

Abstract
Background
Oxidative stress and high salt intake could be independent or intertwined risk factors in the origin of hypertension. Kidneys are the major organ to regulate sodium homeostasis and blood pressure and the renal dopamine system plays a pivotal role in sodium regulation during sodium replete conditions. Oxidative stress has been implicated in renal dopamine dysfunction and development of hypertension, especially in salt‐sensitive animal models. Here we show the nexus between high salt intake and oxidative stress causing renal tubular dopamine oxidation, which leads to mitochondrial and lysosomal dysfunction and subsequently causes renal inflammation and hypertension.
Methods and Results
Male Sprague Dawley rats were divided into the following groups, vehicle (V)—tap water, high salt (HS)—1% NaCl, L‐buthionine‐sulfoximine (BSO), a prooxidant, and HS plus BSO without and with antioxidant resveratrol (R) for 6 weeks. Oxidative stress was significantly higher in BSO and HS+BSO–treated rat compared with vehicle; however, blood pressure was markedly higher in the HS+BSO group whereas an increase in blood pressure in the BSO group was modest. HS+BSO–treated rats had significant renal dopamine oxidation, lysosomal and mitochondrial dysfunction, and increased renal inflammation; however, HS alone had no impact on organelle function or inflammation. Resveratrol prevented oxidative stress, dopamine oxidation, organelle dysfunction, inflammation, and hypertension in BSO and HS+BSO rats.
Conclusions
These data suggest that dopamine oxidation, especially during increased sodium intake and oxidative milieu, leads to lysosomal and mitochondrial dysfunction and renal inflammation with subsequent increase in blood pressure. Resveratrol, while preventing oxidative stress, protects renal function and mitigates hypertension.
Keywords: dopamine, dopamine receptor, hypertension, inflammation, lysosomal enzymes, mitochondrial respiration
Subject Categories: Oxidant Stress, Nephrology and Kidney, High Blood Pressure, Basic Science Research, Inflammation
Clinical Perspective
What Is New?
The study is novel in that it shows that a physiological response like an increase in renal dopamine production in response to high salt intake could be channeled by oxidative stress to trigger a plethora of pathophysiological cascades causing dysregulation of renal sodium handling.
Oxidation of intrinsic renal natriuretic factor dopamine generates byproducts that could result in renal tubular organelle dysfunction and inflammation leading to the development of hypertension.
What Are the Clinical Implications?
The data identify the renal‐specific cause of salt sensitivity as opposed to previously described systemic factors such as increased sympathetic nerve activity and vascular inflammation.
Profound changes at the tubular level may not be detected by current clinical markers such as changes in glomerular filtration rate, underscoring the need for the development of more sensitive plasma and/or urinary markers for better and early diagnosis of detrimental changes in renal tubules.
Introduction
Kidneys play a major role in systemic blood pressure (BP) regulation, primarily by maintaining sodium homeostasis.1, 2, 3, 4 Under normal sodium intake, antinatriuretic factors, such as the renin‐angiotensin‐aldosterone system, play an important role in maintaining sodium balance by facilitating sodium reabsorption throughout the nephron.5 However, during high sodium intake, natriuretic factors (primarily the renal tubular dopamine system) inhibit sodium transporters and enhance salt excretion to maintain sodium homeostasis.6, 7, 8, 9 During sodium replete conditions, renal proximal tubules increase local dopamine synthesis, which acts as an autocrine/paracrine hormone on dopamine receptors.6, 7, 8, 9 The activation of dopamine D1‐like receptors (D1R) inhibits both apical and basolateral sodium transporters to reduce transcellular sodium transport.6, 7, 8, 9, 10, 11 Hypertensive subjects, especially those with essential hypertension, who exhibit salt sensitivity have been shown to have reduced renal D1R function.10, 11, 12, 13, 14, 15, 16, 17 Similarly, animal models of primary, genetic, and salt‐sensitive hypertension also exhibit renal D1R dysfunction.10, 11, 12, 13, 14, 15, 16, 17 The exact mechanisms of D1R dysfunction in hypertension are not settled; however, increased oxidative stress is reported to play an important role in the origin of both D1R dysfunction and hypertension.18, 19, 20, 21 While a link between hypertension, D1R dysfunction, and oxidative stress is well studied, little is known about the role of renal dopamine oxidation, especially during high sodium intake and oxidative stress, in the development of hypertension.
The canonical neuronal dopamine synthesis pathway involves tyrosine hydroxylase–dependent hydroxylation of l‐tyrosine to l‐dihydroxyphenylalanine, which is decarboxylated to dopamine by aromatic l‐amino acid decarboxylase.22, 23 Under normal physiological conditions, dopamine could be enzymatically converted to epinephrine via norepinephrine or metabolized to byproducts such as 3,4‐dihydroxyphenylacetic acid and 3‐methoxytyramine, which are converted to homovanillic acid and eliminated in urine.22, 23 However, during oxidative stress the spontaneous dopamine oxidation could occur, leading to the formation of 3,4‐dopamine‐o‐quinone, which via intramolecular cyclization is converted to aminochrome.22, 23 While the role of oxidized dopamine in neurological disorders, especially Parkinson's disease, is well ascribed, its role in renal diseases is underappreciated despite several studies showing the presence of highly active tyrosine hydroxylase, aromatic l‐amino acid decarboxylase, and other dopamine‐related transporters in renal proximal tubules.22, 23, 24, 25, 26, 27, 28 Therefore, the aim of this study is to elucidate the impact of dopamine oxidation on renal mitochondrial and lysosomal function and inflammation, especially during high sodium intake under oxidant milieu.
Methods
The authors are willing to share the experimental procedure and identify the material sources with any researcher for the purpose of conducting similar or related studies.
Materials
Chemicals, unless otherwise identified, were purchased from Millipore Sigma (St. Louis, MO). An 8‐isoprostane ELISA kit (Cat. No. 516351) and malondialdehyde colorimetric/fluorometric kit (Cat. No. 700870) were purchased from Cayman Chemicals (Ann Arbor, MI). Cathepsin B (ab65300), cathepsin D (ab65302), interleukin 1β (IL‐1β) (Cat. No. CSB‐E08055), interleukin 6 (IL‐6) (Cat. No.CSB‐E04640r), interleukin 10 (IL‐10) (Cat. No. CSB‐E04595r) rat ELISA kits were purchased from Cusabio Technology (Houston, TX). Tumor necrosis factor α (Cat. No. ab236712), monocyte chemoattractant protein 1 (Cat. No. ab100778), granulocyte macrophage colony‐stimulating factor (Cat. No. ab236709) rat ELISA kits, and 2ʹ,7ʹ‐dichlorodihydrofluorescein diacetate assay kit (Cat No. ab113851) were purchased from Abcam (Cambridge, MA).
Animal Treatment
Male Sprague Dawley rats were purchased from Harlan (Indianapolis, IN), acclimatized for a week, and divided into the following 8 experimental groups: (1) vehicle (V)—rats kept on tap/drinking water, (2) high salt (HS)—rats received 1% NaCl in drinking water, (3) l‐buthionine‐sulfoximine (BSO)—rats treated with 30 mmol/L BSO in drinking water, (4) HS+BSO, rats received both high salt and BSO, (5) resveratrol (R)—rats received 50 mg/L in drinking water, (6) HS+R—rats treated with both high salt and resveratrol, (7) BSO+R—rats received BSO plus resveratrol, and (8) HS+BSO+R—rats treated with high salt, BSO, and resveratrol. A stock solution of resveratrol was first prepared in ethanol as detailed previously.29, 30 Rats were treated for 6 weeks; food and water were measured daily while blood and urine samples were collected weekly. BP was measured by radiotelemetry (DSI; St Paul, MN) and glomerular filtration rate (GFR) was measured by creatine clearance as detailed elsewhere.30, 31, 32 Experiments were conducted in compliance with state and federal regulations and initiated after securing approval from Institutional Animal Care and Use Committee (IACUC).
Preparation of Proximal Tubules
A detailed procedure for renal proximal tubular isolation has been previously described.32 Briefly, rats were anesthetized with isoflurane using SomnoSuite Low‐flow Anesthesia System (Kent Scientific, Torrington, CT). After an abdominal incision, the aorta was cannulated with PE50 tubing below the renal arteries and kidneys were perfused with a collagenase/hyaluronidase enzyme solution. Proximal tubules were isolated by using a Ficoll gradient and washed in Krebs buffer. Protein was estimated by using a bicinchoninic acid kit (Thermo Fisher, Waltham, MA).
Oxidative and Inflammatory Biomarkers and Dopamine
Urinary 8‐isoprostane and renal proximal tubular malondialdehyde, IL‐1β, IL‐6, IL‐10, tumor necrosis factor α, monocyte chemoattractant protein 1, and granulocyte macrophage colony‐stimulating factor were measured by commercially available kits as per manufacturers’ protocol. Urinary dopamine was measured by liquid chromatography mass spectrometry as detailed previously27, 33 and dopamine oxidation was measured by near‐infrared fluorescence assay using Li‐Cor Odyssey imager (Lincoln, NE) as described by Mazzulli et al.34
Lysosomal and Mitochondrial Function
Renal proximal tubular β‐glucocerebrosidase activity assay was carried out according to peters et al.35 Briefly, tubules were homogenized and sonicated in 50 mmol/L citric acid/phosphate buffer, pH 5.5 containing 1% (wt/vol) Triton X‐100 and 1.0% (wt/vol) sodium taurocholate. Cellular debris was pelleted by centrifugation and cell extract was mixed with assay buffer containing β‐glucocerebrosidase artificial substrate 4‐methylumbelliferyl‐β‐D‐glucopyranoside (10 mmol/L), sodium taurocholate (1%, wt/vol), bovine serum albumin (0.1%, wt/vol) and incubated for 1 hour at 37°C. The reaction was stopped by adding 1 mol/L glycine‐NaOH (pH 10.5), and 4‐methylumbeliferone fluorescence (excitation/emission: 355/460 nm) was measured using a plate reader. The activities of cathepsin B and D were assayed by using commercially available kits following the manufacturer's instructions. The assay is based on the cleavage of synthetic peptide arginylarginine‐amino‐4‐trifluromethylcoumarin by cathepsin B to release fluorophore amino‐4‐trifluoromethylcoumarin (excitation/emission: 400/505 nm). Cathepsin D utilizes N‐terminal (7‐methoxycoumarin‐4‐acetic acid; excitation/emission: 328/460 nm) labeled substrate sequence GKPILFFRLK(Dnp)‐D‐R‐NH2. The reaction mixtures were incubated for 1 hour at 37°C and enzyme activities were expressed as relative fluorescence units and normalized with protein concentration. For mitochondrial respiration, renal proximal tubules were homogenized in MiR05 medium (0.5 mmol/L EGTA, 3 mmol/L MgCl2, 60 mmol/L potassium lactobionate, 20 mmol/L taurine, 10 mmol/L KH2PO4, 20 mmol/L HEPES, 110 mmol/L sucrose and 0.1% [wt/vol] fatty acid free bovine serum albumin). Respiration was measured by using Oxygraph‐2K (Oroboros Instruments, Austria) as detailed elsewhere.36, 37
Statistical Analysis
Differences between means were evaluated using 1‐way or repeated‐measures ANOVA followed by a post hoc Newman–Keuls multiple test. P<0.05 was considered statistically significant. For physiological parameters, 8 to 12 rats were used per group, whereas 6 to 8 rats per group were used for biochemical experiments. Biochemicals procedures were performed in triplicate or quadruplicate in each rat.
Results
Food and Water Intake and Oxidative Stress
The average food consumption was ≈18.9±3.1 to 21.9±2.9g/day per rat in all the experimental groups (Table). Water intake, on the other hand, was significantly higher in all the animals provided with 1% NaCl (Table). Weight gain was similar in all the experimental groups (data not shown). BSO treatment increased oxidative stress as evidenced by elevated levels of urinary 8‐isoprostane and renal tubular malondialdehyde (Table). Resveratrol per se had no effect on basal oxidative milieu but mitigated BSO‐mediated oxidative stress (Table).
Table 1.
Food and Water Intake and Oxidative Stress Markers
| V | HS | BSO | HS+BSO | R | HS+R | BSO+R | HS+BSO+R | |
|---|---|---|---|---|---|---|---|---|
| Food intake | 20.1±3.1 | 21.2±2.1 | 18.9±3.1 | 20.2±2.2 | 19.3±2.8 | 21.9±2.9 | 19.8±2.8 | 21.1±2.4 |
| Water intake | 29.3±5.2 | 44.8±6.1a | 32.3±4.2 | 43.1±5.3a | 28.9±4.3 | 45.8±4.7a | 30.6±5.1 | 44.3±6.1a |
| 8‐Isoprostane | 1.11±0.11 | 1.23±0.13 | 1.62±0.15a | 2.01±0.14a | 0.92±0.12 | 1.03±0.16 | 1.29±0.15 | 1.33±0.14 |
| Malondialdehyde | 0.23±0.03 | 0.26±0.04 | 0.37±0.04a | 0.48±0.05a | 0.20±0.04 | 0.23±0.03 | 0.25±0.04 | 0.26±0.04 |
Vehicle (V)—tap water, high salt (HS)—1% NaCl in tap water, l‐buthionine‐sulfoximine (BSO) in tap water, resveratrol (R) in tap water. Food intake—g/day per rat, water intake—mL/day per rat, urinary 8‐isoprostane—pg/mg creatine and renal malondialdehyde—nmol/mg protein.
P<0.05 vs V, using 1‐way ANOVA followed by Newman–Keuls post hoc test, n=8 to 12 rats; 8‐isoprostane and malondialdehyde assays were performed in triplicate in each rat. Food and water intake were measured once a day throughout the treatment.
Dopamine Excretion and Oxidation
A robust increase in urinary dopamine excretion was observed in the HS alone group (Figure 1A). The increase in dopamine peaked at 1‐week HS treatment followed by a slight downward trend from weeks 2 through 6 (Figure 1A). BSO per se caused a nonsignificant decline in dopamine excretion compared with vehicle (Figure 1A). However, in HS animals BSO caused a robust decrease in urinary dopamine from weeks 2 to 6 (Figure 1A). Resveratrol had no effect on basal dopamine levels but prevented BSO‐mediated decline in dopamine excretion (Figure 1B). As shown in Figure 1C, in HS rats, BSO caused a robust and linear increase in dopamine oxidation from week 2 onwards. HS and BSO per se showed a nonsignificant but persistent increase in dopamine oxidation (Figure 1C). Resveratrol prevented BSO‐induced dopamine oxidation while having no impact on basal dopamine oxidation (Figure 1D).
Figure 1.
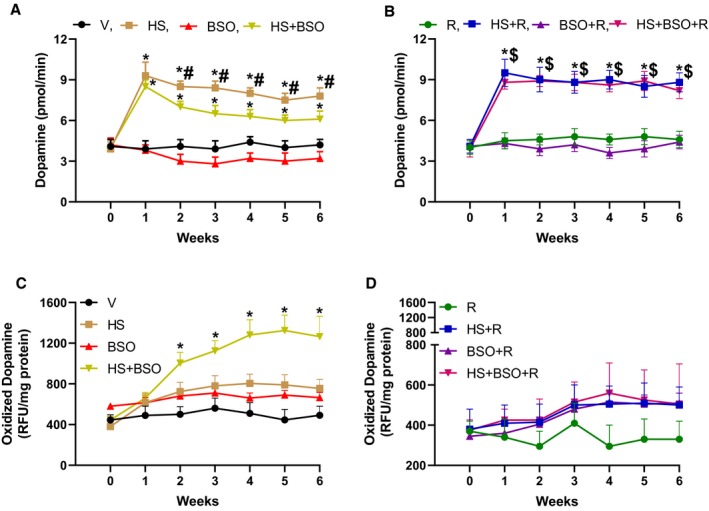
Effect of high salt (HS), l‐buthionine‐sulfoximine (BSO), and resveratrol (R) on urinary dopamine excretion and renal proximal tubular dopamine oxidation. Vehicle (V) rats were kept on tap water. A and B, Rats were treated with HS, BSO, and R for 6 wks and urinary dopamine excretion was measured every wk (n=8–12 rats). C and D, Renal dopamine oxidation was measured in freshly prepared proximal tubules (n=6–8 rats). Zero on the X‐axis indicates the baseline before the initiation of treatment. *P<0.05 vs baseline, $ P<0.05 vs baseline, and # P<0.05 vs corresponding HS+BSO time point by repeated‐measures ANOVA followed by a post hoc Newman–Keuls test. RFU indicates relative fluorescence units.
Mitochondrial Oxidation and Respiration
An increase in mitochondrial oxidation was observed in HS+BSO and BSO‐treated rats when compared with vehicle‐treated rats (Figure 2A). While the increase in mitochondrial oxidants in HS+BSO was robust, a moderate increase was observed in BSO‐treated rats (Figure 2A). The increase in mitochondrial oxidation was paralleled by a decline in mitochondrial respiration in both BSO and HS+BSO rats (Figure 2B). Resveratrol had no effect on basal mitochondrial oxidation or respiration but protected mitochondrial function in both the BSO and HS+BSO rats (Figure 2A and 2B).
Figure 2.
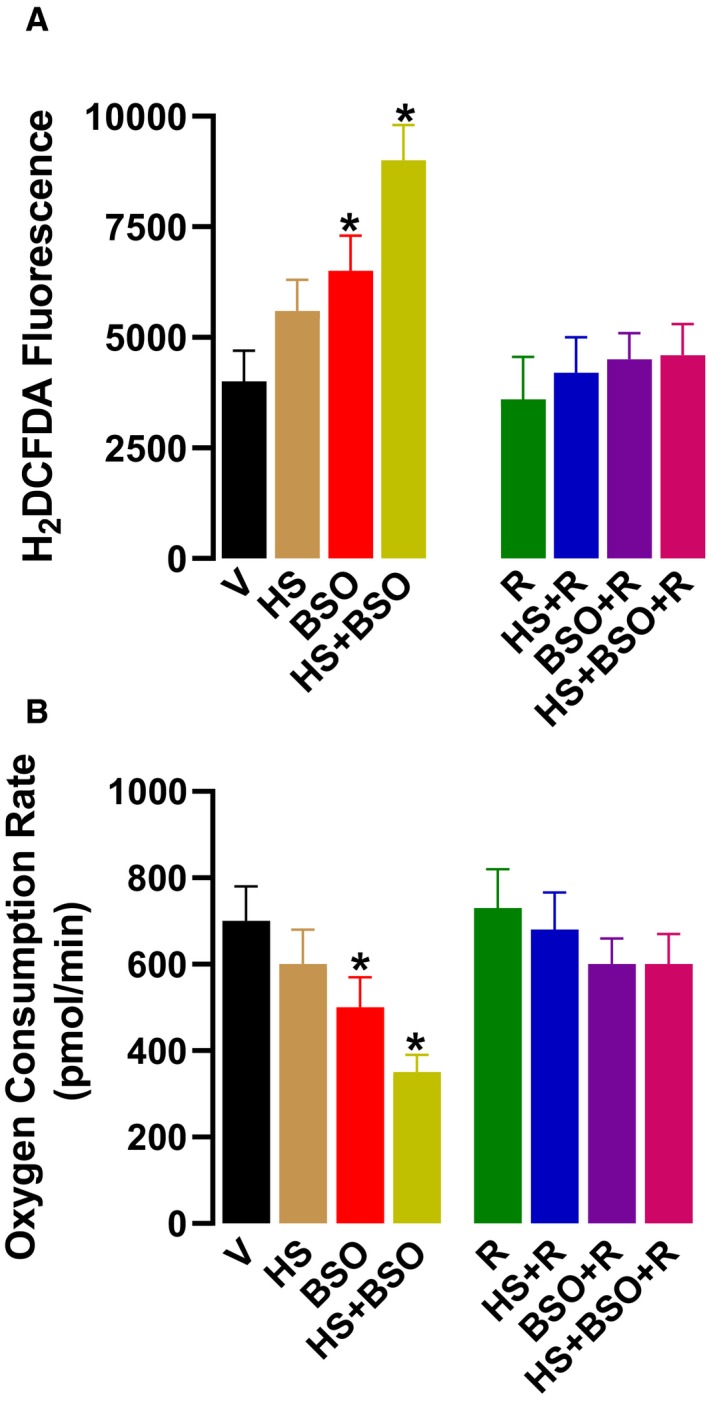
Effect of high salt (HS), l‐buthionine‐sulfoximine (BSO), and resveratrol (R) on renal mitochondria. Vehicle (V) animals were kept on tap water. A, Mitochondrial oxidative stress and respiration were measured in renal proximal tubules after 6 wks of treatment. Oxidant stress was measured by fluorescence using 2,7‐dicholorodihydrofluorescein diacetate in equal amounts of proximal tubular protein. B, Respiration was measured in tubular homogenates. Experiments were performed in triplicate (n=6–8 rats). * P<0.05 vs V by 1‐way ANOVA followed by a post hoc Newman–Keuls test.
Lysosomal Enzyme
Treatment of rats with HS+BSO showed a marked decline in the activities of lysosomal enzymes glucocerebrosidase and cathepsin B and D (Figure 3A through 3C). BSO per se also caused a moderate but significant decrease in the activities of these enzymes, whereas HS‐induced decrease in these enzymes was statistically nonsignificant (Figure 3A through 3C). Resveratrol alone had no effect on lysosomal enzymes but protected lysosomal function in BSO and HS+BSO‐treated rats (Figure 3A through 3C). The activities of cathepsin B and D decreased by ≈70% when enzyme‐specific inhibitor, provided with the kit by the manufacturer, was added to the reaction mixture (Figure 3D), indicating the specificity of the enzyme assays.
Figure 3.
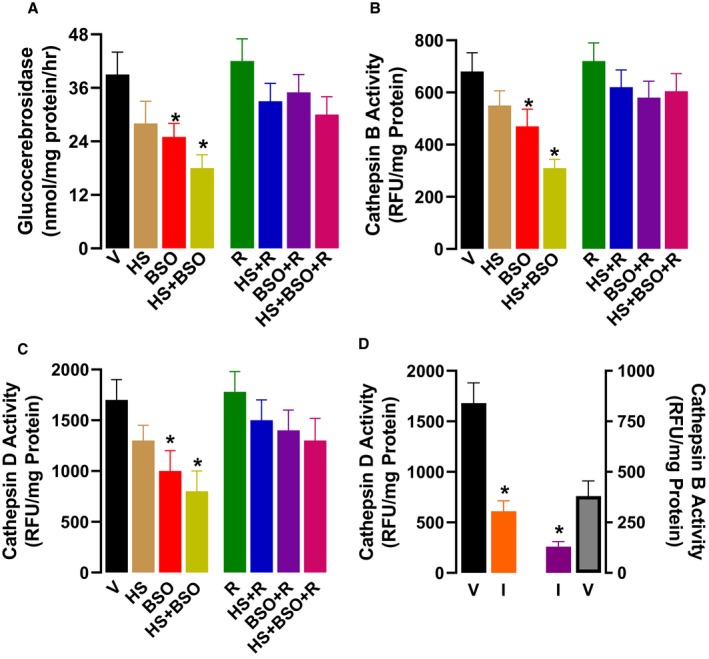
Effect of high salt (HS), l‐buthionine‐sulfoximine (BSO), and resveratrol (R) on renal lysosomal enzymes. Vehicle (V) animals were kept on tap water. A, Glucocerebrosidase and (B through D) cathepsin B and D activities were measured in renal proximal tubules at the end of 6‐wk treatment. Glucocerebrosidase activity was measured by using substrate 4‐methylumbelliferyl‐β‐D‐glucopyrooside. Cathepsin B and D were assayed with commercially available kits and reported as relative fluorescence units (RFU). The specificity of cathepsin B and D were assessed by inhibitors (I) provided with the kit. Experiments were performed in triplicate (n=6–8 rats). *P<0.05 vs V by 1‐way ANOVA followed by a post hoc Newman–Keuls test.
Renal Inflammation
We measured a diverse set of pro‐inflammatory markers in renal proximal tubules. HS+BSO‐treated rats had a profound increase in the levels of renal proximal tubular IL‐1β, IL‐6, tumor necrosis factor α, monocyte chemoattractant protein 1, and granulocyte macrophage colony‐stimulating factor (Figure 4A through 4E). BSO alone also caused a moderate increase in these markers (Figure 4A through 4E). Resveratrol had no impact on the basal levels of these pro‐inflammatory markers but protected both BSO and HS+BSO‐treated rats against inflammation (Figure 4A through 4E). As illustrated in Figure 4F, HS and BSO had no effect on renal IL‐10 levels; however, resveratrol per se and in combination with HS or BSO increased IL‐10 level.
Figure 4.
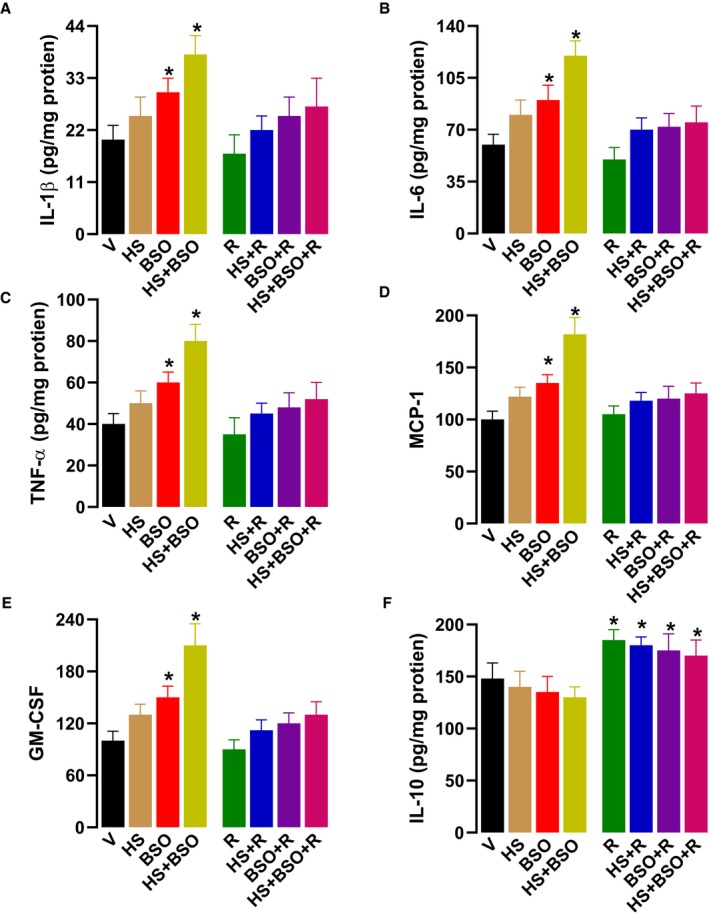
Effect of high salt (HS), l‐buthionine‐sulfoximine (BSO) and resveratrol (R) on renal inflammatory markers. Vehicle (V) rats were kept on tap water. The levels of (A) interleukin 1β (IL‐1β), (B) interleukin 6 (IL‐6), (C) tissue necrosis factor α (TNF‐α), (D) monocyte chemoattractant protein 1 (MCP‐1), (E) granulocyte macrophage colony‐stimulating factor (GM‐CSF), and (F) interleukin 10 (IL‐10) were measured in renal proximal tubules after 6‐wk treatment by commercially available kits. Experiments were performed in quadruplicate (n=6–8 rats). *P<0.05 vs V by 1‐way ANOVA followed by a post hoc Newman–Keuls test.
BP and GFR
A modest increase in BP, reported here as mean arterial pressure, was observed in BSO‐treated rats starting from week 2 onwards (Figure 5A). BP increase in HS+BSO‐treated rats also started after 2 weeks but the rise in BP was significantly higher than any other group (Figure 5A). Resveratrol treatment did not affect the basal BP but mitigated high BP in BSO and HS+BSO‐treated rats (Figure 5B). Rats kept on HS alone or in combination with BSO or resveratrol showed a consistent trend of high GFR, but it remained statistically nonsignificant (Figure 5C and 5D). BSO and resveratrol had no effect on GFR (Figure 5C and 5D).
Figure 5.
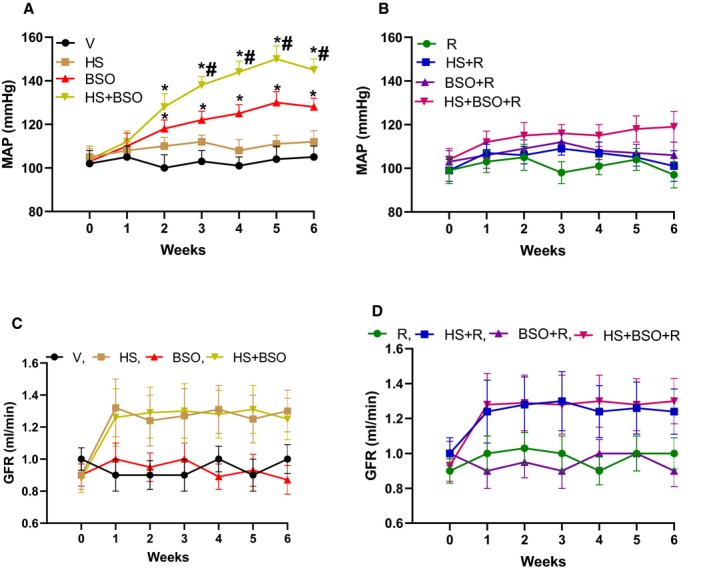
Effect of high salt (HS), l‐buthionine‐sulfoximine (BSO) and resveratrol (R) on mean arterial pressure (MAP) and glomerular filtration rate (GFR). Vehicle (V) animals were kept on tap water. MAP was measured with radiotelemetry throughout the treatment regimen (A and B). GFR was calculated as creatine clearance performed in triplicate (C and D) every week. Day 0 on X‐axis indicates baseline MAP or GFR before the initiation of treatment. (n=10–12 rats for both blood pressure and GFR). * P<0.05 vs baseline and # P<0.05 vs corresponding BSO time point by repeated‐measures ANOVA followed by a post hoc Newman–Keuls test.
Discussion
The major findings of the present study are that during high salt intake, oxidative stress causes profound renal dopamine oxidation, which leads to proximal tubular mitochondrial and lysosomal dysfunction, renal inflammation, and subsequent increase in BP. Oxidative stress alone also induces modest organelle distress and inflammation accompanied by a marginal BP increase. Under normal physiological conditions, high salt intake increases renal dopamine production, which could be responsible for increased sodium excretion and maintenance of BP.
It is well established that during high salt intake or volume expansion, natriuretic factors such as dopamine, nitric oxide, and arterial natriuretic factor play a major role in increased sodium excretion and BP regulation.10, 16, 38, 39, 40, 41, 42 In sodium replete conditions, the renal dopamine system responds by increasing local dopamine production, which via D1R inhibits major renal tubular sodium transporters such as Na/K‐ATPase pump, Na/H‐exchanger 3, Na‐phosphate cotransporter, and Na/HCO3 cotransporter.10, 11, 15, 16 The collective inhibitions of these transporters lead to a decrease in transcellular sodium uptake and a net increase in sodium and water excretion, thereby playing an important role in sodium homeostasis and maintenance of normal systemic pressure.10, 11, 15, 16 As expected, the present data also showed that dopamine production was increased in rats kept on HS alone or in combination with BSO. While rats treated with HS alone exhibited normal BP, a marked increase in BP was observed in the HS+BSO group. BSO‐treated rats had a modest increase in BP. Resveratrol treatment, while having no effect on oxidative stress or BP in the vehicle group, protected BSO and HS+BSO rats against oxidative stress and hypertension. These data reinforce a strong correlation between oxidative stress and hypertension. More importantly, the data show that oxidative stress could be a contributing factor in the cause of salt‐sensitive hypertension, especially in sodium replete condition.
Previously, we have shown that BSO‐induced oxidative stress causes renal D1R dysfunction, resulting in the failure of dopamine to inhibit proximal tubular sodium transporters and induce natriuresis.33, 43 The present study was carried out to investigate whether, in addition to D1R dysfunction, a defect in dopamine metabolism could, at least in part, play a role in the development of hypertension during oxidative stress in sodium replete conditions. It is reported that dopamine oxidation could be a major factor in the cause of Parkinson's disease.22, 44 Interestingly, recent data from our laboratory and others show that renal proximal tubules have an active enzyme system, similar to neural tissue, for de novo dopamine synthesis in addition to their ability to convert filtered l‐dihydroxyphenylalanine to dopamine.26, 27 Our data show that in rats treated with HS+BSO, there was a persistent increase in dopamine oxidation that was paralleled by a significant decrease in dopamine excretion. In rats treated with HS alone, very little increase in dopamine oxidation was found despite a robust increase in dopamine excretion. Resveratrol treatment prevented dopamine oxidation and maintained a high dopamine level in HS+BSO+R rats comparable to HS rats. These data show that a normal physiological response such as an increase in renal dopamine to prevent sodium retention during high salt intake could be detrimental under a high oxidant environment. The data also provide new insight into the disruptive interaction of renal oxidative stress with the renal natriuretic system, especially during high salt intake. Our findings are in agreement, albeit in a different organ system, with the reports showing that dopamine oxidation could be an important factor in the development of various neurological disorders.22, 44
One of the mechanisms by which oxidized dopamine could degenerate cellular function is by interfering with subcellular organelles including mitochondria, lysozymes, or endoplasmic reticulum. Herein we studied mitochondria mainly because renal tubules are rich in active transporters such as Na/K‐ATPase, which consumes ≈70% of cellular ATP.45, 46 We also studied lysosome because it plays an important role in receptor recycling including G protein‐coupled receptors and mitochondrial autophagy.47, 48, 49 Our data show that HS alone had no impact on organelle function, whereas BSO treatment caused a mild increase in mitochondrial oxidation along with a decrease in oxygen consumption and a decline in the activities of lysosomal enzymes. However, in rats treated with HS+BSO, a robust functional decline was observed in both the organelles. Resveratrol treatment protected organelle function in both BSO and HS+BSO‐treated rats. These data suggest that oxidation stress, in addition to being an independent detrimental factor to organelle function, could in complementarity with high salt intake pose a greater risk to renal tubular function, at least in part, via dopamine oxidation.
A decline in mitochondrial and lysosomal function could lead to apoptosis, necrosis, and inflammation.50, 51, 52, 53, 54, 55, 56 In this article, we focused on renal inflammation because recent studies have shown a strong correlation between hypertension and immunity disorders.57, 58, 59, 60, 61 Reflective of this notion, we found a significant increase in renal pro‐inflammatory markers in BSO and HS+BSO‐treated rats, both of which also showed increased BP. Interestingly, the intensity of inflammation was proportional to the increase in BP. Resveratrol reduced the levels of pro‐inflammatory molecules, increased the level of IL‐10, a potent anti‐inflammatory cytokine,62 and abolished hypertension. Taken together, our data lend credit to the notion that renal inflammation could be a contributing factor to a systemic BP increase. Although we did not identify a cellular mechanism that links inflammation and hypertension, it is reported that inflammation could increase sympathetic nerve activity and disrupt renal tubular sodium regulation, both of which could affect systemic pressure.60, 63, 64, 65
As mentioned earlier, organelle dysfunction and inflammation have been associated with tissue necrosis, which in the case of renal tubules could lead to acute or chronic kidney failure.53, 66, 67, 68, 69, 70, 71 Consequent to proximal tubular dopamine oxidation, organelle dysfunction, and inflammation, we expected a decline in creatine clearance. However, the treatment of rats with HS, BSO, or both had no effect on GFR. This discordance between observed versus expected data could be because a significant decline in nephron function is required to observe a clinical manifestation of renal failure.72, 73 Also, this disconnect between GFR and other renal pathological factors observed in this study could be because of the fact that creatine clearance is an insensitive marker to gauge renal pathophysiology at an early stage.74, 75, 76
Limitations
To cement a direct link between renal dopamine oxidation and pathophysiological factors observed in this study, a prudent experiment involving inhibition of renal dopamine synthesis is needed. Unfortunately, administering an inhibitor to block dopamine synthesis will have a systemic impact. In addition, an increase in renal‐specific dopamine synthesis during high salt intake is a protective measure necessitated by the need for increased sodium excretion to maintain normal systemic pressure. Therefore, blocking dopamine production in HS rats could lead to hypertension. Additionally, the beneficial effects of resveratrol could be systemic rather than renal specific. Also, a single dose of NaCl was used and it is not clear whether a different dose of NaCl would have a similar effect on BP. Further studies, which are beyond the scope of this study, are warranted to better understand the renal interaction among oxidative stress, high salt, and resveratrol as it relates to sodium and BP regulation.
In conclusion, our study provides new insight into the role of dopamine oxidation in the decline of proximal tubular mitochondrial and lysosomal function as well as in renal inflammation and subsequent increase in BP. The data also lend credit to the beneficial effects of antioxidant resveratrol as it relates to protecting renal function and mitigating hypertension, especially during high salt intake.
Sources of Funding
This work was supported by the National Institutes of Health (National Heart, Lung, and Blood Institute) grant HL‐139808.
Disclosures
None.
(J Am Heart Assoc. 2020;9:e014977 DOI: 10.1161/JAHA.119.014977.)
References
- 1. Frame AA, Wainford RD. Mechanisms of altered renal sodium handling in age‐related hypertension. Am J Physiol Renal Physiol. 2018;315:F1–F6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qian Q. Salt, water and nephron: mechanisms of action and link to hypertension and chronic kidney disease. Nephrology. 2018;23(suppl 4):44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rucker AJ, Rudemiller NP, Crowley SD. Salt, hypertension, and immunity. Annu Rev Physiol. 2018;80:283–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Titze J, Luft FC. Speculations on salt and the genesis of arterial hypertension. Kidney Int. 2017;91:1324–1335. [DOI] [PubMed] [Google Scholar]
- 5. van Haaster MC, McDonough AA, Gurley SB. Blood pressure regulation by the angiotensin type 1 receptor in the proximal tubule. Curr Opin Nephrol Hypertens. 2018;27:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Armando I, Konkalmatt P, Felder RA, Jose PA. The renal dopaminergic system: novel diagnostic and therapeutic approaches in hypertension and kidney disease. Transl Res. 2015;165:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drozak J, Bryla J. [Dopamine: not just a neurotransmitter]. Postepy Hig Med Dosw (Online). 2005;59:405–420. [Article in Polish] [PubMed] [Google Scholar]
- 8. Rukavina Mikusic NL, Kouyoumdzian NM, Del Mauro JS, Cao G, Trida V, Gironacci MM, Puyo AM, Toblli JE, Fernandez BE, Choi MR. Effects of chronic fructose overload on renal dopaminergic system: alteration of urinary L‐dopa/dopamine index correlates to hypertension and precedes kidney structural damage. J Nutr Biochem. 2018;51:47–55. [DOI] [PubMed] [Google Scholar]
- 9. Zhang MZ, Harris RC. Antihypertensive mechanisms of intra‐renal dopamine. Curr Opin Nephrol Hypertens. 2015;24:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jose PA, Eisner GM, Felder RA. Renal dopamine receptors in health and hypertension. Pharmacol Ther. 1998;80:149–182. [DOI] [PubMed] [Google Scholar]
- 11. Jose PA, Raymond JR, Bates MD, Aperia A, Felder RA, Carey RM. The renal dopamine receptors. J Am Soc Nephrol. 1992;2:1265–1278. [DOI] [PubMed] [Google Scholar]
- 12. Bek MJ, Eisner GM, Felder RA, Jose PA. Dopamine receptors in hypertension. Mt Sinai J Med. 2001;68:362–369. [PubMed] [Google Scholar]
- 13. Felder RA, Eisner GM, Jose PA. D1 dopamine receptor signalling defect in spontaneous hypertension. Acta Physiol Scand. 2000;168:245–250. [DOI] [PubMed] [Google Scholar]
- 14. Jose PA, Eisner GM, Felder RA. Dopaminergic defect in hypertension. Pediatr Nephrol. 1993;7:859–864. [DOI] [PubMed] [Google Scholar]
- 15. Jose PA, Eisner GM, Felder RA. Role of dopamine in the pathogenesis of hypertension. Clin Exp Pharmacol Physiol Suppl. 1999;26:S10–S13. [PubMed] [Google Scholar]
- 16. Jose PA, Eisner GM, Felder RA. Regulation of blood pressure by dopamine receptors. Nephron Physiol. 2003;95:p19–p27. [DOI] [PubMed] [Google Scholar]
- 17. Zeng C, Sanada H, Watanabe H, Eisner GM, Felder RA, Jose PA. Functional genomics of the dopaminergic system in hypertension. Physiol Genomics. 2004;19:233–246. [DOI] [PubMed] [Google Scholar]
- 18. Banday AA, Fazili FR, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor‐kappaB and protein kinase C. J Am Soc Nephrol. 2007;18:1446–1457. [DOI] [PubMed] [Google Scholar]
- 19. Luo H, Wang N, Chen CY, Luo XL, Wang HY, Zeng CY. [Impact of oxidative stress on renal dopamine D(1) receptor dysfunction in offspring of diabetic rat dams]. Zhonghua Xin Xue Guan Bing Za Zhi. 2019;47:393–398. [DOI] [PubMed] [Google Scholar]
- 20. Cuevas S, Villar VA, Jose PA, Armando I. Renal dopamine receptors, oxidative stress, and hypertension. Int J Mol Sci. 2013;14:17553–17572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diao Z, Asico LD, Villar VAM, Zheng X, Cuevas S, Armando I, Jose PA, Wang X. Increased renal oxidative stress in salt‐sensitive human GRK4gamma486V transgenic mice. Free Radic Biol Med. 2017;106:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Badillo‐Ramirez I, Saniger JM, Rivas‐Arancibia S. 5‐S‐cysteinyl‐dopamine, a neurotoxic endogenous metabolite of dopamine: implications for Parkinson's disease. Neurochem Int. 2019;129:104514. [DOI] [PubMed] [Google Scholar]
- 23. Segura‐Aguilar J, Paris I, Munoz P, Ferrari E, Zecca L, Zucca FA. Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem. 2014;129:898–915. [DOI] [PubMed] [Google Scholar]
- 24. Jackson‐Lewis V, Smeyne RJ. MPTP and SNpc DA neuronal vulnerability: role of dopamine, superoxide and nitric oxide in neurotoxicity. Minireview. Neurotox Res. 2005;7:193–202. [DOI] [PubMed] [Google Scholar]
- 25. Zhao J, Yu S, Zheng Y, Yang H, Zhang J. Oxidative modification and its implications for the neurodegeneration of Parkinson's disease. Mol Neurobiol. 2017;54:1404–1418. [DOI] [PubMed] [Google Scholar]
- 26. Taveira‐da‐Silva R, da Silva Sampaio L, Vieyra A, Einicker‐Lamas M. L‐Tyr‐induced phosphorylation of tyrosine hydroxylase at Ser40: an alternative route for dopamine synthesis and modulation of Na+/K+‐ATPase in kidney cells. Kidney Blood Press Res. 2019;44:1–11. [DOI] [PubMed] [Google Scholar]
- 27. Banday AA, Diaz AD, Lokhandwala M. Kidney dopamine D1‐like receptors and angiotensin 1‐7 interaction inhibits renal Na(+) transporters. Am J Physiol Renal Physiol. 2019;317:F949–F956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008;88:249–286. [DOI] [PubMed] [Google Scholar]
- 29. Bhatt SR, Lokhandwala MF, Banday AA. Resveratrol prevents endothelial nitric oxide synthase uncoupling and attenuates development of hypertension in spontaneously hypertensive rats. Eur J Pharmacol. 2011;667:258–264. [DOI] [PubMed] [Google Scholar]
- 30. Javkhedkar AA, Quiroz Y, Rodriguez‐Iturbe B, Vaziri ND, Lokhandwala MF, Banday AA. Resveratrol restored Nrf2 function, reduced renal inflammation, and mitigated hypertension in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2015;308:R840–R846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Javkhedkar AA, Lokhandwala MF, Banday AA. Defective nitric oxide production impairs angiotensin II‐induced Na‐K‐ATPase regulation in spontaneously hypertensive rats. Am J Physiol Renal Physiol. 2012;302:F47–F51. [DOI] [PubMed] [Google Scholar]
- 32. Marwaha A, Banday AA, Lokhandwala MF. Reduced renal dopamine D1 receptor function in streptozotocin‐induced diabetic rats. Am J Physiol Renal Physiol. 2004;286:F451–F457. [DOI] [PubMed] [Google Scholar]
- 33. Banday AA, Lau YS, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and salt‐sensitive hypertension in Sprague‐Dawley rats. Hypertension. 2008;51:367–375. [DOI] [PubMed] [Google Scholar]
- 34. Mazzulli JR, Burbulla LF, Krainc D, Ischiropoulos H. Detection of free and protein‐bound ortho‐quinones by near‐infrared fluorescence. Anal Chem. 2016;88:2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peters SP, Coyle P, Glew RH. Differentiation of beta‐glucocerebrosidase from beta‐glucosidase in human tissues using sodium taurocholate. Arch Biochem Biophys. 1976;175:569–582. [DOI] [PubMed] [Google Scholar]
- 36. Pokkunuri I, Ali Q, Asghar M. Grape powder improves age‐related decline in mitochondrial and kidney functions in fischer 344 rats. Oxid Med Cell Longev. 2016;2016:6135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salvi A, Patki G, Khan E, Asghar M, Salim S. Protective effect of tempol on buthionine sulfoximine‐induced mitochondrial impairment in hippocampal derived HT22 cells. Oxid Med Cell Longev. 2016;2016:5059043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao Y, Stuart D, Takahishi T, Kohan DE. Nephron‐specific disruption of nitric oxide synthase 3 causes hypertension and impaired salt excretion. J Am Heart Assoc. 2018;7:e009236 DOI: 10.1161/JAHA.118.009236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hyndman KA, Mironova EV, Giani JF, Dugas C, Collins J, McDonough AA, Stockand JD, Pollock JS. Collecting duct nitric oxide synthase 1ß activation maintains sodium homeostasis during high sodium intake through suppression of aldosterone and renal angiotensin II pathways. J Am Heart Assoc. 2017;6:e006896 DOI: 10.1161/JAHA.117.006896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ibarra ME, Albertoni Borghese MF, Majowicz MP, Ortiz MC, Loidl F, Rey‐Funes M, Di Ciano LA, Ibarra FR. Concerted regulation of renal plasma flow and glomerular filtration rate by renal dopamine and NOS I in rats on high salt intake. Physiol Rep. 2017;5:e13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Della Penna SL, Cao G, Carranza A, Zotta E, Gorzalczany S, Cerrudo CS, Rukavina Mikusic NL, Correa A, Trida V, Toblli JE, Roson MI, Fernandez BE. Renal overexpression of atrial natriuretic peptide and hypoxia inducible factor‐1alpha as adaptive response to a high salt diet. Biomed Res Int. 2014;2014:936978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanchez RA, Gilbert BH, Masnatta L, Giannone C, Pesiney C, Ramirez AJ. Pro atrial natriuretic peptide (1‐30) and 6‐keto PGF1alpha activity affects Na(+) homeostasis in non‐modulating hypertension. Curr Hypertens Rev. 2015;11:30–37. [DOI] [PubMed] [Google Scholar]
- 43. Banday AA, Lokhandwala MF. Transcriptional regulation of renal dopamine D1 receptor function during oxidative stress. Hypertension. 2015;65:1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monzani E, Nicolis S, Dell'Acqua S, Capucciati A, Bacchella C, Zucca FA, Mosharov EV, Sulzer D, Zecca L, Casella L. Dopamine, oxidative stress and protein‐quinone modifications in Parkinson's and other neurodegenerative diseases. Angew Chem Int Ed Engl. 2019;58:6512–6527. [DOI] [PubMed] [Google Scholar]
- 45. Feraille E, Doucet A. Sodium‐potassium‐adenosinetriphosphatase‐dependent sodium transport in the kidney: hormonal control. Physiol Rev. 2001;81:345–418. [DOI] [PubMed] [Google Scholar]
- 46. Clarke RJ, Catauro M, Rasmussen HH, Apell HJ. Quantitative calculation of the role of the Na(+), K(+)‐ATPase in thermogenesis. Biochim Biophys Acta. 2013;1827:1205–1212. [DOI] [PubMed] [Google Scholar]
- 47. Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gagnon AW, Kallal L, Benovic JL. Role of clathrin‐mediated endocytosis in agonist‐induced down‐regulation of the beta2‐adrenergic receptor. J Biol Chem. 1998;273:6976–6981. [DOI] [PubMed] [Google Scholar]
- 50. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 51. Tomala K, Gabryel B. Lysosomal dysfunction in neurodegenerative diseases. Postepy Hig Med Dosw (Online). 2017;71:291–306. [DOI] [PubMed] [Google Scholar]
- 52. Wang K. Autophagy and apoptosis in liver injury. Cell Cycle. 2015;14:1631–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang K, Lu Y, Xie F, Zou H, Fan X, Li B, Li W, Zhang W, Mei L, Feng SS, Yin Y, Liu Y, Zhang H, Yin C, Zhong Y, Gao J. Cationic liposomes induce cell necrosis through lysosomal dysfunction and late‐stage autophagic flux inhibition. Nanomedicine (Lond). 2016;11:3117–3137. [DOI] [PubMed] [Google Scholar]
- 54. Aflaki E, Moaven N, Borger DK, Lopez G, Westbroek W, Chae JJ, Marugan J, Patnaik S, Maniwang E, Gonzalez AN, Sidransky E. Lysosomal storage and impaired autophagy lead to inflammasome activation in gaucher macrophages. Aging Cell. 2016;15:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de la Mata M, Cotan D, Villanueva‐Paz M, de Lavera I, Alvarez‐Cordoba M, Luzon‐Hidalgo R, Suarez‐Rivero JM, Tiscornia G, Oropesa‐Avila M. Mitochondrial dysfunction in lysosomal storage disorders. Diseases. 2016;4:E31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rocha EM, De Miranda B, Sanders LH. Alpha‐synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson's disease. Neurobiol Dis. 2018;109:249–257. [DOI] [PubMed] [Google Scholar]
- 57. Agita A, Alsagaff MT. Inflammation, immunity, and hypertension. Acta Med Indones. 2017;49:158–165. [PubMed] [Google Scholar]
- 58. Dinh QN, Drummond GR, Sobey CG, Chrissobolis S. Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int. 2014;2014:406960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70:660–667. [DOI] [PubMed] [Google Scholar]
- 60. Kirabo A. A new paradigm of sodium regulation in inflammation and hypertension. Am J Physiol Regul Integr Comp Physiol. 2017;313:R706–R710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Solak Y, Afsar B, Vaziri ND, Aslan G, Yalcin CE, Covic A, Kanbay M. Hypertension as an autoimmune and inflammatory disease. Hypertens Res. 2016;39:567–573. [DOI] [PubMed] [Google Scholar]
- 62. Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32:23–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fan Y, Jiang E, Hahka T, Chen QH, Yan J, Shan Z. Orexin a increases sympathetic nerve activity through promoting expression of proinflammatory cytokines in Sprague Dawley rats. Acta Physiol (Oxf). 2018;222:e12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Khan SA, Sattar MZA, Abdullah NA, Rathore HA, Ahmad A, Abdulla MH, Johns EJ. Improvement in baroreflex control of renal sympathetic nerve activity in obese Sprague Dawley rats following immunosuppression. Acta Physiol (Oxf). 2017;221:250–265. [DOI] [PubMed] [Google Scholar]
- 65. Norlander AE, Madhur MS. Inflammatory cytokines regulate renal sodium transporters: how, where, and why? Am J Physiol Renal Physiol. 2017;313:F141–F144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy‐inflammation‐cell death axis in organismal aging. Science. 2011;333:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee MS. Role of mitochondrial function in cell death and body metabolism. Front Biosci (Landmark Ed). 2016;21:1233–1244. [DOI] [PubMed] [Google Scholar]
- 68. Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25:2689–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda). 2015;30:183–194. [DOI] [PubMed] [Google Scholar]
- 70. Kers J, Leemans JC, Linkermann A. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol. 2016;36:139–152. [DOI] [PubMed] [Google Scholar]
- 71. Martin‐Sanchez D, Poveda J, Fontecha‐Barriuso M, Ruiz‐Andres O, Sanchez‐Nino MD, Ruiz‐Ortega M, Ortiz A, Sanz AB. Targeting of regulated necrosis in kidney disease. Nefrologia. 2018;38:125–135. [DOI] [PubMed] [Google Scholar]
- 72. Zeni L, Norden AGW, Cancarini G, Unwin RJ. A more tubulocentric view of diabetic kidney disease. J Nephrol. 2017;30:701–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chadha V, Alon US. Hereditary renal tubular disorders. Semin Nephrol. 2009;29:399–411. [DOI] [PubMed] [Google Scholar]
- 74. Vaidya VS, Ozer JS, Dieterle F, Collings FB, Ramirez V, Troth S, Muniappa N, Thudium D, Gerhold D, Holder DJ, Bobadilla NA, Marrer E, Perentes E, Cordier A, Vonderscher J, Maurer G, Goering PL, Sistare FD, Bonventre JV. Kidney injury molecule‐1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Waikar SS, Betensky RA, Emerson SC, Bonventre JV. Imperfect gold standards for kidney injury biomarker evaluation. J Am Soc Nephrol. 2012;23:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kampmann J, Siersbaek‐Nielsen K, Kristensen M, Hansen JM. Rapid evaluation of creatinine clearance. Acta Med Scand. 1974;196:517–520. [DOI] [PubMed] [Google Scholar]