

Introduction
Numerous human epidemiological studies have established unequivocally that a high plasma high‐density lipoprotein cholesterol (HDL‐C) level is inversely associated with the risk of developing atherosclerotic cardiovascular disease.1 This has led to the hypothesis that increasing HDL‐C levels may reduce the risk of having a cardiovascular event. However, most therapies that increase plasma HDL‐C levels have not reduced cardiovascular events when tested in large‐scale clinical outcome trials. Despite these discouraging results, there has been an upside suggesting that interventions that increase HDL‐C levels are associated with significantly improved glycemic control in people with type 2 diabetes mellitus (T2DM).2, 3, 4 When this association is considered in light of outcomes from preclinical studies and small randomized clinical trials in which HDL‐C levels were increased in patients with T2DM, as well as in vitro studies that have provided an insight into the molecular basis of the clinical findings, it follows that modulating the individual constituents of HDLs, which may or may not include increasing HDL‐C levels, may be a potential therapeutic target for improving glycemic control in patients with diabetes mellitus.
This review outlines what is known about the antidiabetic properties of HDLs and the main HDL apolipoprotein, apoA‐I. Insights into the mechanistic basis of these properties, and how they are regulated by intracellular and plasma cholesterol levels, is also discussed in the context of the 2 main forms of diabetes mellitus: T2DM, in which insulin resistance leads to pancreatic β‐cell compensation and increased insulin secretion that eventually culminates in β‐cell exhaustion and complete loss of function; and type 1 diabetes mellitus (T1DM), which is characterized by selective autoimmune β‐cell destruction.
Regulation of Glycemic Control by HDLs and apoA‐I: Clinical Aspects
An association between a low plasma HDL‐C level and an increased risk of developing diabetes mellitus has been reported in several human population studies.4, 5, 6, 7, 8 Evidence of an inverse association between HDLs, apoA‐I, and insulin resistance was obtained from the FIELD (Fenofibrate Intervention and Event Lowering in Diabetes) trial in which the homoeostasis model assessment of insulin resistance was used to monitor insulin resistance of patients with T2DM.9 In a smaller study of subjects with impaired glucose tolerance, apoA‐I was further identified as an independent risk factor for glucose tolerance and was also found to be inversely associated with the homoeostasis model assessment of insulin resistance.10 However, this relationship has not been recapitulated in large genome‐wide association studies,11, 12 or in a recent Mendelian randomization study in which it was concluded that the association between genetically determined low HDL‐C levels and incident diabetes mellitus is not causal.13
Despite the negative outcome from the aforementioned Mendelian randomization study, evidence from randomized, placebo controlled clinical trials have indicated that acutely increasing HDL‐C and apoA‐I levels with a single infusion of reconstituted HDLs (rHDLs) consisting of apoA‐I complexed with phosphatidylcholine,2 or chronically increasing HDL‐C and apoA‐I levels by inhibiting activity of CETP (cholesteryl ester transfer protein),3 improves glycemic control in people with T2DM. While the results from these studies are consistent with the observed improvement in glycemic control in these patients being attributable to increased pancreatic β‐cell function and improved insulin sensitivity, they do not provide an insight into whether this benefit is causally related to the increase in HDL and apoA‐I levels, nor do they provide an insight into the extent to which HDL‐C and apoA‐I levels, or other HDL components, must be increased before glycemic control is improved. Nevertheless, support for the notion that increasing HDL‐C and apoA‐I levels may be directly responsible for improving glycemic control in patients with T2DM has emerged from these in vivo studies, as well as from in vitro studies.
Much less is known about the relationship of HDL‐C and apoA‐I levels with glycemic control in patients with T1DM. Although plasma HDL‐C levels tend to be either normal or elevated in patients with T1DM,14 the evidence that these individuals are at increased risk of developing cardiovascular disease at an early age is compelling.15, 16, 17 This suggests that the cardioprotective functions of HDLs may be impaired in patients with T1DM, especially in those with poor glycemic control in whom persistently elevated blood glucose levels can lead to the generation of reactive α‐oxoaldehydes that nonenzymatically glycate HDL apolipoproteins and impair their cardioprotective and antidiabetic functions.18
Support for this hypothesis has been obtained from a small, prospective study of children with T1DM and a case control study of young adults with T1DM, in which cholesterol efflux capacity and HDL–apoA‐I exchange, a surrogate marker of cholesterol efflux capacity, were both significantly impaired.19, 20 Impaired cholesterol efflux, and a reduction in the antioxidant and anti‐inflammatory capacity of HDLs from adult patients with T1DM has also been reported in small cross‐sectional studies.21, 22, 23 The ability of HDLs to improve endothelial function and increase nitric oxide production is also impaired in patients with T1DM.24 Collectively, these studies indicate that at least some of the cardioprotective functions of HDLs are lost in people with T1DM. This is also consistent with the outcome of a recent preclinical study in which macrophage‐to‐feces reverse cholesterol transport was decreased in mouse models of T1DM.25 Whether the antidiabetic functions of HDLs and apoA‐I are also impaired in patients with T1DM is not known, but this is something that is clearly worthy of investigation.
HDLs, apoA‐I, and Insulin Sensitivity
In vitro studies have established that HDLs and lipid‐free apoA‐I improve glycemic control by increasing glucose uptake in skeletal muscle and decreasing insulin resistance. Incubation of primary human skeletal myotubes with conditioned media from cholesterol‐loaded THP‐1 cells, or primary human monocyte‐derived macrophages, reduces insulin‐dependent glucose uptake, and this can be reversed by preincubating the cholesterol‐loaded macrophages with HDLs isolated from the plasma of normal healthy subjects.26
Lipid‐free apoA‐I also improves insulin‐dependent and insulin‐independent glucose uptake in primary human skeletal muscle cells by increasing phosphorylation of the insulin receptor, insulin receptor substrate‐1, phosphoinositide‐3‐K, Akt and Akt substrate of 160 kDa (Figure 1).27 Activation of this signal transduction pathway by HDLs and apoA‐I culminates in the translocation of glucose transporter type 4 to the cell surface and increased uptake of glucose into the cells.27 Activation of this pathway is also dependent on expression of the ATP‐binding cassette transporter A1 (ABCA1), which effluxes cellular cholesterol to lipid‐free/lipid‐poor apoA‐I in the extracellular space,28, 29, 30, 31 and scavenger receptor class B type 1, which selectively removes cholesteryl esters from HDLs and mediates the bidirectional exchange of unesterified cholesterol between cell membranes and HDLs in the extracellular space.32, 33, 34 While it could be argued that the involvement of ABCA1 and scavenger receptor class B type 1 in the apoA‐I–mediated uptake of glucose into skeletal muscle cells is due to apoA‐I effluxing cholesterol from the cells, this was found not to be the case.27
Figure 1.
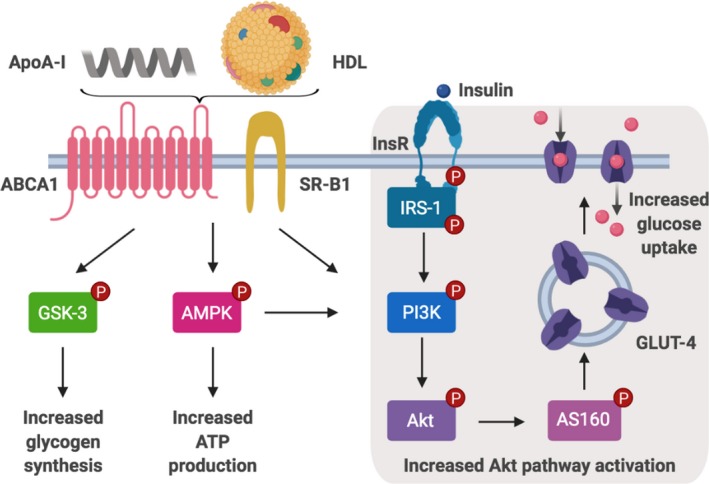
HDL and apoA‐I improve insulin sensitivity and glucose uptake in skeletal muscle. ApoA‐I and HDLs activate GSK‐3 and AMPK. They also increase glucose uptake via increased insulin‐mediated activation of the PI3K/Akt pathway resulting in increased GLUT4 translocation to the cell surface. ABCA1 indicates ATP‐binding cassette transporter A1; AMPK, adenosine monophosphate‐activated protein kinase; apoA‐I, apolipoprotein A‐I; AS160, Akt substrate of 160 kDa; GLUT4, glucose transporter type 4; GSK‐3, glycogen synthase kinase‐3; HDL, high‐density lipoprotein; InsR, insulin receptor; IRS‐1, insulin receptor substrate‐1; PI3K, phosphoinositide‐3‐K; SR‐B1, scavenger receptor class B type 1.
Further insights into the capacity of HDLs and apoA‐I to improve insulin sensitivity have been obtained from incubations of skeletal muscle cells from patients with T2DM with HDLs and apoA‐I (Figure 1). This activates adenosine monophosphate–activated protein kinase (AMPK), an energy‐sensing enzyme that increases ATP production and glucose uptake into skeletal muscle.2, 35 Incubation of skeletal muscle cells with HDLs and apoA‐I also activates glycogen synthase kinase‐3, which promotes glycogen synthesis in response to insulin (Figure 1).36, 37
Long term infusions of rHDLs into db/db mice, a widely used animal model of T2DM, have further been shown to reduce plasma glucose levels and increase phosphorylation of glycogen synthase kinase‐3 and AMPK in skeletal muscle.37 Evidence that this improvement in glycemia is a reflection of increased insulin sensitivity was obtained in a subsequent study in which administration of apoA‐I to db/db mice improved glucose tolerance by increasing glucose uptake into skeletal muscle.38
ApoA‐I treatment also activates AMPK and acetyl‐coenzyme A in apoA‐I–deficient mice, in isolated skeletal muscle from wild‐type mice, and in C2C12 myocytes.35 These results have been recapitulated in incubations of L6 myotubes with rHDLs in a study that additionally established the C‐terminal domain of apoA‐I as a key determinant of increased glucose uptake, AMPK phosphorylation and glucose transporter type 4 translocation to the plasma membrane.39 It should also be noted that these events are independent of Akt phosphorylation.39 Additional mechanistic insights into these observations have been obtained in a study showing that myocytes internalize apoA‐I in a clathrin‐dependent endocytosis process.35 Although the fate of the internalized apoA‐I was not elucidated in that study, the results raise the possibility that apoA‐I may have a previously unrecognized role in metabolic processes in tissue beds that play key roles in the regulation of glycemic control.
A number of the aforementioned in vitro results have been recapitulated in vivo. For example, treatment of high‐fat–fed, insulin‐resistant mice with lipid‐free apoA‐I reduces glucose intolerance, increases insulin sensitivity and improves hepatic glucose metabolism.40 Treatment with lipid‐free apoA‐I also reduces systemic inflammation and attenuates hepatic inflammation by inhibiting activation of nuclear factor‐κB.40 These results have been further confirmed in an in vitro study in which the tumor necrosis factor‐α–induced nuclear translocation of nuclear factor‐κB in the human hepatoma HuH‐7 cell line was inhibited by incubation with apoA‐I–containing rHDLs.40
Systemic and adipose tissue inflammation are also associated with insulin resistance,41 and therapeutic approaches that reduce inflammation can potentially improve insulin sensitivity.42 Incubation of 3T3‐L1 adipocytes with HDLs and apoA‐I has been shown to inhibit the proinflammatory signal transduction pathways that are activated by lipopolysaccharide43 and palmitate.44 Inflammatory markers and macrophage accumulation in adipose tissue of mice transgenic for human apoA‐I are also significantly reduced relative to what has been reported for wild‐type mice.44
Lipid‐free apoA‐I infusions also increase insulin sensitivity and reduce systemic inflammation in rats with pregnancy‐induced insulin resistance.45 In that study, the improvement in insulin sensitivity was attributed specifically to enhanced glucose uptake by white and brown adipose tissue as well as skeletal muscle, while the reduction in systemic inflammation was associated with decreased adipose tissue macrophage content and proinflammatory cytokine production.45 These observations, which raise the possibility that interventions that increase plasma apoA‐I levels may reduce pregnancy‐mediated inflammation and insulin resistance in humans, have important implications for patients at risk of developing gestational diabetes mellitus, the incidence of which is increasing more rapidly than any other form of diabetes mellitus.46
HDLs, apoA‐I, and β‐Cell Function
Although development of insulin resistance leading to a compensatory increase in β‐cell mass and insulin secretory capacity is a key process in the initiation of T2DM, disease progression is driven by β‐cell exhaustion that leads to a reduction in β‐cell mass and function. β‐cell loss in T2DM has been attributed to apoptosis and dedifferentiation into cells that are no longer able to secrete insulin, or express the transcription factors that are essential for maintaining β‐cell identity and survival.47, 48, 49, 50, 51 This makes interventions that improve β‐cell function and promote β‐cell survival highly attractive as therapeutic options for patients with T2DM, as well as for patients who have T1DM with progressive autoimmune‐mediated β‐cell loss. The key caveat for the regeneration of new β cells and the conservation of β cells that have escaped autoimmune destruction in patients with T1DM is that newly regenerated β cells and surviving β cells remain susceptible to autoimmune attack. This suggests that such approaches may need to be implemented in combination with an immune‐based therapy to ensure long‐term efficacy and extended β‐cell survival.
Emerging evidence that apoA‐I may improve β‐cell survival and potentially has the capacity to regenerate new β cells, has come from studies in which treatment with apoA‐I improves glucose tolerance and insulin secretion in high‐fat–fed mice,52, 53 and in mice with conditional deletion of both ABCA1 and ATP‐binding cassette transporter G1(ABCG1) in β cells.54 ABCG1 is a transporter that effluxes cellular cholesterol to HDLs.55, 56
Some mechanistic insights into these observations have been obtained from studies of the Min6 and Ins‐1E clonal β‐cell lines.57, 58 Incubation of Min6 cells with HDLs isolated from normal human plasma, apoA‐I–containing rHDLs or lipid‐free apoA‐I increases transcription of the Ins1 and Ins2 genes, modestly increases insulin secretion under basal conditions and significantly increases glucose‐stimulated insulin secretion (GSIS).58 It is particularly noteworthy that apoA‐I increases insulin secretion in Min6 and Ins‐1E cells without altering intracellular cholesterol levels and that it also increases transcription of the gene encoding for Pdx1, a transcription factor that is essential for maintaining β‐cell identity and survival.57, 58 Importantly, treatment with apoA‐I has recently been shown to increase GSIS in islets from mice in which ABCA1 and ABCG1 are conditionally deleted in β cells.59 The islet cholesterol levels in these mice were approximately double that of control mice, and their GSIS was impaired.59 While it is reasonable to assume that apoA‐I increased GSIS in islets in these mice by acting as an acceptor of the excess cholesterol that effluxed from their β cells, this was not the case, with apoA‐I treatment having no effect on islet cholesterol levels in these animals.54 The mechanistic basis of this unexpected observation is not known and is currently under investigation.
In direct contrast to the in vitro and in vivo results outlined above, HDLs isolated from normal subjects that had been treated for 2 weeks with a CETP inhibitor increased cholesterol efflux and insulin secretion in cholesterol‐loaded Min6 cells.60 The reasons for this discrepant cholesterol efflux result are not entirely clear but may be related to the short duration of CETP inhibitor treatment, which only modestly increased HDL‐C and apoA‐I levels, and the fact that the Min6 cells were cholesterol loaded by incubation with oxidized low‐density lipoproteins (LDLs).60 The cholesterol that accumulates in the islets of mice with conditional β‐cell deletion of ABCA1 and ABCG1 is, by contrast, produced intracellularly and is unlikely to contain significant amounts of oxysterols. In the case of cells that have been cholesterol loaded with oxidized LDLs, oxysterols comprise up to 50% of the total cellular cholesterol.61
Although apoA‐I increases GSIS by a mechanism independent of ABCA1‐mediated cholesterol efflux,54, 58, 59 there is compelling evidence that a direct interaction of apoA‐I with ABCA1 on the β‐cell surface is responsible for increasing GSIS and transcription of the Ins1 and Ins2 genes in Ins‐1E cells (Figure 2).57 This interaction activates a trimeric G‐protein subunit (Figure 2A), which stimulates a transmembrane adenylate cyclase and increases intracellular cAMP levels (Figure 2B). This activates PKA (protein kinase A) (Figure 2C), which phosphorylates and excludes the transcription factor, FoxO1 (forkhead box protein O1) from the Ins‐1E nucleus (Figure 2D), leading to derepression of insulin gene transcription (Figure 2E).57
Figure 2.

Insulin synthesis and secretion is increased in apoA‐I treated pancreatic β cells via a PKA‐FoxO1 dependent mechanism. Interaction between apoA‐I and ABCA1 at the cell surface results in (A) activation of the Gαs subunit of the heterotrimeric G protein and (B) activation of adenylate cyclase which converts ATP to cAMP. Elevated cAMP levels activate PKA (C), which translocates to the nucleus, where it phosphorylates and excludes FoxO1 (D), resulting in derepression of insulin gene transcription (E). Activated PKA also increases intracellular calcium levels (F), and increases insulin secretion. ABCA1 indicates ATP‐binding cassette transporter A1; apoA‐I, apolipoprotein A‐I; FoxO1, forkhead box protein O1; PKA, protein kinase A.
Activated PKA also inactivates K+ channels, opens voltage‐gated Ca2+ channels (Figure 2F), and phosphorylates proteins in the insulin granule surface, leading to an increased Ca2+ response and enhanced secretion of insulin granules from the β‐cell surface (Figure 2G).62, 63 These results offer a potential explanation for the observation that a single rHDL infusion can improve β‐cell function and increase plasma insulin levels in patients with T2DM.2
It is possible that apoA‐I may also improve β‐cell function by regulating intracellular cholesterol trafficking. Internalization of apoA‐I has been reported in endothelial cells,64 skeletal muscle cells,35 and adipocytes.65, 66 Internalization of apoA‐I by β cells has not been reported. Exploration of this possibility would provide insights into whether apoA‐I is able to increase insulin secretion by acting intracellularly as an acceptor of excess cholesterol from insulin granule membranes in islets with elevated cholesterol levels and impaired GSIS.
The HDL‐associated antioxidant enzyme paraoxonase‐1 (PON1) can also increase insulin secretion in both mouse and cell models.67 PON1 knockout mice develop more severe diabetes mellitus when challenged with streptozocin, which selectively destroys β cells, than wild‐type mice. Conversely, streptozotocin‐treated mice transgenic for human PON1 develop less severe diabetes mellitus than control mice.68 Pretreatment with recombinant PON1 before streptozocin administration also protects mice from β‐cell loss, improves glucose tolerance, and increases serum insulin levels.67 These results have been recapitulated in vitro by showing that incubation of the βTC3 cell line with recombinant PON1 increases GSIS and reduces oxidative stress.67 HDLs from wild‐type mice also increase insulin secretion in βTC3 cells under both basal and high‐glucose conditions to a greater extent than HDLs from PON1 knockout mice.67
HDLs also play a pivotal role in maintaining β‐cell survival and protecting against apoptosis. HDLs antagonize the ability of LDLs and very‐low‐density lipoproteins to induce β‐cell apoptosis in isolated rat islets69 and βTC3 cells.70 Isolated human HDLs also prevent oxidized LDLs from reducing insulin and preproinsulin mRNA levels in Min6 cells71, 72 and protect human and murine islets against interleukin‐1β– and glucose‐induced apoptosis.71, 72 Isolated HDLs additionally protect against apoptosis‐induced endoplasmic reticulum (ER) stress in cultured β cells, as well as in human and rat islets.73 In that study, incubation with HDLs restored ER morphology and improved protein folding and export.73 However, in a subsequent study of Min6 cells, HDLs reduced apoptosis by preserving ER morphology but had no effect on protein folding, or the export capacity of the ER.74 This discrepancy may have been attributable to different ER stressors being used in the incubations, which raises the possibility that more than a single mechanism may be responsible for the antiapoptotic effects of HDLs.
Cholesterol Homeostasis and β‐Cell Function
The progressive reduction in β‐cell function and eventual β‐cell loss in patients with T2DM has been attributed to glucotoxicity and elevated free fatty acid levels in association with increased oxidative stress,75 ER stress,76 mitochondrial dysfunction,77 and an elevated inflammatory response, leading to infiltration of inflammatory cells into islets.78 In recent years, mounting evidence has indicated that dysregulation of cholesterol homeostasis in β cells also impairs insulin secretion in response to a glucose challenge and can accelerate the progression of T2DM.59, 79, 80, 81 Elevated cholesterol levels in β cells additionally have the capacity to cause oxidative stress and apoptosis,82 as well as ER stress83 and mitochondrial dysfunction.84
These results are to be expected given that cholesterol homeostasis is a critically important determinant of cell function. Cellular cholesterol is synthesized endogenously or acquired from LDLs that are removed from the circulation via the LDL receptor. Because most peripheral cells lack the necessary machinery for cholesterol catabolism, there is also a need for a mechanism to remove excess cholesterol from cells, a requirement that is fulfilled by ABCA1, which exports cellular cholesterol to lipid‐free/lipid‐poor apoA‐I, and ABCG1, which exports cellular cholesterol to HDLs. Cholesterol also regulates the fluidity and permeability of cell membranes, and it is a key component of the lipid rafts that are located in the outer leaflet of cell membranes and regulate signal transduction pathways.85
Insulin secretory granules are the major site of excess cholesterol accumulation in β cells. This impairs insulin granule maturation, disrupts the insulin secretory machinery,86 and impairs the ability of insulin granules to fuse with the plasma membrane.87 Elevated intracellular β‐cell cholesterol levels can thus reduce insulin secretion by impairing the exocytosis of insulin granules88 and increasing neuronal NO synthase dimerization.79 Direct evidence that cholesterol accumulation causes β‐cell dysfunction emerged from a study by Hao et al,79 who found that GSIS was impaired in cholesterol‐loaded β cells, and normalized by incubation with methyl‐β‐cyclodextrin, which acts as a sink and depletes cells of cholesterol.79 Transgenic mice with selective overexpression of the transcription factor sterol regulatory element‐binding protein‐2 in β cells have normal plasma cholesterol levels but increased islet cholesterol levels attributable to increased transcription of genes encoding for proteins that are rate limiting for cholesterol biosynthesis, including 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase, and the LDL receptor.81 These mice also have impaired GSIS and are glucose intolerant.81
On the other hand, lowering β‐cell cholesterol levels by inhibiting squalene epoxidase, one of the rate‐limiting enzymes in the cholesterol biosynthesis pathway, impairs insulin secretion by reducing activation of voltage‐dependent Ca2+ channels.89, 90 Chronic inhibition of 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase also reduces intracellular cholesterol levels in Ins‐1E cells and impairs insulin secretion by disrupting the structural organization of the plasma membrane.90
Regulation of β‐Cell Function by ABCA1 and ABCG1
Evidence that ABCA1 has a role in β‐cell function in humans comes from studies of patients with Tangier disease that have loss‐of‐function mutations in the gene encoding for ABCA1. These mutations cause cholesterol to accumulate in all cell types, including β cells, where they reduce the first phase of insulin secretion in response to a glucose challenge.91, 92 It is additionally noteworthy that insulin sensitivity is normal in people with loss‐of‐function mutations in ABCA1, which indicates that loss of β‐cell function is likely responsible for the impaired glycemic control that has been reported in these individuals.92, 93
Significant insights into the functional role of ABCA1 and ABCG1 in β cells have been obtained from studies of various mouse models (Figure 3). Mice in which the Abca1 gene is deleted only in β cells have increased islet cholesterol levels and are glucose intolerant.80 They also have impaired GSIS but normal insulin sensitivity.80 These animals additionally exhibit a compensatory upregulation of ABCG1 in β cells, which minimizes the perturbation of β‐cell cholesterol homeostasis caused by the loss of ABCA1.80 These results are consistent with β‐cell dysfunction being directly responsible for the glucose intolerance that has been reported in these animals. This is also consistent with what has been reported in patients with Tangier disease but distinct from what occurs in patients with T1DM and T2DM, where glucose intolerance is driven by autoimmune β‐cell destruction and β‐cell loss subsequent to long‐term insulin resistance, respectively. However, T2DM in the absence of insulin resistance has also been reported in a small number of subjects that are genetically predisposed toward development of the disease.94, 95, 96
Figure 3.

Summary of mouse models used to study the impact of deletion of ABCA1 and/or ABCG1 on β‐cell function. ABCA1 indicates ATP‐binding cassette transporter A1; ABCG1 indicates ATP‐binding cassette transporter G1.
Mice with global ABCG1 deficiency are also glucose intolerant and have impaired GSIS but normal insulin sensitivity (Figure 3).97 The mechanistic basis of this phenotype is distinct from what has been reported for mice with conditional deletion of the Abca1 gene in β cells. Islets isolated from ABCG1 knockout mice have normal cholesterol levels but perturbed cholesterol trafficking that depletes cholesterol from insulin granules. This alters insulin granule morphology and impairs their ability to interact with the insulin secretory machinery, which is a prerequisite for insulin secretion.97, 98
When ABCG1 knockout mice are crossed with mice in which the Abca1 gene is conditionally deleted in β cells, the offspring develop a phenotype that is more pronounced than that reported for either ABCG1 knockout mice or mice with conditional β‐cell deletion of the Abca1 gene (Figure 3).99 In addition to displaying increased islet macrophage infiltration and interleukin‐1β levels, these mice also have more pronounced glucose intolerance, greater cholesterol accumulation in islets, and more severely impaired GSIS than ABCG1 knockout mice or mice with conditional β‐cell ABCA1 deletion.99
Mice in which the Abca1 and Abcg1 genes are both conditionally deleted in β cells have a phenotype that is more complex than what has been reported for the aforementioned models (Figure 3).59 In addition to resembling mice with conditional deletion of ABCA1 in β cells by virtue of having elevated islet cholesterol levels, impaired insulin secretion, and normal insulin sensitivity, mice in which ABCA1 and ABCG1 are both conditionally deleted in β cells also have increased adipose tissue mass, reduced skeletal muscle mass and systemic inflammation.59 When taken together, these studies collectively indicate that cholesterol homeostasis and cholesterol efflux, together with ABCA1 and ABCG1, all play critically important roles in β‐cell function that, if perturbed, can lead to adverse metabolic effects.
Various mechanisms have been proposed to explain β‐cell loss and dysfunction in islets with elevated cholesterol levels. Oxidative stress leading to mitochondrial dysfunction and apoptosis has been reported in Min6 cells with increased cholesterol levels.81, 82, 84, 100, 101, 102 Increased cholesterol levels in Ins1 and βTC‐6 cells also increase ER stress,83 and activate nuclear factor‐κB in Min6 cells, leading to the production of proinflammatory cytokines.101, 102
Cholesterol‐loaded β cells in wild‐type mice and ABCA1 knockout mice also have impaired voltage‐gated Ca2+ channel activity, which reduces glucose‐stimulated Ca2+ influx and insulin secretion.88, 103 In the case of mice with conditional β‐cell deletion of ABCA1, the reduction in GSIS is also associated with ultrastructural changes in the Golgi apparatus, impaired insulin biosynthesis and processing, altered fusion of insulin granules with the plasma membrane, and changes in the organization of proteins that regulate the insulin secretory machinery in the β‐cell membrane.88
Cholesterol Homeostasis and Glucose Disposal in Skeletal Muscle and Adipose Tissue
Caveolae that contain cholesterol, sphingolipids, and caveolin localize to the plasma membrane, where they play a critically important role in the binding of insulin to the insulin receptor and activation of the downstream intracellular insulin signal transduction pathways that mediate glucose uptake by skeletal muscle (Figure 1).104, 105 Caveolae function is highly dependent on plasma membrane cholesterol levels. Disrupting cholesterol homeostasis by depleting cells of cholesterol with β‐cyclodextrin inhibits insulin‐dependent glucose uptake.105 Conversely, increasing cholesterol levels by inducing the hexosamine biosynthesis pathway in 3T3‐L1 adipocytes disrupts the structural organization of the plasma membrane and results in insulin resistance.106
Accumulation of cholesterol in the plasma membrane can also cause insulin resistance in skeletal muscle by decreasing translocation of glucose transporter type 4 to the cell surface.107 This deleterious effect can be reversed by treatment with methyl β‐cyclodextrin, which acts as an acceptor of the excess cholesterol.107, 108
Therapeutic Approaches for Improving Glycemic Control With HDLs and apoA‐I
Emerging evidence indicates that therapies, such as CETP inhibitors and rHDL infusions, that increase plasma HDL‐C and apoA‐I levels have the capacity to slow diabetes mellitus progression, reduce incident diabetes mellitus, and improve glycemic control in patients with established disease.2, 3, 109 However, as these agents were developed to reduce cardiovascular events in at‐risk populations and the outcomes of the clinical trials in which they have been investigated have mostly been negative, the likelihood of any of them being repurposed as a therapy for improving glycemic control in patients with diabetes mellitus is low.
Other HDL‐raising approaches that could be implemented but are likely to improve glycemic control less effectively than CETP inhibition or rHDL infusions include lifestyle interventions such as reducing weight, increasing exercise, and quitting smoking.110, 111, 112 One year of intensive lifestyle intervention that includes calorie restriction and increased physical activity has been reported to improve glycemic control, reduce the use of antidiabetic medications, and increase HDL levels.113 While such interventions may slow disease progression, most likely by improving insulin sensitivity, they fail to address the decline in β‐cell function that drives diabetes mellitus progression. There is thus a major, unmet need to develop new therapies that specifically target the restoration and preservation of β‐cell function in people with prediabetes mellitus or diabetes mellitus.
Cyclodextrins, which accept the excess cell cholesterol that effluxes from cholesterol‐loaded cells may fulfill this need to some extent.114 Cyclodextrin derivatives have shown promising results for treating cardiovascular and neurodegenerative diseases, including atherosclerosis and Niemann‐Pick type C disease.115, 116, 117, 118, 119, 120 Methyl‐β‐cyclodextrin treatment improves glucose tolerance and normalizes fasting glucose levels in mice with diet‐induced obesity.121 It also increases basal and insulin‐stimulated glucose uptake in skeletal muscle,121 and partially restores insulin secretory capacity in isolated islets from apoE‐deficient mice and ob/ob mice.79 As both of these mouse strains have elevated islet cholesterol levels, it follows that this approach may be useful for improving glycemic control in humans with Tangier disease and possibly familial hypercholesterolemia.
Other potential HDL‐targeted options for improving glycemic control include infusion of delipidated HDLs,122 rHDLs,2, 123, 124, 125, 126, 127 and apoA‐I mimetic peptides. The apoA‐I mimetic peptide L‐4F has been shown to reduce adiposity and improve glucose tolerance and insulin sensitivity in ob/ob mice by increasing plasma adiponectin levels, reducing systemic inflammation and phosphorylating AMPK and the insulin receptor.128, 129 The apoA‐I mimetic peptide RG54 also increases glucose uptake in C2C12 myotubes and enhances GSIS in Ins‐1E cells.130 Although considerable effort will be required to develop clinically effective apoA‐I mimetic peptides, they are clearly potential candidates for improving glycemic control, increasing insulin sensitivity, and preventing β‐cell loss in all forms of diabetes mellitus.
Conclusions
Emerging evidence (summarized in Table) indicates that HDL‐ and apoA‐I–targeted therapies are a potential option for conserving residual β‐cell function and improving insulin sensitivity in patients who are progressing toward, or have already developed, T1DM and T2DM. The recent failures of HDL‐raising agents in cardiovascular clinical outcome trials highlight the need to develop novel and innovative HDL‐targeted approaches to achieve these goals. Elucidating the mechanism(s) underlying the antidiabetic functions of HDLs and apoA‐I will also provide opportunities to identify and develop new HDL‐targeted therapies for diabetes mellitus. Achievement of these goals could be particularly advantageous for patients with T1DM for whom treatment options are currently limited to insulin replacement therapy, and for patients with T2DM that are refractory to currently available therapies.
Table 1.
Role of HDL and apoA‐I in Glycemic Control, Insulin Sensitivity and β‐Cell Function
| Topic | Outcome | Reference |
|---|---|---|
| Association of HDL‐C and apoA‐I levels with glycemic control | ||
| Subjects with T2DM | Serum HDL‐C, apoA‐I, and HDL‐C/apoA‐I levels are inversely associated with insulin resistance by HOMA‐IR | 9 |
| Subjects with impaired glucose tolerance | ApoA‐I level is an independent risk factor for glucose tolerance | 10 |
| HDL and apoA‐I in glucose disposal/insulin sensitivity | ||
| Primary human skeletal muscle cells | ApoA‐I improves insulin‐dependent and ‐independent glucose uptake | 27 |
| C2C12 skeletal muscle cells | ApoA‐I increases glucose uptake by phosphorylation of AMPK | 35 |
| High‐fat–fed C57BL/6 mice | ApoA‐I improves insulin sensitivity by reducing systemic and hepatic inflammation | 40 |
| db/db mice | Long‐term HDL infusion improves glucose tolerance by activating GSK‐3 and AMPK in skeletal muscle | 37 |
| Pregnant female Wistar rats | ApoA‐I infusions increase insulin sensitivity, reduces systemic inflammation and protects against pregnancy‐induced insulin resistance | 45 |
| Subjects with T2DM | A single rHDL infusion reduces plasma glucose levels by increasing insulin secretion and promoting glucose uptake in skeletal muscle | 2 |
| HDL and apoA‐I in β‐cell function | ||
| Min6 insulinoma cells | HDLs isolated from normal human plasma, rHDLs, and apoA‐I increase Ins1 and Ins2 gene transcription and GSIS | 58 |
| Ins‐1E insulinoma cells | ApoA‐I increases Pdx1 gene transcription and GSIS | 57 |
| βTC3 insulinoma cells | Incubation with HDL protects βTC3 cells against LDL‐induced apoptosis | 70 |
| C57BL/6 mice | ApoA‐I infusions increase insulin secretion and improve glucose tolerance | 52 |
| High‐fat–fed C57BL/6 mice | Short‐term apoA‐I treatment increases GSIS and improves glucose clearance independent of insulin secretion | 53 |
| Mice with conditional deletion of ABCA1 and ABCG1 in β cells | ApoA‐I infusions increase GSIS in islets isolated from mice with elevated islet cholesterol levels | 54 |
| Healthy subjects and Min6 cells | CETP inhibition increases plasma HDL‐C, apoA‐I, and insulin levels in normal human subjects. Plasma from these subjects also increases GSIS in Min6 cells pretreated with oxidized LDLs | 60 |
| Isolated human islets | HDL protects human islets against oxidized LDL‐induced apoptosis | 71 |
| Isolated human and mouse islets | HDL protects human and mouse islets from interleukin‐1β– and glucose‐induced apoptosis | 72 |
AMPK indicates adenosine monophosphate‐activated protein kinase; apoA‐I, apolipoprotein A‐I; CETP, cholesteryl ester transfer protein; GSIS, glucose‐stimulated insulin secretion; GSK, glycogen synthase kinase‐3; HDL, high‐density lipoprotein; HDL‐C, high‐density lipoprotein cholesterol; HOMA‐IR, Homeostatic model assessment of insulin resistance; LDL, low‐density lipoprotein; rHDL, reconstituted HDL.
Sources of Funding
BM is supported by a University of New South Wales Sydney International Postgraduate Award. BJC and KAR are supported by Grant APP1148468 from the National Health and Medical Research Council of Australia.
Disclosures
None.
J Am Heart Assoc. 2020;9:e013531 . DOI: 10.1161/JAHA.119.013531.
References
- 1. Emerging Risk Factors Collaboration , Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Major Danesh J. lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drew BG, Duffy SJ, Formosa MF, Natoli AK, Henstridge DC, Penfold SA, Thomas WG, Mukhamedova N, de Courten B, Forbes JM, Yap FY, Kaye DM, van Hall G, Febbraio MA, Kemp BE, Sviridov D, Steinberg GR, Kingwell BA. High‐density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation. 2009;119:2103–2111. [DOI] [PubMed] [Google Scholar]
- 3. Barter PJ, Rye KA, Tardif JC, Waters DD, Boekholdt SM, Breazna A, Kastelein JJ. Effect of torcetrapib on glucose, insulin, and hemoglobin A1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Circulation. 2011;124:555–562. [DOI] [PubMed] [Google Scholar]
- 4. Abbasi A, Corpeleijn E, Gansevoort RT, Gans RO, Hillege HL, Stolk RP, Navis G, Bakker SJ, Dullaart RP. Role of HDL cholesterol and estimates of HDL particle composition in future development of type 2 diabetes in the general population: the Prevend Study. J Clin Endocrinol Metab. 2013;98:E1352–E1359. [DOI] [PubMed] [Google Scholar]
- 5. Schmidt MI, Duncan BB, Bang H, Pankow JS, Ballantyne CM, Golden SH, Folsom AR, Chambless LE. Identifying individuals at high risk for diabetes: the Atherosclerosis Risk in Communities Study. Diabetes Care. 2005;28:2013–2018. [DOI] [PubMed] [Google Scholar]
- 6. Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D'Agostino RB Sr. Prediction of incident diabetes mellitus in middle‐aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;167:1068–1074. [DOI] [PubMed] [Google Scholar]
- 7. Hwang YC, Ahn HY, Park SW, Park CY. Association of HDL‐C and apolipoprotein A‐I with the risk of type 2 diabetes in subjects with impaired fasting glucose. Eur J Endocrinol. 2014;171:137–142. [DOI] [PubMed] [Google Scholar]
- 8. Tabara Y, Arai H, Hirao Y, Takahashi Y, Setoh K, Kawaguchi T, Kosugi S, Ito Y, Nakayama T, Matsuda F. Different inverse association of large high‐density lipoprotein subclasses with exacerbation of insulin resistance and incidence of type 2 diabetes: the Nagahama Study. Diabetes Res Clin Pract. 2017;127:123–131. [DOI] [PubMed] [Google Scholar]
- 9. Waldman B, Jenkins AJ, Davis TM, Taskinen MR, Scott R, O'Connell RL, Gebski VJ, Ng MK, Keech AC; FIELD Study Investigators . HDL‐C and HDL‐C/ApoA‐I predict long‐term progression of glycemia in established type 2 diabetes. Diabetes Care. 2014;37:2351–2358. [DOI] [PubMed] [Google Scholar]
- 10. Feng X, Gao X, Yao Z, Xu Y. Low apoA‐I is associated with insulin resistance in patients with impaired glucose tolerance: a cross‐sectional study. Lipids Health Dis. 2017;16:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Muller‐Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stancakova A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutskov K, Langford C, Leander K, Lindholm E, Lobbens S, Mannisto S, Mirza G, Muhleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurethsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvanen AC, Eriksson JG, Peltonen L, Nothen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njolstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen‐Kiukaanniemi SM, Saaristo TE, Korpi‐Hyovalti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jockel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI. Large‐scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia‐Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, Amin N, Barnes D, Cadby G, Hottenga JJ, Ingelsson E, Jackson AU, Johnson T, Kanoni S, Ladenvall C, Lagou V, Lahti J, Lecoeur C, Liu Y, Martinez‐Larrad MT, Montasser ME, Navarro P, Perry JR, Rasmussen‐Torvik LJ, Salo P, Sattar N, Shungin D, Strawbridge RJ, Tanaka T, van Duijn CM, An P, de Andrade M, Andrews JS, Aspelund T, Atalay M, Aulchenko Y, Balkau B, Bandinelli S, Beckmann JS, Beilby JP, Bellis C, Bergman RN, Blangero J, Boban M, Boehnke M, Boerwinkle E, Bonnycastle LL, Boomsma DI, Borecki IB, Bottcher Y, Bouchard C, Brunner E, Budimir D, Campbell H, Carlson O, Chines PS, Clarke R, Collins FS, Corbaton‐Anchuelo A, Couper D, de Faire U, Dedoussis GV, Deloukas P, Dimitriou M, Egan JM, Eiriksdottir G, Erdos MR, Eriksson JG, Eury E, Ferrucci L, Ford I, Forouhi NG, Fox CS, Franzosi MG, Franks PW, Frayling TM, Froguel P, Galan P, de Geus E, Gigante B, Glazer NL, Goel A, Groop L, Gudnason V, Hallmans G, Hamsten A, Hansson O, Harris TB, Hayward C, Heath S, Hercberg S, Hicks AA, Hingorani A, Hofman A, Hui J, Hung J, Jarvelin MR, Jhun MA, Johnson PC, Jukema JW, Jula A, Kao WH, Kaprio J, Kardia SL, Keinanen‐Kiukaanniemi S, Kivimaki M, Kolcic I, Kovacs P, Kumari M, Kuusisto J, Kyvik KO, Laakso M, Lakka T, Lannfelt L, Lathrop GM, Launer LJ, Leander K, Li G, Lind L, Lindstrom J, Lobbens S, Loos RJ, Luan J, Lyssenko V, Magi R, Magnusson PK, Marmot M, Meneton P, Mohlke KL, Mooser V, Morken MA, Miljkovic I, Narisu N, O'Connell J, Ong KK, Oostra BA, Palmer LJ, Palotie A, Pankow JS, Peden JF, Pedersen NL, Pehlic M, Peltonen L, Penninx B, Pericic M, Perola M, Perusse L, Peyser PA, Polasek O, Pramstaller PP, Province MA, Raikkonen K, Rauramaa R, Rehnberg E, Rice K, Rotter JI, Rudan I, Ruokonen A, Saaristo T, Sabater‐Lleal M, Salomaa V, Savage DB, Saxena R, Schwarz P, Seedorf U, Sennblad B, Serrano‐Rios M, Shuldiner AR, Sijbrands EJ, Siscovick DS, Smit JH, Small KS, Smith NL, Smith AV, Stancakova A, Stirrups K, Stumvoll M, Sun YV, Swift AJ, Tonjes A, Tuomilehto J, Trompet S, Uitterlinden AG, Uusitupa M, Vikstrom M, Vitart V, Vohl MC, Voight BF, Vollenweider P, Waeber G, Waterworth DM, Watkins H, Wheeler E, Widen E, Wild SH, Willems SM, Willemsen G, Wilson JF, Witteman JC, Wright AF, Yaghootkar H, Zelenika D, Zemunik T, Zgaga L, Wareham NJ, McCarthy MI, Barroso I, Watanabe RM, Florez JC, Dupuis J, Meigs JB, Langenberg C. A genome‐wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haase CL, Tybjaerg‐Hansen A, Nordestgaard BG, Frikke‐Schmidt R. HDL cholesterol and risk of type 2 diabetes: a Mendelian Randomization Study. Diabetes. 2015;64:3328–3333. [DOI] [PubMed] [Google Scholar]
- 14. Chiesa ST, Charakida M, McLoughlin E, Nguyen HC, Georgiopoulos G, Motran L, Elia Y, Marcovecchio ML, Dunger DB, Dalton RN, Daneman D, Sochett E, Mahmud FH, Deanfield JE. Elevated high‐density lipoprotein in adolescents with Type 1 diabetes is associated with endothelial dysfunction in the presence of systemic inflammation. Eur Heart J. 2019;40:3559–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pinto CS, Lana JM, Gabbay MA, de Sa JR, Dib SA. HDL cholesterol levels and weight are the main determinants of subclinical atherosclerosis in the young with type 1 diabetes and suitable glycaemic control. Diab Vasc Dis Res. 2014;11:125–128. [DOI] [PubMed] [Google Scholar]
- 16. Dabelea D, Stafford JM, Mayer‐Davis EJ, D'Agostino R Jr, Dolan L, Imperatore G, Linder B, Lawrence JM, Marcovina SM, Mottl AK, Black MH, Pop‐Busui R, Saydah S, Hamman RF, Pihoker C. Association of type 1 diabetes vs type 2 diabetes diagnosed during childhood and adolescence with complications during teenage years and young adulthood. JAMA. 2017;317:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gourgari E, Dabelea D, Rother K. Modifiable risk factors for cardiovascular disease in children with type 1 diabetes: can early intervention prevent future cardiovascular events? Curr Diab Rep. 2017;17:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nobécourt E, Tabet F, Lambert G, Puranik R, Bao S, Yan L, Davies MJ, Brown BE, Jenkins AJ, Dusting GJ, Bonnet DJ, Curtiss LK, Barter PJ, Rye KA. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A‐I. Arterioscler Thromb Vasc Biol. 2010;30:766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heier M, Borja MS, Brunborg C, Seljeflot I, Margeirsdottir HD, Hanssen KF, Dahl‐Jorgensen K, Oda MN. Reduced HDL function in children and young adults with type 1 diabetes. Cardiovasc Diabetol. 2017;16:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gourgari E, Playford MP, Campia U, Dey AK, Cogen F, Gubb‐Weiser S, Mete M, Desale S, Sampson M, Taylor A, Rother KI, Remaley AT, Mehta NN. Low cholesterol efflux capacity and abnormal lipoprotein particles in youth with type 1 diabetes: a case control study. Cardiovasc Diabetol. 2018;17:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manjunatha S, Distelmaier K, Dasari S, Carter RE, Kudva YC, Nair KS. Functional and proteomic alterations of plasma high density lipoproteins in type 1 diabetes mellitus. Metabolism. 2016;65:1421–1431. [DOI] [PubMed] [Google Scholar]
- 22. Benghalem I, Meziane W, Hadjidj Z, Ysmail‐Dahlouk L, Belamri A, Mouhadjer K, Aribi M. High‐density lipoprotein immunomodulates the functional activities of macrophage and cytokines produced during ex vivo macrophage‐CD4+ T cell crosstalk at the recent‐onset human type 1 diabetes. Cytokine. 2017;96:59–70. [DOI] [PubMed] [Google Scholar]
- 23. Persegol L, Foissac M, Lagrost L, Athias A, Gambert P, Verges B, Duvillard L. HDL particles from type 1 diabetic patients are unable to reverse the inhibitory effect of oxidised LDL on endothelium‐dependent vasorelaxation. Diabetologia. 2007;50:2384–2387. [DOI] [PubMed] [Google Scholar]
- 24. Jakob P, Luscher TF. Dysfunctional HDL and inflammation: a noxious liaison in adolescents with type 1 diabetes. Eur Heart J. 2019;40:3567–3570. [DOI] [PubMed] [Google Scholar]
- 25. de Boer JF, Annema W, Schreurs M, van der Veen JN, van der Giet M, Nijstad N, Kuipers F, Tietge UJ. Type I diabetes mellitus decreases in vivo macrophage‐to‐feces reverse cholesterol transport despite increased biliary sterol secretion in mice. J Lipid Res. 2012;53:348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carey AL, Siebel AL, Reddy‐Luthmoodoo M, Natoli AK, D'Souza W, Meikle PJ, Sviridov D, Drew BG, Kingwell BA. Skeletal muscle insulin resistance associated with cholesterol‐induced activation of macrophages is prevented by high density lipoprotein. PLoS One. 2013;8:e56601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang S, Tabet F, Cochran BJ, Cuesta Torres LF, Wu BJ, Barter PJ, Rye KA. Apolipoprotein A‐I enhances insulin‐dependent and insulin‐independent glucose uptake by skeletal muscle. Sci Rep. 2019;9:1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch‐Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding ATP‐binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. [DOI] [PubMed] [Google Scholar]
- 29. Francis GA, Knopp RH, Oram JF. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A‐I in Tangier disease. J Clin Invest. 1995;96:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawn RM, Wade DP, Garvin MR, Wang X, Schwartz K, Porter JG, Seilhamer JJ, Vaughan AM, Oram JF. The Tangier disease gene product ABC1 controls the cellular apolipoprotein‐mediated lipid removal pathway. J Clin Invest. 1999;104:R25–R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang N, Silver DL, Costet P, Tall AR. Specific binding of apoA‐I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J Biol Chem. 2000;275:33053–33058. [DOI] [PubMed] [Google Scholar]
- 32. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR‐BI as a high density lipoprotein receptor. Science. 1996;271:518–520. [DOI] [PubMed] [Google Scholar]
- 33. Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein‐mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. [DOI] [PubMed] [Google Scholar]
- 34. Jian B, de la Llera‐Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. [DOI] [PubMed] [Google Scholar]
- 35. Han R, Lai R, Ding Q, Wang Z, Luo X, Zhang Y, Cui G, He J, Liu W, Chen Y. Apolipoprotein A‐I stimulates AMP‐activated protein kinase and improves glucose metabolism. Diabetologia. 2007;50:1960–1968. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Q, Zhang Y, Feng H, Guo R, Jin L, Wan R, Wang L, Chen C, Li S. High density lipoprotein (HDL) promotes glucose uptake in adipocytes and glycogen synthesis in muscle cells. PLoS One. 2011;6:e23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Q, Wan R, Guo R, Jin L, Liu Y, Li S. Long‐term high density lipoprotein infusion ameliorates metabolic phenotypes of diabetic db/db mice. Diabetes Metab Res Rev. 2013;29:130–138. [DOI] [PubMed] [Google Scholar]
- 38. Cochran BJ, Ryder WJ, Parmar A, Tang S, Reilhac A, Arthur A, Charil A, Hamze H, Barter PJ, Kritharides L, Meikle SR, Gregoire MC, Rye KA. In vivo PET imaging with [(18)F]FDG to explain improved glucose uptake in an apolipoprotein A‐I treated mouse model of diabetes. Diabetologia. 2016;59:1977–1984. [DOI] [PubMed] [Google Scholar]
- 39. Dalla‐Riva J, Stenkula KG, Petrlova J, Lagerstedt JO. Discoidal HDL and apoA‐I‐derived peptides improve glucose uptake in skeletal muscle. J Lipid Res. 2013;54:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGrath KC, Li XH, Whitworth PT, Kasz R, Tan JT, McLennan SV, Celermajer DS, Barter PJ, Rye KA, Heather AK. High density lipoproteins improve insulin sensitivity in high‐fat diet‐fed mice by suppressing hepatic inflammation. J Lipid Res. 2014;55:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: review of the underlying molecular mechanisms. J Cell Physiol. 2019;234:8152–8161. [DOI] [PubMed] [Google Scholar]
- 42. Jiang N, Li Y, Shu T, Wang J. Cytokines and inflammation in adipogenesis: an updated review. Front Med. 2019;13:314–329. [DOI] [PubMed] [Google Scholar]
- 43. Sultana A, Cochran BJ, Tabet F, Patel M, Torres LC, Barter PJ, Rye KA. Inhibition of inflammatory signaling pathways in 3T3‐L1 adipocytes by apolipoprotein A‐I. FASEB J. 2016;30:2324–2335. [DOI] [PubMed] [Google Scholar]
- 44. Umemoto T, Han CY, Mitra P, Averill MM, Tang C, Goodspeed L, Omer M, Subramanian S, Wang S, Den Hartigh LJ, Wei H, Kim EJ, Kim J, O'Brien KD, Chait A. Apolipoprotein AI and high‐density lipoprotein have anti‐inflammatory effects on adipocytes via cholesterol transporters: ATP‐binding cassette A‐1, ATP‐binding cassette G‐1, and scavenger receptor B‐1. Circ Res. 2013;112:1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu BJ, Sun Y, Ong KL, Li Y, Tang S, Barter PJ, Rye KA. Apolipoprotein A‐I protects against pregnancy‐induced insulin resistance in rats. Arterioscler Thromb Vasc Biol. 2019;39:1160–1171. [DOI] [PubMed] [Google Scholar]
- 46. Kampmann U, Madsen LR, Skajaa GO, Iversen DS, Moeller N, Ovesen P. Gestational diabetes: a clinical update. World J Diabetes. 2015;6:1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berchtold LA, Prause M, Storling J, Mandrup‐Poulsen T. Cytokines and pancreatic beta‐cell apoptosis. Adv Clin Chem. 2016;75:99–158. [DOI] [PubMed] [Google Scholar]
- 48. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, Yang C, Pannikar A, Doliba N, Zhang T, Stoffers DA, Edlund H, Matschinsky F, Stein R, Stanger BZ. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014;19:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19:872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta‐cells. Nature. 2008;455:627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stenkula KG, Lindahl M, Petrlova J, Dalla‐Riva J, Goransson O, Cushman SW, Krupinska E, Jones HA, Lagerstedt JO. Single injections of apoA‐I acutely improve in vivo glucose tolerance in insulin‐resistant mice. Diabetologia. 2014;57:797–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Domingo‐Espin J, Lindahl M, Nilsson‐Wolanin O, Cushman SW, Stenkula KG, Lagerstedt JO. Dual actions of apolipoprotein A‐I on glucose‐stimulated insulin secretion and insulin‐independent peripheral tissue glucose uptake lead to increased heart and skeletal muscle glucose disposal. Diabetes. 2016;65:1838–1848. [DOI] [PubMed] [Google Scholar]
- 54. Hou L, Tang S, Wu BJ, Ong KL, Westerterp M, Barter PJ, Cochran BJ, Tabet F, Rye KA. Apolipoprotein A‐I improves pancreatic β‐cell function independent of the ATP‐binding cassette transporters ABCA1 and ABCG1. FASEB J. 2019;33:8479–8489. [DOI] [PubMed] [Google Scholar]
- 55. Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP‐binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high‐density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. [DOI] [PubMed] [Google Scholar]
- 57. Cochran BJ, Bisoendial RJ, Hou L, Glaros EN, Rossy J, Thomas SR, Barter PJ, Rye KA. Apolipoprotein A‐I increases insulin secretion and production from pancreatic β‐cells via a G‐protein‐cAMP‐PKA‐FoxO1‐dependent mechanism. Arterioscler Thromb Vasc Biol. 2014;34:2261–2267. [DOI] [PubMed] [Google Scholar]
- 58. Fryirs MA, Barter PJ, Appavoo M, Tuch BE, Tabet F, Heather AK, Rye KA. Effects of high‐density lipoproteins on pancreatic beta‐cell insulin secretion. Arterioscler Thromb Vasc Biol. 2010;30:1642–1648. [DOI] [PubMed] [Google Scholar]
- 59. Cochran BJ, Hou L, Manavalan AP, Moore BM, Tabet F, Sultana A, Cuesta Torres L, Tang S, Shrestha S, Senanayake P, Patel M, Ryder WJ, Bongers A, Maraninchi M, Wasinger VC, Westerterp M, Tall AR, Barter PJ, Rye KA. Impact of perturbed pancreatic β‐cell cholesterol homeostasis on adipose tissue and skeletal muscle metabolism. Diabetes. 2016;65:3610–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Siebel AL, Natoli AK, Yap FY, Carey AL, Reddy‐Luthmoodoo M, Sviridov D, Weber CI, Meneses‐Lorente G, Maugeais C, Forbes JM, Kingwell BA. Effects of high‐density lipoprotein elevation with cholesteryl ester transfer protein inhibition on insulin secretion. Circ Res. 2013;113:167–175. [DOI] [PubMed] [Google Scholar]
- 61. Brown AJ, Mander EL, Gelissen IC, Kritharides L, Dean RT, Jessup W. Cholesterol and oxysterol metabolism and subcellular distribution in macrophage foam cells. Accumulation of oxidized esters in lysosomes. J Lipid Res. 2000;41:226–237. [PubMed] [Google Scholar]
- 62. Song WJ, Seshadri M, Ashraf U, Mdluli T, Mondal P, Keil M, Azevedo M, Kirschner LS, Stratakis CA, Hussain MA. Snapin mediates incretin action and augments glucose‐dependent insulin secretion. Cell Metab. 2011;13:308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu B, Wei S, Petersen N, Ali Y, Wang X, Bacaj T, Rorsman P, Hong W, Sudhof TC, Han W. Synaptotagmin‐7 phosphorylation mediates GLP‐1‐dependent potentiation of insulin secretion from β‐cells. Proc Natl Acad Sci U S A. 2015;112:9996–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fruhwurth S, Pavelka M, Bittman R, Kovacs WJ, Walter KM, Rohrl C, Stangl H. High‐density lipoprotein endocytosis in endothelial cells. World J Biol Chem. 2013;4:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Verghese PB, Arrese EL, Howard AD, Soulages JL. Brefeldin A inhibits cholesterol efflux without affecting the rate of cellular uptake and re‐secretion of apolipoprotein A‐I in adipocytes. Arch Biochem Biophys. 2008;478:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Howard AD, Verghese PB, Arrese EL, Soulages JL. The β‐subunit of ATP synthase is involved in cellular uptake and resecretion of apoA‐I but does not control apoA‐I‐induced lipid efflux in adipocytes. Mol Cell Biochem. 2011;348:155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Koren‐Gluzer M, Aviram M, Meilin E, Hayek T. The antioxidant HDL‐associated paraoxonase‐1 (PON1) attenuates diabetes development and stimulates β‐cell insulin release. Atherosclerosis. 2011;219:510–518. [DOI] [PubMed] [Google Scholar]
- 68. Rozenberg O, Shiner M, Aviram M, Hayek T. Paraoxonase 1 (PON1) attenuates diabetes development in mice through its antioxidative properties. Free Radic Biol Med. 2008;44:1951–1959. [DOI] [PubMed] [Google Scholar]
- 69. Grupping AY, Cnop M, Van Schravendijk CF, Hannaert JC, Van Berkel TJ, Pipeleers DG. Low density lipoprotein binding and uptake by human and rat islet beta cells. Endocrinology. 1997;138:4064–4068. [DOI] [PubMed] [Google Scholar]
- 70. Roehrich ME, Mooser V, Lenain V, Herz J, Nimpf J, Azhar S, Bideau M, Capponi A, Nicod P, Haefliger JA, Waeber G. Insulin‐secreting beta‐cell dysfunction induced by human lipoproteins. J Biol Chem. 2003;278:18368–18375. [DOI] [PubMed] [Google Scholar]
- 71. Abderrahmani A, Niederhauser G, Favre D, Abdelli S, Ferdaoussi M, Yang JY, Regazzi R, Widmann C, Waeber G. Human high‐density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low‐density lipoprotein particles in pancreatic beta cells. Diabetologia. 2007;50:1304–1314. [DOI] [PubMed] [Google Scholar]
- 72. Rutti S, Ehses JA, Sibler RA, Prazak R, Rohrer L, Georgopoulos S, Meier DT, Niclauss N, Berney T, Donath MY, von Eckardstein A. Low‐ and high‐density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta‐cells. Endocrinology. 2009;150:4521–4530. [DOI] [PubMed] [Google Scholar]
- 73. Petremand J, Puyal J, Chatton JY, Duprez J, Allagnat F, Frias M, James RW, Waeber G, Jonas JC, Widmann C. HDLs protect pancreatic β‐cells against ER stress by restoring protein folding and trafficking. Diabetes. 2012;61:1100–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Puyal J, Petremand J, Dubuis G, Rummel C, Widmann C. HDLs protect the MIN6 insulinoma cell line against tunicamycin‐induced apoptosis without inhibiting ER stress and without restoring ER functionality. Mol Cell Endocrinol. 2013;381:291–301. [DOI] [PubMed] [Google Scholar]
- 75. Kaneto H, Fujii J, Seo HG, Suzuki K, Matsuoka T, Nakamura M, Tatsumi H, Yamasaki Y, Kamada T, Taniguchi N. Apoptotic cell death triggered by nitric oxide in pancreatic beta‐cells. Diabetes. 1995;44:733–738. [DOI] [PubMed] [Google Scholar]
- 76. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. [DOI] [PubMed] [Google Scholar]
- 77. Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta‐cell loss in tissue‐specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. [DOI] [PubMed] [Google Scholar]
- 78. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor‐Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, Fontana A, Reinecke M, Homo‐Delarche F, Donath MY. Increased number of islet‐associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–2370. [DOI] [PubMed] [Google Scholar]
- 79. Hao M, Head WS, Gunawardana SC, Hasty AH, Piston DW. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta‐cell dysfunction. Diabetes. 2007;56:2328–2338. [DOI] [PubMed] [Google Scholar]
- 80. Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR. Beta‐cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13:340–347. [DOI] [PubMed] [Google Scholar]
- 81. Ishikawa M, Iwasaki Y, Yatoh S, Kato T, Kumadaki S, Inoue N, Yamamoto T, Matsuzaka T, Nakagawa Y, Yahagi N, Kobayashi K, Takahashi A, Yamada N, Shimano H. Cholesterol accumulation and diabetes in pancreatic beta‐cell‐specific SREBP‐2 transgenic mice: a new model for lipotoxicity. J Lipid Res. 2008;49:2524–2534. [DOI] [PubMed] [Google Scholar]
- 82. Lu X, Liu J, Hou F, Liu Z, Cao X, Seo H, Gao B. Cholesterol induces pancreatic β cell apoptosis through oxidative stress pathway. Cell Stress Chaperones. 2011;16:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kong FJ, Wu JH, Sun SY, Zhou JQ. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol‐induced pancreatic β‐cell injury. Sci Rep. 2017;7:44746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhao YF, Wang L, Lee S, Sun Q, Tuo Y, Wang Y, Pei J, Chen C. Cholesterol induces mitochondrial dysfunction and apoptosis in mouse pancreatic beta‐cell line MIN6 cells. Endocrine. 2010;37:76–82. [DOI] [PubMed] [Google Scholar]
- 85. Silvius JR. Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim Biophys Acta. 2003;1610:174–183. [DOI] [PubMed] [Google Scholar]
- 86. Bogan JS, Xu Y, Hao M. Cholesterol accumulation increases insulin granule size and impairs membrane trafficking. Traffic. 2012;13:1466–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Xu Y, Toomre DK, Bogan JS, Hao M. Excess cholesterol inhibits glucose‐stimulated fusion pore dynamics in insulin exocytosis. J Cell Mol Med. 2017;21:2950–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kruit JK, Wijesekara N, Fox JE, Dai XQ, Brunham LR, Searle GJ, Morgan GP, Costin AJ, Tang R, Bhattacharjee A, Johnson JD, Light PE, Marsh BJ, Macdonald PE, Verchere CB, Hayden MR. Islet cholesterol accumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes. 2011;60:3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xia F, Xie L, Mihic A, Gao X, Chen Y, Gaisano HY, Tsushima RG. Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage‐gated calcium channel function in pancreatic beta‐cells. Endocrinology. 2008;149:5136–5145. [DOI] [PubMed] [Google Scholar]
- 90. Zuniga‐Hertz JP, Rebelato E, Kassan A, Khalifa AM, Ali SS, Patel HH, Abdulkader F. Distinct pathways of cholesterol biosynthesis impact on insulin secretion. J Endocrinol. 2015;224:75–84. [DOI] [PubMed] [Google Scholar]
- 91. Koseki M, Matsuyama A, Nakatani K, Inagaki M, Nakaoka H, Kawase R, Yuasa‐Kawase M, Tsubakio‐Yamamoto K, Masuda D, Sandoval JC, Ohama T, Nakagawa‐Toyama Y, Matsuura F, Nishida M, Ishigami M, Hirano K, Sakane N, Kumon Y, Suehiro T, Nakamura T, Shimomura I, Yamashita S. Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J Atheroscler Thromb. 2009;16:292–296. [DOI] [PubMed] [Google Scholar]
- 92. Vergeer M, Brunham LR, Koetsveld J, Kruit JK, Verchere CB, Kastelein JJ, Hayden MR, Stroes ES. Carriers of loss‐of‐function mutations in ABCA1 display pancreatic beta‐cell dysfunction. Diabetes Care. 2010;33:869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rickels MR, Goeser ES, Fuller C, Lord C, Bowler AM, Doliba NM, Hegele RA, Cuchel M. Loss‐of‐function mutations in ABCA1 and enhanced β‐cell secretory capacity in young adults. Diabetes. 2015;64:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. O'Rahilly SP, Nugent Z, Rudenski AS, Hosker JP, Burnett MA, Darling P, Turner RC. Beta‐cell dysfunction, rather than insulin insensitivity, is the primary defect in familial type 2 diabetes. Lancet. 1986;2:360–364. [DOI] [PubMed] [Google Scholar]
- 95. Pimenta W, Korytkowski M, Mitrakou A, Jenssen T, Yki‐Jarvinen H, Evron W, Dailey G, Gerich J. Pancreatic beta‐cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose‐tolerant individuals with a first‐degree NIDDM relative. JAMA. 1995;273:1855–1861. [PubMed] [Google Scholar]
- 96. Arner P, Pollare T, Lithell H. Different aetiologies of type 2 (non‐insulin‐dependent) diabetes mellitus in obese and non‐obese subjects. Diabetologia. 1991;34:483–487. [DOI] [PubMed] [Google Scholar]
- 97. Sturek JM, Castle JD, Trace AP, Page LC, Castle AM, Evans‐Molina C, Parks JS, Mirmira RG, Hedrick CC. An intracellular role for ABCG1‐mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J Clin Invest. 2010;120:2575–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Churchward MA, Rogasevskaia T, Hofgen J, Bau J, Coorssen JR. Cholesterol facilitates the native mechanism of Ca2+‐triggered membrane fusion. J Cell Sci. 2005;118:4833–4848. [DOI] [PubMed] [Google Scholar]
- 99. Kruit JK, Wijesekara N, Westwell‐Roper C, Vanmierlo T, de Haan W, Bhattacharjee A, Tang R, Wellington CL, LutJohann D, Johnson JD, Brunham LR, Verchere CB, Hayden MR. Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired β‐cell function. Diabetes. 2012;61:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Carrasco‐Pozo C, Gotteland M, Castillo RL, Chen C. 3,4‐dihydroxyphenylacetic acid, a microbiota‐derived metabolite of quercetin, protects against pancreatic β‐cells dysfunction induced by high cholesterol. Exp Cell Res. 2015;334:270–282. [DOI] [PubMed] [Google Scholar]
- 101. Carrasco‐Pozo C, Tan KN, Reyes‐Farias M, De La Jara N, Ngo ST, Garcia‐Diaz DF, Llanos P, Cires MJ, Borges K. The deleterious effect of cholesterol and protection by quercetin on mitochondrial bioenergetics of pancreatic β‐cells, glycemic control and inflammation: in vitro and in vivo studies. Redox Biol. 2016;9:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Carrasco‐Pozo C, Tan KN, Gotteland M, Borges K. Sulforaphane protects against high cholesterol‐induced mitochondrial bioenergetics impairments, inflammation, and oxidative stress and preserves pancreatic β‐cells function. Oxid Med Cell Longev. 2017;2017:3839756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee AK, Yeung‐Yam‐Wah V, Tse FW, Tse A. Cholesterol elevation impairs glucose‐stimulated Ca(2+) signaling in mouse pancreatic β‐cells. Endocrinology. 2011;152:3351–3361. [DOI] [PubMed] [Google Scholar]
- 104. Gustavsson J, Parpal S, Karlsson M, Ramsing C, Thorn H, Borg M, Lindroth M, Peterson KH, Magnusson KE, Stralfors P. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999;13:1961–1971. [PubMed] [Google Scholar]
- 105. Parpal S, Karlsson M, Thorn H, Stralfors P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate‐1, but not for mitogen‐activated protein kinase control. J Biol Chem. 2001;276:9670–9678. [DOI] [PubMed] [Google Scholar]
- 106. Bhonagiri P, Pattar GR, Habegger KM, McCarthy AM, Tackett L, Elmendorf JS. Evidence coupling increased hexosamine biosynthesis pathway activity to membrane cholesterol toxicity and cortical filamentous actin derangement contributing to cellular insulin resistance. Endocrinology. 2011;152:3373–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Habegger KM, Hoffman NJ, Ridenour CM, Brozinick JT, Elmendorf JS. AMPK enhances insulin‐stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology. 2012;153:2130–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Habegger KM, Penque BA, Sealls W, Tackett L, Bell LN, Blue EK, Gallagher PJ, Sturek M, Alloosh MA, Steinberg HO, Considine RV, Elmendorf JS. Fat‐induced membrane cholesterol accrual provokes cortical filamentous actin destabilisation and glucose transport dysfunction in skeletal muscle. Diabetologia. 2012;55:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. The HPS3/TIMI55‐REVEAL Collaborartive Group . Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;377:1217–1227. [DOI] [PubMed] [Google Scholar]
- 110. Thompson PD, Yurgalevitch SM, Flynn MM, Zmuda JM, Spannaus‐Martin D, Saritelli A, Bausserman L, Herbert PN. Effect of prolonged exercise training without weight loss on high‐density lipoprotein metabolism in overweight men. Metabolism. 1997;46:217–223. [DOI] [PubMed] [Google Scholar]
- 111. Enger SC, Herbjornsen K, Erikssen J, Fretland A. High density lipoproteins (HDL) and physical activity: the influence of physical exercise, age and smoking on HDL‐cholesterol and the HDL‐/total cholesterol ratio. Scand J Clin Lab Invest. 1977;37:251–255. [DOI] [PubMed] [Google Scholar]
- 112. Gepner AD, Piper ME, Johnson HM, Fiore MC, Baker TB, Stein JH. Effects of smoking and smoking cessation on lipids and lipoproteins: outcomes from a randomized clinical trial. Am Heart J. 2011;161:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Look ARG, Pi‐Sunyer X, Blackburn G, Brancati FL, Bray GA, Bright R, Clark JM, Curtis JM, Espeland MA, Foreyt JP, Graves K, Haffner SM, Harrison B, Hill JO, Horton ES, Jakicic J, Jeffery RW, Johnson KC, Kahn S, Kelley DE, Kitabchi AE, Knowler WC, Lewis CE, Maschak‐Carey BJ, Montgomery B, Nathan DM, Patricio J, Peters A, Redmon JB, Reeves RS, Ryan DH, Safford M, Van Dorsten B, Wadden TA, Wagenknecht L, Wesche‐Thobaben J, Wing RR, Yanovski SZ. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one‐year results of the look ahead trial. Diabetes Care. 2007;30:1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–17256. [DOI] [PubMed] [Google Scholar]
- 115. Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek A, Hempel C, Heneka MT, Hawxhurst V, Fitzgerald ML, Trebicka J, Bjorkhem I, Gustafsson JA, Westerterp M, Tall AR, Wright SD, Espevik T, Schultze JL, Nickenig G, Lutjohann D, Latz E. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. Chronic cyclodextrin treatment of murine Niemann‐Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 2009;4:e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann‐Pick C disease. Life Sci. 2001;70:131–142. [DOI] [PubMed] [Google Scholar]
- 118. Matsuo M, Togawa M, Hirabaru K, Mochinaga S, Narita A, Adachi M, Egashira M, Irie T, Ohno K. Effects of cyclodextrin in two patients with Niemann‐Pick Type C disease. Mol Genet Metab. 2013;108:76–81. [DOI] [PubMed] [Google Scholar]
- 119. Matsuo M, Shraishi K, Wada K, Ishitsuka Y, Doi H, Maeda M, Mizoguchi T, Eto J, Mochinaga S, Arima H, Irie T. Effects of intracerebroventricular administration of 2‐hydroxypropyl‐β‐cyclodextrin in a patient with Niemann‐Pick Type C disease. Mol Genet Metab Rep. 2014;1:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Berry‐Kravis E, Chin J, Hoffmann A, Winston A, Stoner R, LaGorio L, Friedmann K, Hernandez M, Ory DS, Porter FD, O'Keefe JA. Long‐term treatment of Niemann‐Pick Type C1 disease with intrathecal 2‐hydroxypropyl‐β‐cyclodextrin. Pediatr Neurol. 2018;80:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Llanos P, Contreras‐Ferrat A, Georgiev T, Osorio‐Fuentealba C, Espinosa A, Hidalgo J, Hidalgo C, Jaimovich E. The cholesterol‐lowering agent methyl‐β‐cyclodextrin promotes glucose uptake via GLUT4 in adult muscle fibers and reduces insulin resistance in obese mice. Am J Physiol Endocrinol Metab. 2015;308:E294–E305. [DOI] [PubMed] [Google Scholar]
- 122. Waksman R, Torguson R, Kent KM, Pichard AD, Suddath WO, Satler LF, Martin BD, Perlman TJ, Maltais JA, Weissman NJ, Fitzgerald PJ, Brewer HB Jr. A first‐in‐man, randomized, placebo‐controlled study to evaluate the safety and feasibility of autologous delipidated high‐density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55:2727–2735. [DOI] [PubMed] [Google Scholar]
- 123. Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin‐Dusting J, Hoang A, Sviridov D, Celermajer DS, Kingwell BA. Reconstituted high‐density lipoprotein increases plasma high‐density lipoprotein anti‐inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009;53:962–971. [DOI] [PubMed] [Google Scholar]
- 124. Nieuwdorp M, Vergeer M, Bisoendial RJ, op‘t Roodt J, Levels H, Birjmohun RS, Kuivenhoven JA, Basser R, Rabelink TJ, Kastelein JJ, Stroes ES. Reconstituted HDL infusion restores endothelial function in patients with type 2 diabetes mellitus. Diabetologia. 2008;51:1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nasr H, Torsney E, Poston RN, Hayes L, Gaze DC, Basser R, Thompson MM, Loftus IM, Cockerill GW. Investigating the effect of a single infusion of reconstituted high‐density lipoprotein in patients with symptomatic carotid plaques. Ann Vasc Surg. 2015;29:1380–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tricoci P, D'Andrea DM, Gurbel PA, Yao Z, Cuchel M, Winston B, Schott R, Weiss R, Blazing MA, Cannon L, Bailey A, Angiolillo DJ, Gille A, Shear CL, Wright SD, Alexander JH. Infusion of reconstituted high‐density lipoprotein, CSL112, in patients with atherosclerosis: safety and pharmacokinetic results from a phase 2a randomized clinical trial. J Am Heart Assoc. 2015;4:e002171 DOI: 10.1161/JAHA.115.002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Michael Gibson C, Korjian S, Tricoci P, Daaboul Y, Yee M, Jain P, Alexander JH, Steg PG, Lincoff AM, Kastelein JJ, Mehran R, D'Andrea DM, Deckelbaum LI, Merkely B, Zarebinski M, Ophuis TO, Harrington RA. Safety and tolerability of CSL112, a reconstituted, infusible, plasma‐derived apolipoprotein A‐I, after acute myocardial infarction: the AEGIS‐I trial (ApoA‐I Event Reducing in Ischemic Syndromes I). Circulation. 2016;134:1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, Abraham NG. L‐4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res. 2008;49:1658–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Peterson SJ, Kim DH, Li M, Positano V, Vanella L, Rodella LF, Piccolomini F, Puri N, Gastaldelli A, Kusmic C, L'Abbate A, Abraham NG. The L‐4F mimetic peptide prevents insulin resistance through increased levels of HO‐1, pAMPK, and pAKT in obese mice. J Lipid Res. 2009;50:1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Edmunds SJ, Liebana‐Garcia R, Nilsson O, Domingo‐Espin J, Gronberg C, Stenkula KG, Lagerstedt JO. ApoAI‐derived peptide increases glucose tolerance and prevents formation of atherosclerosis in mice. Diabetologia. 2019;62:1257–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]