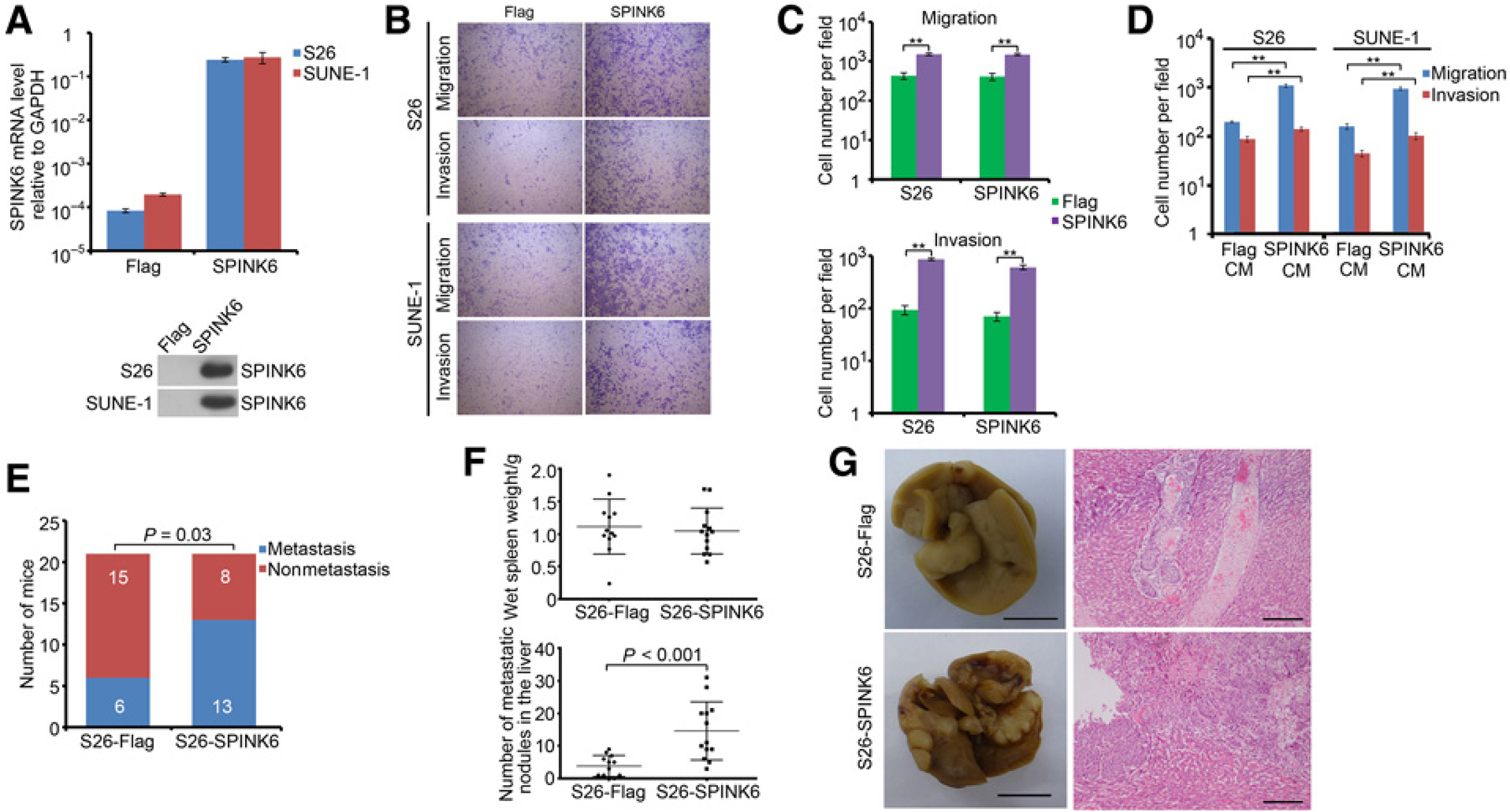
Secreted SPINK6 enhanced migration, invasion, and metastasis of low metastasis nasopharyngeal carcinoma cells. A, SPINK6 expression in stable SPINK6-overexpressing cells was shown. Top, mRNA level; bottom, protein level in condition medium. B, Overexpression of SPINK6 enhanced migration and invasion of S26 and SUNE-1 cells. C, Statistical results for B. Column, mean; bar, SEM (from triplicates). **, P < 0.01 by Student t test. D, Stimulation of wild-type cells with SPINK6-overexpressing condition medium (CM) resulted in enhanced migration and invasion. Column, mean; bar, SEM. *, P < 0.05; **, P < 0.01 by Student t test. E, Overexpression of SPINK6 promoted in vivo metastatic rate of S26 cells. P value, result of χ2 test. F, Overexpression of SPINK6 increased liver metastasis of S26 cells. P value, result of Student t test. G, Representative picture from F. Left, representative picture of livers; scale bar, 1 cm; right, representative H&E staining of livers; scale bar, 100 μm.
Suppression of nasopharyngeal carcinoma cell migration, invasion, and metastasis by knocking down SPINK6 expression
We also constructed stable SPINK6 knockdown S18 and 5–8F cells (S18-SPINK6-sh1 and S18-SPINK6-sh2, 5–8F-SPINK6-sh-1 and 5–8F-SPINK6-sh-2, respectively; Fig. 3A). In S18 and 5–8F cells, knocking down SPINK6 significantly inhibited cellular migration, invasion, and wound-healing ability (Fig. 3B and C and Supplementary Fig. S3). Moreover, ectopic expression of SPINK6 in S18-SPINK6-sh2 cells could restore the cellular migration and invasion ability (Supplementary Fig. S4A). In murine lymph node metastasis model, SPINK6 knockdown significantly decreased the metastatic rate of S18 and 5–8F cells (Fig. 3D). Furthermore, in murine spleen–liver metastasis model, SPINK6 knockdown dramatically inhibited S18 cellular metastasis with a reduced number of metastasis nodules in the liver, without changing primary tumor growth in the spleen (Fig. 3E and F). These results revealed that knockdown of SPINK6 could inhibit nasopharyngeal carcinoma migration, invasion, and metastasis.
Figure 3.
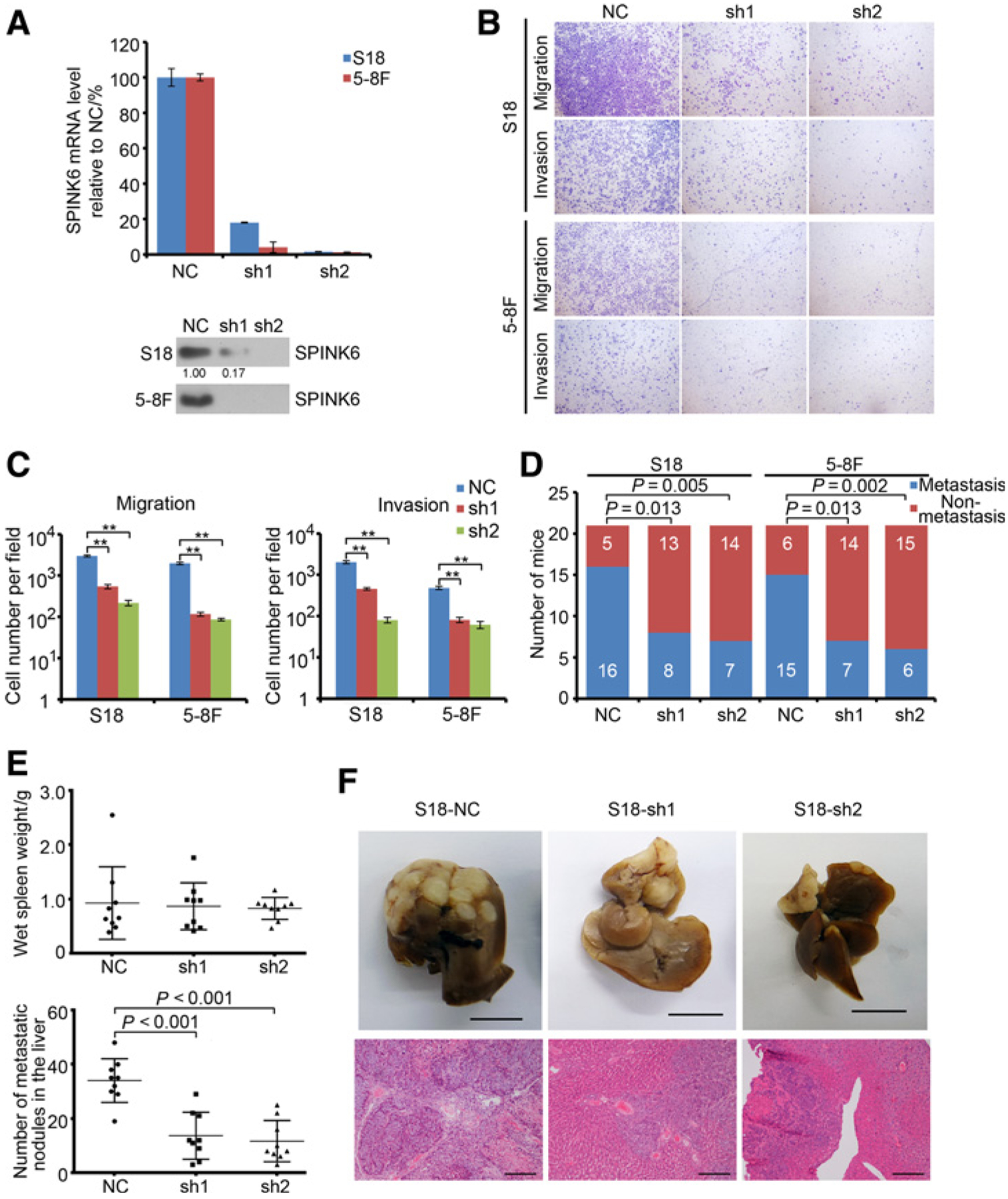
Knockdown of SPINK6 inhibited migration, invasion, and metastasis of highly metastatic nasopharyngeal carcinoma cells. A, SPINK6 expression in stable SPINK6 knockdown cells is shown. Top, mRNA level; bottom, protein level in condition medium with relative gradation shown below the band. B, Knockdown of SPINK6 suppressed migration and invasion of S18 and 5–8F cells. C, Statistical results from B. Column, mean; bar, SEM (from triplicates). **, P < 0.01 by Student t test. D, Knockdown of SPINK6 decreased in vivo metastatic rate of S18 and 5–8F cells. Cells (1× 105) were subcutaneously injected into the left hind footpad of nude mice, and metastasis to the left popliteal LN was measured. P value, result of χ2 test. E, Knockdown of SPINK6 impaired in vivo liver metastasis of S18 cells. P value, result of Student t test. F, Representative picture from E. Top, representative picture of livers; scale bar, 1 cm; bottom, representative H&E staining of livers; scale bar, 100 μm. NC, negative control, with detail shown in Supplementary Table S2.
Binding and activation of EGFR by SPINK6
To examine the role of KLKs in SPINK6-promoted nasopharyngeal carcinoma cellular motility, we used siRNA to knock down KLKs expression in nasopharyngeal carcinoma cells. We found no difference in the expression of KLK5, KLK12, KLK13, or KLK14 in nasopharyngeal carcinoma cells, and knockdown of SPINK6 did not alert the expression of the KLKs (Supplementary Fig. S4B and S4C). Moreover, silencing KLKs did not restore migration and invasion inhibited by SPINK6 knockdown in S18 cells (Supplementary Fig. S4D–S4G). In addition, we found that ectopic expression of a SPINK6 mutant without serine protease–inhibitory activity (ΔSPI) could still promote migration and invasion of S26 cells, whereas this was not found in the SPINK6 mutant missing the three disulfide bonds in the Kazal domain (ΔDSB; Fig. 4A), suggesting that SPINK6 could promote nasopharyngeal carcinoma motility independent of its well-defined serine protease–inhibitory activity.
Figure 4.
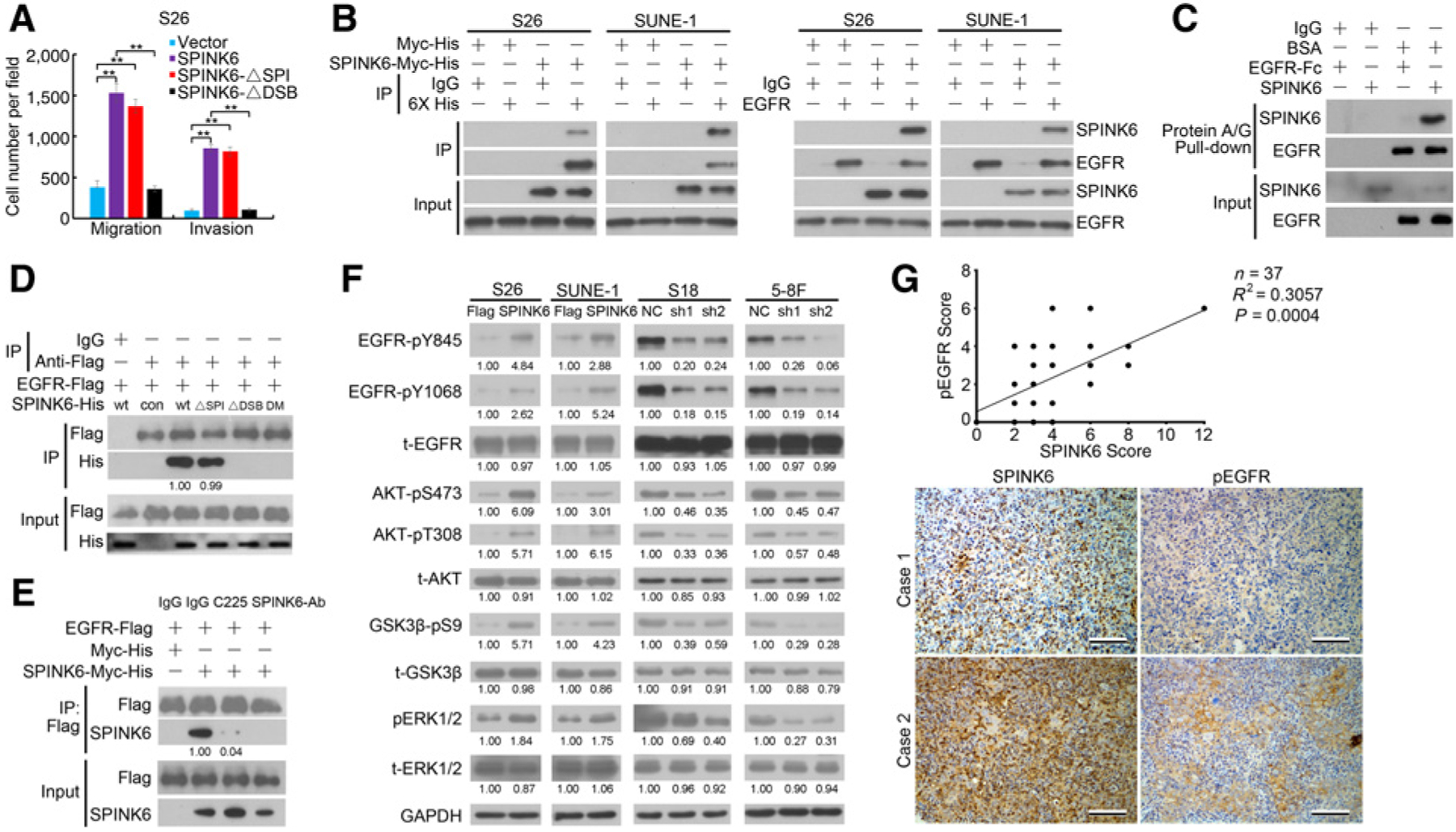
SPINK6 mediated its oncogenic effects through EGFR. A, Migration and invasion of S26 cells transfected with SPINK6 or mutant plasmids was measured. DSPI, mutant without serine protease inhibitory activity; ΔDSB, mutant with disruption of intrachain disulfide bridges. **, P < 0.01 by Student t test. B, SPINK6 was coprecipitated with endogenous EGFR. C, In vitro pull-down assay revealed direct binding of SPINK6 and EGFR. D, EGFR could bind to SPINK6 mutant without serine protease–inhibitory activity. DM, mutant with both ΔSPI and ΔDSB mutation; wt, wild type, con, control. Relative gradation corrected by IP-Flag is shown below the band. E, Antibodies against EGFR or SPINK6 impaired binding of EGFR and SPINK6. Cell-free–expressed 3× Flag-tagged EGFR and Myc-His–tagged SPINK6 were mixed and incubated with EGFR antibody (C225, 10 μg/mL) or SPINK6 antibody (2 mg/mL), followed by coimmunoprecipitation assays. Relative gradation corrected by IP-Flag is shown below the band. F, Phosphorylation of EGFR and downstream signaling in nasopharyngeal carcinoma cells is shown. Relative gradation corrected by GAPDH is shown below each band. G, SPINK6 expression was positively corrected with EGFR phosphorylation in nasopharyngeal carcinoma tissues. Top, a significant positive correlation between SPINK6 expression and EGFR phosphorylation was found; bottom, representative images of two nasopharyngeal carcinoma cases are shown. Scale bar, 100 μm.
SPINK6 has a Kazal domain that has a similar structure to EGF with a high-sequence homology (Supplementary Fig. S5A and S5B). We observed a strong interaction between SPINK6-Myc-His and endogenous EGFR (Fig. 4B). Moreover, in vitro pull-down assay using recombinant SPINK6 protein (Supplementary Fig. S5C and S5D) and a recombinant EGFR extracellular domain-Fc fusion protein revealed that rSPINK6 could directly bind to the extracellular domain of EGFR (Fig. 4C). The EGFR extracellular domain possessed four subdomains. To further investigate the interaction between EGFR and SPINK6, we generated individual subdomains and mutants with subdomain deletions and carried out cell-free expression (Supplementary Fig. S6A). Coimmunoprecipitation assay revealed that SPINK6 could bind to the EGFR extracellular domain via subdomain 1 and 3 (Supplementary Fig. S6B), and the SPINK6 mutant lacking serine protease–inhibitory activity could still bind to EGFR but not the mutant missing the three disulfide bonds in the Kazal domain (Fig. 4D). Importantly, an antibody against EGFR (C225) or a polyclonal antibody against SPINK6 inhibited the binding between EGFR and SPINK6 (Fig. 4E). Coimmunoprecipitation assay also revealed that SPINK6 could compete with EGF for EGFR binding in a dose-dependent manner (Supplementary Fig. S6C). EGFR can undergo dimerization and internalization upon ligand binding. DTSSP (21578, Thermo Fisher Scientific) cross-linking assay and immunofluorescence staining showed that SPINK6 could also induce EGFR dimerization and internalization (Supplementary Fig. S6D and S6E). We also found that SPINK6 induced EGFR activation and downstream ERK1/2 and AKT phosphorylation in S26 and SUNE-1 cells, whereas SPINK6 knockdown inhibited their phosphorylation in S18 and 5–8F cells (Fig. 4F and Supplementary Fig. S6F). However, no change in STAT3 phosphorylation was found (Supplementary Fig. S7A). EGFR and downstream signaling were highly activated in S18 and 5–8F cells with a high level of SPINK6 (Supplementary Fig. S7B), suggesting its possible involvement in nasopharyngeal carcinoma cellular mobility. Importantly, we also found a significant correlation between SPINK6 expression and EGFR phosphorylation in nasopharyngeal carcinoma tissues (Fig. 4G). In summary, these results revealed that SPINK6 could bind to EGFR and activate EGFR signaling.
Suppression of SPINK6-promoted nasopharyngeal carcinoma cellular migration, invasion, and metastasis by EGFR/AKT inhibition
To examine whether SPINK6 induced nasopharyngeal carcinoma motility via EGFR activation, we tested the motility of nasopharyngeal carcinoma cells following inhibition of EGFR and downstream ERK1/2 and AKT pathway. We found that the antibodies against EGFR or SPINK6 could impair migration and invasion of S26-SPINK6 cells (Fig. 5A). The EGFR inhibitor erlotinib and the PI3K/AKT pathway inhibitor LY294002 also attenuated migration and invasion of S26-SPINK6 and SUNE-1-SPINK6 cells but not the MEK/ERK1/2 pathway inhibitor U0126 (Fig. 5B). LY294002 treatment also reduced the wound-healing ability of S26-SPINK6 and SUNE-1-SPINK6 cells (Supplementary Fig. S8). These results suggested that SPINK6 promoted nasopharyngeal carcinoma cell motility via EGFR/AKT signaling pathway. Furthermore, we also found that erlotinib treatment decreased EGFR and downstream ERK1/2 and AKT phosphorylation in SPINK6-overexpressing S26 and SUNE-1 cells (Fig. 5C). Importantly, in murine liver metastasis model, treatment with erlotinib dramatically reduced metastasis in mice implanted with S26-SPINK6 cells, with reduced metastasis nodules in the liver (Fig. 5D and E). These results showed that inhibition of EGFR pathway can inhibit migration, invasion, and metastasis of SPINK6-overexpressing nasopharyngeal carcinoma cells.
Figure 5.
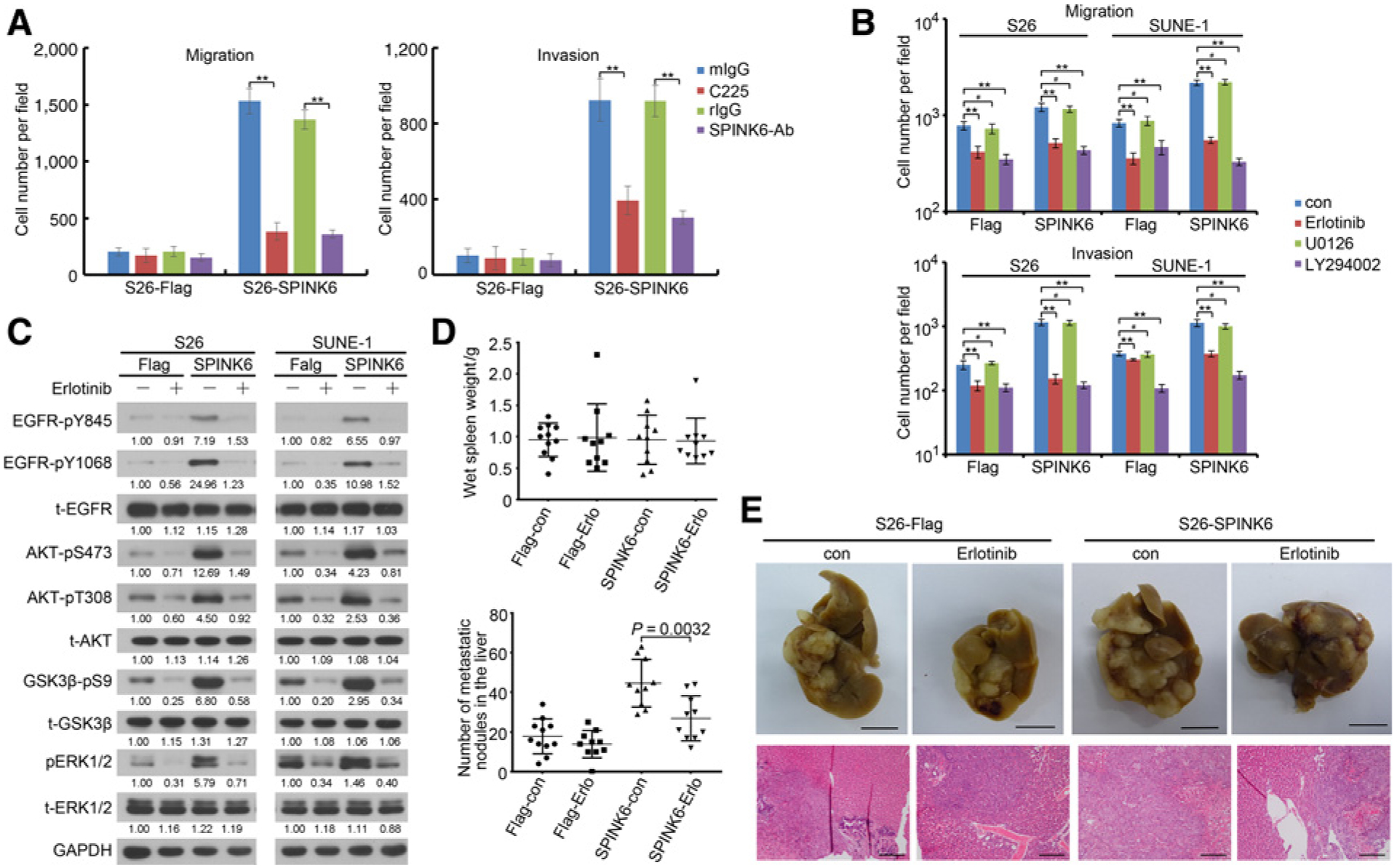
Suppression of EGFR impaired migration, invasion, and metastasis of SPINK6-overexpressing nasopharyngeal carcinoma cells. A, Neutralizing antibodies reduced migration and invasion of S26-SPINK6 cells. Cells were starved for 16 hours and pretreated with C225 (10 μg/mL) or SPINK6 (2 μg/mL) antibody for 16 hours and subjected to migration and invasion assays in the presence of C225 or SPINK6 antibody. Column, mean; bar, SEM. **, P < 0.01 by Student t test. B, Inhibitors of EGFR and AKT impaired migration and invasion of SPINK6-overexpressing cells. con, control. Cells were treated with 1 μmol/L erlotinib, 2 μmol/L U0126, 20 μmol/L LY294002, or DMSO for 12 hours and subjected to migration and invasion assays. Column, mean; bar, SEM. **, P < 0.01; #, P > 0.05, by Student t test. C, Erlotinib inhibited phosphorylation of EGFR and downstream signaling. After treatment with 1 μmol/L erlotinib for 12 hours, cell lysateswere harvested for immunoblotting detection. Relative gradation corrected by GAPDH is shown below each band. D, Erlotinib impaired liver metastasisof S26-SPINK6 cells. P value, result of Student t test. E, Representative picture from D. Top, representative picture of livers; scale bar, 1 cm; bottom, representative H&E staining of liver; scale bar, 100 μm.
Promotion of EMT by SPINK6 via EGFR/AKT signaling
EMT is a crucial step for cancer cells to achieve metastasis (27). We found that SPINK6 expression resulted in downregulation of the epithelial markers E-cadherin and desmoplakin, whereas the mesenchymal markers vimentin, N-cadherin, and fibronectin were upregulated as well as the EMT-promoting transcription factors Snail, Slug, and Twist1 in S26 and SUNE-1 cells (Fig. 6A). Conversely, SPINK6 knockdown resulted in upregulation of E-cadherin and desmoplakin and downregulation of vimentin, N-cadherin, fibronectin, Snail, Slug, and Twist1 in S18 and 5–8F cells (Fig. 6A and Supplementary Fig. S9). Importantly, we also found a positive correlation between SPINK6 and vimentin expression in nasopharyngeal carcinoma tissues (Fig. 6B). Furthermore, LY294002 treatment restored the expression of epithelial markers and decreased the expression of mesenchymal markers and EMT-promoting transcription factors in SPINK6-overexpressing S26 and SUNE-1 cells (Fig. 6C). Taken together, these data suggest that SPINK6 can induce EMT in nasopharyngeal carcinoma cells and inhibition of AKT signaling can reverse this process.
Figure 6.
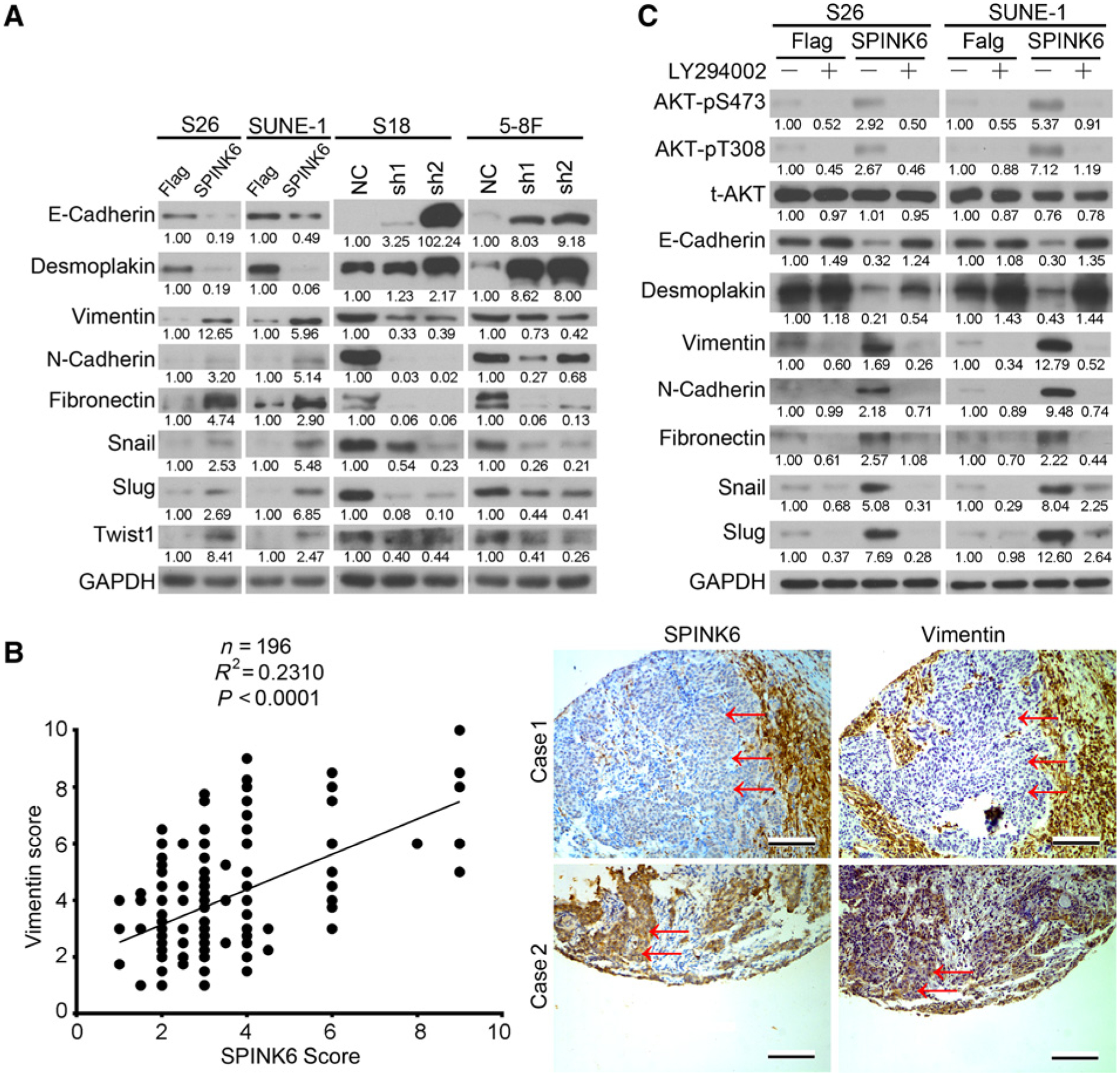
SPINK6 promoted EMT of nasopharyngeal carcinoma cells. A, The expression of epithelial markers, mesenchymal makers, and EMT-promoting transcription factors in nasopharyngeal carcinoma cells was detected by immunoblotting. Relative gradation corrected by GAPDH is shown below each band. B, SPINK6 expression was positively corrected with vimentin expression in nasopharyngeal carcinoma tissues. Top, a positive correction between SPINK6 expression and vimentin expression was found; bottom, representative images of two nasopharyngeal carcinoma cases are shown. Scale bar, 100 μm. To distinguish from noncancerous cells, the representative cancer regions are labeled by red arrowheads. C, The expression of epithelial and mesenchymal markers and EMT-promoting transcription factors in SPINK6-overexpressing S26 and SUNE-1 cells after LY294002 treatment was detected by immunoblotting. After treatment with 20 μmol/L LY294002 or DMSO for 12 hours, cell lysates were harvested for Western blot detection. Relative gradation corrected by GAPDH is shown below each band.
Discussion
Metastasis is the major cause of treatment failure in nasopharyngeal carcinoma, and thus, preventing, predicting, and inhibiting metastasis is critical to improve treatment outcomes. In the current study, we reported the high expression of SPINK6 in highly metastatic cells in contrast to their parental and other poorly metastatic nasopharyngeal carcinoma cells. Moreover, elevated SPINK6 expression in primary nasopharyngeal carcinoma was found to be an independent unfavorable prognostic factor for patients (Fig. 1; Table 1).
SPINK6 belongs to the Kazal-type serine protease inhibitor family, several members of which have been reported to be involved in tumor progression. However, their roles in cancer are ambiguous. SPINK1 was reported to be upregulated in colorectal cancer, prostate cancer, and pancreatic cancer (32–34), and it was reported to promote tumor growth, migration, invasion, and angiogenesis (25, 35, 36). Conversely, SPINK7 has been found to inhibit cancer cell migration, invasion, and metastasis (37–39). These differences may be explained by the different substrate protease specificity of the SPINKs and the contradistinctive roles of substrate proteases in cancer. SPINK6 inhibits the protease activity of KLKs, but the roles of KLKs as tumor markers and/or tumor progression regulators remain ambiguous in different types of tissues and different tumor microenvironments (20–22). For example, the KLK5 protease suppresses breast cancer by repressing the mevalonate pathway (40), whereas KLK14 can promote colon cancer cell proliferation through proteinase-activated receptor 2 activation followed by increased ERK1/2 phosphorylation (41). However, there was no difference in the expression of known SPINK6 target KLK5, KLK12, KLK13, or KLK14 between nasopharyngeal carcinoma tissues and noncancerous nasopharyngeal tissues (Supplementary Fig. S10A–S10D).
Functionally, we found that the overexpression of SPINK6 could induce nasopharyngeal carcinoma cell migration and invasion in vitro, as well as metastasis in vivo, via autocrine and paracrine mechanisms, whereas knockdown of SPINK6 inhibited migration, invasion, and metastasis of highly metastatic nasopharyngeal carcinoma cells (Figs. 2 and 3). However, silencing the SPINK6 targets KLK5, KLK12, KLK13, or KLK14 did not restore the migration and invasion of SPINK6 knockdown nasopharyngeal carcinoma cells. Moreover, the migration and invasion potential could be enhanced by overexpression of a SPINK6 mutant without protease-inhibitory activity (Fig. 4A and Supplementary Fig. S4). These results suggested that SPINK6 could promote nasopharyngeal carcinoma motility independent of its serine protease–inhibitory activity.
By applying whole-exome and targeted deep sequencing of nasopharyngeal carcinoma combined with integrated analysis, Lin and colleagues found that ErbB–PI3K signaling is enriched even when no EGFR mutation or amplification was found (7). EGFR is overexpressed in 73% to 89% of nasopharyngeal carcinoma patients (42, 43), contributing to an increased risk of metastasis and decreased OS, but not DMFS (42, 44). Meta-analysis revealed that elevated EGFR expression in nasopharyngeal carcinoma is correlated with poorer OS, DFS, and locoregional control, while the association between EGFR overexpression and DMFS was not statistically significant (45, 46). We also reported that EGFR and pEGFR expression was upregulated in nasopharyngeal carcinoma tissues, and elevated pEGFR expression was related to shorter metastasis-free survival in patients (47). These findings indicate that EGFR activation, but not just EGFR expression, is correlated with distant metastasis of nasopharyngeal carcinoma patients. In the current study, we also found that EGFR and downstream signaling pathway were more active in highly metastatic S18 and 5–8F cells, compared with poorly metastatic S26 and 5–8F cells, whereas there was no difference in EGFR expression between highly and poorly metastatic nasopharyngeal carcinoma cells (Supplementary Fig. S7B). All these findings confirm the important roles of EGFR signaling in nasopharyngeal carcinoma progression and metastasis. Therefore, EGFR signaling could be a potential therapeutic target for nasopharyngeal carcinoma. However, EGFR mutations were rarely detected, and no EGFR amplification was found in nasopharyngeal carcinoma (6, 7, 48), suggesting that EGFR amplification and mutation might not be critical reasons for EGFR activation in nasopharyngeal carcinoma. Besides, in spite of overexpression of EGFR in nasopharyngeal carcinoma tissues, there was no difference in EGF expression between nasopharyngeal carcinoma tissues and noncancerous nasopharyngeal tissues (Supplementary Fig. S10E and S10F). These findings indicate that there may be other factor(s) to activate EGFR signaling in nasopharyngeal carcinoma.
SPINK6 contains a Kazal repeat, which includes a long extended chain, two short alpha helices and a three-stranded antiparallel beta sheet, similar to that of the EGF-like domain (40). In the current study, we demonstrated that SPINK6 could bind to EGFR through subdomain 1 and 3 of the EGFR extracellular domain, which also forms a pocket for EGF and TGFα binding (49, 50), resulting in the activation of EGFR and its downstream signaling pathway (Fig. 4 and Supplementary Fig. S6). Furthermore, treatment with SPINK6 antibody or EGFR neutralizing antibody disrupted the binding and impaired migration and invasion of SPINK6-stimulated nasopharyngeal carcinoma cells (Figs. 4E and 5A). More importantly, treatment of the EGFR inhibitor erlotinib efficiently decreased migration and invasion in vitro, as well as metastasis in vivo, in SPINK6-overexpressing nasopharyngeal carcinoma cells (Fig. 5). These results confirmed that SPINK6 could trigger EGFR activation, and inhibition of EGFR can inhibit nasopharyngeal carcinoma cellular motility. In regard of no amplification and mutation of EGFR and no EGF overexpression in nasopharyngeal carcinoma, as well as the positive correction of SPINK6 and pEGFR expression, we presume that the overexpression of SPINK6 seems to be a candidate factor responsible for the activation of EGFR signaling in nasopharyngeal carcinoma.
In summary, our study is the first to elucidate the critical roles of SPINK6 in promoting cancer metastasis and predicting unfavorable patient prognosis. Moreover, SPINK6 severs as another effective ligand that binds to EGFR in nasopharyngeal carcinoma cells and activates EGFR signaling. We believe that SPINK6 is a potential molecular target for predicting, preventing, and treating nasopharyngeal carcinoma metastasis.
Supplementary Material
Grant Support
This work was supported by grants from the National Natural Science Foundation of China (nos. 81672872, 81272340, and 81472386 to C.-N. Qian; 81572901 to B.-J. Huang; 81572848 to L. Guo; and 81402248 toD.-H. Luo), the National High Technology Research and Development Program of China (863 Program; no. 2012AA02A501 to C.-N. Qian), the Provincial Natural Science Foundation of Guangdong, China (no. 2016A030311011 to C.-N. Qian), and the Science and Technology Planning Project of Guangdong Province, China (nos. 2014B020212017, 2014B050504004, and 2015B050501005 to C.-N. Qian; and 2014A020209024 to B.-J. Huang).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Chua ML, Wee JT, Hui EP, Chan AT. Nasopharyngeal carcinoma. Lancet 2015;387:1012–24. [DOI] [PubMed] [Google Scholar]
- 2.Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev 2006;15:1765–77. [DOI] [PubMed] [Google Scholar]
- 3.Tao Q, Chan AT. Nasopharyngeal carcinoma: molecular pathogenesis and therapeutic developments. Expert Rev Mol Med 2007;9:1–24. [DOI] [PubMed] [Google Scholar]
- 4.Lee AW, Ma BB, Ng WT, Chan AT. Management of nasopharyngeal carcinoma: current practice and future perspective. J Clin Oncol 2015;33: 3356–64. [DOI] [PubMed] [Google Scholar]
- 5.Razak AR, Siu LL, Liu FF, Ito E, O’Sullivan B, Chan K. Nasopharyngeal carcinoma: the next challenges. Eur J Cancer 2010;46:1967–78. [DOI] [PubMed] [Google Scholar]
- 6.Janvilisri T Omics-based identification of biomarkers for nasopharyngeal carcinoma. Dis Markers 2015;2015:762128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin DC, Meng X, Hazawa M, Nagata Y, Varela AM, Xu L, et al. The genomic landscape of nasopharyngeal carcinoma. Nat Genet 2014;46: 866–71. [DOI] [PubMed] [Google Scholar]
- 8.Zeng Z, Zhou Y, Xiong W, Luo X, Zhang W, Li X, et al. Analysis of gene expression identifies candidate molecular markers in nasopharyngeal carcinoma using microdissection and cDNA microarray. J Cancer Res Clin Oncol 2007;133:71–81. [DOI] [PubMed] [Google Scholar]
- 9.Zeng ZY, Zhou YH, Zhang WL, Xiong W, Fan SQ, Li XL, et al. Gene expression profiling of nasopharyngeal carcinoma reveals the abnormally regulated Wnt signaling pathway. Hum Pathol 2007;38:120–33. [DOI] [PubMed] [Google Scholar]
- 10.Szeto CY, Lin CH, Choi SC, Yip TT, Ngan RK, Tsao GS, et al. Integrated mRNA and microRNA transcriptome sequencing characterizes sequence variants and mRNA-microRNA regulatory network in nasopharyngeal carcinoma model systems. FEBS Open Bio 2014;4:128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang LF, Li YH, Xie SH, Ling W, Chen SH, Liu Q, et al. Incidence trend of nasopharyngeal carcinoma from 1987 to 2011 in Sihui County, Guangdong Province, South China: an age-period-cohort analysis. Chin J Cancer 2015;34:350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Lv X, Chen J, Xie C, Xia W, Jiang C, et al. CCL2-CCR2 axis promotes metastasis of nasopharyngeal carcinoma by activating ERK1/2-MMP2/9 pathway. Oncotarget 2016;7:15632–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li XJ, Ong CK, Cao Y, Xiang YQ, Shao JY, Ooi A, et al. Serglycin is a theranostic target in nasopharyngeal carcinoma that promotes metastasis. Cancer Res 2011;71:3162–72. [DOI] [PubMed] [Google Scholar]
- 14.Li XJ, Peng LX, Shao JY, Lu WH, Zhang JX, Chen S, et al. As an independent unfavorable prognostic factor, IL-8 promotes metastasis of nasopharyngeal carcinoma through induction of epithelial-mesenchymal transition and activation of AKT signaling. Carcinogenesis 2012;33:1302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin L, Yin YT, Zheng FJ, Peng LX, Yang CF, Bao YN, et al. WNT5A promotes stemness characteristics in nasopharyngeal carcinoma cells leading to metastasis and tumorigenesis. Oncotarget 2015;6:10239–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian CN, Berghuis B, Tsarfaty G, Bruch M, Kort EJ, Ditlev J, et al. Preparing the “soil”: the primary tumor induces vasculature reorganization in the sentinel lymph node before the arrival of metastatic cancer cells. Cancer Res 2006;66:10365–76. [DOI] [PubMed] [Google Scholar]
- 17.Meyer-Hoffert U, Wu Z, Kantyka T, Fischer J, Latendorf T, Hansmann B, et al. Isolation of SPINK6 in human skin: selective inhibitor of kallikrein-related peptidases. J Biol Chem 2010;285:32174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kantyka T, Fischer J, Wu Z, Declercq W, Reiss K, Schroder JM, et al. Inhibition of kallikrein-related peptidases by the serine protease inhibitor of Kazal-type 6. Peptides 2011;32:1187–92. [DOI] [PubMed] [Google Scholar]
- 19.Furio L, Hovnanian A. When activity requires breaking up: LEKTI proteolytic activation cascade for specific proteinase inhibition. J Invest Dermatol 2011;131:2169–73. [DOI] [PubMed] [Google Scholar]
- 20.Avgeris M, Mavridis K, Scorilas A. Kallikrein-related peptidase genes as promising biomarkers for prognosis and monitoring of human malignancies. Biol Chem 2010;391:505–11. [DOI] [PubMed] [Google Scholar]
- 21.Oikonomopoulou K, Diamandis EP, Hollenberg MD. Kallikrein-related peptidases: proteolysis and signaling in cancer, the new frontier. Biol Chem 2010;391:299–310. [DOI] [PubMed] [Google Scholar]
- 22.Mavridis K, Scorilas A. Prognostic value and biological role of the kallikrein-related peptidases in human malignancies. Future Oncol 2010;6: 269–85. [DOI] [PubMed] [Google Scholar]
- 23.Jung S, Fischer J, Spudy B, Kerkow T, Sonnichsen FD, Xue L, et al. The solution structure of the kallikrein-related peptidases inhibitor SPINK6. Biochem Biophys Res Commun 2016;471:103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong KJ, Hsu MC, Hou MF, Hung WC. The tumor suppressor RECK interferes with HER-2/Neu dimerization and attenuates its oncogenic signaling. FEBS Lett 2011;585:591–5. [DOI] [PubMed] [Google Scholar]
- 25.Ateeq B, Tomlins SA, Laxman B, Asangani IA, Cao Q, Cao X, et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci Transl Med 2011;3:72ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer J, Koblyakova Y, Latendorf T, Wu Z, Meyer-Hoffert U. Cross-linking of SPINK6 by transglutaminases protects from epidermal proteases. J Invest Dermatol 2013;133:1170–7. [DOI] [PubMed] [Google Scholar]
- 27.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871–90. [DOI] [PubMed] [Google Scholar]
- 28.Sang Y, Chen MY, Luo D, Zhang RH, Wang L, Li M, et al. TEL2 suppresses metastasis by down-regulating SERPINE1 in nasopharyngeal carcinoma. Oncotarget 2015;6:29240–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song LB, Yan J, Jian SW, Zhang L, Li MZ, Li D, et al. [Molecular mechanisms of tumorgenesis and metastasis in nasopharyngeal carcinoma cell sub-lines]. Ai Zheng 2002;21:158–62. [PubMed] [Google Scholar]
- 30.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PloS One 2009;4:e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001;410:50–6. [DOI] [PubMed] [Google Scholar]
- 32.Gaber A, Nodin B, Hotakainen K, Nilsson E, Stenman UH, Bjartell A, et al. Increased serum levels of tumour-associated trypsin inhibitor independently predict a poor prognosis in colorectal cancer patients. BMC Cancer 2010;10:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohmuraya M, Yamamura K. Roles of serine protease inhibitor Kazal type 1 (SPINK1) in pancreatic diseases. Exp Anim 2011;60:433–44. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Wang L, Su B, Lu N, Song J, Yang X, et al. Serine protease inhibitor Kazal type 1 promotes epithelial-mesenchymal transition through EGFR signaling pathway in prostate cancer. Prostate 2014;74:689–701. [DOI] [PubMed] [Google Scholar]
- 35.Gouyer V, Fontaine D, Dumont P, de Wever O, Fontayne-Devaud H, Leteurtre E, et al. Autocrine induction of invasion and metastasis by tumor-associated trypsin inhibitor in human colon cancer cells. Oncogene 2008;27:4024–33. [DOI] [PubMed] [Google Scholar]
- 36.Lu X, Lamontagne J, Lu F, Block TM. Tumor-associated protein SPIK/TATI suppresses serine protease dependent cell apoptosis. Apoptosis 2008;13: 483–94. [DOI] [PubMed] [Google Scholar]
- 37.Cheng X, Lu SH, Cui Y. ECRG2 regulates ECM degradation and uPAR/FPRL1 pathway contributing cell invasion/migration. Cancer Lett 2010;290:87–95. [DOI] [PubMed] [Google Scholar]
- 38.Cheng X, Shen Z, Yin L, Lu SH, Cui Y. ECRG2 regulates cell migration/invasion through urokinase-type plasmin activator receptor (uPAR)/beta1 integrin pathway. J Biol Chem 2009;284:30897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang G, Hu Z, Li M, Cui Y, Li Y, Guo L, et al. ECRG2 inhibits cancer cell migration, invasion and metastasis through the down-regulation of uPA/plasmin activity. Carcinogenesis 2007;28:2274–81. [DOI] [PubMed] [Google Scholar]
- 40.Pampalakis G, Obasuyi O, Papadodima O, Chatziioannou A, Zoumpourlis V, Sotiropoulou G. The KLK5 protease suppresses breast cancer by repressing the mevalonate pathway. Oncotarget 2014;5:2390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung H, Hamza M, Oikonomopoulou K, Gratio V, Saifeddine M, Virca GD, et al. Kallikrein-related peptidase signaling in colon carcinoma cells: targeting proteinase-activated receptors. Biol Chem 2012;393:413–20. [DOI] [PubMed] [Google Scholar]
- 42.Chua DT, Nicholls JM, Sham JS, Au GK. Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy. Int J Radiat Oncol Biol Phys 2004;59:11–20. [DOI] [PubMed] [Google Scholar]
- 43.Sheen TS, Huang YT, Chang YL, Ko JY, Wu CS, Yu YC, et al. Epstein-Barr virus-encoded latent membrane protein 1 co-expresses with epidermal growth factor receptor in nasopharyngeal carcinoma. Jpn J Cancer Res 1999;90:1285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma BB, Poon TC, To KF, Zee B, Mo FK, Chan CM, et al. Prognostic significance of tumor angiogenesis, Ki 67, p53 oncoprotein, epidermal growth factor receptor and HER2 receptor protein expression in undifferentiated nasopharyngeal carcinoma–a prospective study. Head Neck 2003;25:864–72. [DOI] [PubMed] [Google Scholar]
- 45.Ooft ML, Braunius WW, Heus P, Stegeman I, van Diest PJ, Grolman W, et al. Prognostic significance of the EGFR pathway in nasopharyngeal carcinoma: a systematic review and meta-analysis. Biomarkers Med 2015;9: 997–1010. [DOI] [PubMed] [Google Scholar]
- 46.Sun W, Long G, Wang J, Mei Q, Liu D, Hu G. Prognostic role of epidermal growth factor receptor in nasopharyngeal carcinoma: a meta-analysis. Head Neck 2014;36:1508–16. [DOI] [PubMed] [Google Scholar]
- 47.Yuan TZ, Li XX, Cao Y, Qian CN, Zeng MS, Guo X. [Correlation of epidermal growth factor receptor activation to metastasis-free survival of nasopharyngeal carcinoma patients]. Ai Zheng 2008;27:449–54. [PubMed] [Google Scholar]
- 48.Lee SC, Lim SG, Soo R, Hsieh WS, Guo JY, Putti T, et al. Lack of somatic mutations in EGFR tyrosine kinase domain in hepatocellular and nasopharyngeal carcinoma. Pharmacogenet Genomics 2006;16:73–4. [DOI] [PubMed] [Google Scholar]
- 49.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ecto-domain dimerization. Mol Cell 2003;11:507–17. [DOI] [PubMed] [Google Scholar]
- 50.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002;110: 763–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.