

Abstract
Background
Impaired neurological function affects 85% to 90% of cardiac arrest (CA) survivors. Pulsatile blood flow may play an important role in neurological recovery after CA. Cerebral blood flow (CBF) pulsatility immediately, during, and after CA and resuscitation has not been investigated. We characterized the effects of asphyxial CA on short‐term (<2 hours after CA) CBF and femoral arterial blood pressure (ABP) pulsatility and studied their relationship to cerebrovascular resistance (CVR) and short‐term neuroelectrical recovery.
Methods and Results
Male rats underwent asphyxial CA followed by cardiopulmonary resuscitation. A multimodal platform combining laser speckle imaging, ABP, and electroencephalography to monitor CBF, peripheral blood pressure, and brain electrophysiology, respectively, was used. CBF and ABP pulsatility and CVR were assessed during baseline, CA, and multiple time points after resuscitation. Neuroelectrical recovery, a surrogate for neurological outcome, was assessed using quantitative electroencephalography 90 minutes after resuscitation. We found that CBF pulsatility differs significantly from baseline at all experimental time points with sustained deficits during the 2 hours of postresuscitation monitoring, whereas ABP pulsatility was relatively unaffected. Alterations in CBF pulsatility were inversely correlated with changes in CVR, but ABP pulsatility had no association to CVR. Interestingly, despite small changes in ABP pulsatility, higher ABP pulsatility was associated with worse neuroelectrical recovery, whereas CBF pulsatility had no association.
Conclusions
Our results reveal, for the first time, that CBF pulsatility and CVR are significantly altered in the short‐term postresuscitation period after CA. Nevertheless, higher ABP pulsatility appears to be inversely associated with neuroelectrical recovery, possibly caused by impaired cerebral autoregulation and/or more severe global cerebral ischemia.
Keywords: cardiac arrest, cerebral autoregulation, cerebral blood flow, electroencephalography, laser speckle imaging, perfusion, pulsatile
Subject Categories: Imaging, Blood Pressure, Cardiopulmonary Arrest, Cardiopulmonary Resuscitation and Emergency Cardiac Care, Hemodynamics
Clinical Perspective
What Is New?
We have demonstrated that our multimodal platform, which combines laser speckle imaging, arterial blood pressure (BP), and electroencephalography, can monitor and quantify BP pulsatility and cerebral blood flow pulsatility in regard to short‐term neuroelectrical recovery after cardiac arrest and resuscitation.
Alterations in cerebral blood flow pulsatility occur during the short‐term (<2 hours) postresuscitation period, yet surprisingly may not be associated with short‐term neuroelectrical recovery.
Although BP pulsatility changes little from baseline, higher BP pulsatility after resuscitation may be associated with worse short‐term neuroelectrical recovery.
What Are the Clinical Implications?
Our data suggest that monitoring BP pulsatility (eg, pulse pressure) and related waveform analyses of BP within 2 hours of cardiac arrest and resuscitation may be important to help inform the neurological recovery of patients.
A combined effort to monitor and modify mean arterial pressure and pulse pressure could lead to more specific BP guidelines and the development of therapeutic interventions that possibly target pulsatile features of BP.
Introduction
Annually, >550 000 people in the United States experience cardiac arrest (CA), and initial survival rates remain poor (10%–30%).1 Approximately 85% to 90% of survivors have impaired cognitive function, which leads to decreased quality of life for survivors, increased burden on caregivers, and direct costs of >$6 billion/year.1, 2 To improve neurological recovery and understand how the brain recovers after CA, a critical need exists to characterize physiological processes that impact the brain, such as cerebral blood flow (CBF), arterial blood pressure (ABP), and brain electrophysiology.
A key physiological process that has been shown to be extremely important is pulsatile blood flow because of its impact on oxygen uptake, vascular tone, and cellular metabolism.3 Studies have shown that pulsatile flow (compared with nonpulsatile flow) during cardiac bypass surgery and extracorporeal membrane life support is associated with improved neurological and physiological outcomes.4, 5, 6, 7 Furthermore, the use of mechanically controlled devices to induce pulsatile blood flow is associated with reduced cerebrovascular resistance (CVR) and improved oxygen uptake during postischemic reperfusion, which can improve neurological outcome.8 These studies show that in the binary case of pulsatile versus nonpulsatile flow, significant neurological benefits occur with pulsatile flow.4, 5, 6, 7, 8 However, aging,9, 10, 11 stroke,12 acute brain injury,13 small‐vessel disease,14 and CA15, 16 all modulate pulsatile blood flow to different degrees. Results from these studies suggest conflicting associations between whether increased or decreased pulsatility results in better neurological outcome.
To better understand the relationship between pulsatility and neurological outcome after CA, several gaps in the knowledge base must be addressed. First, low‐frequency (<0.4‐Hz) pulsatile studies have typically examined survivors versus nonsurvivors of CA, with little analysis of neurological function or outcome after CA.17 Second, studies of the pulsatile waveform attributable to each heartbeat typically focus on CBF of the middle cerebral artery15 and not cerebral microvasculature or peripheral hemodynamics (eg, aorta or femoral artery). Third, the influence of CVR on CBF and its association with the peripheral pulsatile waveform has not been investigated. Finally, clinical studies begin on hospital admission, followed by measurements on subsequent days after CA, and hence do not enable study of immediate pulsatile signal changes after resuscitation (during ischemia‐induced hyperemia and hypoperfusion) and their relationship to neurological outcome.
To address these gaps, we used an established, translational preclinical model of asphyxial CA (ACA) that mimics the hospital setting.18, 19, 20 Using our novel preclinical platform, we continuously monitored and quantified pulsatile femoral ABP and, for the first time, pulsatile CBF of the cerebral microvasculature using high‐speed optical imaging during CA and resuscitation. We hypothesized that higher CBF pulsatility would be associated with improved short‐term neuroelectrical recovery. However, we show that higher ABP pulsatility is associated with worse short‐term neuroelectrical recovery, whereas CBF pulsatility is not associated with short‐term neuroelectrical recovery.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. The ACA model and study protocol (No. 2013‐3098) were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. Additional methods and results can be found in Data S1.
Animal Preparation
We used our ACA model18, 19, 20, 21, 22 to perform experiments on 11 adult male Wistar rats (Charles River). Baseline characteristics are shown in Table 1. Rats were calorically restricted 12 to 16 hours before onset of ACA. On the day of ACA induction, rats were anesthetized, intubated, and connected to a mechanical ventilator via tubing (Kent Scientific, Torrington, CT). During surgery, isoflurane was maintained at 2% with a 50/50 N2/O2 mixture at 2 LPM (liters per minute). The ventilation settings were maintained at 70 beats per minute, with 12 to 14 cm H2O peak inspiration pressure and 3 cm H2O peak end expiratory pressure. Animal temperature was continuously measured with a rectal thermometer (Kent Scientific) and maintained between 36.5°C and 37.5°C.
Table 1.
Baseline Characteristics of Study Subjects
| Characteristics | 5‐min ACA (n=6) | 7‐min ACA (n=5) | P Value |
|---|---|---|---|
| Preparation time, h | 6.28 (5.29–6.64) | 5.23 (5.13–5.27) | 0.537 |
| Weight, g | 333 (328–378) | 355 (341–374) | 0.937 |
| Temperature, °C | 37.0 (36.7–37.3) | 37.0 (37.0–37.2) | 0.944 |
| pH | 7.40 (7.40–7.40) | 7.43 (7.42–7.45) | 0.346 |
| pco 2, mm Hg | 39.7 (37.2–41.4) | 34.8 (34.0–39.2) | 0.247 |
| po 2, mm Hg | 159.5 (144.0–175.0) | 139.0 (128.0–141.0) | 0.247 |
| , mmol/L | 25.4 (24.5–25.7) | 25.4 (23.1–25.6) | 0.662 |
| Hemoglobin, g/dL | 12.6 (11.2–13.4) | 12.2 (10.9–12.9) | 0.771 |
Data are presented as median (interquartile range). The P value was calculated with a 2‐sided Wilcoxon ranked sum test. ACA indicates asphyxial cardiac arrest.
The intubated rat was positioned on a stereotactic frame (Kopf Instruments, Tujunga, CA). A midline incision was performed over the scalp, and the scalp was retracted to expose the skull for electroencephalographic (EEG) electrode implantation (Plastics One Inc, Roanoke, VA). Two frontal electrodes were implanted 2 mm anterior and 2.5 mm lateral to bregma, and a third electrode was implanted over the visual cortex 5.5 mm posterior and 4 mm left of bregma. A reference electrode was implanted 3 mm posterior to lambda. After electrode implantation, a 4×6‐mm craniectomy was created over the right sensory and visual cortices to create a window for optical imaging. Saline was applied periodically to ensure the brain remained hydrated. After the craniectomy, the femoral artery and vein were cannulated. Invasive ABP from the femoral artery was measured continuously at ≈200 Hz. Baseline arterial blood gas measurements (Abaxis, Union City, CA) were obtained within 30 minutes before initiation of ACA.
Experimental Protocol
At experiment start time, rats were placed on 0.5% to 1.0% isoflurane (balance, 100% O2). After 2 minutes, inhaled air was changed to room air (21% O2), isoflurane administration was turned off to wash out anesthesia, and neuromuscular blockade was initiated with 1 mL of IV vecuronium (2 mg/kg), flushed with 1 mL of heparinized saline. At 5 minutes, asphyxia was initiated by turning the ventilator off and clamping the tubing. Rats were subjected to a period of either 5‐ or 7‐minute asphyxia to mimic mild or moderate severity of CA, respectively. Thirty seconds before the end of the asphyxial period, mechanical ventilation was reinitiated at 100% O2 with respiratory rate of 75 to 85 beats per minute, peak inspiration pressure of 17.5 to 18.5 cm H2O, and peak end expiratory pressure of 3 cm H2O at 2.5 LPM. Epinephrine (0.01 mg/kg) and sodium bicarbonate (1 mmol/kg), followed by 2 mL of heparinized saline, were administered intravenously. Chest compressions were started at the end of the asphyxial period until return of spontaneous circulation (ROSC) was achieved. Arterial blood gases were obtained 10 minutes after ROSC and every 40 minutes thereafter, to assess and modify ventilator settings as necessary.
Data Acquisition
As previously described in detail,18 we used a multimodal approach that combined laser speckle imaging (LSI), ABP, and electroencephalography to monitor CBF, femoral ABP, and brain electrophysiology before, during, and after ACA and resuscitation. Briefly, LSI data were obtained using an 809‐nm laser (Ondax Inc, Monrovia, CA) as the excitation source, and images were acquired at ≈60 fps (frames per second) using a Point Grey camera with a 10‐ms exposure time. To measure ABP, systolic and diastolic blood pressures were recorded from the femoral artery at ≈200 Hz. The mean arterial pressure (MAP) was calculated at 1 Hz from the extracted systolic and diastolic blood pressure waveforms. EEG data acquisition was recorded from each implanted electrode at 1526 Hz with a first‐order high‐pass filter (0.35 Hz). EEG and ABP data were collected via a Tucker Davis Technologies (Alachua, FL) RZ5D platform and digitally time synchronized. The LSI data were manually synchronized to EEG and ABP data using the experiment start time (time=0 minutes).
Initial Data Processing
LSI processing used custom‐written MATLAB (The Mathworks Inc, Natick, MA) code to process each raw speckle image and obtain CBF data. A 5×5 sliding window was used to convert each raw speckle image to a corresponding speckle contrast (K) image using the equation K=σ/<I>, where σ is the SD of the intensity and <I> is the average intensity within each sliding window position. Each speckle contrast image was then converted to a CBF map using a simplified speckle imaging equation CBF=1/2TK2, where T is the exposure time of the camera in seconds.23 A representative region of interest was then selected within the craniectomy to obtain an average CBF value and create time‐resolved pulsatile CBF waveforms. To create pulsatile relative CBF (rCBF) time courses, we normalized the CBF at each time point to the baseline CBF. The baseline CBF was defined as the mean CBF calculated over a 1‐minute interval immediately before the onset of asphyxia.
Raw EEG data were filtered using custom‐written MATLAB code. Data were detrended, and common average referencing was performed using each electrode.24 A finite impulse response notch filter at 60 Hz was used, followed by a finite impulse response bandpass filter from 1 to 150 Hz. Signals were then downsampled to 600 Hz to reduce computational time.
Pulsatile Signal Analysis
To quantify CBF and ABP pulsatile waveform changes in response to ACA, 2 previously published methods were used. The first method used the maximum value of the power spectrum (peak power), and the second calculated the difference between the peak and trough values (peak to trough) of the pulsatile waveform signal25, 26, 27 (shown in Figure 1B). CBF and ABP data from each experiment were analyzed. Time periods during signal artifact caused by bleeding, movement, and noise, arterial blood gas measurements, and the absence of pulsatile data during the later periods of CA were manually removed from further analysis.
Figure 1.
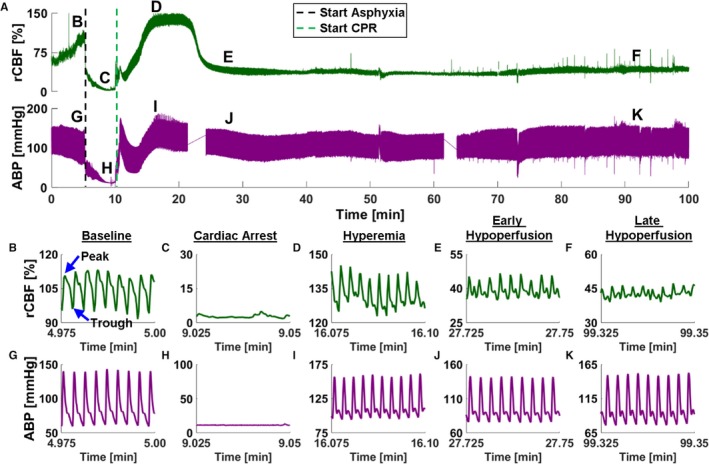
Pulsatile cerebral blood flow (CBF) and arterial blood pressure (ABP) dynamics after resuscitation from asphyxial cardiac arrest (ACA). A, Relative CBF (rCBF) (green; top) and ABP (purple; bottom) time courses during a representative ACA and resuscitation experiment. B through F, Representative 1.5‐second periods of rCBF data from phases labeled in A (top). The y axis is rescaled for each figure (B through F) to show the same CBF range. B, Baseline pulsatile CBF. C, Cardiac arrest (CA) pulsatile CBF. D, Hyperemia pulsatile CBF. E, Early hypoperfusion pulsatile CBF. F, Late hypoperfusion pulsatile CBF. G through K, Representative 1.5 seconds of ABP data from phases labeled in A (bottom). The y axis is rescaled for each figure (G through K) to show the same blood pressure range. G, Baseline pulsatile ABP. H, CA pulsatile ABP. I, Hyperemia pulsatile ABP. J, Early hypoperfusion phase of pulsatile ABP. K, Late hypoperfusion phase of pulsatile ABP. CPR indicates cardiopulmonary resuscitation.
To use the peak power from the power spectrum, an accurate measurement of heart rate is needed. Previous articles have used LSI to quantify the heart rate and pulsatile blood flow waveform flow from the skin26, 27, 28 and teeth.29 However, to the best of our knowledge, LSI has never been used to obtain and quantify pulsatile CBF data during CA and resuscitation. To ensure accurate CBF peak power quantification from the power spectrum, the CBF heart rate was compared with the ABP heart rate as the gold standard. See Results (Figure 2) for comparison.
Figure 2.
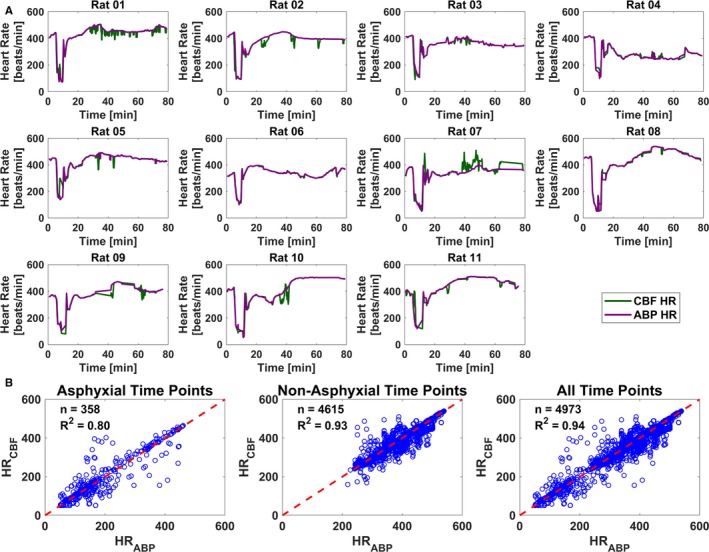
Heart rate calculations from cerebral blood flow (CBF) data agree well with those from arterial blood pressure (ABP) data during all experimental phases. A, Time courses from each of the 11 subjects that compare ABP and CBF heart rate (HR) calculations. Data from laser speckle imaging to measure CBF are in green, and data from peripheral ABP are in purple. B, Comparison of ABP and CBF HR calculations from all experiments (n=11) during asphyxial (n=358; R 2=0.80; left), nonasphyxial (n=4615; R 2=0.93; middle), and all experimental time points (n=4973; R 2=0.94; right).
To obtain the heart rate and extract the peak power from the power spectrum, the following steps were performed for both the pulsatile CBF and ABP time courses. First, a bandpass filter from 0.5 to 10 Hz was used to remove the DC component and low‐ and high‐frequency content. Next, the time‐resolved power spectrum was computed using 10‐second intervals with 2 seconds of overlap. During nonasphyxial periods, the maximum power in the 4‐ to 9‐Hz (240–540 beats/minute, typical rat heart rate values) frequency band was extracted within each interval. During the asphyxial period, the maximum power from all frequencies was extracted within each interval. Time courses of peak power were then normalized to the baseline peak power, which is defined as the mean peak power during a 1‐minute period immediately preceding the onset of asphyxia. The heart rate was computed by extracting the frequency that corresponded to the peak power. The heart rate calculated from the CBF data was compared with the heart rate calculated from the ABP data.
To calculate the peak‐to‐trough time courses from the pulsatile CBF and ABP signals, the following steps were performed. First, a bandpass filter from 0.5 to 10 Hz was used. Next, local peaks and troughs were determined over 1‐second intervals using the pulsatile signals for the entire time course. A 10‐second sliding average filter was applied to the peak and trough time courses separately. The difference between the filtered peak and trough time courses was calculated to obtain the peak‐to‐trough data. Time courses of peak to trough were then normalized to the baseline peak to trough, which is defined as the mean peak to trough during a 1‐minute period immediately preceding the onset of asphyxia to minimize effects caused by isoflurane.
Cerebrovascular Resistance
Relative CVR versus time [rCVR(t)] is defined as rCVR(t)=rMAP(t)/rCBF(t),30 where rMAP is the MAP normalized by the baseline MAP, which is defined as the mean MAP calculated over a 1‐minute interval immediately before onset of asphyxia. rCBF was calculated as follows. A 10‐second sliding median filter was applied to the pulsatile CBF time courses to remove the pulsatile component. The filtered waveform was then normalized to the mean median‐filtered CBF calculated over a 1‐minute interval immediately before onset of asphyxia. The rCBF data were then downsampled to 1 Hz to match the rMAP data, and the rCVR was calculated as the quotient of the 2 waveforms.
Electroencephalographic Information Quantity
To obtain a measurement of short‐term neuroelectrical recovery, we used a qEEG (quantitative EEG) method called information quantity (IQ).31, 32 The following steps were performed to obtain EEG IQ as a function of time. Using a temporal sliding window of 10 seconds with 20% overlap, a 5‐level discrete wavelet transform was applied to the filtered EEG data to extract discrete wavelet transform coefficients. One minute before the onset of asphyxia, 20 fixed microstates were calculated that encompassed the mean±3 SDs of the discrete wavelet transform coefficients. A histogram of the continuous EEG recording was created using the discrete wavelet transform coefficients with 20 microstates. The probability distribution function was calculated, from the histogram, and the Shannon entropy (SE) was calculated using the equation . M is the number of microstates, and pdf is the probability distribution function from the histogram. We used this equation within each 10‐second window to calculate EEG IQ as a function of time. The EEG IQ was then normalized to the baseline EEG IQ, which is defined as the mean EEG IQ calculated over a 1‐minute interval immediately before onset of asphyxia. An EEG IQ of 0 indicates no brain electrical activity, whereas an EEG IQ of 1 indicates normal brain electrical activity.
Extracted Parameters and Statistical Analysis
To assess changes in CBF and ABP pulsatility, the peak power and peak‐to‐trough data were extracted over 1‐minute intervals during baseline, CA, hyperemia (≈10 minutes after ROSC), early hypoperfusion (≈20 minutes after ROSC), and late hypoperfusion (≈60 minutes after ROSC). These periods are labeled in Figure 1 as B through F and G through K for the CBF and ABP data, respectively. Similarly, rCVR was extracted during the same time intervals. To determine the effects of asphyxial duration on peak power, peak to trough, and rCVR, a 2‐sided Wilcoxon ranked sum test was used to compare 5‐ and 7‐minute ACA at baseline, CA, hyperemia, early hypoperfusion, and late hypoperfusion. To determine the response of peak power, peak to trough, and rCVR after ACA, a 2‐sided, paired Wilcoxon signed rank test was used to compare baseline levels with subsequent experimental time periods. To determine the association between pulsatility and rCVR, a Spearman ranked correlation was used.
To obtain a metric of short‐term neuroelectrical recovery, a surrogate for neurological outcome, the median EEG IQ from 90 to 100 minutes after ROSC was determined. To assess the relationship between CBF and ABP pulsatility to short‐term neuroelectrical recovery, a Spearman ranked correlation was used.
Results
Arterial blood gas measurements taken 10, 50, and 90 minutes after ROSC are shown in Table 2. There is no significant difference between 5‐ and 7‐minute ACA for pH, pco 2, po 2, , and hemoglobin at all time points (P>0.05).
Table 2.
Arterial Blood Gas Characteristics After ROSC
| Characteristics | 5‐min ACA (n=6) | 7‐min ACA (n=5) | P Value |
|---|---|---|---|
| 10 min after ROSC | |||
| pH | 7.28 (7.25–7.29) | 7.29 (7.27–7.30) | 0.537 |
| pco 2, mm Hg | 50.4 (49.4–58.2) | 38.0 (36.7–44.7) | 0.177 |
| po 2, mm Hg | 136.0 (83.5–221.5) | 237.0 (146.0–250.0) | 0.537 |
| , mmol/L | 23.3 (22.3–25.6) | 18.4 (18.0–20.5) | 0.082 |
| Hemoglobin, g/dL | 10.5 (9.0–12.3) | 10.9 (10.5–11.6) | 0.792 |
| 50 min after ROSC | |||
| pH | 7.32 (7.26–7.36) | 7.42 (7.37–7.43) | 0.329 |
| pco 2, mm Hg | 51.6 (44.8–66.2) | 38.6 (33.3–39.3) | 0.177 |
| po 2, mm Hg | 282.5 (137.3–360.3) | 382.0 (341.0–385.0) | 0.329 |
| , mmol/L | 27.1 (25.7–31.0) | 23.0 (22.5–26.0) | 0.195 |
| Hemoglobin, g/dL | 11.8 (10.8–12.7) | 10.9 (10.9–11.2) | 0.758 |
| 90 min after ROSC | |||
| pH | 7.44 (7.39–7.48) | 7.23 (7.18–7.27) | 0.052 |
| pco 2, mm Hg | 39.0 (33.8–46.0) | 60.3 (59.8–64.6) | 0.177 |
| po 2, mm Hg | 324.0 (190.0–458.8) | 133.0 (117.0–319.0) | 0.126 |
| , mmol/L | 28.8 (24.4–29.4) | 25.1 (24.8–25.8) | 0.537 |
| Hemoglobin, g/dL | 10.9 (9.3–11.7) | 11.2 (10.2–11.9) | 0.740 |
Data are presented as median (interquartile range). The P value was calculated with a 2‐sided Wilcoxon ranked sum test. ACA indicates asphyxial cardiac arrest; ROSC, return of spontaneous circulation.
After Resuscitation From ACA, CBF and Femoral Artery Pulsatility Characteristics Vary Considerably
Figure 1A shows representative time courses of CBF (top) and ABP (bottom) after an ACA and resuscitation experiment. ABP decreases and CBF increases during the baseline period (0–5 minutes). We speculate that the administration of vecuronium causes relaxation of muscles (ie, intercostal and limb muscles) that can lead to peripheral pooling of blood that results in a decrease in ABP, whereas arousal and brain activation from washout of isoflurane in our paradigm may cause an increase in rCBF, despite isoflurane being known to increase CBF. Figure 1B through 1F shows representative 1.5‐second periods of CBF data during specific phases labeled in Figure 1A (top). The y axis of Figure 1B through 1F is rescaled to keep the same CBF range in each subfigure. Figure 1B shows baseline pulsatile CBF waveform with an example of the peak and trough labeled. Figure 1C shows CBF pulsatility during CA with low amplitude and overall low flow. Figure 1D shows CBF pulsatility during hyperemia. Compared with baseline, the CBF waveform in this phase is higher in amplitude and consists of sharper features. Figure 1E shows CBF pulsatility during early hypoperfusion. The CBF pulsatility has a lower amplitude than during both hyperemia and baseline, with overall lower flow. Figure 1F shows CBF pulsatility during late hypoperfusion. The CBF pulsatility has a lower amplitude than during both hyperemia and baseline, with overall lower flow. During baseline, hyperemia, and early hypoperfusion, respiratory variations are observed as a low‐frequency oscillation in the CBF data that is not observed during CA and is difficult to visualize during late hypoperfusion.
Figure 1G through 1K shows representative 1.5‐second periods of ABP data during the same phases labeled in Figure 1A (bottom). The y axis of Figure 1G through 1K is rescaled to keep the same blood pressure range in each subfigure. Figure 1G shows the pulsatile ABP waveform during baseline. Figure 1H shows the pulsatile ABP during CA, with a nearly featureless waveform. Figure 1I shows ABP pulsatility during hyperemia, with a return in pulsatility and a slightly higher amplitude than during baseline. Figure 1J shows ABP pulsatility during early hypoperfusion. Unlike the decreased CBF pulsatility during early hypoperfusion, the ABP pulsatility is near baseline levels. Figure 1K shows ABP pulsatility during late hypoperfusion, with similar pulsatility to early hypoperfusion. During baseline, hyperemia, early hypoperfusion, and late hypoperfusion, respiratory variations are observed as a slow frequency oscillation in the ABP data that is not observed during CA.
LSI Enables Accurate Quantification of Heart Rate Measured From the Rodent Brain
To determine if LSI is able to quantitatively resolve CBF heart rate changes, heart rate calculations measured from LSI data were compared with the ABP heart rate calculations as the gold standard. Figure 2A shows time courses of heart rate calculations measured from each experiment using CBF data (green) and ABP data (purple). The data collectively show excellent agreement across all experiments (n=11). Figure 2B quantifies the linear correlation between the heart rate calculated using all CBF and ABP data. The linear correlation was examined in 3 periods: (1) only asphyxial time points (left), (2) only nonasphyxial time points (middle), and (3) combined asphyxial and nonasphyxial time points (right). Using only asphyxial time points (n=358), the R 2=0.80; using only nonasphyxial time points (n=4615), the R 2=0.93; and using all experimental time points (n=4973), the R 2=0.94.
CBF Pulsatility Dynamically Changes From Baseline, Whereas ABP Pulsatility Remains Fairly Stable
After pooling the 5‐ and 7‐minute ACA, there was no significant difference between CBF and ABP pulsatility during hyperemia (peak to trough: P=0.15; peak power: P=0.12), but there were significant deficits in CBF pulsatility during early hypoperfusion (peak to trough: P=0.003; peak power: P<0.001) and late hypoperfusion (peak to trough: P<0.001; peak power: P<0.001) in comparison to ABP pulsatility metrics (n=11) (Figure 3).
Figure 3.

Resuscitation after asphyxial cardiac arrest (ACA) leads to deficits in cerebral blood flow (CBF) pulsatility, whereas arterial blood pressure (ABP) pulsatility minimally changes. Combined 5‐ and 7‐minute ACA comparison between CBF (green, circle) pulsatility and ABP (purple, square) pulsatility (n=11) using normalized peak‐to‐trough metric (A) and normalized peak power metric (B). Experimental phases are baseline (B), cardiac arrest (CA), hyperemia (H), early hypoperfusion (EH), and late hypoperfusion (LH). *P<0.05, **P<0.01, and ***P<0.001.
We then compared baseline CBF and ABP pulsatility metrics with their respective metrics during CA, hyperemia, early hypoperfusion, and late hypoperfusion (n=11). CBF pulsatility during CA was significantly less than baseline CBF pulsatility (peak to trough: P<0.001; peak power: P<0.001). On the other hand, CBF pulsatility during hyperemia was significantly higher than CBF pulsatility at baseline (peak to trough: P=0.02; peak power: P=0.03). However, CBF pulsatility during early hypoperfusion (peak to trough: P=0.003; peak power: P<0.001) and late hypoperfusion (peak to trough: P<0.001; peak power: P<0.001) had significant deficits in comparison to baseline CBF pulsatility.
Meanwhile, CA ABP pulsatility was significantly less than baseline ABP pulsatility (peak to trough: P<0.001; peak power: P<0.001). ABP pulsatility only showed deficits in peak power (not peak to trough) during early hypoperfusion (peak power: P=0.01) compared with baseline ABP pulsatility. During hyperemia and late hypoperfusion, ABP pulsatility was not significantly different from baseline ABP pulsatility (n=11).
We also tested if CBF pulsatility (normalized peak to trough) followed overall CBF changes, during hyperemia, early hypoperfusion, and late hypoperfusion (n=11). There was a strong significant positive correlation during hyperemia (R=0.86; P=0.001). However, there was no significant correlation during early (R=0.32; P=0.34) and late hypoperfusion (R=0.37; P=0.26) (Figure S1).
Longer Asphyxial Duration Leads to Higher ABP Pulsatility Only During the Late Hypoperfusion Phase, But Does Not Significantly Influence Deficits in CBF Pulsatility
To assess the effects of asphyxial duration on CBF and ABP pulsatility, 5‐minute (n=6) and 7‐minute (n=5) ACA values were compared during CA, hyperemia, early hypoperfusion, and late hypoperfusion. There were no significant differences between either the normalized peak power and peak‐to‐trough CBF pulsatility metrics for 5‐ and 7‐minute ACA experiments during each experiment phase (P>0.05) (Figure 4A and 4B). However, during hyperemia, there was a nonsignificant trend toward increased CBF pulsatility for 7‐minute ACA experiments. There was no significant difference between either the normalized peak power and peak‐to‐trough ABP pulsatility metrics for 5‐ and 7‐minute ACA experiments during CA, hyperemia, and early hypoperfusion (P>0.05) (Figure 4C and 4D). Similar to CBF pulsatility, during hyperemia, there was a nonsignificant trend toward increased ABP pulsatility for 7‐minute ACA experiments. However, during late hypoperfusion, 7‐minute ACA experiments had significantly more ABP pulsatility than 5‐minute ACA experiments, using both the normalized peak power (P=0.03) and normalized peak‐to‐trough (P=0.03) pulsatility metrics (Figure 4C and 4D).
Figure 4.

Longer asphyxial duration leads to higher arterial blood pressure (ABP) pulsatility, but does not significantly influence deficits in cerebral blood flow (CBF) pulsatility. A and B, Two methods to calculate CBF pulsatility (normalized peak to trough and peak power) demonstrate that there is no significant difference in CBF pulsatility between 5‐minute (n=6) and 7‐minute (n=5) asphyxial cardiac arrest (ACA) experiments during cardiac arrest (CA), hyperemia (H), early hypoperfusion (EH), and late hypoperfusion (LH). C and D, Two methods to calculate ABP pulsatility (normalized peak to trough and peak power) demonstrate that there is no significant difference between 5‐minute (n=6) and 7‐minute (n=5) ACA experiments during CA, H, and EH (P>0.05). During LH, 7‐minute ACA experiments have significantly greater pulsatility than 5‐minute experiments (P=0.03). Experimental phases are baseline (B), CA, H, EH, and LH. (*P<0.05).
CVR Is Correlated to CBF Pulsatility, But Not ABP Pulsatility
Figure 5A illustrates a representative experimental rCVR time course. Analyses for rCVR pooled 5‐ and 7‐minute ACA (n=11). Figure 5B demonstrates that rCVR increases significantly during CA (P<0.001), early hypoperfusion (P=0.002), and late hypoperfusion (P<0.001) in comparison to baseline rCVR (n=11). However, rCVR during hyperemia remains similar to baseline rCVR. Interestingly, there was a significant negative correlation between CBF pulsatility and rCVR during hyperemia (R=−0.68; P=0.025) and late hypoperfusion (R=−0.78; P=0.007). There was also a modest trend (nonsignificant) toward a negative correlation between CBF pulsatility and rCVR, during early hypoperfusion (R=−0.42; P=0.20). In contrast, there was no significant correlation between ABP pulsatility and rCVR during hyperemia (R=0.04; P=0.92), early hypoperfusion (R=0.34; P=0.31), and late hypoperfusion (R=0.16; P=0.63). Interestingly, when rCVR and overall CBF were compared, there was no significant association during hyperemia (R=−0.52; P=0.107), early hypoperfusion (R=−0.35; P=0.286), and late hypoperfusion (R=−0.58; P=0.066) (Figure S2). Nevertheless, there was a trend toward significance during late hypoperfusion.
Figure 5.
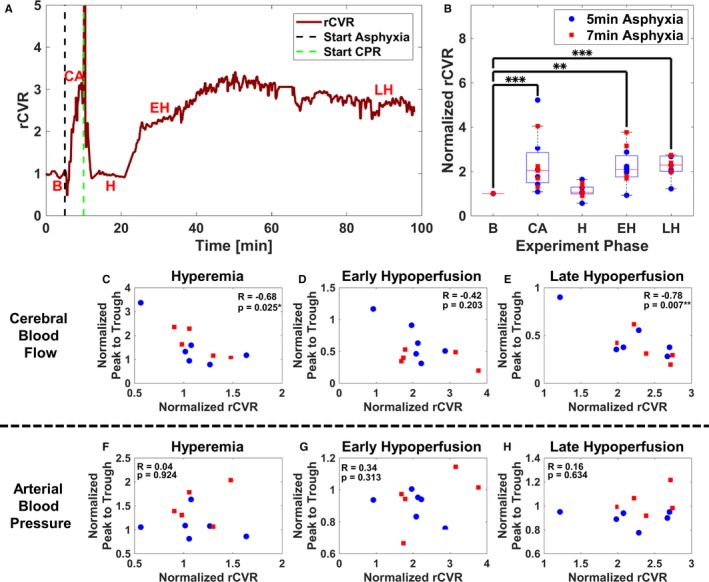
Relative cerebrovascular resistance (rCVR) increases after resuscitation and is inversely correlated to cerebral blood flow (CBF) pulsatility but not arterial blood pressure (ABP) pulsatility. A, Representative experiment illustrating temporal changes in rCVR. B, Combined 5‐ and 7‐minute asphyxial cardiac arrest (ACA) shows rCVR is significantly elevated during cardiac arrest (CA) (P<0.001), early hypoperfusion (EH) (P=0.002), and late hypoperfusion (LH) (P<0.001), whereas rCVR remains at near baseline (B) levels during hyperemia (H) (P=0.17) (n=11). Experimental phases are B, CA, H, EH, and LH. C through E, Combined 5‐ and 7‐minute ACA comparison of CBF normalized peak‐to‐trough data and rCVR during H, EH, and LH (n=11). C, Comparison of CBF pulsatility and normalized rCVR during H (R=−0.68; P=0.025). D, Comparison of CBF pulsatility and normalized rCVR during EH (R=−0.42; P=0.20). E, Comparison of CBF pulsatility and normalized rCVR during LH (R=−0.78; P=0.007). F through H, Combined 5‐ and 7‐minute ACA comparison of ABP normalized peak‐to‐trough data and normalized rCVR during H, EH, and LH (n=11). F, Comparison of ABP pulsatility and normalized rCVR during H (R=0.04; P=0.92). G, Comparison of ABP pulsatility and normalized rCVR during EH (R=0.34; P=0.31). H, Comparison of ABP pulsatility and normalized rCVR during LH (R=0.16; P=0.63). (*P<0.05, **P<0.01, ***P<0.001).
Higher ABP Pulsatility, But Not CBF Pulsatility, Is Associated With Worse Short‐Term Neuroelectrical Recovery
Figure 6A illustrates a representative experimental EEG IQ time course. We first analyzed whether overall CBF and MAP were correlated to short‐term neuroelectrical recovery (data not shown). Because of excessive bleeding that corrupted EEG signal acquisition, data from one subject were removed from further consideration. Analyses pooled 5‐ and 7‐minute ACA (n=10). There was no significant correlation between overall MAP and EEG IQ 90 minutes after ROSC (a surrogate for short‐term neurological recovery)31 during hyperemia (R=−0.53; P=0.12), early hypoperfusion (R=−0.31; P=0.39), or late hypoperfusion (R=−0.32; P=0.37). Similarly, there was no significant correlation between overall CBF and EEG IQ 90 minutes after ROSC during hyperemia (R=−0.2; P=0.58), early hypoperfusion (R=−0.21; P=0.56), or late hypoperfusion (R=−0.13; P=0.73).
Figure 6.
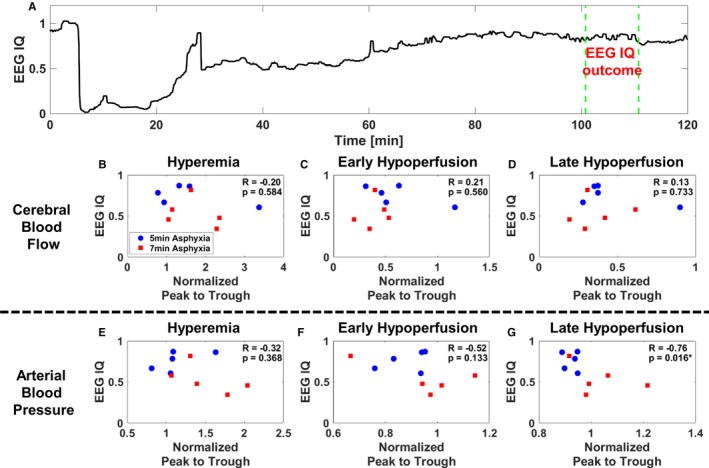
Worse short‐term neuroelectrical recovery is associated with higher arterial blood pressure (ABP) pulsatility, whereas changes in cerebral blood flow (CBF) pulsatility are not associated with short‐term neuroelectrical recovery. A, Representative experiment that shows temporal changes in electroencephalographic (EEG) information quantity (IQ). B through D, Combined 5‐ and 7‐minute asphyxial cardiac arrest (ACA) comparisons of CBF normalized peak‐to‐trough data during hyperemia (H), early hypoperfusion (EH), and late hypoperfusion (LH) to EEG IQ 90 minutes after return of spontaneous circulation (ROSC) (n=10). Experimental phases are baseline, cardiac arrest, H, EH, and LH. B, Comparison of CBF pulsatility during H and EEG IQ 90 minutes after ROSC (R=−0.20; P=0.58). C, Comparison of CBF pulsatility during EH and EEG IQ 90 minutes after ROSC (R=0.21; P=0.56). D, Comparison of CBF pulsatility during LH and EEG IQ 90 minutes after ROSC (R=0.13; P=0.73). E through G, Combined 5‐ and 7‐minute ACA comparisons of ABP normalized peak‐to‐trough data during H, EH, and LH to EEG IQ 90 minutes after ROSC (n=10). E, Comparison of ABP pulsatility during H and EEG IQ 90 minutes after ROSC (R=−0.32; P=0.37). F, Comparison of ABP pulsatility during EH and EEG IQ 90 minutes after ROSC (R=−0.52; P=0.13). G, Comparison of ABP pulsatility during LH and EEG IQ 90 minutes after ROSC (R=−0.76; P=0.016).
We then investigated the relationship between pulsatility metrics and EEG IQ 90 minutes after ROSC. As seen in Figure 6B through 6D, there was no significant trend between EEG IQ 90 minutes after ROSC and CBF peak to trough during hyperemia (R=−0.20; P=0.58), early hypoperfusion (R=0.21; P=0.56), and late hypoperfusion (R=0.13; P=0.73). There was no significant trend between EEG IQ 90 minutes after ROSC and ABP peak to trough during hyperemia (R=−0.32; P=0.37) (Figure 6E) and early hypoperfusion (R=−0.52; P=0.13) (Figure 6F). However, there was a significant negative trend comparing EEG IQ 90 minutes after ROSC and ABP peak to trough during late hypoperfusion (R=−0.76; P=0.016) (Figure 6G).
Discussion
Pulsatile blood flow is critical in oxygen uptake, vascular tone, cellular metabolism,3 and the clearance of metabolites through the glymphatics.33 However, measuring pulsatility remains challenging, especially in the brain. Transcranial Doppler approaches probe the middle cerebral artery and measure CBF pulsatility in humans, but lack information about the cerebral microvasculature, which plays a vital role in oxygen metabolism.34 Another essential mechanism for brain function, health, and homeostasis is cerebral autoregulation, which is the ability of the brain to maintain CBF when peripheral blood pressure changes. One way to assess cerebral autoregulation is measuring CVR. Simultaneously measuring CBF pulsatility, ABP pulsatility, and CVR has been challenging because of a lack of multimodal platforms. These measurements have been equally difficult in preclinical models because of a lack of high‐speed tools that are needed for the increased heart rate in animal models. However, these measurements in a controlled, preclinical setting are essential to understand the complex physiological characteristics in brain disease and trauma, which drive the need for the development and use of high‐speed tools.
One area of interest where there is a critical need to measure high‐speed changes is CA and resuscitation. We previously published on the relationship between overall CBF and ABP, and how we can use this information to predict the resumption of EEG activity after resuscitation.18 In this study, we improved on our platform to show, for the first time, simultaneous measurement of CBF pulsatility, ABP pulsatility, and CVR and relate these changes to short‐term neuroelectrical recovery in a preclinical model of CA and resuscitation. Our results reveal that the response of CBF and ABP pulsatility to ACA and resuscitation differs (Figures 3 and 4); and that CBF pulsatility demonstrates persistent deficits concomitant with increases in rCVR (Figure 5) that appear independent of overall CBF changes (Figures S1 and S2) within 2 hours after resuscitation. Interestingly, these deficits in CBF pulsatility do not appear to be associated with short‐term neuroelectrical recovery, whereas higher ABP pulsatility is associated with worse short‐term neuroelectrical recovery (Figure 6).
Poor neurological recovery remains a common occurrence for patients with acute brain injury. CA35, 36, 37 and non‐CA14, 38, 39, 40 studies have quantified CBF and ABP pulsatility attributable to each heartbeat. On the basis of the collective published results, controversy exists about whether higher or lower pulsatility is related to better neurological outcome. For example, the following results from CA studies that examined the middle cerebral artery have reported the following: (1) lower pulsatility index (PI) is associated with good outcome,37 (2) there is no relationship between PI and survivability up to 72 hours after admission to the intensive care unit,36 and (3) good neurological outcome had higher PIs in the initial 48 hours after admission to the intensive care unit.35 Results by van den Brule et al15 and Lemiale et al36 suggest that post‐ROSC changes in pulsatile CBF are caused by increased level of vasoconstrictors and cerebral arterial resistance, but that mortality depends on the presence of luxury perfusion. However, these 2 CA studies do not comment on how CBF pulsatility impacts neurological outcome. Non‐CA studies showed that patients with higher pulse pressure, which is analogous to ABP peak to trough measured in this study, have the following: (1) increased likelihood to develop restenosis after percutaneous transluminal coronary angioplasty,38 (2) higher prevalence of subcortical infarcts and worse cognitive function in the aging population,39 (3) more severe forms of leukoaraiosis,14 and (4) higher incidence of stroke.40 Mitchell et al39 and Webb et al14 reported that increased stiffening of the aorta is associated with reduced wave reflection and, therefore, transmission of excessive pulsatility into the brain, which can lead to structural brain damage of the microvasculature and impaired cognitive function.14, 39 Our data support this result, as we showed that higher ABP pulsatility is associated with worse short‐term neuroelectrical recovery (Figure 6G). A key difference is that the findings in previous studies were caused by long‐term increases in pulsatility, which may lead to cerebral microvascular damage. The results presented in our study are related to short‐term changes in ABP pulsatility that occur over the course of <2 hours after global cerebral ischemia.
One potential reason that short‐term changes in increased ABP pulsatility may lead to more cerebral microvascular damage is impaired cerebral autoregulation. After CA, cerebral autoregulation is impaired41, 42 and overall CBF diminished, but ABP recovers to baseline levels.18, 32, 43, 44 Several studies reported that higher variability in low‐frequency CBF and MAP signals 72 hours after admission to the intensive care unit was associated with increased chance of survival.17 These studies suggested that decreased low‐frequency variability and higher mortality are associated with impaired myogenic response and autonomic regulation.17 However, previous studies did not compare cerebral autoregulation measures, such as CVR, to CBF and ABP pulsatility. Our imaging approach provides the requisite capabilities to monitor these hyperdynamic changes in relation to short‐term neuroelectrical recovery. Our data suggest that CVR is elevated during the hypoperfusion phases after resuscitation (Figure 5B), and is associated with CBF pulsatility but not ABP pulsatility (Figure 5C through 5H). These results suggest that CVR may be more dependent on pulsatile cerebral hemodynamics than pulsatile peripheral hemodynamics. However, in cases of impaired cerebral autoregulation, excessive peripheral pulsatility and overall hemodynamics may lead to damage of cerebral microvasculature. We did not observe a significant difference between CVR for 5‐ and 7‐minute ACA experiments during any of the time points (P>0.05). However, during late hypoperfusion, subjects experiencing 7‐minute ACA had significantly more ABP pulsatility than those experiencing 5‐minute ACA (P=0.03). Thus, we postulate that more severe CA affects cerebral autoregulation and increases ABP pulsatility. Hence, the brain is unable to modulate the increased ABP pulsatility, which may lead to increased reperfusion‐mediated cerebral microvascular damage. On the basis of the demonstrated association between CVR and CBF pulsatility (Figure 5C through 5E), we hypothesize that a normal CVR, or less impaired cerebral autoregulation, enables the brain to minimize the potential harm that an elevated pulsatile ABP may cause the cerebral microvasculature, which in turn may lead to better neurological recovery.
However, another potential explanation of the association between higher ABP pulsatility and worse short‐term neuroelectrical recovery could be that the subjects with higher ABP pulsatility experienced more severe ACA (Figure 4). The more severe ACA and increase in ABP pulsatility may lead not only to more damage to the cerebral microvasculature, but also to the cardiac and systemic microvasculature. Furthermore, the increase in ABP pulsatility may also be a compensatory mechanism after more severe global ischemia to potentially offset the observed deficits in CBF pulsatility (Figure 3). To address these gaps, mechanistic studies are needed to determine causality between severity of ACA, CBF pulsatility, and ABP pulsatility and how these affect the brain and neurological outcome. A better understanding of how cerebral, cardiac, and systemic vasculature is affected after CA pulsatility may also help target future development and use of therapeutic agents after CA.
Hemodynamic targets for patients experiencing CA rely on targeting systolic blood pressure and MAP of >90 and 65 mm Hg, respectively.45 These targets typically do not consider ABP pulsatility. Studies have shown that exceeding a threshold of MAP is associated with good neurological recovery46, 47; however, these studies did not examine the relationship between pulse pressure and elevated MAP on neurological outcome. Our results, which show that higher ABP pulsatility is associated with worse short‐term neuroelectrical recovery parameters, suggest that monitoring acute ABP pulsatility may help inform neurological recovery after CA. We postulate that a combined effort to monitor MAP and pulse pressure could potentially lead to discoveries that can allow for minimization of cerebral microvascular damage and possibly improve good neurological outcome after CA. Epinephrine administration is the standard of clinical practice during cardiopulmonary resuscitation.48 However, recent studies demonstrated that epinephrine alone can lead to decreased survivability and worse neurological outcome than when epinephrine is combined with vasopressin or sodium nitroprusside.49, 50, 51, 52, 53, 54 It is not clear how these drugs affect pulsatility of the peripheral arteries versus cerebral arteries. Future studies that determine how these drugs affect pulsatility, and therapeutic modulation of pulsatility during the short‐term period after CA (within 2 hours), may be critical to achieving an overall improvement in neurological outcome.
Limitations
Although the findings of this study warrant further investigations, there are several limitations. First, global regions of interest within the craniectomy were chosen to calculate CBF data, and therefore we did not distinguish between arteriolar and venular pulsatility, which has recently been shown with LSI.25 In separate analyses of our data, regions of interest that were taken exclusively from arterioles were analyzed. Our data resulted in poor signal/noise ratio of the pulsatile waveform, which did not enable the use of the quantifiable metrics presented in this study. Furthermore, another limitation of using LSI to quantify pulsatility is that it cannot calculate PI, a well‐accepted measure of pulsatility. Absolute velocity is needed to calculate PI; however, the LSI signal is more of a perfusion measurement. Also, although the data analyses presented herein produce statistical significance and trends, the power of these findings is limited because of the small sample size and complexity of experiments. Specifically, there appears to be a trend toward decreased CBF pulsatility and longer duration of ACA, but we did not see statistically significant differences (Figure 4A and 4B). Furthermore, these experiments used a craniectomy, which may affect pulsatile CBF changes because of differences in intracranial pressure and brain pulsations. A chronic cranial window may mitigate such effects. To address this limitation, we performed 2 ACA experiments using a chronic cranial window procedure55 that showed similar changes in CBF pulsatility compared with the open craniectomy procedure (Figure S3). Moreover, we used isoflurane to induce anesthesia, which may impact CBF and ABP pulsatility. Isoflurane was turned off 3 minutes before onset of asphyxia to minimize isoflurane effects on pulsatility. In addition, the correlations in this study combined the 5‐ and 7‐minute ACA groups because of the small sample size. Moreover, because we used young, healthy animals, these data should be reinvestigated in aged animals with heart disease and decreased cardiac function to better mimic the human population at highest risk of CA. It may be possible that ABP pulsatility and compensatory cardiac function after CA are highly different in the diseased state. Thus, future work should use larger sample sizes in varying severities of ACA using young and aged animals and use linear regression models. Our metric of neurological outcome (EEG IQ), although it is an objective end point that has been previously validated,31 is not based on cognitive,56 behavioral,21 or histopathological outcomes.37, 57 Finally, the results in this study only measured acute pulsatility changes. Use of a wearable imaging device27, 55, 58 over a chronic cranial window will enable longer‐term monitoring of CBF pulsatility.
Conclusions
We used our translational preclinical model of ACA and a multimodal measurement platform to demonstrate that CBF pulsatility dynamically changes after resuscitation, whereas ABP pulsatility changes little. rCVR appears to be associated to changes in CBF pulsatility, but not ABP pulsatility. We found that our observed changes in CBF pulsatility are not correlated with short‐term neuroelectrical recovery, whereas higher ABP pulsatility is associated with worse short‐term neuroelectrical recovery. We postulate that impaired cerebral autoregulation after CA may contribute to higher ABP pulsatility, leading to worse short‐term neuroelectrical recovery. However, this may also be attributable to higher ABP pulsatility being associated with more severe ACA. Clinical translation of these findings has the potential to inform us on the development of novel interventions that modulate cerebral autoregulation and ABP pulsatility within the first 2 hours after ROSC, which could potentially improve neurological outcome for CA survivors.
Sources of Funding
This work was supported by the Arnold and Mabel Beckman Foundation, the Roneet Carmell Memorial Endowment Fund (Dr Akbari), the US National Institutes of Health (NIH) (P41EB015890), the National Science Foundation Graduate Research Fellowship Program (DGE‐1321846) (Dr Crouzet), and the National Center for Research Resources and National Center for Advancing Translational Sciences, NIH, through the following grants: TL1TR001415‐01 (Dr Wilson), R21 EB024793 (Dr Akbari), and 5KL2TR001416 (Dr Akbari) via UL1 TR001414. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Disclosures
None.
Supporting information
Data S1. Relationship among cerebral blood flow pulsatility, cerebrovascular resistance, and overall cerebral blood flow, during various stages of cardiac arrest and resuscitation.
Figure S1. Cerebral blood flow (CBF) pulsatility and overall CBF are associated during hyperemia, but not early and late hypoperfusion.
Figure S2. Overall cerebral blood flow (CBF) and relative cerebrovascular resistance (rCVR) are not significantly associated post‐cardiac arrest (CA).
Figure S3. Chronic cranial window cerebral blood flow (CBF) pulsatility agrees with open craniectomy CBF pulsatility.
Acknowledgments
We would like to thank the Beckman Laser Institute Microvascular Therapeutics and Imaging laboratory and The Akbari Lab for their continual support and input in this work.
(J Am Heart Assoc. 2020;9:e012691 DOI: 10.1161/JAHA.119.012691.)
Contributor Information
Yama Akbari, Email: yakbari@uci.edu.
Bernard Choi, Email: choib@uci.edu.
References
- 1. Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 2. Lick CJ, Aufderheide TP, Niskanen RA, Steinkamp JE, Davis SP, Nygaard SD, Bemenderfer KK, Gonzales L, Kalla JA, Wald SK, Gillquist DL, Sayre MR, Oski Holm SY, Oakes DA, Provo TA, Racht EM, Olsen JD, Yannopoulos D, Lurie KG. Take Heart America: a comprehensive, community‐wide, systems‐based approach to the treatment of cardiac arrest. Crit Care Med. 2011;39:26–33. [DOI] [PubMed] [Google Scholar]
- 3. Wilkens H, Regelson W, Hoffmeister FS. The physiologic importance of pulsatile blood flow. N Engl J Med. 1962;267:443–446. [DOI] [PubMed] [Google Scholar]
- 4. Anstadt MP, Stonnington MJ, Tedder M, Crain BJ, Brothers MF, Hilleren DJ, Rahija RJ, Menius JA, Lowe JE. Pulsatile reperfusion after cardiac arrest improves neurologic outcome. Ann Surg. 1991;214:478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Neil MP, Fleming JC, Badhwar A, Guo LR. Pulsatile versus nonpulsatile flow during cardiopulmonary bypass: microcirculatory and systemic effects. Ann Thorac Surg. 2012;94:2046–2053. [DOI] [PubMed] [Google Scholar]
- 6. Salameh A, Kühne L, Grassl M, Gerdom M, von Salisch S, Vollroth M, Bakhtiary F, Mohr F‐W, Dähnert I, Dhein S. Protective effects of pulsatile flow during cardiopulmonary bypass. Ann Thorac Surg. 2015;99:192–199. [DOI] [PubMed] [Google Scholar]
- 7. Aykut K, Albayrak G, Guzeloglu M, Hazan E, Tufekci M, Erdoǧan I. Pulsatile versus non‐pulsatile flow to reduce cognitive decline after coronary artery bypass surgery: a randomized prospective clinical trial. J Cardiovasc Dis Res. 2013;4:127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allen BS, Ko Y, Buckberg GD, Tan Z. Studies of isolated global brain ischaemia, III: influence of pulsatile flow during cerebral perfusion and its link to consistent full neurological recovery with controlled reperfusion following 30 min of global brain ischaemia. Eur J Cardiothorac Surg. 2012;41:1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsao CW, Seshadri S, Beiser AS, Westwood AJ, DeCarli C, Au R, Himali JJ, Hamburg NM, Vita JA, Levy D, Larson MG, Benjamin EJ, Wolf PA, Vasan RS, Mitchell GF. Relations of arterial stiffness and endothelial function to brain aging in the community. Neurology. 2013;81:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pase MP, Himali JJ, Mitchell GF, Beiser A, Maillard P, Tsao C, Larson MG, Decarli C, Vasan RS, Seshadri S. Association of aortic stiffness with cognition and brain aging in young and middle‐aged adults the Framingham Third Generation Cohort Study. Hypertension. 2016;67:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Montgolfier O, Pinçon A, Pouliot P, Gillis M‐A, Bishop J, Sled JG, Villeneuve L, Ferland G, Lévy BI, Lesage F, Thorin‐Trescases N, Thorin É. High systolic blood pressure induces cerebral microvascular endothelial dysfunction, neurovascular unit damage, and cognitive decline in mice. Hypertension. 2019;73:217–228. [DOI] [PubMed] [Google Scholar]
- 12. Gasecki D, Rojek A, Kwarciany M, Kubach M, Boutouyrie P, Nyka W, Laurent S, Narkiewicz K. Aortic stiffness predicts functional outcome in patients. Stroke. 2012;43:543–544. [DOI] [PubMed] [Google Scholar]
- 13. Papaioannou V, Giannakou M, Maglaveras N, Sofianos E, Giala M. Investigation of heart rate and blood pressure variability, baroreflex sensitivity, and approximate entropy in acute brain injury patients. J Crit Care. 2008;23:380–386. [DOI] [PubMed] [Google Scholar]
- 14. Webb AJS, Simoni M, Mazzucco S, Kuker W, Schulz U, Rothwell PM. Increased cerebral arterial pulsatility in patients with leukoaraiosis: arterial stiffness enhances transmission of aortic pulsatility. Stroke. 2012;43:2631–2636. [DOI] [PubMed] [Google Scholar]
- 15. van den Brule JMD, Vinke E, van Loon LM, van der Hoeven JG, Hoedemaekers CWE. Middle cerebral artery flow, the critical closing pressure, and the optimal mean arterial pressure in comatose cardiac arrest survivors‐an observational study. Resuscitation. 2017;110:85–89. [DOI] [PubMed] [Google Scholar]
- 16. Lin J‐J, Hsia S‐H, Wang H‐S, Chiang M‐C, Lin K‐L. Transcranial Doppler ultrasound in therapeutic hypothermia for children after resuscitation. Resuscitation. 2015;89:182–187. [DOI] [PubMed] [Google Scholar]
- 17. van den Brule JMD, Vinke EJ, van Loon LM, van der Hoeven JG, Hoedemaekers CWE. Low spontaneous variability in cerebral blood flow velocity in non‐survivors after cardiac arrest. Resuscitation. 2017;111:110–115. [DOI] [PubMed] [Google Scholar]
- 18. Crouzet C, Wilson RH, Bazrafkan A, Farahabadi MH, Lee D, Alcocer J, Tromberg BJ, Choi B, Akbari Y. Cerebral blood flow is decoupled from blood pressure and linked to EEG bursting after resuscitation from cardiac arrest. Biomed Opt Express. 2016;7:4660–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson RH, Crouzet C, Torabzadeh M, Bazrafkan A, Farahabadi MH, Jamasian B, Donga D, Alcocer J, Zaher SM, Choi B, Akbari Y, Tromberg BJ. High‐speed spatial frequency domain imaging of rat cortex detects dynamic optical and physiological properties following cardiac arrest and resuscitation. Neurophotonics. 2017;4:045008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee DE, Lee LG, Siu D, Bazrafkan AK, Farahabadi MH, Dinh TJ, Orellana J, Xiong W, Lopour BA, Akbari Y. Neural correlates of consciousness at near‐electrocerebral silence in an asphyxial cardiac arrest model. Brain Connect. 2017;7:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang Y‐J, Tian G, Bazrafkan A, Farahabadi MH, Azadian M, Abbasi H, Shamaoun B, Steward O, Akbari Y. Recovery from coma post‐cardiac arrest is dependent on the orexin pathway. J Neurotrauma. 2017;34:2823–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin HZ, Wang HL, Ji SG, Medvedeva YV, Tian G, Bazrafkan AK, Maki NZ, Akbari Y, Weiss JH. Rapid intramitochondrial Zn2+ accumulation in CA1 hippocampal pyramidal neurons after transient global ischemia: a possible contributor to mitochondrial disruption and cell death. J Neuropathol Exp Neurol. 2019;78:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramirez‐San‐Juan JC, Ramos‐García R, Guizar‐Iturbide I, Martínez‐Niconoff G, Choi B. Impact of velocity distribution assumption on simplified laser speckle imaging equation. Opt Express. 2008;16:3197–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ludwig KA, Miriani RM, Langhals NB, Joseph MD, Anderson DJ, Kipke DR. Using a common average reference to improve cortical neuron recordings from microelectrode arrays. J Neurophysiol. 2009;101:1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Postnov DD, Erdener SE, Kilic K, Boas DA. Cardiac pulsatility mapping and vessel type identification using laser speckle contrast imaging. Biomed Opt Express. 2018;9:6388–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dunn CE, Lertsakdadet B, Crouzet C, Bahani A, Choi B. Comparison of speckleplethysmographic (SPG) and photoplethysmographic (PPG) imaging by Monte Carlo simulations and in vivo measurements. Biomed Opt Express. 2018;9:4306–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghijsen M, Rice TB, Yang B, White SM, Tromberg BJ. Wearable speckle plethysmography (SPG) for characterizing microvascular flow and resistance. Biomed Opt Express. 2018;9:3937–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dunn CE, Monroe DC, Crouzet C, Hicks JW, Choi B. Speckleplethysmographic (SPG) estimation of heart rate variability during an orthostatic challenge. Sci Rep. 2019;9:14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Regan C, Yang BY, Mayzel KC, Ramirez‐San‐Juan JC, Wilder‐Smith P, Choi B. Fiber‐based laser speckle imaging for the detection of pulsatile flow. Lasers Surg Med. 2015;47:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parthasarathy AB, Gannon KP, Baker WB, Favilla CG, Balu R, Kasner SE, Yodh AG, Detre JA, Mullen MT. Dynamic autoregulation of cerebral blood flow measured non‐invasively with fast diffuse correlation spectroscopy. J Cereb Blood Flow Metab. 2018;38:230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen B, Chen G, Dai C, Wang P, Zhang L, Huang Y, Li Y. Comparison of quantitative characteristics of early post‐resuscitation EEG between asphyxial and ventricular fibrillation cardiac arrest in rats. Neurocrit Care. 2018;28:247–256. [DOI] [PubMed] [Google Scholar]
- 32. Wang Q, Miao P, Modi HR, Garikapati S, Koehler RC, Thakor NV. Therapeutic hypothermia promotes cerebral blood flow recovery and brain homeostasis after resuscitation from cardiac arrest in a rat model. J Cereb Blood Flow Metab. 2019;39:1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiviniemi V, Wang X, Korhonen V, Keinanen T, Tuovinen T, Autio J, LeVan P, Keilholz S, Zang Y‐F, Hennig J, Nedergaard M. Ultra‐fast magnetic resonance encephalography of physiological brain activity—glymphatic pulsation mechanisms? J Cereb Blood Flow Metab. 2016;36:1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park CS, Payne SJ. Modelling the effects of cerebral microvasculature morphology on oxygen transport. Med Eng Phys. 2016;38:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doepp (Connolly) F, Reitemeier J, Storm C, Hasper D, Schreiber SJ. Duplex sonography of cerebral blood flow after cardiac arrest—a prospective observational study. Resuscitation. 2014;85:516–521. [DOI] [PubMed] [Google Scholar]
- 36. Lemiale V, Huet O, Vigué B, Mathonnet A, Spaulding C, Mira JP, Carli P, Duranteau J, Cariou A. Changes in cerebral blood flow and oxygen extraction during post‐resuscitation syndrome. Resuscitation. 2008;76:17–24. [DOI] [PubMed] [Google Scholar]
- 37. Wennervirta JE, Ermes MJ, Tiainen SM, Salmi TK, Hynninen MS, Särkelä MOK, Hynynen MJ, Stenman UH, Viertiö‐Oja HE, Saastamoinen KP, Pettilä VY, Vakkuri AP. Hypothermia‐treated cardiac arrest patients with good neurological outcome differ early in quantitative variables of EEG suppression and epileptiform activity. Crit Care Med. 2009;37:2427–2435. [DOI] [PubMed] [Google Scholar]
- 38. Ueda H, Nakayama Y, Tsumura K, Yoshimaru K, Hayashi T, Yoshikawa J. Inflection point of ascending aortic waveform is a powerful predictor of restenosis after percutaneous transluminal coronary angioplasty. Am J Hypertens. 2002;15:823–826. [DOI] [PubMed] [Google Scholar]
- 39. Mitchell GF, Van Buchem MA, Sigurdsson S, Gotal JD, Jonsdottir MK, Kjartansson Ó, Garcia M, Aspelund T, Harris TB, Gudnason V, Launer LJ. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility‐Reykjavik Study. Brain. 2011;134:3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chuang SY, Cheng HM, Bai CH, Yeh WT, Chen JR, Pan WH. Blood pressure, carotid flow pulsatility, and the risk of stroke: a community‐based study. Stroke. 2016;47:2262–2268. [DOI] [PubMed] [Google Scholar]
- 41. Sundgreen C, Larsen FS, Herzog TM, Knudsen GM, Boesgaard S, Aldershvile J. Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke. 2001;32:128–132. [DOI] [PubMed] [Google Scholar]
- 42. Lee JK, Brady KM, Mytar JO, Kibler KK, Carter EL, Hirsch KG, Hogue CW, Easley RB, Jordan LC, Smielewski P, Czosnyka M, Shaffner DH, Koehler RC. Cerebral blood flow and cerebrovascular autoregulation in a swine model of pediatric cardiac arrest and hypothermia. Crit Care Med. 2011;39:2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buunk G, van der Hoeven JG, Meinders AE. Cerebral blood flow after cardiac arrest. Neth J Med. 2000;57:106–112. [DOI] [PubMed] [Google Scholar]
- 44. Iordanova B, Li L, Clark RSB, Manole MD. Alterations in cerebral blood flow after resuscitation from cardiac arrest. Front Pediatr. 2017;5:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Callaway CW, Donnino MW, Fink EL, Geocadin RG, Golan E, Kern KB, Leary M, Meurer WJ, Peberdy MA, Thompson TM, Zimmerman JL. Part 8: post‐cardiac arrest care: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2015;132:S465–S482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Topjian AA, Berg RA, Taccone FS. Haemodynamic and ventilator management in patients following cardiac arrest. Curr Opin Crit Care. 2015;21:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roberts BW, Kilgannon JH, Hunter BR, Puskarich MA, Shea L, Donnino M, Jones C, Fuller BM, Kline JA, Jones AE, Shapiro NI, Abella BS, Trzeciak S. Association between elevated mean arterial blood pressure and neurologic outcome after resuscitation from cardiac arrest. Crit Care Med. 2019;47:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Attaran RR, Ewy GA. Epinephrine in resuscitation: curse or cure? Future Cardiol. 2010;6:473–482. [DOI] [PubMed] [Google Scholar]
- 49. Yannopoulos D, Bartos JA, George SA, Sideris G, Voicu S, Oestreich B, Matsuura T, Shekar K, Rees J, Aufderheide TP. Sodium nitroprusside enhanced cardiopulmonary resuscitation improves short term survival in a porcine model of ischemic refractory ventricular fibrillation. Resuscitation. 2017;110:6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schultz JC, Segal N, Caldwell E, Kolbeck J, Mcknite S, Lebedoff N, Zviman M, Aufderheide TP, Yannopoulos D. Sodium nitroprusside‐enhanced cardiopulmonary resuscitation improves resuscitation rates after prolonged untreated cardiac arrest in two porcine models*. Crit Care Med. 2011;39:2705–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yannopoulos D, Segal N, Matsuura T, Sarraf M, Thorsgard M, Caldwell E, Rees J, Mcknite S, Santacruz K, Lurie KG. Ischemic post‐conditioning and vasodilator therapy during standard cardiopulmonary resuscitation to reduce cardiac and brain injury after prolonged untreated ventricular fibrillation. Resuscitation. 2013;84:1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Q, Liu B, Zhao L, Qi Z, Shao H, An L, Li C. Efficacy of vasopressin‐epinephrine compared to epinephrine alone for out of hospital cardiac arrest patients: a systematic review and meta‐analysis. Am J Emerg Med. 2017;35:1555–1560. [DOI] [PubMed] [Google Scholar]
- 53. Mentzelopoulos SD, Malachias S, Chamos C, Konstantopoulos D, Ntaidou T, Papastylianou A, Kolliantzaki I, Theodoridi M, Ischaki H, Makris D, Zakynthinos E, Zintzaras E, Sourlas S, Aloizos S, Zakynthinos SG. Vasopressin, steroids, and epinephrine and neurologically favorable survival after in‐hospital cardiac arrest a randomized clinical trial. J Am Med Assoc. 2013;310:270–279. [DOI] [PubMed] [Google Scholar]
- 54. Yannopoulos D, Matsuura T, Schultz J, Rudser K, Halperin HR, Lurie KG. Sodium nitroprusside enhanced cardiopulmonary resuscitation improves survival with good neurological function in a porcine model of prolonged cardiac arrest. Crit Care Med. 2011;39:1269–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sigal I, Koletar MM, Ringuette D, Gad R, Jeffrey M, Carlen PL, Stefanovic B, Levi O. Imaging brain activity during seizures in freely behaving rats using a miniature multi‐modal imaging system. Biomed Opt Express. 2016;7:3596–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cohan CH, Neumann JT, Dave KR, Alekseyenko A, Binkert M, Stransky K, Lin HW, Barnes CA, Wright CB, Perez‐Pinzon MA. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle‐aged rats. PLoS One. 2015;10:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Julio‐Amilpas A, Montiel T, Soto‐Tinoco E, Gerónimo‐Olvera C, Massieu L. Protection of hypoglycemia‐induced neuronal death by β‐hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J Cereb Blood Flow Metab. 2015;35:851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Senarathna J, Yu H, Deng C, Zou AL, Issa JB, Hadjiabadi DH, Gil S, Wang Q, Tyler BM, Thakor NV, Pathak AP. A miniature multi‐contrast microscope for functional imaging in freely behaving animals. Nat Commun. 2019;10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Relationship among cerebral blood flow pulsatility, cerebrovascular resistance, and overall cerebral blood flow, during various stages of cardiac arrest and resuscitation.
Figure S1. Cerebral blood flow (CBF) pulsatility and overall CBF are associated during hyperemia, but not early and late hypoperfusion.
Figure S2. Overall cerebral blood flow (CBF) and relative cerebrovascular resistance (rCVR) are not significantly associated post‐cardiac arrest (CA).
Figure S3. Chronic cranial window cerebral blood flow (CBF) pulsatility agrees with open craniectomy CBF pulsatility.