

Abstract
Background
The regulation of sodium excretion is important in the pathogenesis of hypertension and salt sensitivity is predictive of cardiovascular events and mortality. C57Bl/6 and BALB/c mice have different blood pressure sensitivities to salt intake. High salt intake increases blood pressure in some C57Bl/6J mouse strains but not in any BALB/c mouse strain.
Methods and Results
We determined the cause of the difference in salt sensitivity between C57Bl/6 and BALB/c mice. Basal levels of superoxide and H2O2 were higher in renal proximal tubule cells (RPTCs) from BALB/c than C57Bl/6J mice. High salt diet increased H2O2 production in kidneys from BALB/c but C57Bl/6J mice. High sodium concentration (170 mmol/L) in the incubation medium increased H2O2 levels in BALB/c‐RPTCs but not in C57Bl/6J‐RPTCs. H2O2 (10 μmol/L) treatment decreased sodium transport in RPTCs from BALB/c but not C57Bl/6J mice. Overexpression of catalase in the mouse kidney predisposed BALB/c mice to salt‐sensitive hypertension.
Conclusions
Our data show that the level of salt‐induced H2O2 production negatively regulates RPTC sodium transport and determines the state of salt sensitivity in 2 strains of mice. High concentrations of antioxidants could prevent H2O2 production in renal proximal tubules, which would result in sodium retention and increased blood pressure.
Keywords: catalase, hydrogen peroxide, hypertension, oxidative stress, salt‐sensitivity
Subject Categories: High Blood Pressure, Basic Science Research, Mechanisms, Oxidant Stress, Gene Expression & Regulation
Clinical Perspective
What Is New
To our knowledge, this is the first report on the strain‐dependence of the beneficial role of renal H2O2 production on blood pressure regulation when sodium intake is high.
What Are the Clinical Implications?
This study shows that antioxidants may have deleterious effects on blood pressure regulation when salt intake is excessive, in the pertinent genetic background.
These results, if confirmed in humans, could explain why some antioxidants have no effect or deleterious effects on blood pressure levels and could have a significant effect on the rationale for the use of vitamins and others antioxidants that are consumed in excessive amounts.
Introduction
About one third of the US population, aged 40 to 59 years, have hypertension.1 Salt sensitivity is present when the blood pressure changes by 5% to 10% or at least 5 mm Hg, in response to a change in NaCl intake. Fifty‐one percent of the hypertensive and 26% of the normotensive population are salt‐sensitive.2 Salt sensitivity is predictive of cardiovascular events and mortality, irrespective of blood pressure3 and is associated with other diseases, such as asthma, gastric cancer, and insulin resistance, among others.4 However, the mechanisms involved in the expression of the salt‐sensitive phenotype are still unclear.
Genetic background contributes, in part, to salt sensitivity of blood pressure.2, 5 The C57Bl/6 and BALB/c mouse strains are 2 of the most commonly used mouse strains in cardiovascular research, including those used for the generation of transgenic mice. Several studies have shown that a high NaCl diet increases blood pressure in some6, 7 but not all8, 9, 10 C57Bl/6J mice. The concomitant feeding of a low potassium diet and a high sodium diet amplifies the increase in blood pressure caused by the high sodium diet in C57Bl/6J mice from the Jackson Laboratory but not the C57Bl/6 mice from Taconic Biosciences.6 By contrast, SJL/J7 and BALB/c mice11, 12 are salt‐resistant. However, the high salt diet‐ and hypertension‐associated renal injury is less in salt‐sensitive C57Bl/6J than salt‐resistant SJL/J mice.7 A high salt diet also increased the microalbuminuria in SJL/J but not C57Bl/6J mice.7
Oxidative stress increases blood pressure.13 However, a high salt diet increased the urinary excretion of 8‐isoprostane, a product of lipid peroxidation, to a much greater extent in salt‐resistant SJL/J than salt‐sensitive C57Bl/6J mice.7 The effect of reactive oxygen species (ROS) on renal sodium transport varies according to the particular ROS and nephron segment. For example, superoxide and H2O2 stimulate sodium transport (NKCC2 [Na‐K‐2Cl cotransporter]) in the thick ascending limb of Henle's loop, and cause salt sensitivity.14, 15 Superoxide and H2O2 also stimulate sodium channel activity in principal cells of the collecting duct.16, 17 However, superoxide has also been reported to inhibit sodium‐hydrogen exchanger 3 (NHE3) and Na+/K+‐ATPase activities in renal proximal tubules,18 under certain circumstances. H2O2 has also been shown to inhibit Na+/K+‐ATPase in a concentration dependent manner14, 18, 19, 20, 21 Thus, the effect of ROS on renal sodium transport is not clear and additional studies are needed to clarify these apparent inconsistencies.
In this report, we examined the role of H2O2 on the effect of high salt diet on blood pressure in salt‐resistant BALB/c11, 12 and salt‐sensitive C57Bl/66, 7 mice. We also describe a possible mechanism to explain the differences in the blood pressure response to a high salt diet in these 2 strains of mice.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mouse Renal Proximal Tubule Cells
Mouse renal proximal tubule cells (mRPTCs) were isolated from BALB/c mice kidneys using the Miltenyi Biotec's Magnetic‐Activated Cell Sorting technology. We used biotinylated Lotus tetragonolobus lectin (Vector #B‐1325) for the isolation of mRPTCs. The purity of the isolated cells was assessed by immunofluorescence staining using an antibody to SGLT2 (a marker protein of S1 and S2 segments of renal proximal tubule), as used previously.22 mRPTCs from C5Bl/6J mice were cultured from progenitor kidney cells isolated from mouse embryo kidneys of C57Bl/6J mice (kindly supplied by Dr. Ulrich Hopfer, Case Western Reserve University, School of Medicine), as reported by Woost et al.23
Determination of H2O2 and Superoxide Production
Intracellular ROS were assayed through the oxidation of 2′, 7′‐dichlorofluorescein diacetate (DCFDA, Molecular Probes).24 Briefly, mRPTCs were incubated with fresh DCFDA (10 μmol/L) in serum‐free medium for 30 minutes at 37°C. DCFDA fluorescence was measured, in 96‐well plates, using a microplate reader, at an excitation wavelength of 485 nm and emission wavelength of 530 nm. ROS production was expressed in arbitrary units, corrected for protein concentration (arbitrary units/per mg protein).
ROSstar 550 (LI‐COR Biosciences) is a cell‐permeable hydrocyanine probe which is initially non‐fluorescent but becomes fluorescent after oxidation by ROS.25 The ROSstar 550 assay is specific for oxygen radicals, in particular for superoxide and hydroxyl radicals. The mRPTCs were incubated with 50 μmol/L ROSstar 550 for 30 minutes at 37°C, washed 2 times with PBS, and the fluorescence was immediately read, in the plate reader at excitation of 540 nm and emission of 560 nm. The data were normalized by the protein concentration in each well.
Determination of H2O2 Production in Live Cells
HyPer (pHyPer‐cyto, Evrogen Inc) is a mutant green fluorescent protein. The fluorescence intensity of HyPer can measure submicromolar amounts of intracellular H2O2. 25 mRPTCs from C57Bl/6 and BALB/c mice were transfected with Hyper plasmid. Three micrograms of DNA plasmid and 12 μL of FuGENE HD Transfection Reagent (Promega Inc) were added to each well in a 6‐well plate. After 48 hours, the mRPTCs were incubated in 90, 145, and 170 mmol/L sodium with the osmolalities adjusted to 340 mOsm/L with mannitol. We chose these concentrations of sodium because in neurons, the threshold for the sensitivity of sodium channel for extracellular sodium concentration in vitro is about 150 mmol/L [Na+].26, 27 The renal outer medulla, in which the proximal straight tubule is located,28, 29 can have an osmolarity of up to 450 mOsm/L30; the sodium concentration in the outer stripe of the outer medulla has been reported to be 190±19 mEq/kg wet weight.31 The sodium concentration in the interstitial fluid at the beginning of the renal proximal straight tubule has been calculated to be 160 mmol/L and increases to 225 mmol/L at the end of the renal proximal straight tubule.32
Sodium Transport Assay
mRPTCs were cultured in Transwells and transfected with the plasmids pcDNA (empty vector) or pCMVCAT (vector carrying catalase). Two days later, the cells were serum‐starved overnight and treated with vehicle or H2O2 (1, 5, 10, 20, 50, 100, 1000, or 10 000 μmol/L) on the basolateral side for 30 minutes before the sodium accumulation assay.33 In some studies, the serum‐starved mRPTCs were treated, on the basolateral side, with the Na+/K+‐ATPase inhibitor ouabain (50 μmol/L) for 30 minutes and used as positive control.34 Ouabain was also added to the cells treated with H2O2 to determine if the effect of H2O2 is via Na+/K+‐ATPase. The cells were loaded with the sodium dye, sodium green (5 μmol/L) (Molecular Probes, Eugene, OR) with Pluronic 127 for 30 minutes, then washed twice with PBS. Intracellular sodium was measured from the emission at 510 nm when excited at 340 or 380 nm using a fluorimeter. Results are expressed as arbitrary values.
In Vivo Mouse Studies
All animal studies were conducted in accordance with the National Institutes of Health Guidelines, approved by the Institutional Animal Care and Use Committee at the George Washington University. BALB/c mice (8–10 weeks old weighing ≈20 g) were purchased from the Jackson Laboratory (Bar Harbor, ME). A right uninephrectomy was performed under pentobarbital anesthesia. One week later, the renal‐selective overexpression of catalase was performed by the retrograde infusion into the ureter of the remaining left kidney of adeno‐associated virus (AAV) vector35 carrying catalase; AAV vector carrying cDNA served as control. The AAV vector harboring the cytomegalovirus promoter driving the expression of catalase promoter was constructed, using the plasmid pACS, as described previously.35 The mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg), placed in a supine position, and the legs taped down on a heated board to maintain their body temperature at 37°C. For all procedures, the depth of anesthesia was monitored by foot‐pinch reflex. An abdominal incision was made between the point of the xyphoid cartilage and the navel, and then, the ureter was located and gently dissected out. The distal portion of the ureter closest to the bladder and the renal artery supplying the target kidney were clamped off with micro‐venous clips. Using a tuberculin syringe fitted with a 33‐gauge needle, the ureter was then punctured. The needle was temporarily and snugly ligated in place using a 6‐0 silk suture to prevent leakage. After the urine was gently aspirated out, the tuberculin syringe was replaced with another containing ≈100 μL of the AAV vector (1011 viral genome particles) and the solution was slowly and retrogradely injected towards the kidney, via the ureter. The needle was withdrawn and a micro‐venous clip was placed proximal to the injection site on the ureter to prevent leakage. The arterial and the ureteral clips were maintained for 15 minutes to attain maximum exposure to the infusion. The arterial and ureteral clips were then sequentially removed and the ureter was inspected for any evidence of leakage. The abdominal contents were replaced in reverse order and the incision site was closed using a double layer of 6‐0 silk sutures for the muscle and skin. The mice were fed normal (0.8%) or high (4%) NaCl diet for 14 days after AAV infection. Systolic and diastolic blood pressures were measured, under pentobarbital anesthesia, at baseline (before AAV infusion) and 14 days after AAV treatment through the femoral or carotid artery using Cardiomax II (Columbus Instruments). The kidneys were harvested before the mice were euthanized with an overdose of pentobarbital (100 mg/kg).
Statistical Analyses
Data are presented as mean±SEM. Statistical analyses were performed using Sigma Plot 11.0 software (Systat Software, Inc, San Jose, CA). Normality Test Shapiro‐Wilk test was performed to evaluate the statistical distribution of all data in the study. Non‐parametric test, Mann‐Whitney Rank Sum Test, was used in data that were not in a normal distribution. ANOVA and Student t test were used to compare the data that were in a normal distribution. Comparisons between 2 groups used the Student t test. One‐way ANOVA, followed by post‐hoc analysis using Tukey multiple comparison tests in groups with normal distribution and n≥5 per group, was used to assess significant differences among ≥3 groups. Kruskal‐Wallis test was used for multiple comparison tests to assess significant differences among ≥3 groups with n≤4. P<0.05 was considered statistically significant.
Results
High Sodium Concentration in the Incubation Medium Increases ROS Production to a Greater Extent in RPTCs From BALB/c Than C57Bl/6J Mice
To determine the role of superoxide/hydroxyl radicals and H2O2 in the salt sensitivity of the C57Bl/6J mouse and salt resistance of the BALB/c mouse, we measured superoxide/hydroxyl radicals and H2O2 production in primary cultures of mRPTCs isolated from the 2 strains, salt‐sensitive C57Bl/6J mice6, 7 and salt‐resistant BALB/c mice.11, 12 H2O2 was measured using 2′, 7′‐dichlorodihydrofluorescein diacetate (DCFDA)24 and superoxide/hydroxyl radicals, using ROSstar 550, a hydrocyanine‐based probe.25 Figure 1A shows that H2O2 and superoxide productions were 42% and 61% higher, respectively, in BALB/c‐RPTCs than C57Bl/6J‐RPTCs. The lower ROS production in mRPTCs from salt‐sensitive C57Bl/6J mice, relative to RPTCs from salt‐resistant BALB/c mice, agrees with the decreased urinary 8‐isoprostane in C57Bl/6J mice, relative to salt‐resistant SJL/J mice.7
Figure 1.
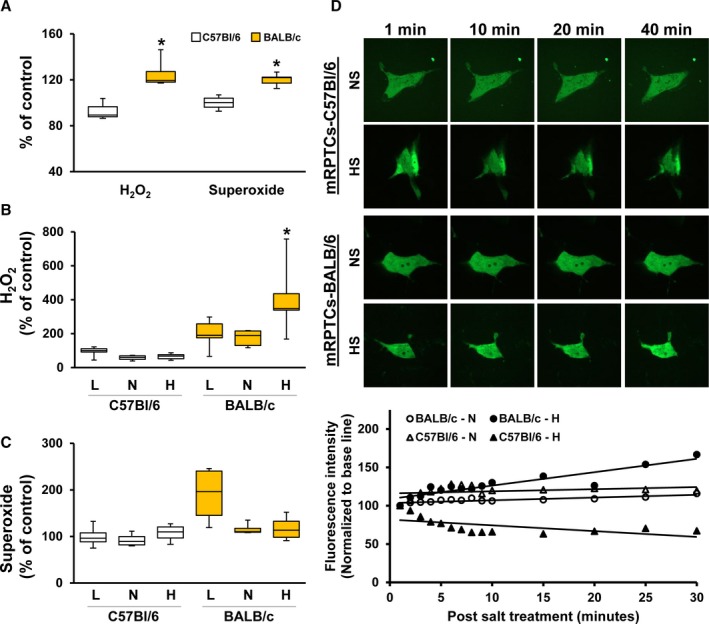
Reactive oxygen species production in mouse renal proximal tubule cells (mRPTCs) from BALB/c and C57Bl/6 mice under different Na+ concentrations. The production of reactive oxygen species in mRPTCs from C57Bl/6 and BALB/c mice at baseline and in response to different sodium concentrations were measured. H2O2 production was measured using dichlorofluorescein diacetate and superoxide/hydroxyl radical was measured using ROSstar 550. A, This figure shows basal H2O2 and superoxide production in mRPTCs from salt‐resistant BALB/c mice and salt‐sensitive C57Bl/6 mice. Results were normalized to H2O2 and superoxide levels in mRPTCs from C57Bl/6 mice. Data are expressed as mean±SE. H2O2 production: *P<0.05 BALB/c vs C57Bl/6 mice, t test, n=8 to 10 per group. Superoxide production: *P<0.05 BALB/c vs C57Bl/6 mice, Mann‐Whitney Rank Sum Test (Normality Test, Shapiro‐Wilk failed), n=8 per group. B and C, RPTCs from the C57Bl/6 and BALB/c mice were treated for 24 hours with 90 mmol/L (low), 145 mmol/L (normal) or 170 mmol/L (high) sodium concentrations; the osmolalities were adjusted to 340 mOsm/L with mannitol, as necessary. H2O2 (B) and superoxide (C) productions were normalized to H2O2 or superoxide produced in the mRPTCs from C57Bl/6 treated with 90 mmol/L sodium; data are expressed as mean±SE H2O2 production: *P<0.05 BALB/c 170 mmol/L vs BALB/c 145 mmol/L, one‐way ANOVA (Tukey post‐hoc test) (group 5), n=5 per group. D, H2O2 production was measured by live‐cell imaging using HyPer‐cyto vector system as described in the methods section. mRPTCs from the C57Bl/6 and BALB/c mice were treated were with 90 mmol/L (low), 145 mmol/L (normal) or 170 mmol/L (high) sodium concentrations with the osmolality adjusted to 340 mOsm/L with mannitol, as necessary. Time‐lapse images of the live cells were captured at 1‐minute intervals for 30 minutes at a single confocal section. Images were taken using a spinning disk confocal microscope (Carl Zeiss) and processed using Velocity 6.3 software (PerkinElmer). Fluorescence images of the cells captured at 1, 10, 20, and 40 minutes are shown. Fluorescence intensities were normalized to their respective basal levels plotted against time. mRPTCs indicates mouse renal proximal tubule cells; NS, normal salt; HS, high salt.
We determined the consequences of incubation of mRPTCs from BALB/c and C57Bl/6J mice in 3 different concentrations of sodium, as sodium chloride, with all the osmolalities adjusted to 340 mOsm/L using mannitol: low=90 mmol/L, normal=145 mmol/L, and high=170 mmol/L. H2O2 production, measured by DCFDA, was higher in BALB/c‐RPTCs than C57Bl/6J‐RPTCs in all 3 sodium concentrations (Figure 1B). There was a tendency for H2O2 production to be decreased by an increase in sodium concentration in C57/Bl/6J‐RPTCs. By contrast, the high sodium concentration, 170 mmol/L, significantly increased the H2O2 production in BALB/c‐RPTCs, (Figure 1B). Superoxide/hydroxyl radical production was not different between BALB/c and C57Bl/6J RPTCs. Although there was a tendency of the high sodium concentration to decrease superoxide production in BALB/c‐RPTCs, which would go along with the increased H2O2 production in these cells, the difference was not statistically significant (Figure 1C).
We used a second method to measure H2O2, using pHyPer‐cyto vector system (Evrogen JSC, Moscow, Russia) to confirm the increased basal production of H2O2 and response to 170 mmol/L Na+ in BALB/c‐RPTCs, relative to C57Bl/6J RPTCs. The pHyPer‐cyto vector codes for protein HyPer that exhibits fluorescence directly proportional to an increase in intracellular H2O2; HyPer is recommended as the primary assay for H2O2.25 Relative to the normal sodium concentration (145 mmol/L), H2O2 production, using pHyPer‐cyto live‐cell imaging, high concentration (170 mmol/L) increased H2O2 production in BALB/c‐RPTCs but decreased it in C57Bl/6J RPTCs, confirming the results using DCFDA (Figure 1D). In rats, an increase in sodium delivery to the medullary thick ascending limb of Henle has been reported to increase H2O2 production by the mitochondria and cell membrane NADPH oxidase.36 The increase in H2O2 production may be responsible for the increase in renal sodium transport that serves as negative feedback mechanism to maintain normal sodium balance and blood pressure. However, H2O2 may not always increase sodium transport and drugs that decrease oxidative stress may impair sodium excretion and increase of blood pressure.37
H2O2 Inhibits Sodium Transport in a Concentration‐Dependent Manner in RPTCs From BALB/c Mice
We, next, tested the effect of H2O2 on sodium transport in mRPTCs from BALB/c mice, incubated in normal sodium (145 mmol/L) concentration. H2O2 (μmol/L) had a biphasic effect on sodium accumulation, increasing it at 5 to 50 μmol/L, with 1.62‐fold increase, relative vehicle treatment (Figure 2A); an increase in sodium accumulation indicates inhibition of sodium transport at the basolateral membrane (see below). Higher concentrations of H2O2 (100 and 1000 μmol/L) showed no significant effect. However, treatment with 10 mmol/L H2O2 decreased sodium accumulation by 65% (Figure 2A). In C57Bl/6J mice, the renal concentration of H2O2 can be as high as 3 to 4 mmol/L38, 39, 40 that can increase to 10 mmol/L in kidneys subject to ischemia‐reperfusion injury.40 The mRPTCs were grown in polarized state in Transwells; inhibition of sodium transport at the basolateral surface increases intracellular sodium. Therefore, a decrease in intracellular sodium is the result of an increase in sodium transport from the inside of the cell to outside of the cell by the basolateral membrane. These effects were blocked by ouabain, indicating that the effects were on Na+/K+‐ATPase.34 Overexpression of catalase, which decomposes H2O2 to H2O and O2, in BALB/c‐RPTCs decreased sodium accumulation by 38% compared with BALB/c‐RPTCs transfected with pcDNA empty vector (Figure 2B). Treatment of BALB/c‐RPTCs transfected with pcDNA empty vector with H2O2 (10 μmol/L) confirmed (Figure 2C) the increase in sodium accumulation shown in Figure 2A. Overexpression of catalase in BALB/c‐RPTCs decreased sodium accumulation in vehicle‐treated cells (Figure 2C), confirming the results in Figure 2B. In the studies in catalase overexpressing BALB/c‐RPTCs, H2O2 (10 μmol/L) was no longer able to increase sodium accumulation (Figure 2C). This result indicates that decreasing H2O2 levels impairs the inhibition of basolateral sodium transport in BALB/c‐RPTCs exposed to normal (145 mmol/L) sodium concentration.
Figure 2.
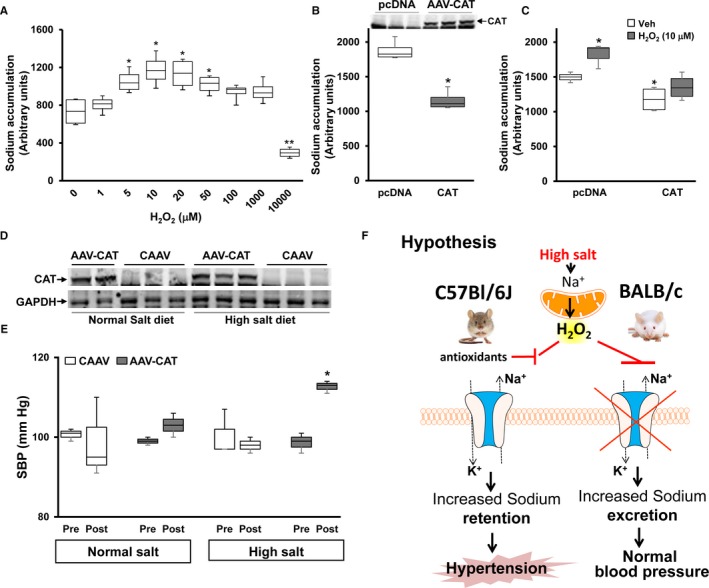
H2O2 regulates Na+/K+‐ATPase activity and blood pressure in BALB/c mice on high salt diet. A, BALB/c‐renal proximal tubule cells were treated with the indicated concentration of H2O2 before the sodium accumulation assay. H2O2 (30 minutes) was added to the basolateral side of polarized renal proximal tubule cells grown in Transwells. The figure shows the sodium accumulation in the cells which is inversely related to Na+/K+‐ATPase activity (measured with ouabain); an increase in sodium accumulation occurs with inhibition of Na+/K+‐ATPase activity. Data are expressed as mean± SE (A) *P<0.05 vs control, **P<0.05 vs rest of the groups, Kruskal‐Wallis test, n=3 per group. B, BALB/c‐renal proximal tubule cells were transfected with plasmid pcDNA or plasmid carrying cDNA coding for catalase (adeno‐associated virus [AAV]‐vector carrying catalase). Two days following transfection, sodium accumulation was measured. Protein from the same cells were immunoblotted for the expression of catalase. Data are expressed as mean±SE. *P<0.05 vs pcDNA, t test, n=3 per group. C, BALB/c‐renal proximal tubule cells, transfected as in (B) were treated with vehicle or H2O2 (10 μmol/L) for 30 minutes on the basolateral side before sodium accumulation assay. Data are expressed as mean±SE. *P<0.05 vs Vehicle‐treated pcDNA, t test, n=3 per group. D, Renal homogenates from BALB/c mice fed normal or high salt diet, whose kidneys were infected with AAV vector carrying catalase or empty vector (AAV vector carrying cDNA), were immunoblotted using rabbit anti‐catalase antibody. Catalase expression was normalized to GAPDH. E, BALB/c mice treated with AAV vector carrying cDNA or AAV vector carrying catalase were fed normal (0.8%) or high (4%) NaCl diet for 14 days following treatment with AAV. Systolic blood pressure was measured from the carotid artery under pentobarbital anesthesia. Data are expressed as mean±SE. *P<0.05 vs others, one‐way Kruskal‐Wallis test, n=3 per group. F, Hypothesis: In salt‐resistant BALB/c mice, high salt diet increases H2O2 production in renal proximal tubule cells, which inhibits the Na+/K+‐ATPase activity resulting in increased sodium excretion. However, in salt‐sensitive C57Bl/6J mice, high salt diet does not increase H2O2 production; renal Na+/K+‐ATPase activity is not inhibited which results in sodium retention and eventually causes hypertension. mRPTCs indicates mouse renal proximal tubule cells.
Catalase Overexpression Converts BALB/c Mice to Hypertensive in Response to High Salt
To test the effect of the elimination of H2O2 and superoxide generated in the kidney, in response to a high sodium diet, on the blood pressure, we overexpressed catalase, selectively in renal tubules using AAV9 vector in BALB/c mice.22, 35 We have reported a high efficiency and efficacy of the expression of the gene of interest in the whole tubule or specific segments of the tubules of the target kidney by the retrograde ureteral infusion of AAV carrying the gene of interest with no or minimal systemic spill‐over.22, 35 Thus, the left ureteral retrograde infusion of AAV carrying the firefly luciferase showed bioluminescence in the left kidney that received the injection but not the right kidney that did not receive the injection or the rest of the body indicating no or minimum systemic spill‐over.35
The retrograde ureteral infusion of AAV increased renal catalase expression by 8.2±1.4‐ and 7.45±1.8‐fold in the normal and high salt diet groups, respectively, compared with the AAV vector carrying cDNA, empty vector‐treated group, with the corresponding diet (Figure 2D). Systolic blood pressure was measured under pentobarbital anesthesia,7, 35 14 days following AAV vector administration. In the normal salt diet group, catalase overexpression did not alter the systolic blood pressures (99±1.4 versus 103±4.2 mm Hg) (in agreement with the sodium transport data). However, in the high salt diet group, systolic blood pressure was significantly higher in the catalase‐overexpressed than the non‐overexpressed group (98± 1.1 versus 112±1.4 mm Hg). This affirms the in vitro studies in which overexpression of catalase in mRPTCs prevented the ability of H2O2 to increase sodium accumulation, ie, inhibit basolateral sodium transport, presumably by impairing Na+/K+‐ATPase activity at the basolateral membrane (Figure 2C). These results indicate that the degradation of H2O2 by overexpression of catalase predisposes the salt‐resistant BALB/c mice to salt‐sensitive hypertension (Figure 2E).
Discussion
We report for the first time that salt‐sensitive hypertension is related to a lack of an increase in renal H2O2 production in response to a high salt diet. This conclusion is based on 4 observations: (1) high NaCl (170 mmol/L) concentration increases H2O2 production in mRPTCs from salt‐resistant BALB/c but not in mRPTCs from salt‐sensitive C57Bl/6 mice; (2) low concentrations of H2O2 negatively regulate basolateral sodium transport in mRPTCs from salt‐resistant BALB/c mice; (3) overexpression of catalase in mRPTCs from salt‐resistant BALB/c mice prevents the ability of H2O2 to inhibit basolateral sodium transport; and 4) overexpression of catalase, selectively in the kidney, transforms salt‐resistant BALB/c mice to salt‐sensitive mice.
The role of ROS in the regulation of renal sodium transport in the renal nephron is not yet settled.37 Previous studies have shown that superoxide can enhance sodium transport in the renal proximal tubule, loop of Henle, and collecting duct.14, 15, 16, 17 H2O2 also increases alanine transport in RPTCs from spontaneously hypertensive rats.41 H2O2, produced by Nox4, has also been shown to stimulate sodium transport in mouse renal collecting duct cells.17 By contrast, H2O2, superoxide, and hydroxyl radical have been shown to inhibit Na+/K+‐ATPase in several tissues, including the kidney.19, 20, 21, 39 Using mRPTCs from salt‐resistant BALB/c mice, we show a biphasic effect of H2O2 on basolateral sodium transport (Figure 2A). While 5 to 50 μmol/L H2O2 in the incubation medium inhibits basolateral sodium transport, a high concentration (10 mmol/L) of H2O2 activates sodium transport. In BALB/c RPTCs, the reduction of endogenous H2O2 by overexpression of catalase increases basolateral sodium transport, resulting in a decrease in sodium accumulation (Figure 2B and 2C). However, the overexpression of catalase in these BALB/c‐RPTCs prevents the inhibitory effect of 10 μmol/L H2O2 on sodium transport (Figure 2C). We also show that the renal‐restricted overexpression of catalase increases the blood pressure of salt‐resistant BALB/c mice fed a high sodium diet (Figure 2E).
H2O2 can serve as a second messenger, at intracellular concentrations of 1 to 100 nmol/L, which may be equivalent to extracellular concentrations of 100 nmol/L to 10 μmol/L,39 the concentration in the incubation medium in our studies. However, higher extracellular concentrations of H2O2 can cause inflammation and fibrogenesis, and 0.2 to 1 mmol/L extracellular concentrations of H2O2 can lead to growth arrest and cell death.39 In our studies, the stimulatory effect of H2O2 of sodium accumulation (ie, decreased sodium transport at the basolateral membrane) was noted at 5 to 50 μmol/L H2O2, while 10 mmol/L H2O2 decreased sodium accumulation (increased sodium transport at the basolateral membrane). The concentration of 10 mmol/L H2O2 is in the range of renal H2O2 production 24 hours after renal ischemia‐reperfusion injury in mice.40 The hypertensinogenic effect of angiotensin II, infused subcutaneously in rats, has been shown to be associated with increased production of H2O2 in the kidney, especially in the renal medulla. Reducing the amount of H2O2 production with polyethylene glycol catalase temporarily normalized the blood pressure in these rats.42 Increased levels of superoxide or H2O2 in the rat renal medulla increase sodium transport in the thick ascending limb of Henle and cause medullary ischemia.14 These effects and the impairment of the pressure‐natriuresis response contribute to the pathogenesis of hypertension in these rats that are associated with over and unregulated H2O2 expression. All the aforementioned studies suggest that the timing and duration of activation, type, amount, and cellular localization of ROS production are critical in determining the cellular effects of ROS.36, 37, 38, 43 These effects of ROS are also influenced genetics. Certain strains of C57Bl/6J mice are predisposed to develop hypertension when fed a high salt diet6, 7 but appear to be resistant to renal injury, in spite of the hypertension caused by the high salt diet or chronic infusion of angiotensin II that is related to decreased production of ROS.7, 44 Whether the kidney injury in C57Bl/6J mice with hypertension caused by the combination of deoxycorticosterone acetate, salt, and angiotensin II infusion is related to increased production of ROS remains to be determined.45 We propose that physiological concentrations of H2O2 may have a beneficial biological function, in part, by the inhibition of renal Na+/K+‐ATPase activity. By contrast, higher concentrations of H2O2 or other prooxidants, such us superoxide, induce oxidative stress, increase renal sodium transport, and cause hypertension, among other deleterious effects.
Sodium reabsorption in the nephron is regulated by various factors, via the modification of α1 subunit of Na+/K+‐ATPase. Carboxylation of the α1 subunit of Na+/K+‐ATPase is one such critical step in proximal tubular signaling.21 Thiol groups (SH) of several proteins are easily oxidized by molecules such as H2O2, producing the oxidative state of these SH groups (sulfenic acid [SOH], sulfinic acid [SO2H] or sulfonic acid [SO3H]) or induce interaction with other groups of the protein (R‐S‐S‐R) to induce conformational changes in the structure of the target proteins.46 Several SH groups in Na+/K+‐ATPase are associated with its inhibition,19 suggesting that SH groups in the α1 subunit of Na+/K+‐ATPase protein could be essential for its enzymatic activity and antioxidants may neutralize its oxidation preventing the formation of hydroxyl radicals.20
We propose a new mechanism for the sodium retention in hypertension. In salt‐resistant BALB/c mice, high salt diet increases H2O2 production in RPTCs, which inhibits Na+/K+‐ATPase activity, resulting in increased sodium excretion. However, in salt‐sensitive C57Bl/6J mice, high salt diet does not increase H2O2 production; renal Na+/K+‐ATPase activity is not inhibited which results in increased sodium retention and eventually causes hypertension (Figure 2F). We speculate that this mechanism could explain some of the inconsistent effects of some antioxidants on blood pressure regulation. For example, the intake of vitamin E, which has antioxidant properties47 decreases systolic but not diastolic blood pressure.48, 49 Vitamin C, which can be both an antioxidant and prooxidant,50 can also decrease the risk of uncontrolled hypertension.50, 51 Vitamin D has antioxidant properties52 but may not decrease blood pressure.53 The dietary antioxidant index has also been reported to be associated negatively with carotid intima media thickness in women but not in men.54 The effects of vitamins can also be different depending on the population subgroups, such as age, obesity, and cardiovascular risk, among others.55 Adverse consequences of antioxidant supplementation have also been reported, with vitamin E increasing the blood pressure of individuals with type 2 diabetes mellitus.56Therefore, several variables have to be taken into consideration in determining the beneficial or deleterious effect of antioxidants, including salt‐intake and genetic background, as shown in our current report.
Conclusions
In summary, to our knowledge, we show for the first time: 1) the beneficial effect of physiological concentrations of prooxidants on blood pressure; 2) chronic reduction of the prooxidant, H2O2, by overexpression of an antioxidant enzyme, catalase, increases blood pressure; and 3) the effects of H2O2 on blood pressure are strain‐ and salt intake‐dependent. If studies in humans confirm these observations, then “excessive” intake of antioxidants may have deleterious effects on blood pressure regulation that may be observed only when salt intake is excessive, in genetically predisposed individuals.
Sources of Funding
These studies were supported in part by NIH T32 DK007545 to CDM and NIH/NHLBI 5P01 HL074940‐10 and NIH/NIDDK 7R01 DK039308‐31 to Jose and R56DK116828 to Konkalmatt.
Disclosures
None.
(J Am Heart Assoc. 2020;9:e013818 DOI: 10.1161/JAHA.119.013818.)
References
- 1. Fryar CD, Ostchega Y, Hales CM, Zhang G, Kruszon‐Moran D. Hypertension prevalence and control among adults: United states, 2015–2016. NCHS Data Brief. 2017;1–8. [PubMed] [Google Scholar]
- 2. Felder RA, White MJ, Williams SM, Jose PA. Diagnostic tools for hypertension and salt sensitivity testing. Curr Opin Nephrol Hypertens. 2013;22:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–432. [DOI] [PubMed] [Google Scholar]
- 4. He FJ, MacGregor GA. Reducing population salt intake worldwide: from evidence to implementation. Prog Cardiovasc Dis. 2010;52:363–382. [DOI] [PubMed] [Google Scholar]
- 5. Frame AA, Wainford RD. Renal sodium handling and sodium sensitivity. Kidney Res Clin Pract. 2017;36:117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Combe R, Mudgett J, El Fertak L, Champy MF, Ayme‐Dietrich E, Petit‐Demouliere B, Sorg T, Herault Y, Madwed JB, Monassier L. How does circadian rhythm impact salt sensitivity of blood pressure in mice? A study in two close C57Bl/6 substrains. PLoS One. 2016;11:e0153472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Escano CS, Armando I, Wang X, Asico LD, Pascua A, Yang Y, Wang Z, Lau YS, Jose PA. Renal dopaminergic defect in C57Bl/6J mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1660–R1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kopkan L, Hess A, Huskova Z, Cervenka L, Navar LG, Majid DS. High‐salt intake enhances superoxide activity in enos knockout mice leading to the development of salt sensitivity. Am J Physiol Renal Physiol. 2010;299:F656–F663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mangrum AJ, Gomez RA, Norwood VF. Effects of AT(1A) receptor deletion on blood pressure and sodium excretion during altered dietary salt intake. Am J Physiol Renal Physiol. 2002;283:F447–F453. [DOI] [PubMed] [Google Scholar]
- 10. Hartner A, Cordasic N, Klanke B, Veelken R, Hilgers KF. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant. 2003;18:1999–2004. [DOI] [PubMed] [Google Scholar]
- 11. Suzuki J, Otsuka F, Matsumoto Y, Inagaki K, Miyoshi T, Takeda M, Tsukamoto N, Nakamura E, Ogura K, Makino H. Enhanced expression of bone morphogenetic protein system in aldosterone‐treated mouse kidneys. Hypertens Res. 2012;35:312–317. [DOI] [PubMed] [Google Scholar]
- 12. Lorenz JN, Nieman M, Sabo J, Sanford LP, Hawkins JA, Elitsur N, Gawenis LR, Clarke LL, Cohen MB. Uroguanylin knockout mice have increased blood pressure and impaired natriuretic response to enteral NaCl load. J Clin Invest. 2003;112:1244–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sorriento D, De Luca N, Trimarco B, Iaccarino G. The antioxidant therapy: new insights in the treatment of hypertension. Front Physiol. 2018;9:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cowley AW Jr, Abe M, Mori T, O'Connor PM, Ohsaki Y, Zheleznova NN. Reactive oxygen species as important determinants of medullary flow, sodium excretion, and hypertension. Am J Physiol Renal Physiol. 2015;308:F179–F197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saez F, Hong NJ, Garvin JL. NADPH oxidase 4‐derived superoxide mediates flow‐stimulated NKCC2 activity in thick ascending limbs. Am J Physiol Renal Physiol. 2018;314:F934–F941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou L, Linck V, Zhai YJ, Galarza‐Paez L, Li L, Yue Q, Al‐Khalili O, Bao HF, Ma HP, Thai TL, Jiao J, Eaton DC. Knockout of mitochondrial voltage‐dependent anion channel type 3 increases reactive oxygen species (ROS) levels and alters renal sodium transport. J Biol Chem. 2018;293:1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu X, Wang F, Liu M, Yang KT, Nau A, Kohan DE, Reese V, Richardson RS, Yang T. Activation of ENaC in collecting duct cells by prorenin and its receptor PRR: involvement of Nox4‐derived hydrogen peroxide. Am J Physiol Renal Physiol. 2016;310:F1243–F1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan Y, Shapiro AP, Mopidevi BR, Chaudhry MA, Maxwell K, Haller ST, Drummond CA, Kennedy DJ, Tian J, Malhotra D, Xie ZJ, Shapiro JI, Liu J. Protein carbonylation of an amino acid residue of the Na/K‐ATPase alpha1 subunit determines Na/K‐ATPase signaling and sodium transport in renal proximal tubular cells. J Am Heart Assoc. 2016;5:e003675 DOI: 10.1161/JAHA.116.003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang WH, Wang Y, Askari A. (Na+ + K+)‐atpase: inactivation and degradation induced by oxygen radicals. Int J Biochem. 1992;24:621–626. [DOI] [PubMed] [Google Scholar]
- 20. Kurella EG, Tyulina OV, Boldyrev AA. Oxidative resistance of Na/K‐ATPase. Cell Mol Neurobiol. 1999;19:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah PT, Martin R, Yan Y, Shapiro JI, Liu J. Carbonylation modification regulates Na/K‐ATPase signaling and salt sensitivity: a review and a hypothesis. Front Physiol. 2016;7:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asico LD, Cuevas S, Ma X, Jose PA, Armando I, Konkalmatt PR. Nephron segment‐specific gene expression using AAV vectors. Biochem Biophys Res Commun. 2018;497:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woost PG, Kolb RJ, Finesilver M, Mackraj I, Imboden H, Coffman TM, Hopfer U. Strategy for the development of a matched set of transport‐competent, angiotensin receptor‐deficient proximal tubule cell lines. Vitro Cell Dev Biol Anim. 2006;42:189–200. [DOI] [PubMed] [Google Scholar]
- 24. Kalyanaraman B, Darley‐Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ II, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A; American Heart Association Council on Basic Cardiovascular S . Measurement of reactive oxygen species, reactive nitrogen species, and redox‐dependent signaling in the cardiovascular system: a scientific statement from the american heart association. Circ Res. 2016;119:e39–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hiyama TY, Watanabe E, Ono K, Inenaga K, Tamkun MM, Yoshida S, Noda M. Na(x) channel involved in CNS sodium‐level sensing. Nat Neurosci. 2002;5:511–512. [DOI] [PubMed] [Google Scholar]
- 27. Noda M, Hiyama TY. Sodium‐level‐sensitive sodium channel and salt‐intake behavior. Chem Senses. 2005;30(suppl 1):i44–i45. [DOI] [PubMed] [Google Scholar]
- 28. Beck FX, Sone M, Dörge A, Thurau K. Effect of loop diuretics on organic osmolytes and cell electrolytes in the renal outer medulla. Kidney Int. 1992;42:843–850. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y, Zhang X, Chen L, Wu J, Su D, Lu WJ, Hwang MT, Yang G, Li S, Wei M, Davis L, Breyer MD, Guan Y. Liver X receptor agonist TO‐901317 upregulates SCD1 expression in renal proximal straight tubule. Am J Physiol Renal Physiol. 2006;290:F1065–F1073. [DOI] [PubMed] [Google Scholar]
- 30. Chessa F, Mathow D, Wang S, Hielscher T, Atzberger A, Porubsky S, Gretz N, Burgdorf S, Gröne HJ, Popovic ZV. The renal microenvironment modifies dendritic cell phenotype. Kidney Int. 2016;89:82–94. [DOI] [PubMed] [Google Scholar]
- 31. Beck FX, Ohno A, Dörge A, Thurau K. Ischemia‐induced changes in cell element composition and osmolyte contents of outer medulla. Kidney Int. 1995;48:449–457. [DOI] [PubMed] [Google Scholar]
- 32. Layton AT, Edwards A. Tubuloglomerular feedback signal transduction in a short loop of henle. Bull Math Biol. 2010;72:34–62. [DOI] [PubMed] [Google Scholar]
- 33. Gildea JJ, Xu P, Kemp BA, Carlson JM, Tran HT, Bigler Wang D, Langouet‐Astrie CJ, McGrath HE, Carey RM, Jose PA, Felder RA. Sodium bicarbonate cotransporter NBCE2 gene variants increase sodium and bicarbonate transport in human renal proximal tubule cells. PLoS One. 2018;13:e0189464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blaustein MP. The pump, the exchanger, and the holy spirit: origins and 40‐year evolution of ideas about the ouabain‐Na(+) pump endocrine system. Am J Physiol Cell Physiol. 2018;314:C3–C26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Konkalmatt PR, Asico LD, Zhang Y, Yang Y, Drachenberg C, Zheng X, Han F, Jose PA, Armando I. Renal rescue of dopamine D2 receptor function reverses renal injury and high blood pressure. JCI Insight. 2016;1:e85888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohsaki Y, O'Connor P, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, Cowley AW Jr. Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol. 2012;302:F95–F102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez‐Vicente A, Hong NJ, Garvin JL. Effects of reactive oxygen species on renal tubular transport. Am J Physiol Renal Physiol. 2019. Available at: https://www.physiology.org/doi/abs/10.1152/ajprenal.00604.2018. Accessed December 28, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. 2014;289:8735–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 2017;11:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J, Kim KY, Jang HS, Yoshida T, Tsuchiya K, Nitta K, Park JW, Bonventre JV, Park KM. Role of cytosolic NADP+‐dependent isocitrate dehydrogenase in ischemia‐reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2009;296:F622–F633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pinto V, Pinho MJ, Jose PA, Soares‐da‐Silva P. Role of h(2)o(2) on the kinetics of low‐affinity high‐capacity na(+)‐dependent alanine transport in shr proximal tubular epithelial cells. Biochem Biophys Res Commun. 2010;398:553–558. [DOI] [PubMed] [Google Scholar]
- 42. Sousa T, Oliveira S, Afonso J, Morato M, Patinha D, Fraga S, Carvalho F, Albino‐Teixeira A. Role of H(2)O(2) in hypertension, renin‐angiotensin system activation and renal medullary disfunction caused by angiotensin II. Br J Pharmacol. 2012;166:2386–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci. 2011;32:82–89. [DOI] [PubMed] [Google Scholar]
- 44. Wesseling S, Ishola DA Jr, Joles JA, Bluyssen HA, Koomans HA, Braam B. Resistance to oxidative stress by chronic infusion of angiotensin II in mouse kidney is not mediated by the AT2 receptor. Am J Physiol Renal Physiol. 2005;288:F1191–F1200. [DOI] [PubMed] [Google Scholar]
- 45. Kirchhoff F, Krebs C, Abdulhag UN, Meyer‐Schwesinger C, Maas R, Helmchen U, Hilgers KF, Wolf G, Stahl RA, Wenzel U. Rapid development of severe end‐organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int. 2008;73:643–650. [DOI] [PubMed] [Google Scholar]
- 46. Trivedi MV, Laurence JS, Siahaan TJ. The role of thiols and disulfides on protein stability. Curr Protein Pept Sci. 2009;10:614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti‐inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014;72:76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuwabara A, Nakade M, Tamai H, Tsuboyama‐Kasaoka N, Tanaka K. The association between vitamin E intake and hypertension: results from the re‐analysis of the National Health and Nutrition Survey. J Nutr Sci Vitaminol (Tokyo). 2014;60:239–245. [DOI] [PubMed] [Google Scholar]
- 49. Emami MR, Safabakhsh M, Alizadeh S, Asbaghi O, Khosroshahi MZ. Effect of vitamin E supplementation on blood pressure: a systematic review and meta‐analysis. J Hum Hypertens. 2019;33:499–507. [DOI] [PubMed] [Google Scholar]
- 50. Yuan X, Li X, Ji Z, Xiao J, Zhang L, Zhang W, Su H, Kaliannan K, Long Y, Shao Z. Effects of vitamin C supplementation on blood pressure and hypertension control in response to ambient temperature changes in patients with essential hypertension. Clin Exp Hypertens. 2019;41:414–442. [DOI] [PubMed] [Google Scholar]
- 51. Juraschek SP, Guallar E, Appel LJ, Miller ER III. Effects of vitamin C supplementation on blood pressure: a meta‐analysis of randomized controlled trials. Am J Clin Nutr. 2012;95:1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murdaca G, Tonacci A, Negrini S, Greco M, Borro M, Puppo F, Gangemi S. Emerging role of vitamin D in autoimmune diseases: an update on evidence and therapeutic implications. Autoimmun Rev. 2019;18:102350. [DOI] [PubMed] [Google Scholar]
- 53. Beveridge LA, Struthers AD, Khan F, Jorde R, Scragg R, Macdonald HM, Alvarez JA, Boxer RS, Dalbeni A, Gepner AD, Isbel NM, Larsen T, Nagpal J, Petchey WG, Stricker H, Strobel F, Tangpricha V, Toxqui L, Vaquero M, Wamberg L, Zittermann A, Witham MD; D‐PRESSURE Collaboration . Effect of Vitamin D supplementation on blood pressure: a systematic review and meta‐analysis incorporating individual patient data. JAMA Intern Med. 2015;175:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maugeri A, Hruskova J, Jakubik J, Kunzova S, Sochor O, Barchitta M, Agodi A, Bauerova H, Medina‐Inojosa JR, Vinciguerra M. Dietary antioxidant intake decreases carotid intima media thickness in women but not in men: a cross‐sectional assessment in the Kardiovize study. Free Radic Biol Med. 2019;131:274–281. [DOI] [PubMed] [Google Scholar]
- 55. Ashor AW, Brown R, Keenan PD, Willis ND, Siervo M, Mathers JC. Limited evidence for a beneficial effect of vitamin C supplementation on biomarkers of cardiovascular diseases: an umbrella review of systematic reviews and meta‐analyses. Nutr Res. 2019;61:1–12. [DOI] [PubMed] [Google Scholar]
- 56. Ward NC, Wu JH, Clarke MW, Puddey IB, Burke V, Croft KD, Hodgson JM. The effect of vitamin e on blood pressure in individuals with type 2 diabetes: a randomized, double‐blind, placebo‐controlled trial. J Hypertens. 2007;25:227–234. [DOI] [PubMed] [Google Scholar]