

Abstract
Background
Activation of the YAP (Yes‐associated protein) pathway has been demonstrated to be related to smooth muscle cells (SMCs) phenotypic modulation and vessel restenosis. The aim of this study was to illustrate the molecular mechanisms that regulate the expression of YAP during the process of SMCs phenotypic switch. Whether the molecular basis identified in the study could be a potential therapeutic target for drug‐eluting stents is further tested.
Methods and Results
In cell culture and in rat carotid arterial injury models, Sp‐1 (specificity protein 1) expression was significantly induced, and correlated with SMCs proliferative phenotype. Overexpression of Sp‐1 promoted SMCs proliferation and migration. Conversely, siSp‐1 transfection or Sp‐1 inhibitor Mithramycin A treatment attenuates SMC proliferation and migration. Through gain‐ and loss‐function assays, we demonstrated that YAP was involved in Sp‐1‐mediated SMC phenotypic switch. Mechanistically, activated Sp‐1 regulated YAP transcriptional expression through binding to its promoter. Moreover, we fabricated a Sp‐1 inhibitor Mithramycin A‐eluting stent and further tested it. In the rabbit carotid model, Mithramycin A‐eluting stent inhibited YAP transcription and attenuated in‐stent restenosis through regulating YAP‐mediated SMC phenotypic switch.
Conclusions
Sp‐1 controls phenotypic modulation of SMC by regulating transcription factor YAP. Drug‐eluting stent targeting Sp‐1 might represent a novel therapeutic strategy to prevent in‐stent restenosis.
Keywords: drug‐eluting stent, phenotypic modulation, restenosis, smooth muscle cell, transcription factor specificity protein 1, Yes‐associated protein
Subject Categories: Animal Models of Human Disease, Basic Science Research, Smooth Muscle Proliferation and Differentiation
Clinical Perspective
What Is New?
Sp‐1 (specificity protein 1) is upregulated during neointimal formation and Sp‐1 expression correlates with the smooth‐muscle cell synthetic phenotype.
Sp‐1 modulates smooth‐muscle cell phenotypic modulation by increasing Yes‐associated protein promoter activity.
Sp‐1 inhibitor‐eluting stent attenuated restenosis through targeting smooth‐muscle cell phenotypic modulation.
What Are the Clinical Implications?
Targeting Sp‐1–mediated smooth‐muscle cell phenotypic modulation by drug‐eluting stent will be an attractive therapeutic approach for the treatment of occlusive vascular diseases.
Introduction
Recent advances in drug‐eluting stent (DES) fabrication to attenuate in‐stent restenosis are among the encouraging success stories in vascular intervention. DESs such as paclitaxel‐eluting stents and sirolimus‐eluting stents have substantially reduced the rate of angiographic and clinical restenosis.1 Nevertheless, in‐stent neointimal formation still occurs in ≈10% to 15% of all patients.2, 3
Until recently, the underlying molecular mechanism of in‐stent neointimal formation has not been fully understood. The phenotypic switch of vascular smooth‐muscle cells (SMCs) plays a major role in the progression of vascular stenosis. Fully differentiated SMCs are almost quiescent with little proliferation and are programmed for contraction with relatively high expression of SM myosin heavy chain (SMMHC), SM22α, and calponin. However, in response to local vascular injury, SMCs dedifferentiate from contractile phenotype toward synthetic state, which was characterized by a decreased expression of contractile SMC marker genes and increased rates of migration and proliferation.4, 5, 6, 7 An accumulation of synthetic SMCs in the stented artery contribute to in‐stent restenosis. To the best of our knowledge, no commercial DES has been specifically designed to target SMC phenotypic modulation to inhibit restenosis. No researchers have tried this strategy to fabricate DES before.
Unraveling the molecular mechanisms involved in SMC phenotypic modulation is a vital step toward understanding the pathology of restenosis and ultimately designing of therapeutic DES. The switch between the contractile and proliferative SMC phenotypes is regulated by a complex network of signaling pathways that converge on a few conserved transcriptional factors (eg SRF, Krüppel‐like factor 4: KLF 4, c‐MYB, DOCK 2, Olfm 2, etc.).8, 9, 10, 11, 12, 13 Recent studies have identified the hippo‐YAP (Yes‐associated protein) pathway as a key transcriptional factor that drives the expression of SMC signature genes.14, 15, 16, 17 Downregulation of YAP can promote the SMC contractile phenotype through upregulating myocardin expression and increasing the association of the SRF–myocardin complex with SMC contractile gene promoters.18 Despite the fact that YAP plays an important role in SMC contractile marker gene repression, the precise mechanisms responsible for activating YAP expression in the process of SMC dedifferentiation are unknown.
The transcription factor Sp‐1 (specificity protein 1) was recently identified as a promoter binding factor, which plays important roles in various diseases. Sp‐1 contains C2H2‐type zinc fingers and can preferentially bind to the GC consensus site [5′‐(G/T)GGGCGG(G/A)(G/A)(G/T)‐3′].19, 20 Sp‐1 was found to play a critical role in the regulation of histone deacetylase 7 gene transcriptions in SMC differentiation from embryonic stem cells,21 and Sp‐1 is a transcription factor of KLF4, which is a well‐known negative regulator of SMC marker expression.22 After detailed analyses of YAP gene promoter, several GC boxes in the promoter were noted. The presence of such typical characteristics within the YAP promoter might suggest that Sp‐1 regulates YAP gene expression and YAP‐mediated SMC phenotypic modulation. In particular, whether Sp‐1 could be a potential candidate for DES to attenuate in‐stent restenosis through regulating SMC phenotypic modulation is still unknown.
In this study, upregulation of Sp‐1 expression was found during SMC phenotypic modulation, which was induced by balloon injury and platelet‐derived growth factor BB (PDGF‐BB) administration. Conversely, the knockdown of Sp‐1 expression led to impaired proliferation of SMCs and enhanced expression of contractile gene expression. Sp‐1 affected SMC phenotypic modulation through increasing the activity of the YAP promoter. Furthermore, we fabricated DES targeting Sp‐1 in vitro. This DES attenuated restenosis while not affecting re‐endothelialization through regulating SMC phenotypic switch in a rabbit carotid model. This preliminary study provides new insights into the molecular mechanisms controlling phenotypic switch of SMC and identifies a novel potential target for DES.
Methods
The authors declare that all supporting data are available within the article and its online supplementary file.
Rat Aortic SMC Culture
We used the enzyme digestion method to prepare rat aortic SMCs from thoracic aorta. SMCs at passages 6 to 12 were used in the experiments.
Knockdown and Overexpression of Genes
The siRNA or overexpression plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) for knockdown or overexpression of Sp‐1.The sequence of the siSp‐1 oligos were 5′‐UUG AGU CAC CCA AUG AGAA‐3′. YAP overexpression was achieved by transduction with a lentivirus encoding YAP or YAP1S. Mutation of YAP serine 127 to alanine (YAP1S) induces YAP resistance to Lats1/2‐mediated phosphorylation, resulting in a more powerful YAP function.
Real‐Time Quantitative–Polymerase Chain Reaction
Real‐time polymerase chain reaction was performed according to our laboratory workflow. The primer sequences for the involved genes were listed as follows (forward, reverse): Sp‐1: GCATGCACCTGCCCCTACTGTAAAGAC, CGTTTGTGCCTCTGTAGCTCATCC; SM22α: 5′‐CGGCAGATCATCAGT TAGAAAG‐3′, 5′‐GGGCTGAGGCTGAGGATAGGT‐3′; calponin: 5′‐ATG GGCACCAATAAGTTT GC‐3′, 5′‐GACCTGGCTCAAAGATCTGC‐3′; SMMHC: 5′‐ATGCTGGGAAGGTGGACTACAA‐3′, 5′‐TGTGCAGGGCTGTGG TTGA‐3′; YAP: 5′‐TCGGCAGGCAATACGGAATA‐3′, 5′‐CATGCTGAG GCCACTGTCTGT‐3′;18S RNA: 5′‐GGAAGGGCACCACCAGGAGT‐3′,5′‐TGCAGCCCCGGACATCTAAG‐3′.
Western Blot Analysis
We used radioimmunoprecipitation assay buffer to extract protein from SMCs. Twenty micrograms of protein were loaded in 6% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel. Antibodies used were as follows: anti‐Sp‐1 (CST; catalog no. #9389), anti‐YAP (CST; catalog no. #14074), anti‐SM22α (Abcam; catalog no. ab10135), anti‐calponin (Abcam; catalog no. ab46794), anti‐SMMHC (Abcam; catalog no. ab52319), anti‐Cyclin D (CST; catalog no. #2978), anti‐GAPDH (CST; catalog no. #5174). A horseradish peroxidase‐conjugated goat anti‑rabbit secondary antibody (Abcam; catalog no. ab6721) was then added to the membranes at room temperature for 2 hours. Image J software 1.51b (National Institutes of Health, Bethesda, MD, USA) version was used to quantify the protein bands.
Immunofluorescent Staining
SMCs were treated with PDGF‐BB (20 ng/mL) after starving for 24 hours. SMCs were fixed with 4% paraformaldehyde for 10 minutes following permeabilization in 0.2% Triton X‐100 for 20 minutes. Primary antibodies used were rabbit anti‐Sp‐1 antibody (Abcam, catalog no. ab227383) and mouse anti‐SM22α antibody (Abcam, catalog no. ab212857). After incubation with primary antibody, secondary antibodies including anti‐rabbit IgG (Alexa Fluor 488‐conjugate) and anti‐mouse IgG (Alexa Fluor 594‐conjugate) were applied. Cell nucleus was stained with 4′,6‐diamidino‐2‐phenylindole (Molecular Probe).
MTT Assay
Cell viability was measured using the MTT assay. SMCs were transfected with siSp‐1 oligos or treated with Mithramycin A (catalogue no. ab142723, Abcam) 24 hours after seeding in a 96‐well plate. Fifty microliter MTT solution was added to each well and incubated for 4 hours. To dissolve the formazan crystals, dimethyl sulfoxide (100 μL) was added. Absorbance was measured at 570 nm using a microplate reader.
Transwell Invasion Assays and Scratch Wound Assay
We used a 24‐well chamber to perform transwell invasion assays (Costar 3422; Corning Inc., Corning, NY). SMCs were cultured in the upper chamber into DMEM. In the lower chamber, additional 10% FBS was added to DMEM. After 24 hours, SMCs were allowed to migrate from the upper chamber to the underside of the membrane. Migrated SMCs on the lower membrane were counted using an Olympus light microscope and analyzed using the Image J. For scratch wound assay, we used a 100‐μL pipette tip to scratch SMCs and cultured for 36 hours. We performed scratch wound assay in the presence of 25 μg/mL Mitomycin C in order to inhibit SMC proliferation. SMCs were visualized using a microscope and captured images were assessed using Image‑Pro Plus software.
Cell Cycle Assessment
After SMCs were treated with PDGF‐BB, siSp‐1, or Mithramycin A, they were harvested and immersed in 70% ethanol. After washing with PBS, the pellet was fixed with cold propidium iodide solution (50 μg/mL) containing RNase A (0.1 mg/mL). We used a FACS‐Calibur cytometer to analyze at least 1×104 cells for each sample. The percentage of SMCs in different cycle phases was determined using CellQuest software (BD Biosciences).
Luciferase Reporter Assay
After culturing on 12‐well plates for 3 days, SMCs were transfected with reporter plasmids pGL3‐YAP‐Luc (containing 2200, 1150, 450, or 50 bp of proximal YAP promoter region), using FuGENE 6 reagent (Roche). The YAP p450 Sp‐1 mutant pGL3 plasmid was generated using basic restriction enzyme cloning methods as previously reported,22 and we confirmed mutations by sequencing. Renilla activities were further detected as the control. The definition for relative luciferase unit was the ratio of Firefly versus Renilla. The control was set as 1.0.
Chromatin Immunoprecipitation Assay
SMCs were treated with 1% formaldehyde for 10 minutes at room temperature, and were quenched with glycine at normal temperature. SMCs were cultivated for sonication after the medium was removed. Immunoprecipitations consisted of 2 μg of anti‐Sp‐1 antibody (catalog no. ab13370; Abcam), 20 μg of chromatin, and protein G magnetic beads in 200 μL buffer incubated for 24 hours at 4°C on a bottle roller. We added proteinase K solution to a total elution volume of 300 μL and incubated at 60°C overnight in order to reverse the cross‐linking. Semiquantitative polymerase chain reaction was performed to amplify YAP promoter regions containing Sp‐1 binding sites in GC box. In vivo chromatin immunoprecipitation (ChIP) assay was performed on normal and stented aorta. Chromatin–protein complexes were immunoprecipitated with 3 μg anti‐Sp1 antibody using normal IgG as a control. Semiquantitative real‐time polymerase chain reaction was performed following the manufacturer's protocol.
Fabrication of DES
We purchased bare‐metal stents (BMS) (length, 20 mm; diameter, 2 mm) from Dalian Yinyi Biomaterials Development Co., Ltd. (Dalian, China). Sp‐1 inhibitor Mithramycin A (catalog no. ab142723; Abcam)‐eluting stents were fabricated in our laboratory as previously described.15, 23 We used a scanning electron microscope (SEM) to examine the surface morphology of the Mithramycin A‐eluting stent (MES). Mithramycin A release was measured using high‐performance liquid chromatography (Agilent 1100; Agilent Technologies, Inc., Santa Clara, CA) as previously described.23
Rat Balloon Injury Model and Stent Implantation
The current study was approved by Nantong University and Nanjing University's ethical research committee, and all handling and care of the animals was performed in accordance with the guidelines of the laboratory. For rat balloon injury model, rats were anesthetized by an intraperitoneal injection of ketamine (80 mg/kg). A 1.5‐mm balloon catheter was then introduced into the carotid artery through a 0.014‐in guidewire. The balloon was inflated with 10 atm to cause injury to the carotid artery. For the stent implantation model, New Zealand white rabbits (male, body weight between 2.0 kg and 2.5 kg) were randomly divided into 2 groups, and were implanted with BMS (n=12) and MES (n=12) and were followed for 1 month and 6 months.
Three days before the procedure started, rabbits were given 10 mg of aspirin. The carotid artery was isolated from the surrounding tissue with the rabbits under anesthesia. A 0.014‐in run‐through guidewire was advanced into the rabbit carotid artery through a small incision. We then deployed the balloon‐expanded stent by inflation with 12 atm, and closed the cut‐down site with 9‐0 Prolene suture (Ethicon, Inc., Somerville, NJ).
Tissue Harvest and Histomorphological Analyses
After 1 and 6 months following stent deployment, the stents with surrounding arteries were harvested after rabbits were euthanized through injection of potassium chloride. We evenly divided the stented arteries into 3 segments as we did in the previous study.15, 23 The first segment was stained with hematoxylin and eosin for quantitative histomorphological parameters. The second segment was used for SEM. The re‑endothelialized area was assessed using SEM photomicrographs. The third segment was used for ChIP assay and Western blot analysis.
Statistical Analysis
Data are expressed as the mean±SD. Data were analyzed using the Wilcoxon rank sum test. Statistical analyses were performed in SPSS version 13.0 (SPSS, Inc., Chicago, IL). P<0.05 was considered to indicate a statistically significant difference.
Results
Sp‐1 Expression Correlates With SMC Synthetic Phenotype
PDGF‐BB is a potent mediator of the SMC phenotypic modulation from a contractile to a synthetic state by promoting SMC proliferation as well as repressing SMC marker gene expression, which was demonstrated in previous publications.24 Our data revealed that the protein and mRNA level of Sp‐1 in SMCs stimulated by PDGF‐BB were significantly elevated in a dose‐dependent manner compared with the vehicle‐treated control, whereas expression of contractile SMC markers including SM22α, SMMHC, and calponin was significantly reduced (Figure 1A and 1B). Pro‐proliferation gene cyclin D expression was enhanced in parallel with Sp‐1 expression. Consistent with this finding, immunohistochemical staining determined that Sp‐1 localized in the nuclei of the SMCs, and is induced in the synthetic SMCs (Figure 1C). In the in vitro study, the expression of Sp‐1 is upregulated in the balloon injury model, which resembles endovascular angioplasty in humans, both 3 and 14 days following injury. Sp‐1 overexpression coincides with downregulation of SMC contractile genes (Figure 1D and 1E). These data suggest that upregulation of Sp‐1 is positively correlated with the synthetic SMC phenotype both in vitro and in vivo.
Figure 1.
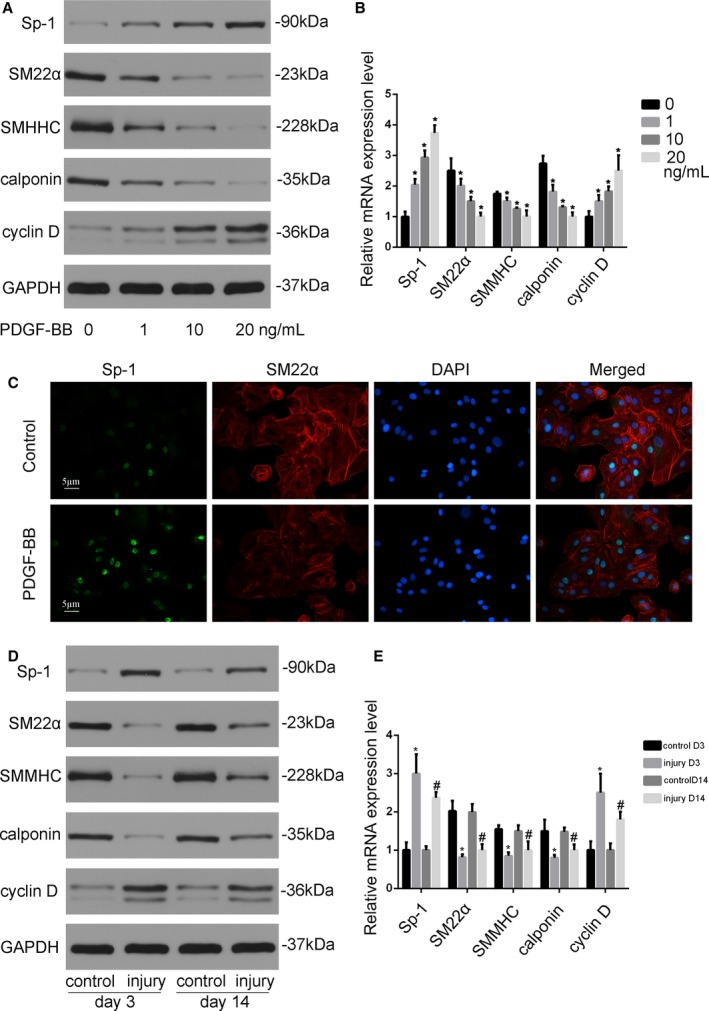
Sp‐1 (specificity protein 1) was upregulated in smooth muscle cell (SMC) phenotypic modulation both in vitro and in vivo. A, Platelet‐derived growth factor‐BB (PDGF‐BB) induced Sp‐1 expression and attenuated contractile protein expression in SMCs. SMCs were starved in DMEM without fetal bovine serum (FBS) for 24 h before being treated with vehicle or PDGF‐BB (1, 10, and 20 ng/mL). Western blotting was performed to examine the expression of Sp‐1, SM22α, SM myosin heavy chain (SMMHC), calponin, and proliferation marker cyclin D. B, Quantitative analysis of mRNA expression. *P<0.05 vs vehicle group (n=3). C, Sp‐1 (green) and SM22α (red) expression were detected by immunostaining. 4′,6‐Diamidino‐2‐phenylindole stained the nuclei. Sp‐1 was highly expressed in the synthetic SMCs. D and E, Three or 14 d after rat carotid artery injury, control or injured arterial vessels were harvested for Western blotting (D) and reverse transcriptase–polymerase chain reaction (E) to assess protein and mRNA expression as indicated. *P<0.05 vs control group at d 3. n=3. #P<0.05 vs control group at d 14 (n=3).
Sp‐1 Promotes SMC Proliferation and Migration
Proliferation of vascular SMCs plays a vital role in the development of neointimal formation, and SMC migration from the medial layer is another key event to build neointima. Since Sp‐1 was induced in phenotypically modulated SMCs and these cells are known to be proliferative and migratory, we directly examined the effects of Sp‐1 on SMC proliferation and migration. Using MTT assays, we found that overexpression of Sp‐1 by plasmid significantly increased SMC proliferation in a variety of cell culture media (Figure 2A and 2B). Knockdown of Sp‐1 by siSp‐1 and Mithramycin A in SMCs reduced cell proliferation compared with controls as shown in Figure 2C. Consistent with impaired SMC proliferation, protein levels of the proliferative marker cyclin D decreased (Figure 2D). Cell cycle analysis further demonstrated that PDGF‐BB‐induced Sp‐1 upregulation resulted in an increase of the percentage of cells in the S phase (from 12±2% to 25±6%). In contrast, siSp‐1 and Mithramycin A treatment significantly blocked cell cycle progression (Figure 2E and 2F). Moreover, PDGF‐BB promoted SMC migration as assessed by transwell invasion assays and scratch wound assay, and silencing Sp‐1 attenuated PDGF‐BB‐induced SMC migration (Figure 2G through 2J). Together, data from these experiments revealed that the rates of SMC proliferation and migration were stimulated by Sp‐1 overexpression while inhibited by silencing endogenous Sp‐1 expression, supporting a positive role for the Sp‐1 in regulating vascular SMC proliferation and migration.
Figure 2.
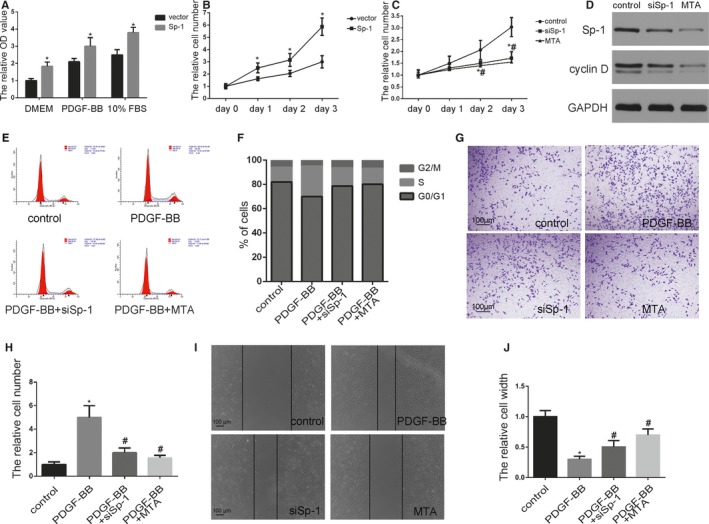
Sp‐1 (specificity protein 1) promoted smooth muscle cell (SMC) proliferation and migration. A, SMCs were transfected with Sp‐1 plasmid which expressed a high level of Sp‐1. Proliferation of SMCs was measured in a variety of cell culture media using 4‐5‐dimethyl‐2‐thiazolyl‐2‐5‐diphenyl‐2H‐tetrazolium bromide assay as indicated. *P<0.05 vs vector group (n=3). B and C, Plasmid Sp‐1 (B), siSp‐1 transfected SMCs (C), or SMC treated by Mithramycin A (MTA) (10 nmol/L) were plated at equal density in 10% fetal bovine serum (FBS) medium and cells were then counted at each time point as indicated. *P<0.05 Sp‐1 or siSp‐1 vs control group (n=3). # P<0.05 MTA treatment vs control group (n=3). D, SMCs were transfected siSp‐1 or treated by MTA. Expression of Sp‐1 and cyclin D were analyzed by Western blotting. E and F, Cell cycle was measured by flow cytometric DNA analysis. The percentage of cells within the sub‐G0/G1, S, and G2/M phases of the cell cycle was determined (S phase control: 12±2%, PDGF‐BB: 25±6%, PDGF‐BB+siSp‐1: 15±2.3%, PDGF‐BB+MTA: 13±2.6%, P<0.05 PDGF‐BB vs control group. n=3). PDGF‐BB induced Sp‐1 upregulation resulted in an increase of the percentage of cells in the S phase. In contrast, siSp‐1 and MTA treatment significantly blocked cell cycle progression. G and H, Cellular invasion ability was analyzed by transwell assay. *P<0.05 vs control group (n=3). # P<0.05 vs PDGF‐BB‐treated group (n=3). I and J, Cellular migration ability was analyzed by cell scratch assay. *P<0.05 vs control group (n=3). # P<0.05 vs PDGF‐BB‐treated group (n=3). OD indicates optical density; PDGF‐BB, platelet‐derived growth factor BB.
Sp‐1 Modulates SMC Phenotypic Modulation by Increasing YAP Promoter Activity
Yap is one of the well‐known factors regulating SMC contractile protein expression. In order to determine the mechanisms underlying Sp‐1 function in SMC phenotypic modulation, we first tested whether Sp‐1 regulates YAP expression. We found that PDGF‐BB increased Sp‐1 expression in parallel with YAP expression. YAP expression level was significantly attenuated by the knockdown of Sp‐1 (siSp‐1 and Mithramycin A treatment) at both the protein and mRNA level (Figure 3A and 3B). In contrast, overexpression of Sp‐1 by plasmid induced YAP expression at the protein and mRNA level (Figure 3C and 3D). Next, we investigated the function of YAP in Sp‐1‐mediated phenotypic modulation using a gain‐of‐function assay. Lentivirus encoding an empty control vector, wild‐type YAP, and active YAP 1S were transduced into SMC. Overexpression of YAP or YAP 1S blocked Sp‐1 inhibitor (Figure 3E and 3F) or siSp‐1 (Figure S1) mediated SMC phenotypic modulation. Furthermore, overexpression of YAP by lentivirus is able to reverse the effect of Mithramycin A on SMC proliferation (Figure 3G). These data provided compelling evidence that Sp‐1 might play an important role in mediating the expression of YAP during the process of SMC phenotypic modulation.
Figure 3.
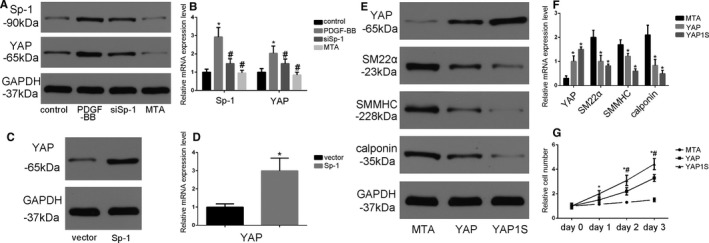
YAP (Yes‐associated protein) was involved in Sp‐1 (specificity protein 1)‐mediated SMC phenotypic modulation. A and B, Platelet‐derived growth factor‐BB (PDGF‐BB, 20 ng/mL for 24 h) induced YAP protein (A) and mRNA (B) expression in parallel with Sp‐1 expression. Additional siSp‐1 transfected or Mithramycin A (MTA) treatment could reverse this process. *P<0.05 vs control group. n=3. # P<0.05 vs PDGF‐BB treatment group. n=3. C and D, Plasmid‐induced Sp‐1 overexpression increased YAP gene expression at both the protein (C) and mRNA (D) level. *P<0.05 vs vector‐treated group. n=3. E and F, SMCs were transduced with control lentivirus or lentivirus encoding wild‐type or 1S mutant YAP, and treated with MTA (20 nmol/L) for 48 h. Sp‐1 inhibitor MTA regulated SMC phenotypic modulation and cyclin D expression could be partially reversed by overexpression of YAP. *P<0.05 vs MTA‐treated group. n=3. G, Cell number was counted at the time points as indicated to determine the effects of MTA, MTA+YAP, and MTA+YAP 1S on proliferation. *P<0.05 YAP vs MTA‐treated group. n=3. # P<0.05 YAP 1S vs MTA‐treated group. n=3. SMMHC indicates SM myosin heavy chain.
To investigate the mechanisms of Sp‐1 on YAP expression in SMC, we used a series of promoter‐reporter plasmids containing various lengths of YAP promoter (depicted in Figure 4A). Cultured SMCs were transfected with the different YAP promoter‐reporter plasmids and either control plasmid or Sp‐1 plasmid. Promoter activity was analyzed by luciferase assay. The results showed that overexpression of Sp‐1 increased the activity of the 2200‐, 1150‐, and 450‐bp promoters but was unable to increase the activity of the 50‐bp promoter (Figure 4B). SMCs were then cotransfected with control or siSp‐1 plasmids and 1 of the YAP promoter‐reporter plasmids. After knockdown, SMCs were treated with vehicle or PDGF‐BB. The knockdown of Sp‐1 prevented PDGF‐BB‐induced activation of the 2200‐, 1150‐, and 450‐bp YAP promoters (Figure 4C). These results suggest that the regulatory elements necessary for the PDGF‐BB‐induced increase in YAP expression are found within −450 and −50 bp of the 5′ region of the YAP promoter. We analyzed the −450/−50 region using the PROMO platforms (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promo.cgi?). Interestingly, we found 2 potential Sp‐1 binding GC boxes located in this region of YAP promoter (Figure 4D). Whether the 2 Sp‐1 sites within the YAP promoter were responsible for YAP transcriptional activity was further tested. The sequence of each of these 2 sites was mutated from GGGCGG to GTTTTG (as depicted in Figure 4E), which has previously been shown to abolish Sp‐1 binding.22, 25 The mutation of the 2 Sp‐1 sites drastically decreased the baseline activity of the 450‐bp YAP promoter compared with the wild‐type (Figure 4F). In addition, the mutation of the 2 Sp‐1 sites prevented Sp‐1 plasmid‐induced increases in YAP promoter activity. ChIP assays were performed to confirm Sp‐1 binding to the −450/−50 region. We observed that this region in SMCs overexpressing Sp‐1 promoted Sp‐1 binding to YAP promoter. The opposite effects were observed upon Mithramycin A treatment (Figure 4G through 4H). These data indicated that Sp‐1 bound the YAP promoter and increased YAP transcription in the process of SMC phenotypic modulation, and Sp‐1 inhibitor Mithramycin A attenuated these effects.
Figure 4.
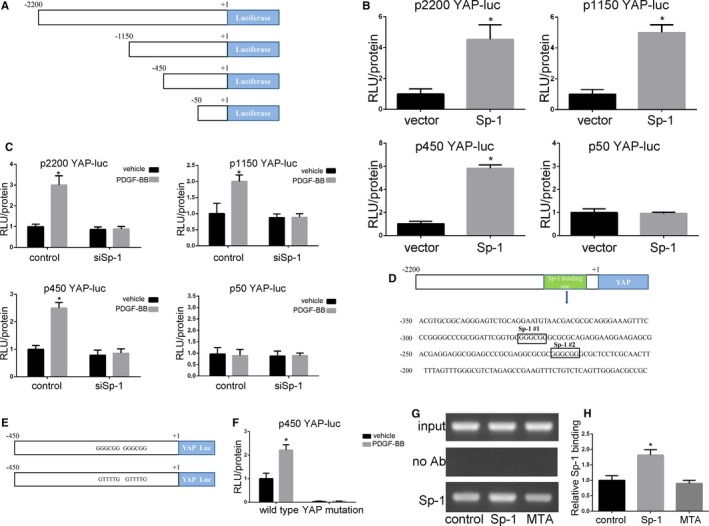
Sp‐1 (specificity protein 1) increased YAP (Yes‐associated protein) promoter activity in the process of SMC phenotypic modulation. A, Schematic showing the series of YAP promoter‐reporter plasmids used. B, Smooth muscle cells (SMCs) were transfected with the 2200‐, 1150‐, 450‐, or 50‐bp YAP promoter‐luciferase (Luc) plasmid and further transfected with vector or Sp‐1 plasmid before determining relative changes in luciferase expression. Data indicated that Sp‐1 overexpression‐mediated increases in YAP promoter activity required sequences between −450 and −50 bp. *P<0.05 vs vector group. n=3. C, siRNA knockdown of Sp‐1 abolished platelet‐derived growth factor‐BB (PDGF‐BB)‐induced activation of the YAP promoter. SMCs were cotransfected with the 2200‐, 1150‐, 450‐, or 50‐bp YAP promoter reporter plasmid and either control siRNA plasmid or siSp‐1 for 36 h, and treated with PDGF‐BB (20 ng/mL) or vehicle for 4 h before demonstrating relative changes in luciferase expression. *P<0.05 vs vehicle group. n=3. D, Sequence of the YAP promoter region from −450 to −50 showing the 2 consensus Sp‐1 binding sites. E and F, Mutation of the 2 Sp‐1 binding sites with the 450‐bp YAP promoter abolished basal and PDGF‐BB (20 ng/mL)‐induced activity. E indicates that 2 consensus Sp‐1 binding sites in the 450‐bp YAP promoter were mutated from GGGCGG to GTTTTG to generate a 450‐bp Sp‐1 mutant YAP promoter‐reporter plasmid. F, SMCs were transfected with either wild‐type or Sp‐1 mutant YAP promoter and treated with vehicle or PDGF‐BB (20 ng/mL) to demonstrate changes in luciferase activity. *P<0.05 vs vehicle group. n=3. G and H, SMCs were transfected with Sp‐1 plasmid to make Sp‐1 overexpression or treated with Mithramycin A after Sp‐1 plasmid transfection. Changes in binding of Sp‐1 to the region of the YAP promoter containing the 2 putative Sp‐1 sites were determined by semiquantitative in vivo ChIP assay. *P<0.05 vs control group. n=3. ChIP indicates chromatin immunoprecipitation; RLU, relative luciferase units.
Fabrication of Sp‐1 Inhibitor‐Eluting Stent and Stent Implantation
Stents were crimped onto a shrunken catheter balloon to fabricate balloon‐expanded stents. A pressure of 12 atm was applied to make the stent expand fully. Incubation of BMS in Mithramycin A jelly solution resulted in coating with this compound, and 35.2±5.1 μg Mithramycin A was bound to the stent. The SEM images of BMS and MES are shown in Figure 5A. For MES, there is no doubt that a uniform coating is successfully constructed on the stent with no cracking and debonding. After balloon expansion, no delamination or other effects were observed on the surface, confirming that the coating was able to withstand the compressive strains. The results of the in vitro release of Mithramycin A from MES are shown in Figure 5B. We note that ≈80% of Mithramycin A was released in 7 days and almost of it was released in 2 weeks. Stents were successfully deployed in the rabbit carotid arteries (as depicted in Figure 5C). Postoperative radiography indicated that no stent migration and fracture occurred in the following time (Figure 5D).
Figure 5.
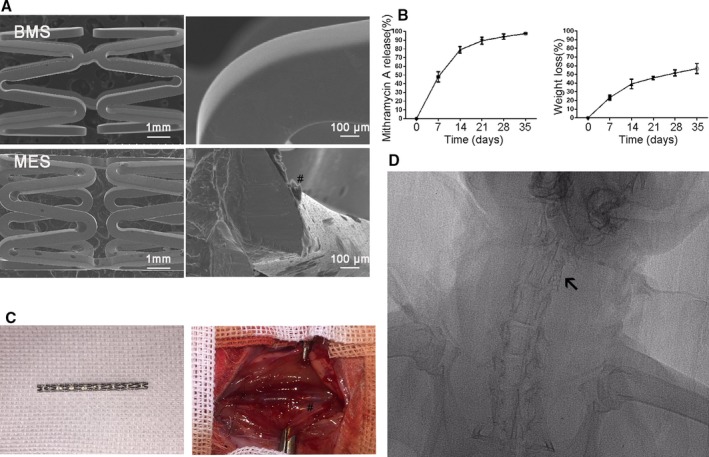
Fabrication of Sp‐1 (specificity protein 1) inhibitor Mithramycin A (MTA)‐eluting stent and stent implantation. A, Scanning electron microscope (SEM) images of BMS and MTA‐eluting stent (MES). #Indicates the coating surface of the stents, which was smooth and uniform. B, Cumulative release profile of Mithramycin A from MES. C, Stents were successfully implanted in the rabbit carotid model. #Indicates the location of the stent. D, Postoperative radiograph demonstrates no stent migration and fracture in the following time. Arrow in the figure indicates the location of the stent.
Sp‐1 Inhibitor‐Eluting Stent Attenuated Restenosis Through Targeting SMC Phenotypic Modulation
Histological and morphometric analyses were performed to evaluate restenosis. At 6 months poststent implantation, the average intimal thickness varied (BMS: 0.36±0.05 mm, MES: 0.15±0.01 mm). Mithramycin A‐eluting stent had lower average intimal thickness than BMS. In addition, the neointimal areas were 1.33±0.08 mm2 in the BMS group, and 0.36±0.04 mm2 in the MES group (P<0.05, Figure 6A and 6B).
Figure 6.
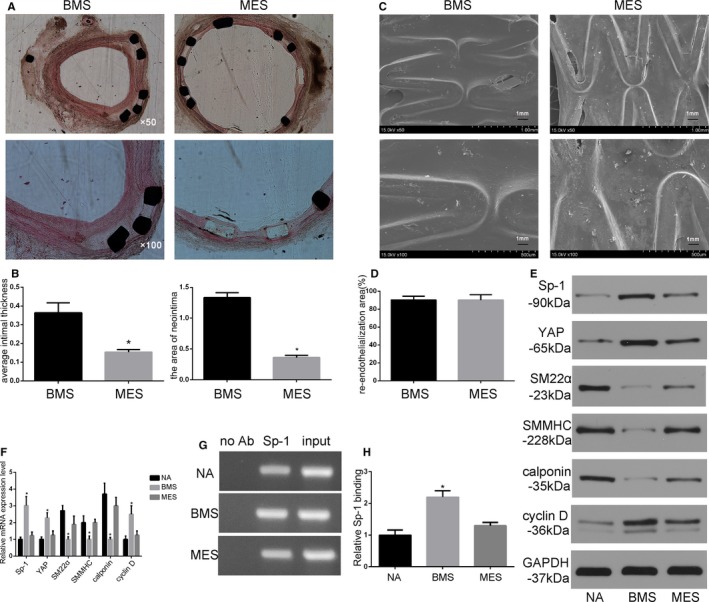
Sp‐1 (specificity protein 1) inhibitor‐eluting stent attenuated restenosis through targeting smooth muscle cell phenotypic modulation. A, Representative images of hematoxylin and eosin staining of cross sections 6 mo after implantation. Mithramycin A‐eluting stent (MES) had significantly reduced neointimal formation compared with bare metal stent (BMS). B, Statistical analysis of average intimal thickness and areas of neointima of the stents. *P<0.05 vs BMS. n=6. C and D, Re‐endothelialization assessed by scanning electron microscope of the stented arteries 4 weeks following stent implantation. The BMS and MES were efficient at inducing re‐endothelialization. E and F, Protein expression (E) and mRNA (F) from normal artery (NA), BMS, and MES. *P<0.05 vs NA. n=6. G and H, Change in binding of Sp‐1 to the region of YAP (Yes‐associated protein) promoter was determined by semiquantitative in vivo ChIP assay. Quantitative graph (H) showing the change in Sp‐1 binding to the YAP promoter based on densitometry measurements from 3 independent in vivo ChIP assay experiment. *P<0.05 vs NA. n=3. ChIP indicates chromatin immunoprecipitation; SMMHC, SM myosin heavy chain.
We evaluated the re‐endothelialization area by SEM at 1 month. Re‐endothelialization was almost complete in the present study 1 month following implantation. The re‐endothelialization areas for the BMS group and the MES group were as follows: BMS group: 90.3± 4.2% versus MES group: 90.0± 6.1% (Figure 6C and 6D, P<0.05). MES was equally efficient at increasing re‐endothelialization compared with BMS. These data indicated that MES attenuated in‐stent restenosis without interfering with re‐endothelialization.
Protein and mRNA expression of neointimal formation were further analyzed. Sp‐1 and YAP expression were significantly higher in BMS than in the Sp‐1 inhibitor‐eluting stent. BMS reduced the expression of contractile SMC marker, including SM22α, calponin, and SMMHC, whereas cyclin D was dramatically induced in parallel with the expression of Sp‐1 and YAP compared with normal artery. Contractile SMC markers were induced in the Sp‐1 inhibitor‐eluting stent, suggesting that the Sp‐1 inhibitor‐eluting stent attenuated neointimal formation through regulation of SMC phenotype modulation (Figure 6E and 6F). To further illustrate the potential transcriptional effects of Sp‐1 on YAP in vivo, we examined whether Sp‐1 binding to GC box of the YAP promoter was attenuated in the MES group. For these experiments, we performed in vivo ChIP assays on chromatin isolated from normal and stented arteries. As shown in Figure 6G, the Sp‐1 binding to the endogenous YAP promoter was enhanced 2.2‐fold in response to BMS implantation compared with normal artery. However, this effect was significantly attenuated in the MES group. Taken together, these in vivo data provide evidence that the Sp‐1 inhibitor‐eluting stent inhibited YAP transcription and attenuated restenosis through regulating YAP‐mediated SMC phenotypic modulation.
Discussion
DESs have been demonstrated to be more effective than BMSs in reducing in‐stent restenosis. However, the antiproliferative agents released by DESs also suppressed endothelial cell growth, resulting in delayed endothelialization, which raised concerns over the future of clinical application of DES.26 SMC phenotypic modulation and vascular remodeling have recently been proved to contribute to the development of atherosclerosis and restenosis after angioplasty. Regulating phenotypic modulation of SMC after stenting might be a promising but untested therapeutic target. Eluted drugs from stents that target SMC phenotypic modulation would decrease the growth and migration of SMC, and thereby reduce restenosis.15 In this study, we tested the hypothesis that the Sp‐1 inhibitor‐eluting stent could attenuate in‐stent restenosis through regulating SMC phenotypic modulation (Figure 7).
Figure 7.
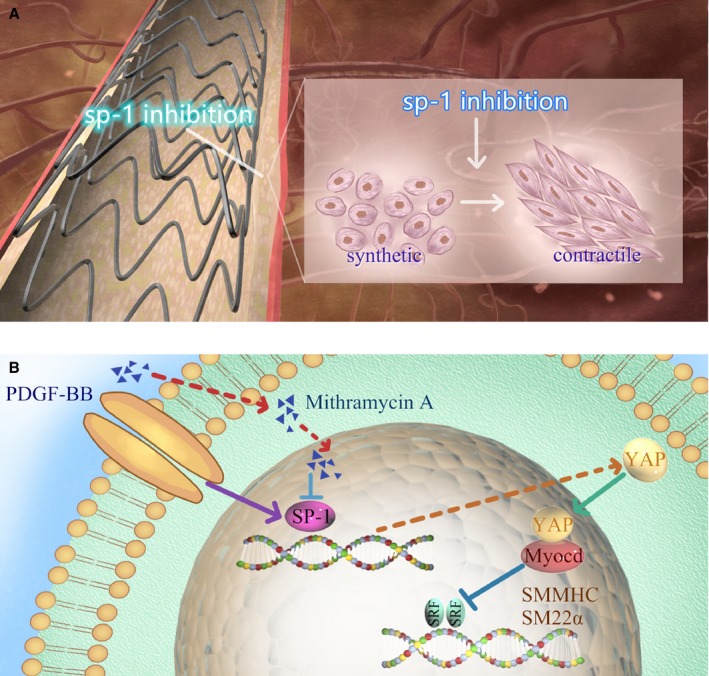
Working hypothesis whereby drug‐eluting stent targeting Sp‐1 (specificity protein 1) inhibited neointimal formation via regulating YAP (Yes‐associated protein)‐mediated smooth muscle cell (SMC) phenotypic switch. A, An accumulation of synthetic SMCs in the stented artery contributed to in‐stent restenosis. Sp‐1 inhibitor eluted from the stent regulates SMC from synthetic to contractile state. B, Mithramycin A downregulates YAP expression through inhibiting transcription factor Sp‐1. Reduced YAP expression facilitates MyocD/SRF interaction and thereby maintains contractile gene expression (SMMHC, SM22α). PDGF‐BB indicates platelet‐derived growth factor BB; SMMHC, SM myosin heavy chain.
Several promising therapeutic DESs are being tested in models of restenosis after stent implantation. Some researchers identified miRNAs as therapeutic targets of DES in restenosis.27, 28 Vascular gene therapy was also thought to be a successful alternative for improving clinical outcomes of stent deployment.29 We previously fabricated DES (sunitinib‐eluting stent) to reduce the level of the PDGF pathway, and this DES inhibited neointimal formation in a rabbit carotid model.23 Nevertheless, despite the number of strategies currently available to reduce restenosis, the complex multifactorial nature of this pathophysiological process frequently leads to a failure of its prevention in patients. We believe that efforts should be concentrated in particular on the critical pathological and signaling molecules that control the progression of in‐stent restenosis. Detailed studies have revealed the existence of different subtypes of SMCs (contractile type and synthetic type) contributing to restenosis. Neointimal hyperplasia implies SMC switching from a contractile to a synthetic phenotype, followed by their proliferation and migration toward the strut of stents. We fabricated a new DES in this study to regulate SMC phenotypic switching, anticipating its potential effect in attenuating restenosis. However, before fabricating new DES, an effort should be made to identify key regulators of SMC phenotypic modulation.
Herein, Sp‐1 was identified as a regulator in SMC phenotypic modulation and vascular remodeling. There is low expression of Sp‐1 in normal SMC and vascular tissue. However, it is induced during SMC phenotypic modulation induced by serum or PDGF‐BB in vitro and vascular injury in vivo. Knockdown of Sp‐1 by siRNA or Mithramycin A increased the expression of SMC marker proteins downregulated by PDGF‐BB in vitro, which demonstrated that Sp‐1 promoted SMC phenotypic modulation, leading to vascular remodeling and restenosis. Consistent with these findings, Sp‐1 deficiency also suppressed PDGF‐BB‐induced SMC proliferation and migration. Importantly, stent implantation induced Sp‐1 expression, which is likely attributed to injury‐triggered serum factors such as PDGF‐BB. Blocking the initial expression of Sp‐1 immediately after stent implantation using eluted drug Mithramycin A effectively blocked injury‐induced in‐stent restenosis, indicating that stent‐based inhibition of the Sp‐1 pathway attenuated neointimal formation through regulating SMC phenotypic modulation. To the best of our knowledge, no commercially available DES has been specifically designed to target SMC phenotypic modulation to inhibit neointimal formation. No researchers have tried this strategy to fabricate DES before. We tried this idea in our preliminary study and found this new DES could attenuate restenosis in a rabbit carotid model.
We demonstrate that Sp‐1 modulates SMC phenotypic switch by increasing YAP gene transcription. Using a truncated YAP promoter fragment, we confirmed that Sp‐1 does bind to the region of YAP promoter containing the 2 Sp‐1 sites within intact chromatin, and that knockdown of Sp‐1 prevented PDGF‐BB‐induced increases in endogenous YAP expression. Overexpression of YAP or YAP 1S blocked Sp‐1 inhibitor‐medicated smooth muscle phenotypic modulation compared with the empty control vector. Of critical significance, the results from in vivo ChIP assays showed an increased binding of Sp‐1 to the YAP promoter in rabbit aorta in response to stent implantation. This strongly suggests that the role of Sp‐1‐dependent increase in YAP expression is not an artifact of cell culture but represents a universal mechanism to increase YAP expression, resulting in the phenotypic modulation of SMC associated with in‐stent restenosis. YAP can physically interact with Myocd in SMC, and increased YAP‐Myocd interaction could attenuate Myocd binding to SRF, thereby downregulating smooth‐specific gene transcription.18 Previous study has shown that Sp‐1 could bind to the KLF4 promoter and regulate SMC phenotypic modulation in an aortic balloon injury model.22 Our current data have identified an additional mechanism through which Sp‐1 mediates the repression of SMC marker genes. Whether Sp‐1‐mediated YAP and KLF4 can cooperatively regulate SMC phenotypic modulation needs further investigation. Theoretically, Sp‐1 may alter the expression of multiple genes that contribute to phenotypic switching. Indeed, Sp‐1 has been shown to increase the expression of genes involved in promoting SMC proliferation (such as PDGF‐BB), and migration (such as matrix metalloproteinase‐9).30 Despite our incomplete knowledge of the precise mechanisms of Sp‐1‐mediated repression, it is clear that it involves multiple mechanisms that significantly contribute to SMC phenotypic switching.
A number of important SMC marker genes have been identified to control SMC differentiation and phenotypic switch. Drugs have been designed and discovered recently to reverse SMC phenotypic modulation, and thus attenuate restenosis in animal models. Most of the studies chose oral administration of drugs, which required a relatively high dose and might cause adverse effects. We identified Sp‐1 as a vital regulator for SMC phenotypic switch and fabricated the Sp‐1 inhibitor‐eluting stent. The estimated dose of Sp‐1 inhibitor in our DES was 35.2±5.1 μg per stent, which is lower than the drug dose used in oral application. Approximately 80% of Mithramycin A was released in 7 days and almost of it was released in 2 weeks. The sustained release is undoubtedly because of the hydrophobicity of Mithramycin A. Mithramycin A‐eluting stent inhibited in‐stent restenosis through regulating SMC phenotypic modulation in a rabbit model. It is likely that the inhibition of neointimal formation is mediated by longer retention and slow release of Mithramycin A at our DES implantation site. In vivo molecular biology data on YAP transcription, SMC marker, Sp‐1 gene, and YAP gene expression further confirmed the inhibitory effect of our DES on neointimal formation.
Delayed healing following implantation is a predictable feature of DES, which may be the result of adverse effects of the nonspecific antiproliferative agent used.31, 32In our study, SEM revealed almost complete re‐endothelialization in BMS and MES, suggesting that Sp‐1 inhibitor has no toxic effects on vascular endothelial cells. Inoue et al demonstrated that stent can trigger their MMP‐9 release, possibly leading to the mobilization of bone marrow–derived stem cells. These reactions were substantially inhibited by sirolimus‐eluting stents.33 Another considerable factor is that DES implanted in arteries always encounter long, heavily calcified, or chronically occluded lesions. Therefore, rapid re‑endothelialization is difficult in humans. Certain pro‐healing strategies might be developed to promote endothelialization. Stents with CD34 antibodies have shown excellent results in animals.34 In our previous study, we found that systemic application of hepatocyte growth factor was an efficient and logical strategy for the prevention of impaired re‐endothelialization.35 Thus, the combination of SMC phenotypic modulation and pro‐healing strategy may be promising in fabrication of new DES.
Neo‐atherosclerosis is another concern of DES, causing late or very late stent thrombosis. This pathological process was marked by foamy macrophages infiltration, followed by in‐stent atherosclerotic plaque development.36 Neo‐atherosclerosis might represent a potential target for DES. Implanted stents can cause a chronic foreign body reaction, and subsequently release pro‐inflammatory factors (NF‐κB, IL‐1, IL‐6, etc.). Inflammation is an important driver of neo‐atherosclerosis. Previous study has identified an Sp‐1 binding site in IL‐1 promoter, and Sp‐1 has been demonstrated to markedly enhance IL‐1 synthesis.37 Thus, it is natural to speculate that DES targeting Sp‐1 may regulate inflammation in the stented artery, and therefore attenuate neo‐atherosclerosis. Future study will be mandatory to confirm this hypothesis.
In conclusion, this study focused on the potential target of DES to attenuate neointimal formation. We demonstrate that Sp‐1 is a highly regulated transcriptional factor that is involved in regulating SMC phenotypic modulation that contributes to restenosis. Inhibition of Sp‐1 suppresses both SMC proliferation and migration. Before Sp‐1 was exploited as a target for DES, mechanistic understanding of its activities on the YAP promoter was illustrated. Our data indicate that Sp‐1 bound the YAP promoter and increased YAP transcription in the process of SMC phenotypic modulation. In a rabbit model, Sp‐1 inhibitor‐eluting stent inhibits in‐stent neointimal formation without affecting re‐endothelialization. Therefore, targeting Sp‐1‐mediated SMC phenotypic modulation by DES will be an attractive therapeutic approach for the treatment of occlusive vascular diseases.
Sources of Funding
This study was funded by grants from National Nature Science Foundation of China (81600374).
Disclosures
None.
Supporting information
Figure S1. siSp‐1 regulated smooth muscle cell (SMC) phenotypic modulation and cyclin D expression could be partially reversed by overexpression of Yes‐associated protein (YAP). *P<0.05 vs siSp‐1‐treated group. n=3.
(J Am Heart Assoc. 2020;9:e014103 DOI: 10.1161/JAHA.119.014103.)
References
- 1. Piccolo R, Bonaa KH, Efthimiou O, Varenne O, Baldo A, Urban P, Kaiser C, Remkes W, Räber L, de Belder A, van ‘t Hof AWJ, Stankovic G, Lemos PA, Wilsgaard T, Reifart J, Rodriguez AE, Ribeiro EE, Serruys PWJC, Abizaid A, Sabaté M, Byrne RA, de la Torre Hernandez JM, Wijns W, Jüni P, Windecker S, Valgimigli M; Coronary Stent Trialists’ Collaboration . Drug‐eluting or bare‐metal stents for percutaneous coronary intervention: a systematic review and individual patient data meta‐analysis of randomised clinical trials. Lancet. 2019; 393:2503–2510. [DOI] [PubMed] [Google Scholar]
- 2. Räber L, Yamaji K, Kelbæk H, Engstrøm T, Baumbach A, Roffi M, von Birgelen C, Taniwaki M, Moschovitis A, Zaugg S, Ostojic M, Pedrazzini G, Karagiannis‐Voules DA, Lüscher TF, Kornowski R, Tüller D, Vukcevic V, Heg D, Windecker S. Five‐year clinical outcomes and intracoronary imaging findings of the COMFORTABLE AMI trial: randomized comparison of biodegradable polymer‐based biolimus‐eluting stents with bare‐metal stents in patients with acute ST‐segment elevation myocardial infarction. Eur Heart J. 2019;40:1909–1919. [DOI] [PubMed] [Google Scholar]
- 3. Baan J Jr, Claessen BE, Dijk KB, Vendrik J, van der Schaaf RJ, Meuwissen M, van Royen N, Gosselink ATM. A randomized comparison of paclitaxel‐eluting balloon versus everolimus‐eluting stent for the treatment of any in‐stent restenosis: the DARE trial. JACC Cardiovasc Interv. 2018;11:275–283. [DOI] [PubMed] [Google Scholar]
- 4. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 5. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. [DOI] [PubMed] [Google Scholar]
- 6. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;95:194–204. [DOI] [PubMed] [Google Scholar]
- 8. Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292:C70–C81. [DOI] [PubMed] [Google Scholar]
- 9. Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Krüppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. J Biol Chem. 2005;280:9719–9727. [DOI] [PubMed] [Google Scholar]
- 10. Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of KLF4, pELK‐1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo X, Shi N, Cui XB, Wang JN, Fukui Y, Chen SY. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ Res. 2015;116:e71–e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng WL, She ZG, Qin JJ, Guo JH, Gong FH, Zhang P, Fang C, Tian S, Zhu XY, Gong J, Wang ZH, Huang Z, Li H. Interferon regulatory factor 4 inhibits neointima formation by engaging krüppel‐like factor 4 signaling. Circulation. 2017;136:1412–1433. [DOI] [PubMed] [Google Scholar]
- 13. Shi N, Li CX, Cui XB, Tomarev SI, Chen SY. Olfactomedin 2 regulates smooth muscle phenotypic modulation and vascular remodeling through mediating runt‐related transcription factor 2 binding to serum response factor. Arterioscler Thromb Vasc Biol. 2017;37:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Hu G, Gao X, Wang Y, Zhang W, Harmon EY, Zhi X, Xu Z, Lennartz MR, Barroso M, Trebak M, Chen C, Zhou J. The induction of yes‐associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arterioscler Thromb Vasc Biol. 2012;32:2662–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang C, Zhou M, Zheng X. RhoA inhibitor‐eluting stent attenuates restenosis by inhibiting YAP signaling. J Vasc Surg. 2019;69:1581–1589. [DOI] [PubMed] [Google Scholar]
- 16. Yu Q, Li W, Jin R, Yu S, Xie D, Zheng X, Zhong W, Cheng X, Hu S, Li M, Zheng Q, Li G, Song Z. PI3Kγ (Phosphoinositide 3‐Kinase γ) regulates vascular smooth muscle cell phenotypic modulation and neointimal formation through CREB (Cyclic AMP‐response element binding protein)/YAP (Yes‐Associated Protein) Signaling. Arterioscler Thromb Vasc Biol. 2019;39:e91–e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Hu G, Liu F, Wang X, Wu M, Schwarz JJ, Zhou J. Deletion of yes‐associated protein (YAP) specifically in cardiac and vascular smooth muscle cells reveals a crucial role for YAP in mouse cardiovascular development. Circ Res. 2014;114:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xie C, Guo Y, Zhu T, Zhang J, Ma PX, Chen YE. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. 2012;287:14598–14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagaoka M, Shiraishi Y, Sugiura Y. Selected base sequence outside the target binding site of zinc finger protein Sp1. Nucleic Acids Res. 2001;29:4920–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beishline K, Azizkhan‐Clifford J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015;282:224–258. [DOI] [PubMed] [Google Scholar]
- 21. Yang G, Pei Y, Teng H, Cao Q, Wang R. Specificity protein‐1 as a critical regulator of human cystathionine gamma‐lyase in smooth muscle cells. J Biol Chem. 2011;286:26450–26460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deaton RA, Gan Q, Owens GK. Sp1‐dependent activation of KLF4 is required for PDGF‐BB‐induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol. 2009;296:H1027–H1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang C, Mei H, Zhou M, Zheng X. A novel PDGF receptor inhibitor‐eluting stent attenuates in‐stent neointima formation in a rabbit carotid model. Mol Med Rep. 2017;15:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Halterman JA, Kwon HM, Zargham R, Bortz PD, Wamhoff BR. Nuclear factor of activated T cells 5 regulates vascular smooth muscle cell phenotypic modulation. Arterioscler Thromb Vasc Biol. 2011;31:2287–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deacon K, Onion D, Kumari R, Watson SA, Knox AJ. Elevated SP‐1 transcription factor expression and activity drives basal and hypoxia‐induced vascular endothelial growth factor (VEGF) expression in non‐small cell lung cancer. J Biol Chem. 2012;287:39967–39981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Colombo A, Chandrasekhar J, Aquino M, Ong TK, Sartori S, Baber U, Lee M, Iniguez A, Hajek P, Borisov B. Safety and efficacy of the COMBO bio‐engineered stent in an all‐comer PCI cohort: 1‐Year final clinical outcomes from the MASCOT post‐marketing registry. Int J Cardiol. 2019;283:67–72. [DOI] [PubMed] [Google Scholar]
- 27. Moore KJ, Rayner KJ. Local anti‐miR delivery: the latest in the arsenal of drug‐eluting stents. Arterioscler Thromb Vasc Biol. 2015;35:1905–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang D, Deuse T, Stubbendorff M, Chernogubova E, Erben RG, Eken SM, Jin H, Li Y, Busch A, Heeger CH, Behnisch B, Reichenspurner H, Robbins RC, Spin JM, Tsao PS, Schrepfer S, Maegdefessel L. Local MicroRNA modulation using a novel anti‐miR‐21‐eluting stent effectively prevents experimental in‐stent restenosis. Arterioscler Thromb Vasc Biol. 2015;35:1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hytönen JP, Taavitsainen J, Laitinen JTT, Partanen A, Alitalo K, Leppänen O, Ylä‐Herttuala S. Local adventitial anti‐angiogenic gene therapy reduces growth of vasa‐vasorum and in‐stent restenosis in WHHL rabbits. J Mol Cell Cardiol. 2018;121:145–154. [DOI] [PubMed] [Google Scholar]
- 30. Shin SS, Park SS, Hwang B, Moon B, Kim WT, Kim WJ, Moon SK. MicroRNA‐892b influences proliferation, migration and invasion of bladder cancer cells by mediating the p19ARF/cyclin D1/CDK6 and Sp‐1/MMP‐9 pathways. Oncol Rep. 2016;36:2313–2320. [DOI] [PubMed] [Google Scholar]
- 31. Arbustini E, Favalli V, Narula J. Functionally Incomplete Re‐endothelialization of stents and neoatherosclerosis. JACC Cardiovasc Interv. 2017;10:2388–2391. [DOI] [PubMed] [Google Scholar]
- 32. Inoue T, Croce K, Morooka T, Sakuma M, Node K, Simon DI. Vascular inflammation and repair: implications for re‐endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc Interv. 2011;4:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Inoue T, Taguchi I, Abe S, Toyoda S, Nakajima K, Sakuma M, Node K. Activation of matrix metalloproteinase‐9 is associated with mobilization of bone marrow‐derived cells after coronary stent implantation. Int J Cardiol. 2011;152:332–336. [DOI] [PubMed] [Google Scholar]
- 34. Chen F, Yang F, Zhao Q, Feng S, Li W, Bi Y, Zhang S, Wang Y, Feng BO. Long‐term effects of novel combination coating anti‐CD34 antibody applied on sirolimus‐eluting stents. J Interv Cardiol. 2015;28:257–263. [DOI] [PubMed] [Google Scholar]
- 35. Huang C, Zheng X, Mei H, Zhou M. Rescuing impaired re‐endothelialization of drug‐eluting stents using the hepatocyte growth factor. Ann Vasc Surg. 2016;36:273–282. [DOI] [PubMed] [Google Scholar]
- 36. Borovac JA, D'Amario D, Vergallo R, Porto I, Bisignani A, Galli M, Annibali G, Montone RA, Leone AM, Niccoli G. Neoatherosclerosis after drug‐eluting stent implantation: a novel clinical and therapeutic challenge. Eur Heart J Cardiovasc Pharmacother. 2019;5:105–116. [DOI] [PubMed] [Google Scholar]
- 37. Husmann M, Jehnichen P, Jahn B, Schlosshan D, Romahn E, Marx J. A novel SP‐1 site in the human interleukin‐1 beta promoter confers preferential transcriptional activity in keratinocytes. Eur J Immunol. 1996;26:3008–3014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. siSp‐1 regulated smooth muscle cell (SMC) phenotypic modulation and cyclin D expression could be partially reversed by overexpression of Yes‐associated protein (YAP). *P<0.05 vs siSp‐1‐treated group. n=3.