

Abstract
Background
CardioChimeras produced by fusion of murine c‐kit+ cardiac interstitial cells with mesenchymal stem cells promote superior structural and functional recovery in a mouse model of myocardial infarction compared with either precursor cell alone or in combination. Creation of human CardioChimeras (hCCs) represents the next step in translational development of this novel cell type, but new challenges arise when working with c‐kit+ cardiac interstitial cells isolated and expanded from human heart tissue samples. The objective of the study was to establish a reliable cell fusion protocol for consistent optimized creation of hCCs and characterize fundamental hCC properties.
Methods and Results
Cell fusion was induced by incubating human c‐kit+ cardiac interstitial cells and mesenchymal stem cells at a 2:1 ratio with inactivated Sendai virus. Hybrid cells were sorted into 96‐well microplates for clonal expansion to derive unique cloned hCCs, which were then characterized for various cellular and molecular properties. hCCs exhibited enhanced survival relative to the parent cells and promoted cardiomyocyte survival in response to serum deprivation in vitro.
Conclusions
The generation of hCC is demonstrated and validated in this study, representing the next step toward implementation of a novel cell product for therapeutic development. Feasibility of creating human hybrid cells prompts consideration of multiple possibilities to create novel chimeric cells derived from cells with desirable traits to promote healing in pathologically damaged myocardium.
Keywords: cardiac, cardiac interstitial cells, cell fusion, human, mesenchymal stromal cells
Subject Categories: Basic Science Research, Cell Therapy, Cellular Reprogramming, Stem Cells, Myocardial Biology
Clinical Perspective
What Is New?
“Next‐generation” cell therapeutics will build on initial findings that demonstrate enhanced reparative action of combining distinct cell types for treatment of cardiomyopathic injury.
Differential biological properties of various cell types are challenging for optimization of delivery, engraftment, persistence, and synergistic action when used in combination.
Creation of a novel hybrid cell called a CardioChimera overcomes limitations inherent to use of multiple cell types, and they exhibit unique properties relative to either parental cell anticipated to be advantageous in cellular therapeutic applications.
What Are the Clinical Implications?
CardioChimeras have now been created and characterized using cells derived from human heart tissue, advancing initial proof of concept previously demonstrated with mice.
CardioChimeras represent an engineered solution that can be implemented as a path forward for improving the outcome of myocardial cell therapy.
Introduction
Beneficial, albeit modest, effects of cell therapy for treatment of myocardial damage have been established in extensive preclinical studies1, 2 as well as early clinical trial testing.3, 4, 5 Improvement of efficacy remains a primary focus of the cell therapy field with ongoing studies attempting to define whether there is an optimal cell type or even a combination of cell types delivered simultaneously to promote reparative remodeling.6, 7 The concept of creating an “enhanced” cell with augmented biological properties to promote healing remains a longstanding interest of our group. Simply stated, we have sought to create “unnatural solutions” to the natural limitations of myocardial repair by ex vivo modification resulting in engineered cell products derived from c‐kit+ cardiac interstitial cells (cCICs) possessing enhanced survival, persistence, proliferation, and many other desirable characteristics that deliver greater recovery from myocardial injury than corresponding originating parental cells.8, 9, 10, 11 One example of this philosophy is the CardioChimera (CC), named for the fusion of cCICs and mesenchymal stem cells (MSCs) together into a single hybrid cell that can be expanded in culture and used for adoptive transfer therapy to mitigate myocardial infarction injury.8 The seminal study of CCs was performed using a homotypic murine model system with delivery murine CCs, and results demonstrated superiority of the CCs over either cCICs alone, MSCs alone, or the combination of cCICs and MSCs delivered together as a single cell suspension. These promising findings prompted subsequent studies to translate the murine findings into the human context; however, working with myocardial‐derived human cells presents a new set of challenges to be overcome.
As opposed to murine cCICs and MSCs obtained from 2‐ to 3‐month‐old hearts with high yield quantities,8 human cardiac cells are derived from small ventricular biopsy specimens.12 Limited source material results in lower yield of human myocardial‐derived cells, and the relatively slow proliferation of myocardial‐derived human cells relative to mouse counterparts12, 13 further hampers therapeutic manipulations. Furthermore, replicative senescence occurs much earlier in culture passaging of human cells, whereas murine cardiac cells demonstrate significantly extended passaging capability.8, 14, 15 Another technical challenge posed by inherent species‐specific biology are differences between murine and human cells in response to experimental interventions.16 Murine cells readily fuse and form polyploid hybrid cells,8 whereas human cells are much less tolerant of polyploidy leading to genomic instability17, 18 that necessitates rigorous optimization of culture conditions to overcome impaired expansion following fusion. Efficient and reproducible creation of CCs is facilitated by use of murine cardiac cell populations from syngeneic healthy young donors8 unlike the individual patient‐specific nature of human myocardial‐derived cells with unavoidable sample characteristic variability.14 Collectively, these challenges prompted this study to demonstrate feasibility and reproducibility of human CC (hCC) generation.
Concurrent isolation of 3 distinct cardiac interstitial cell (CIC) types comprising cCIC, MSC, and endothelial progenitor cells from a single human myocardial tissue sample with optimized culture condition was developed by our group.12 Therefore, this protocol was employed to obtain human cCIC and MSC for subsequent fusion proof of concept using low passage neonatal human myocardial‐derived cells. The rationale for using neonatal cells for feasibility testing is based on their more youthful biological phenotype of enhanced proliferation and survival, preservation of telomere length, and decreased level of senescence relative to cells derived from patients with heart failure.14, 15, 19
hCCs created in the present study represent novel hybrid cells with phenotypic properties consistent with our previous observations of murine CCs. Having overcome the multiple aforementioned challenges of human myocardial‐derived cell utilization, cell fusion is a feasible promising engineering approach to enhance functional properties of human cardiac cells and pave the way for “designer” hCCs exhibiting enhanced reparative properties.
Methods
The authors declare that all supporting data are available within the article (and its online supplementary files). The data that support the findings of this study are available from the corresponding author upon reasonable request.
cCIC and MSC Isolation
The study was designed in accordance with and approved by the institutional review committees at San Diego State University along with the institutional review board (#120686) and no informed consent was required. Neonatal cells were derived from nonsurgically obtained postmortem cardiac tissue. Cells were isolated as previously published.12 Briefly, heart samples were mechanically minced into 1‐mm3 pieces and digested in collagenase II (150 U mg/mL, Worthington, LS004174) followed by brief low‐speed (850 rpm for 2 minutes) centrifugation to remove cardiomyocytes and tissue debris. The supernatant was subjected to magnetic‐activated cell sorting using c‐kit–conjugated microbeads (Miltenyi Biotec, #130‐091‐332), and c‐kit–enriched cells were plated in human c‐kit CIC media. The c‐kit–negative population was further purified by magnetic‐activated cell sorting for MSC surface markers CD90 and CD105. c‐kit CICs and MSCs were incubated at 37°C in 5% CO2 and used for cellular fusion between passages 5 and 10. Media used in the study are listed in Table S1.
Lentiviral Constructs and Stem Cell Infection
Lentiviral plasmids and viral particles were created as previously described.8 c‐kit CICs and MSCs were stably transduced at passage 6 with PGK (phosphoglycerate kinase)‐EGFP (enhanced green fluorescent protein)‐ Puro and PGK‐mCherry‐Bleo, respectively, at a multiplicity of infection of 40. Expression of EGFP and mCherry fluorescent proteins in c‐kit CICs and MSCs was confirmed by fluorescent microscopy as well as flow cytometry. Lentivirally modified cells were stabilized through multiple rounds of passaging and freeze/thaw cycles before fusion experiments.
Cell Fusion and Creation of hCCs
Cell fusion was performed with GenomeONETM‐CF EX Sendai Virus (Hemagglutinating virus of Japan envelope or HVJ‐E) Envelope Cell Fusion Kit (Cosmo Bio USA) using the suspension method according to the manufacturer's protocol. Briefly, cCIC‐EGFP and MSC‐mCherry populations were combined in 25 μL of 1X cell fusion buffer at a ratio of 2:1 (total cell number of 100 000). The 2:1 ratio yielded the maximum number of double‐positive fused cells (hCCs) and therefore was chosen for the study. Inactivated Sendai virus was added to the cell mixture and incubated on ice for 5 minutes. Cell suspension was transferred to the 37°C incubator for 30 minutes with intermittent shaking every 5 minutes. Next, cells were plated in 2‐mL fusion media on a well of a 6‐well dish. Media was changed after 24 hours and cells were then cultured in fusion media for 4 days. Hybrid cells were identified and isolated by fluorescence‐activated cell sorter (FACS) sorting using parent cells in pure as well as mixed (but not fused) as controls. hCCs were plated on a 96‐well microplate (1 cell per well) for clonal expansion. Clone nomenclature was based on well number in the plate. All hCCs were used between passages 4 to 6 for experiments.
Light Microscopy and Measurement of Cell Morphology
Bright field images of hCCs and the parent cells were obtained using a LEICA DM IL microscope and cell boundaries were outlined by image analysis using ImageJ software (National Institutes of Health). Cell surface area and length to width ratio were determined as previously described.20
Proliferation and Doubling Time
c‐kit CICs, MSCs, and hCCs were plated on a 6‐well dish at a density of 20 000 cells per well. At 3 time points (days 1, 3, and 5) cells were trypsinized and counted manually using a hemocytometer. Cell proliferation rate was determined for each group as fold change over day 1. Cell doubling times were calculated using online population doubling time software (http://www.doubling-time.com/compute_more.php).
Immunocytochemistry
c‐kit CICs, MSCs, and hCCs were plated on a 2‐well chamber slide (15 000 per well). Staining was performed following the protocol previously described.20 Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole diluted in 1X PBS at room temperature for 15 minutes. Cells were imaged using a Leica TCS SP8 confocal microscope. Antibodies and dilutions are listed in Table S2.
Immunoblot Analysis
c‐kit CICs, MSCs, and hCCs were plated on 100‐mm plates. Protein cell lysates were collected using 200 μL of SDS‐PAGE protein sample buffer. Proteins were separated on a 4% to 12% NuPage Novex Bis Tris gel (Invitrogen) and transferred onto a polyvinylidene fluoride membrane. Nonspecific binding sites were blocked using Odyssey blocking buffer (LI‐COR, Inc) and proteins were labeled with primary antibodies in 0.2% Tween in Odyssey blocking buffer overnight. After washing, blots were incubated with secondary antibodies in 0.2% Tween 20 in Odyssey blocking buffer for 1.5 hours at room temperature and scanned using the LICOR Odyssey CLx scanner. Quantification was performed using ImageJ software. Antibodies and their dilutions are listed in Table S2.
Real‐Time Polymerase Chain Reaction
c‐kit CICs, MSCs, and hCCs were plated on 100‐mm plates. RNA lysate was collected from the culture and purified using Quick‐RNA MiniPrep (Zymo Research). cDNA synthesis was conducted using iScript cDNA Synthesis Kit (Bio‐Rad Laboratories, Inc). Quantitative polymerase chain reaction was performed on a Bio‐Rad CFX real‐time cycler using iQ SYBR Green (Bio‐Rad Laboratories, Inc) and gene‐specific primers. Signals were normalized to 18S for analysis. Data were calculated using the ∆∆C(t) method. Primers are listed in Table S3.
Ploidy and Cytogenic Analysis
c‐kit CICs, MSCs, and hCCs were plated on a 6‐well plate at a density of 50 000 cells per well. Cells were collected the following day and centrifuged at 305g for 5 minutes, the pellet was resuspended in 70% ethanol, and stored at −20°C for at least 24 hours before use. After centrifugation at 350g for 5 minutes, cell pellet was resuspended in 350 μL of propidium iodide incubated at 37°C for 15 minutes before flow cytometry analysis. Cytogenetic analysis of c‐kit CICs, MSCs, and hCCs (G4) plated at a density of 300 000 cells on 2500 mm2 flasks was performed by KaryoLogic, Inc (http://www.karyologic.com).
Cell Death Assay
For reactive oxygen injury, c‐kit CICs, MSCs, and hCCs were plated on a 6‐well plate at a density of 60 000 cells per well. Cells were subjected to low serum media for 24 hours (depleted to 25% of growth media serum level) followed by 4 hours of hydrogen peroxide (350 μmol/L) treatment. Annexin V and Sytox Blue staining was performed to label apoptotic and necrotic cells and cell death was measured using FACS Aria (BD Biosciences).
For ischemia‐reperfusion injury, cCICs, MSCs, and hCCs were seeded on 6‐well plates at a density of 60 000 cells per well. The following day, media was replaced with Krebs‐Heinsleit buffer to induce glucose starvation, and cells were transferred to a hypoxic incubator with 1% oxygen tension for 3 hours to simulate ischemia. Cells were re‐exposed to regular growth media and incubated in a standard cell culture incubator with ambient (21%) oxygen for 24 hours to simulate reperfusion. Annexin V and Sytox Blue staining was performed to label apoptotic and necrotic cells, and cell death was measured using FACS Aria (BD Biosciences). Cells cultured in growth media in normoxic conditions and cells subjected to Krebs‐Heinsleit buffer in hypoxic condition were used as the controls of the experiment to measure basal and hypoxia‐induced cell death, respectively. Krebs‐Heinsleit buffer and the respective media used inside the hypoxic glove box were equilibrated in hypoxia overnight before starting the experiment.
NRCM Co‐Culture With Stem Cells
Neonatal rat cardiomyocytes (NRCMs) were isolated as previously described21, 22 and seeded in a 6‐well dish at a density of 200 000 per well in M199 media with 15% fetal bovine serum. The following day, cells were incubated in media with 10% fetal bovine serum for 8 hours followed by 24‐hour serum depletion in serum‐free media. Stem cells (cCICs, MSCs, combination of cCICs and MSCs, hCCs) were added to the culture at a 1:5 ratio. The slow‐growing clone B3 was excluded from this experiment because of a low expansion rate. After 24 hours in co‐culture, cells were stained with Annexin V and Sytox Blue. Unlike CCs or their parent cells, the NRCMs were nontransduced allowing for separation by FACS of negative cells (NRCMs) versus green fluorescent protein+, mCherry+, or green fluorescent protein+/mCherry+ cells. Thus, parental and CC cells were removed from the population for survival analysis of NRCMs, which was completed by flow cytometry using the FACS Aria. Controls for the NRCMs included: (1) culture in serum‐free media alone; (2) “rescue” by replenishment with M199 media + 10% serum; or (3) constant culture in M199 media + 10% serum for the duration of the experiment.
Statistical Analysis
All data are expressed as mean±SEM. Statistical significance was assessed using 1‐way ANOVA or 2‐way ANOVA for multiple comparisons, with the Dunnett and Tukey tests as post hoc tests to compare groups with a control group in GraphPad Prism version 5.0 or Microsoft excel. P<0.05 was considered statistically significant.
Results
hCCs Creation From c‐kit CIC and MSC Cell Fusion
Human c‐kit CICs expressing green fluorescent protein and human MSCs expressing mCherry, incubated in the presence of inactivated RNA Sendai virus, underwent cellular fusion to form mononuclear hybrid cells23, 24 (Figure 1A). A group of mixed c‐kit CICs and MSCs without the viral treatment was used as the negative control for the fusion experiment (Figure S1E). Readily detectable uniform fluorescence labeling of parental c‐kit CICs and MSCs is required for optimal yield of double fluorescent hybrids. Flow cytometry analysis shows 96.6% of c‐kit CICs and 92% of MSCs expressed EGFP and mCherry, respectively (Figure S1A through S1D). Double fluorescent hybrids were sorted into 96‐well microplates for clonal expansion to derive hybrid clones called hCCs. Five unique hCCs named D6, F1, G4, D2, and B3 were derived from 5 independent fusion experiments with 1% to 4% efficiency (Table). Dual fluorescent positivity of hCCs was validated by fluorescence microscopy (Figure S2). Immunocytochemistry with respective EGFP and mCherry antibodies confirmed integration of genomic content from both parent cells into hCC that was maintained after clonal expansion and passaging in culture (Figure 1B). Parent cells immunolabeled for EGFP and mCherry exhibited appropriate single wavelength fluorescence signal, validating antibody specificity for their respective fluorophores (Figure S3). Immunoblotting results corroborate flow cytometry and microscopy observations for expression of both fluorescent marker proteins (Figure 1C). Collectively, these findings indicate successful cellular fusion and stability of the chimeric state after clonal expansion.
Figure 1.
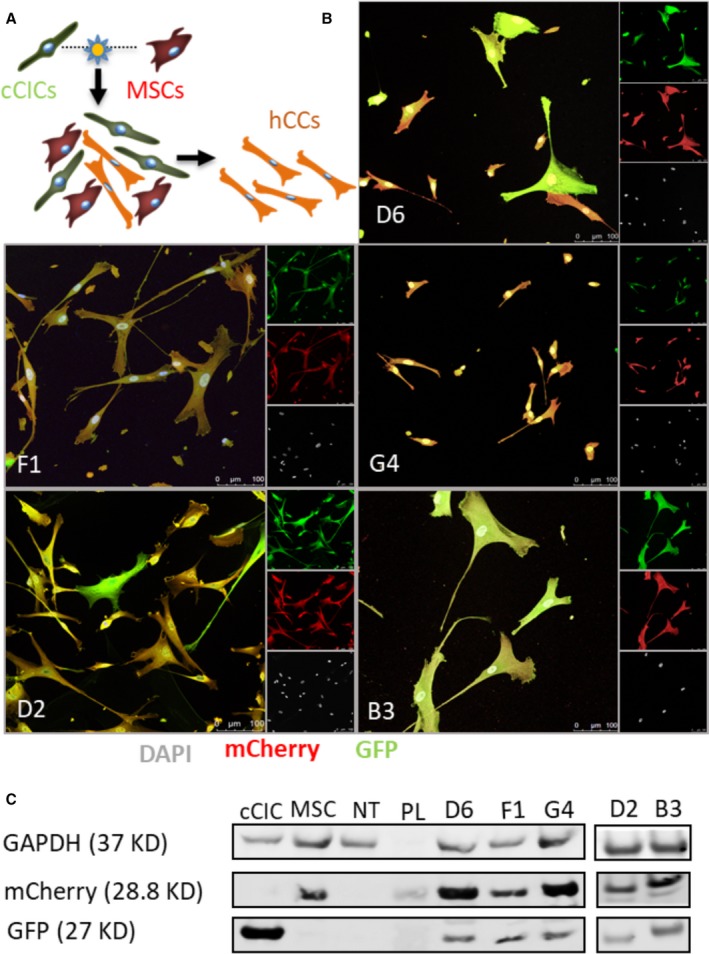
Human CardioChimeras (hCCs) are created from c‐kit+ cardiac interstitial cell (cCIC) and mesenchymal stem cell (MSC) fusion. A, Fusion‐suspension of cCICs and MSCs. Initial fused cells undergo a mitotic event to combine chromatin content followed by sorting of successfully fused cells (hCC). B, Intraclass correlation of the hCCs for GFP (green fluorescent protein) and mCherry. C, Immunoblot analysis of cCIC‐GFP, MSC‐mCherry, nontransduced (NT) MSC, and hCCs. GAPDH is used as the loading control. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; PL, protein ladder.
Table 1.
List of Fusions
| Fusions | Yield, % | hCC Analyzed |
|---|---|---|
| 1 | 4 | G4 |
| 2 | 3.1 | D6 |
| 3 | 1.5 | F1 |
| 4 | 1.3 | B3 |
| 5 | 1.2 | D2 |
hCC indicates human CardioChimeras.
hCCs Possess Diploid DNA Content
Fusion of 2 parental lines resulted in elevated ploidy in murine CCs DNA content.8 In comparison, hCCs possess diploid (2n) DNA content comparable to the parent cells as assessed using flow cytometry analysis (Figure 2A and Figure S4). Since all hCCs exhibited 2n ploidy status, a representative line (hCC G4) was chosen for karyotypic analysis that confirmed normal male karyotype with 46 chromosomes including XY as observed in G‐banded spreads (Figure 2B). Therefore, hCCs maintain normal 2n ploidy content relative to the parent cells, in contrast to increased ploidy of murine CCs.8
Figure 2.
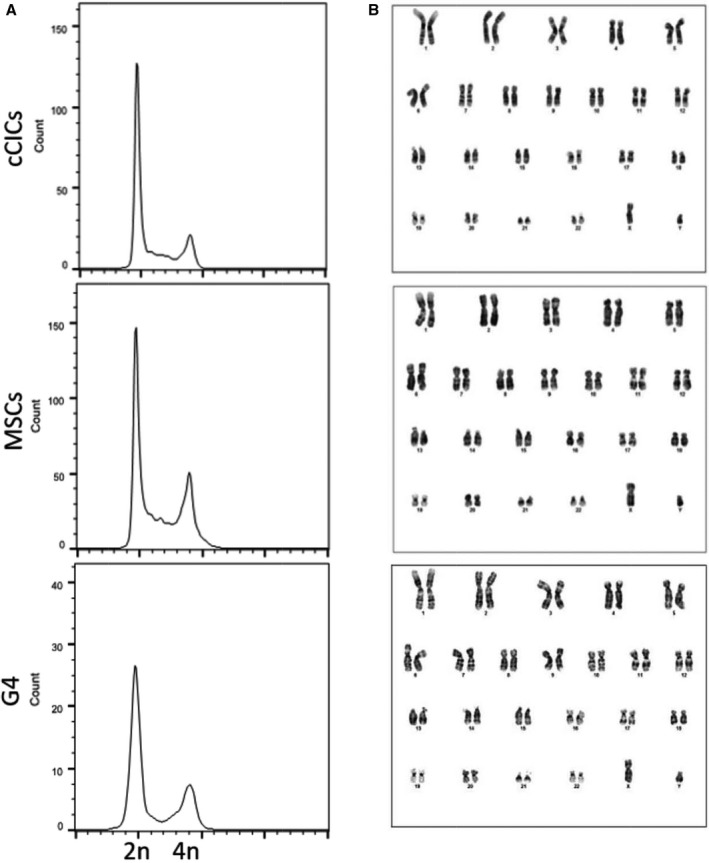
Human CardioChimeras (hCCs) reveal ploidy status and DNA content corresponding to the parent cells. A, Flow cytometry plots for propidium iodide /RNAse staining of parent cells and hCCs, G4. N=3 independent experiments. B, Cytogenetic analysis of parent cells and hCC, G4. cCICs indicates c‐kit+ cardiac interstitial cells; MSCs, mesenchymal stem cells.
hCCs Phenotypic and Proliferative Characteristics Resemble Parent Cells
Murine CCs characteristics of survival and proliferation were comparable to parental lines albeit with increased DNA content.8 Cellular properties of select hCCs were assessed for phenotypic traits of morphology and growth rate relative to parental lines, chosen to represent typical examples for variability of hCC characteristics. Cellular morphology measured using bright‐field images of the hCCs and parent cells reveals a range of cell surface area and length to width ratio (Figure 3). hCCs D6, F1, and G4 exhibited cell surface area similar to cCICs that was significantly lower than MSCs (Figure 3A). Length to width ratio of these clones was significantly greater compared with MSCs and was comparable to that of cCICs (Figure 3B). The proliferation rate of these hCCs was increased relative to parental cells with a doubling time of ≈24 hours and was therefore characterized as fast growth (Figure 3C and 3D). Cell surface area of hCC D2 was significantly greater than the cCICs and similar to the MSCs (Figure 3A). Length to width ratio for hCC D2 was significantly decreased relative to cCICs, instead resembling that of MSCs (Figure 3B). Proliferation rate of hCC D2 was intermediate compared with parent cells with a doubling time of 32 hours and was identified as medium growth (Figure 3C and 3D). Last, hCC B3 cell surface area was increased, length to width ratio was reduced, and proliferation rate was slow compared with either of the parent cells, resulting in designation of slow growth (Figure 3A through 3D). Collectively, these data profile phenotypic properties of hCCs with cell surface areas and length to width ratios that correspond to their proliferative rates, consistent with previous findings for murine CCs.8
Figure 3.
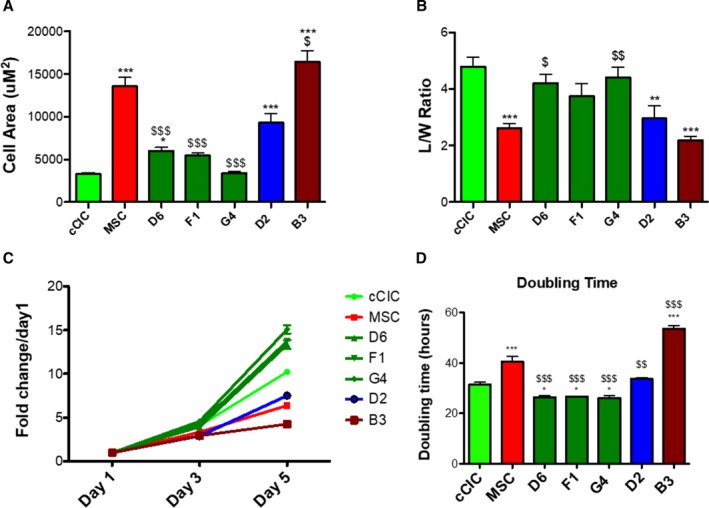
Morphometric and proliferative characteristics of human CardioChimeras (hCCs). A, Cell surface area and (B) length to width (L/W) ratio of the parent cells and hCCs. C, Proliferation rate of parent cells and hCCs represented as fold change over day 1, and (D) cell doubling time represented in hours. N=3 independent experiments. Statistical analysis was performed by 1‐way ANOVA multiple comparison with Dunnett. *P<0.05 vs c‐kit+ cardiac interstitial cell (cCIC), **P<0.001 vs cCIC, ***P<0.0001 vs cCIC, $ P<0.05 vs mesenchymal stem cell (MSC), $$ P<0.001 vs MSC, $$$ P<0.0001 vs MSC. Error bars are ±SEM.
Profiling for Commitment and Secretory Gene Expression Reveals hCC Variability
Gene expression profiling revealed significant heterogeneity between various murine CCs,8 therefore commitment and secretory gene profiles for hCCs were assessed to find whether similar diversity was present. Cardiac lineage markers including cardiac type troponin T2, GATA4 (GATA binding protein 4), PECAM1 (platelet and endothelial cell adhesion molecule 1), and smooth muscle actin were investigated by measuring mRNA expression level by quantitative polymerase chain reaction analysis. Early cardiac transcription factor GATA4 was significantly upregulated in all hCCs except B3 in which GATA4 expression was not detected when compared with MSCs. When measured relative to cCICs, GATA4 was ≈1‐fold higher in F1, unchanged in D6 and G4, and downregulated in D2 (Figure 4A). Cardiomyocyte marker cardiac type troponin T2 level was reduced in fast‐growing hCCs D6, F1, and G4 as opposed to medium‐ and slow‐growing clones D2 and B3 (Figure 4B). PECAM1 level was higher in F1, G4, and D2 clones than in MSCs, but lower than cCICs. PECAM1 was not detected in B3 (Figure 4C). All hCCs significantly downregulated smooth muscle actin relative to MSCs resembling closely to cCICs (Figure 4D). Collectively, these data demonstrate that fast‐growing hCCs upregulate expression of early cardiac transcription factor GATA4 and downregulate expression of maturational cardiac marker cardiac type troponin T2, reflective of their cardiac progenitor‐like phenotype. In contrast, medium‐ and slow‐growing hCCs exhibit a more committed mRNA profile from this cursory assessment with select transcripts.
Figure 4.
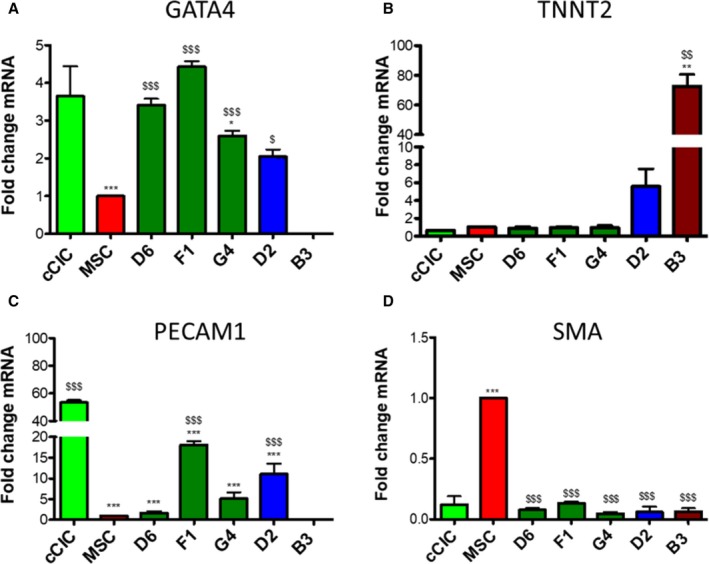
Human Cardio Chimeras (hCCs) exhibit variable expression level of cardiomyogenic commitment markers. mRNA expression level of (A) GATA4 (GATA binding protein), (B) TNNT2 (cardiac type troponin T2), (C) PECAM1 (platelet and endothelial cell adhesion molecule 1), and (D) smooth muscle actin (SMA) for parent cells and hCCs. N=3 independent experiments, 3 replicates (wells) per group per experiment. All genes expression is normalized to ribosomal 18s and represented as a fold change relative to mesenchymal stem cells (MSCs). Statistical analysis was performed by 1‐way ANOVA multiple comparison with Dunnett. *P<0.05 vs c‐kit+ cardiac interstitial cell (cCIC), **P<0.001 vs cCIC, ***P<0.0001 vs cCIC, $ P<0.05 vs MSC, $$ P<0.001 vs MSC, $$$ P<0.0001 vs MSC. Error bars are ±SEM.
hCCs were also analyzed for expression of prosurvival paracrine factors that mediate protective effects. Heparin‐binding epidermal growth factor–like growth factor, a secreted glycoprotein involved in wound healing and cardiac development,25, 26 was significantly upregulated in hCC F1. In comparison, hCCs D6, G4, and D2 expressed heparin‐binding epidermal growth factor–like growth factor at levels intermediate between the 2 parent cell ranges. Heparin‐binding epidermal growth factor–like growth factor expression was undetectable in hCC B3 clone (Figure 5A). Hepatocyte growth factor, a paracrine growth, motility, and morphogenic factor27, 28 was expressed at low levels in hCC D6, F1, G4, and D2, but at a comparable level to MSCs in B3 (Figure 5B). hCCs F1 and D2 expressed high levels of chemotactic factor stromal‐derived factor relative to cCICs but lower than in MSCs. Stromal‐derived factor expression was downregulated in D6 and G4 and undetectable in B3 (Figure 5C). FGF2, an important growth factor for wound healing, angiogenesis, and cellular proliferation29 was highly upregulated in B3. hCC F1 and D2 increased FGF2 expression relative to cCICs and were more reminiscent of MSCs, while hCC D6 and G4 expressed lower levels of FGF2 compared with both parent cells (Figure 5D). In summary, similar to what was previously observed with murine CC,8 profiling of hCC reveals distinct transcriptome profiling signatures between the clones.
Figure 5.
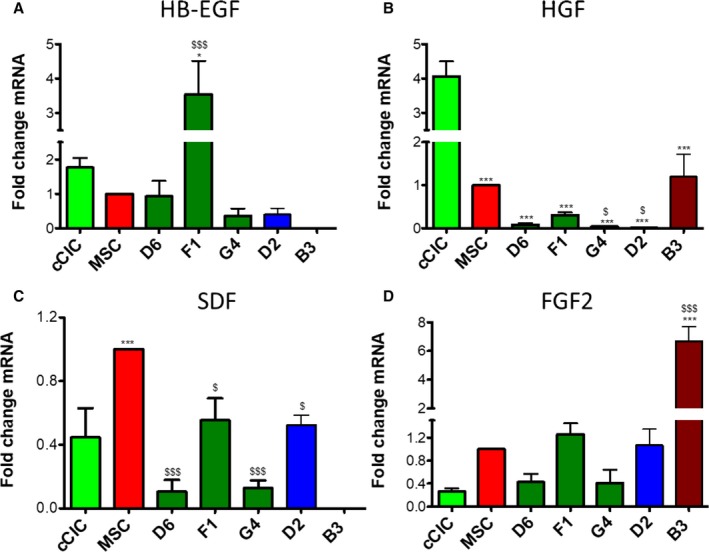
Human CardioChimeras exhibit variable secretory gene profiles. mRNA expression level of (A) HB‐EGF (heparin‐binding epidermal growth factor–like growth factor), (B) HGF (hepatocyte growth factor), (C) SDF (stromal‐derived factor), and (D) FGF2 (fibroblast growth factor 2). N=3 independent experiments, 3 replicates (wells) per group per experiment. All genes expression is normalized to ribosomal 18s and represented as a fold change relative to mesenchymal stem cells (MSCs). Statistical analysis was performed by 1‐way ANOVA multiple comparison with Dunnett. *P<0.05 vs c‐kit+ cardiac interstitial cell (cCIC), **P<0.001 vs cCIC, ***P<0.0001 vs cCIC, $ P<0.05 vs MSC, $$ P<0.001 vs MSC, $$$ P<0.0001 vs MSC. Error bars are ±SEM.
Survival in Response to Environmental Stress is Enhanced in hCC
hCCs response to stress was assessed by serum deprivation with cell death quantitation by flow cytometric measurement of necrosis and apoptosis. Cell death rates were similar between hCCs and MSCs but lower than in cCICs (Figure 6A). Response to oxidative stress induced by hydrogen peroxide treatment (350 μmol/L) in serum‐free conditions reveals significantly lower rates of apoptosis and necrosis compared with either parental line with a correspondingly higher rate of survival (Figure 6B), consistently present even when media formulations for parental lines (cCIC or MSC) are used for hCC culture (Figure S5; D2 line). Simulated ischemia‐reperfusion injury was also used as an environmental stress with cells subjected to Krebs‐Heinsleit buffer in hypoxia, and growth media in normoxia as experimental controls. Lower necrotic cell death was found for all hCCs compared with parent cells. Similar apoptosis rates were observed comparing hCCs to the parental lines with the exception of D2, which exhibited higher apoptotic cell death (Figure 6C). Correspondingly, survival of all of the hCCs except D2 was not significantly different from either parental cell. Similar percentages of apoptotic, necrotic, and live cells were observed in experimental controls (Figure S6). In conclusion, unlike susceptibility to environmental stress in murine CCs similar to parent lines,8 hCCs exhibit enhanced resistance to oxidative stress.
Figure 6.
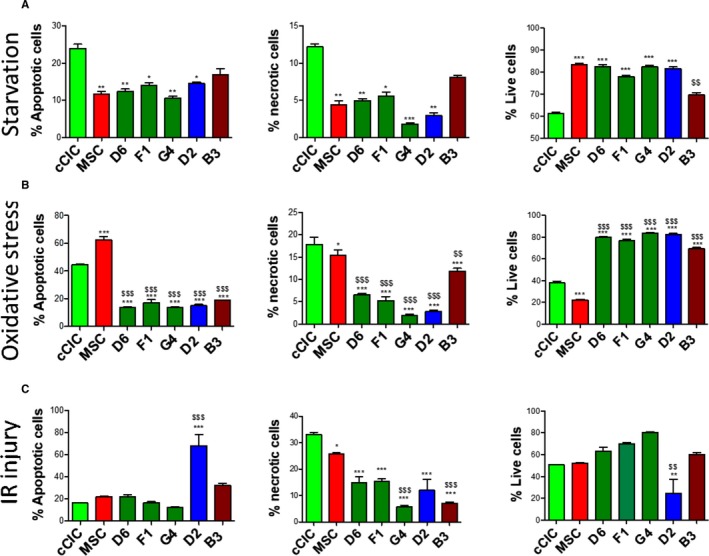
Human Cardio Chimeras demonstrate enhanced survival in response to stress. Percentage of apoptotic, necrotic, and live cells following (A) serum starvation, (B) oxidative stress, and (C) ischemia/reperfusion (IR) injury. N=3 independent experiments. Statistical analysis was performed by 2‐way ANOVA multiple comparison with Tukey. *P<0.05 vs c‐kit+ cardiac interstitial cell (cCIC), **P<0.001 vs cCIC, ***P<0.0001 vs cCIC, $ P<0.05 vs mesenchymal stem cell (MSC), $$ P<0.001 vs MSC, $$$ P<0.0001 vs MSC. Error bars are ±SEM.
Cardiomyocyte Survival is Enhanced by hCC Co‐Culture
Protective effects in response to serum deprivation challenge of NRCMs in vitro are similar between murine CCs or their corresponding parental cells.8 The protective effect of hCCs was similarly assessed, with serum deprivation challenge of NRCMs that prompted significantly increased cell death, which was partially mitigated by addition of serum for 24 hours (Figure 7). Clone B3 was excluded from further experiments because of slow expansion rate, undergoing replicative senescence after passage 10 in culture. In comparison, lines D6, F1, and G4 expansion rate slowed after passage 10, yet had not reached replicative senescence. Line D2 maintained consistent proliferation rate for more than 10 passages, and currently expanded more than 27 passages without arrest (Figure S7). NRCMs showed improved survival mediated by each hCC in response to serum deprivation, with co‐culture with hCC D2 yielding the highest rate of survival (Figure 7). NRCMs co‐cultured with cCICs exhibited a high level of survival in response to serum deprivation. Co‐culture of MSCs and hCCs G4 partially rescued NRCMs resembling the effect of serum addition for 24 hours. hCCs D6 and F1 exerted intermediate protective effect on NRCMs compared with parental cells, while hCCs D2 provided the highest protective effect on NRCMs, exceeding even that of NRCMs in high serum. Except for hCC G4, which exhibited a protective effect similar to MSCs, most hCCs resembled cCICs in co‐culture effect upon NRCMs. Therefore, in contrast to murine CCs with a protective effect similar to their corresponding parent cells, hCCs exhibit individually distinct variability in protective action.
Figure 7.
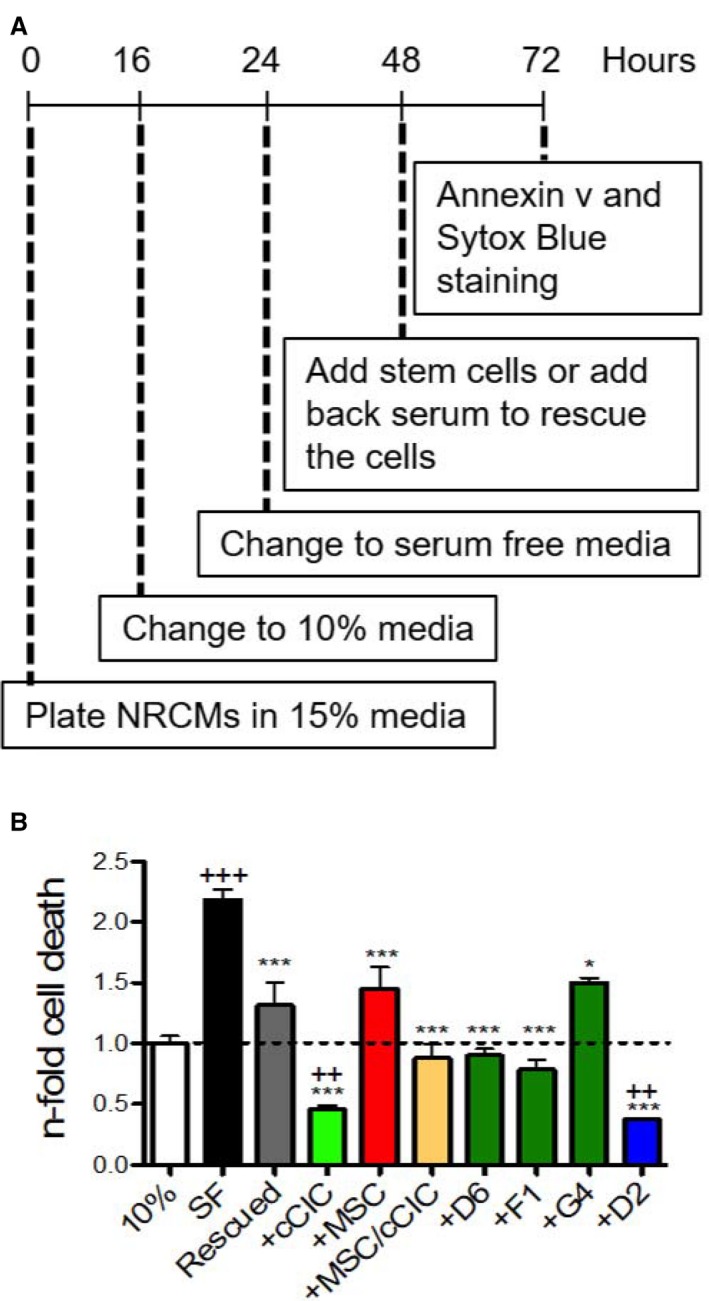
Human CardioChimeras promote cardiomyocytes survival in response to serum deprivation. A, Neonatal rat cardiomyocyte (NRCM) co‐culture protocol. B, Cardiomyocytes cell death in 10% serum media (10%), serum‐free (SF) media, rescued (Resc) and in the presence of c‐kit+ cardiac interstitial cells (cCICs), mesenchymal stem cells (MSCs), combinatorial group of MSCs and cCICs (+MSCs/cCICs), D6, F1, G4, and D2. Values presented are fold change of Annexin v+ Sytox Blue+ cells relative to 10% serum media (also represented as dashed line, 1.0). N=3 independent experiments. Statistical analysis was performed by 1‐way ANOVA multiple comparison with Dunnett. *P<0.05 vs SF, ***P<0.001 vs SF, ++ P<0.01 vs 10%, +++ P<0.001 vs 10%.
Discussion
Myocardial repair and regeneration research continues to progress on multiple fronts supported by over a decade of cumulative studies despite a recent “whipsaw” movement of controversies related to the terminology and biology of “cardiac stem cells.”30, 31 Although cardiogenic potential of cardiac stem cells remains debatable,32, 33 cardioprotective effects have been repeatedly demonstrated by our group and others13, 34, 35, 36 with cardiac stem cells now referred to as cCIC in this article to avoid being misconstrued as cardiogenic cells. Furthermore, cardioprotective effects of cCICs are enhanced by combinatorial delivery with MSCs.6 These findings are encouraging, but efficacy of such adoptive transfer cell therapies is inherently limited by multiple factors including poor cell survival, engraftment, and persistence that can be overcome to some extent by ex vivo cell engineering. Our group has focused on potentiation of myocardial repair through use of modified cells created by genetic engineering,9, 10, 14, 37 fusion,8 and combinatorial culture/delivery38 (M. Monsanto, unpublished data, 2019). Implementation of these “unnatural” approaches to create engineered cell types and combinations is deliberately intended and rationally designed to overcome inherent limitations of normal mammalian myocardial that lacks endogenous cellular reservoirs to repair and restore myocardial structure and function following pathological damage. The rationale for CC in our initial study was to avoid issues related to variable survival, engraftment, persistence, and proliferation rates of cCIC versus MSC when mixing these 2 distinct populations together for cell therapy administration. We posited that creating a novel fused hybrid CC would deliver significantly enhanced reparative activity compared with each parental cell type used either alone or in mixed single cell suspensions; a postulate that was validated in a syngeneic murine infarction model.8 The next logical advance of this patented technology (US Patent #20160346330A1) was to apply a comparable approach in the context of human cCIC and MSC. Species‐specific characteristics between murine versus human biology result in differences in the creation and phenotypic properties of CC, as reinforced by results in this study.
Comparison between human versus murine CC reveal both similarities and important distinctions. Specifically with respect to similarities between mouse CC and hCC, there were a few notable parallels including: (1) variable proliferative characteristics (slow, medium, fast); (2) variable CC size; (3) comparable PECAM 1 expression (intermediate level in both mouse and hCCs compared with the parent cells); and (4) cardiac troponin T expression at a high level relative to parental lines in hCC D2 and B3 (medium and slow grower lines, respectively) similar to mouse CC versus parental lines. In comparison, differences between mouse CC and hCC were numerous and significant as demonstrated by: (1) higher survival level compared with parental cells in response to oxidative stress by hCCs, whereas murine CC survival was either similar or lower than parental cells; (2) normal diploid DNA content of hCC compared with tetraploidy in murine CC; (3) lower smooth muscle actin expression in hCC than either parental cell, but higher in mouse CCs than the cCIC parental cell; (4) similar cardiac troponin T expression level in hCC to fast‐growing parental lines versus higher level expression in mouse CC; and (5) variable protective effects in NRCM co‐culture studies of hCC relative to parental cells versus similar protective effects between murine CC and parental cells. Even with an admittedly small sampling size of 2 murine CCs and 5 hCCs there are demonstrable biological and phenotypic differences between species‐specific CC warranting further characterization.
Polyploidy observed in mouse CC was notably absent from hCC lines. The diploid (2n) DNA content of hCC (Figure 2) contrasts with that of murine CCs, which had increased DNA content.8 Cytogenic analysis of hCC G4 further confirmed a normal karyotype with 46 chromosomes. The most straightforward interpretation of this finding is that human cell fusions do not tolerate polyploidy as a permissive state for mitotic growth, unlike their murine counterparts. hCCs may acquire 1 copy of each chromosome complement from either of the parent cells leading to no change in ploidy content, despite being successfully fused. This cytogenetic state eliminates concerns regarding fusion‐induced increased DNA content and aneuploidy, which can lead to genomic instability and cellular senescence.39 All hCCs retained cellular properties corresponding to cCIC and MSCs consistent with chromosomal mosaicism in somatic cells without significantly altering stem cell behavior.40
MSCs mediate cell survival through secretion of prosurvival chemokines and cytokines including stromal‐derived factor 1 and insulin‐like growth factor 1.41, 42 hCCs expressed growth factors and cardioprotective cytokines including epidermal growth factor and stromal‐derived factor and were resistant against starvation‐induced cell death (Figure 5A and 5C and Figure 6A). Similarly, hCCs demonstrated enhanced survival in response to oxidative stress (Figure 6B) and attenuated necrotic cell death in response to ischemia/reperfusion injury (Figure 6B and 6C). When co‐cultured with NRCMs, hCC G4 exhibited a cardioprotective effect similar to MSCs, whereas hCCs F1, D6, and D2 exhibited a cardioprotective effect similar to cCIC with hCC D2 exhibiting the highest cardioprotective activity (Figure 7). Cellular crosstalk survival signals might be facilitated in CC, since exosome contents such as MSC‐secreted micro‐RNA 21 protects c‐kit+ stem cells from oxidative injury through the PTEN/PI3K/Akt signaling pathway.43 Paracrine signaling activity has been suggested as the mechanistic basis for cardioprotection using cell therapy,44, 45, 46 so the relevance of the secretome from hCCs deserves further detailed analyses to establish a putative role in enhancing survival signaling. The potential contribution of cell fusion to heart regeneration is essentially unexplored despite the established role of cell fusion as a reprogramming mechanism leading to enhanced proliferation and growth rate in differentiated cells.47 The findings presented here suggest cell fusion as a reliable and stable technique to generate human hybrid cells. cCICs and MSCs are ideal cellular candidates for cell fusion. They can be consistently isolated and expanded in laboratory settings and they are well‐established cell candidates for cellular therapy based on the results of basic biological studies and clinical trials.4, 48 In the present study, cCICs and MSCs were used at a ratio of 2:1, but cell ratios used for fusion can influence the fate and functional potential of resultant hybrids. Therefore, modifying the cell number ratio before fusion could be a promising future direction leading to the design of hybrids with individualized phenotypic characteristics. Moreover, adapting cell fusion ratios for future studies may promote creation of hCCs with more consistent biological profiles.
Conclusions
Cell fusion is a tractable engineering approach to generate a novel cell population incorporating characteristics of 2 different human cardiac–derived interstitial cell populations into a single cell. Hybrid CCs possess molecular and phenotypic characteristics similar to previously established murine CCs with enhanced myocardial repair potential.8 However, cell engraftment and cardiomyogenic and reparative potential of hCCs need to be determined and are subject of future investigations that could support the next steps toward translational application of hCCs.
Sources of Funding
Sussman is supported by National Institutes of Health (NIH) grants: R01HL067245, R37HL091102, R01HL105759, R01HL113647, R01HL117163, P01HL085577, and R01HL122525, as well as an award from the Foundation Leducq. Firouzi is supported by a Grace Hoover Sciences Scholarship. Broughton is supported by NIH grant F32HL136196.
Disclosures
Sussman is a founding member of CardioCreate, Inc. The remaining authors have no disclosures to report.
Supporting information
Table S1. List of Media
Table S2. List of Antibodies
Table S3. qRT PCR Primer List
Figure S1. Flow cytometry plots of one representative fusion experiment including. A, Naive c‐kit+ cardiac interstitial cells (cCICs), (B) cCIC‐GFP (green fluorescent protein), (C) naive mesenchymal stem cells (MSCs), (D) MSC‐mCherry, (E) the combinatorial cCIC and MSC group as the negative control, and (F) Sendai virus–induced fused cells.
Figure S2. Live native fluorescent images of human CardioChimeras (hCCs) illustrating double positivity of the fused cells.
Figure S3. Immunocytochemistry images of (A) c‐kit+ cardiac interstitial cell (cCIC)‐GFP (green fluorescent protein), (B) MSC‐mCherry, and (C) nontransduced MSCs stained for GFP and mCherry. Sytox green was used as the nuclear stain.
Figure S4. Flow cytometry plots for propidium iodide/RNAse staining of human CardioChimeras (hCCs) D6 and F1.
Figure S5. Percentage of survival (live cells) of D2 clones in c‐kit+ cardiac interstitial cell (cCIC) media, cCICs in cCIC media, D2 clones in mesenchymal stem cell (MSC) media, and MSCs in MSC media (from left to right) after treatment with hydrogen peroxide (350 μmol/L). Error bars are ±SEM. ***P<0.001.
Figure S6. Percentage of apoptotic, necrotic, and live cells. A, Cultured in growth media in normoxic condition and (B) subjected to Krebs‐Heinsleit buffer in hypoxic condition. Error bars are ±SEM.
Figure S7. Cell doubling time of human CardioChimeras (hCCs) up to and after passage 10 in culture represented in hours. Error bars are ±SEM.
Acknowledgments
We gratefully acknowledge Dr Christopher Glembotski and Erik Blackwood for providing the Sussman lab with the NRCMs used for co‐culture experiment, as well as Dr Roland Wolkowicz, Cameron Smurthwaite, and Ruby Cotton for their assistance with cell sorting and flow cytometry analysis. All people named in the Acknowledgments section have provided the corresponding author with permission to be named in the article.
Author contributions: Firouzi, Choudhury, and Sussman designed the experiments. Firouzi, Choudhury, Broughton, and Salazar performed the experiments and analyzed data. Bailey assisted with the statistical analysis of the results. Firouzi and Sussman wrote the article. All of the authors read and approved the article.
(J Am Heart Assoc. 2020;9:e013452 DOI: 10.1161/JAHA.119.013452.)
Preprint posted on BioRxiv October 07, 2019. doi: https://doi.org/10.1101/796870.
References
- 1. Li Q‐H, Cao P, Vajravelu BN, Guo Y, Du J, Bolli R, Hong KU, Zhu X, Bhatnagar A, Book MJ, Nong Y, Al‐Maqtari T. c‐kit+ cardiac stem cells alleviate post‐myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PLoS One. 2014;9:e96725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanganalmath SK, Bolli R. Cell therapy for heart failure. Circ Res. 2013;113:810–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY, Tracy M, Ghersin E, Johnston PV, Brinker JA, Breton E, Davis‐Sproul J, Schulman IH, Byrnes J, Mendizabal AM, Lowery MH, Rouy D, Altman P, Wong Po Foo C, Ruiz P, Amador A, Da Silva J, McNiece IK, Heldman AW. Comparison of allogeneic vs autologous bone marrow‐derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. J Am Med Assoc. 2012;308:2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Slaughter MS, Chugh AR, Rokosh DG, D'Amario D, Kajstura J, Solankhi NK, Sanada F, Hosoda T, Ikram S, Bolli R, Anversa P, Stoddard MF, Fahsah I, Elmore JB, Loughran JH, Goichberg P, Beache GM, Wagner SG, Leri A, Cappetta D. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5. Gerstenblith G, Johnston PV, Marbán L, Bonow RO, Marbán E, Mendizabal A, Lardo AC, Malliaras K, Wu E, Smith RR, Cingolani E, Makkar RR, Schuleri KH, Cheng K. Intracoronary cardiosphere‐derived cells after myocardial infarction. J Am Coll Cardiol. 2013;63:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Williams AR, Hatzistergos KE, Addicott B, McCall F, Carvalho D, Suncion V, Morales AR, Da Silva J, Sussman MA, Heldman AW, Hare JM. Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation. 2013;127:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitrani RD, Traverse JH, Chugh A, DiFede DL, March KL, Willerson JT, Bolli R, Sayre SL, Khan A, Bettencourt J, Lima JAC, Hare JM, Vojvodic RW, Perin EC, Simari RD, Moyé L, Hernandez‐Schulman I, Henry TD, Loughran J, Cohen M, Ebert RF, Taylor DA, Yang PC, Pepine CJ. Rationale and design of the CONCERT‐HF Trial (Combination of Mesenchymal and c‐kit + Cardiac Stem Cells As Regenerative Therapy for Heart Failure). Circ Res. 2018;122:1703–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quijada P, Salunga HT, Hariharan N, Cubillo JD, El‐Sayed FG, Moshref M, Bala KM, Emathinger JM, De La Torre A, Ormachea L, Alvarez R, Gude NA, Sussman MA. Cardiac stem cell hybrids enhance myocardial repair. Circ Res. 2015;117:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fischer KM, Cottage CT, Wu W, Din S, Gude NA, Avitabile D, Quijada P, Collins BL, Fransioli J, Sussman MA. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing pim‐1 kinase. Circulation. 2009;120:2077–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohsin S, Khan M, Toko H, Bailey B, Cottage CT, Wallach K, Nag D, Lee A, Siddiqi S, Lan F, Fischer KM, Gude N, Quijada P, Avitabile D, Truffa S, Collins B, Dembitsky W, Wu JC, Sussman MA. Human cardiac progenitor cells engineered with Pim‐I kinase enhance myocardial repair. J Am Coll Cardiol. 2012;60:1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Korski KI, Kubli DA, Wang BJ, Khalafalla FG, Monsanto MM, Firouzi F, Echeagaray OH, Kim T, Adamson RM, Dembitsky WP, Gustafsson ÅB, Sussman MA. Hypoxia prevents mitochondrial dysfunction and senescence in human c‐kit+ cardiac progenitor cells. Stem Cells. 2019;37:555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Monsanto MM, Dembitsky WP, Ilves K, Kim T, White KS, Mohsin S, Fisher K, Sussman MA, Broughton K, Casillas A, Wang BJ, Khalafalla FG. Concurrent isolation of 3 distinct cardiac stem cell populations from a single human heart biopsy. Circ Res. 2017;121:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gude NA, Firouzi F, Broughton KM, Ilves K, Nguyen KP, Payne CR, Sacchi V, Monsanto MM, Casillas AR, Khalafalla FG, Wang BJ, Ebeid DE, Alvarez R, Dembitsky WP, Bailey BA, van Berlo J, Sussman MA. Cardiac c‐kit biology revealed by inducible transgenesis. Circ Res. 2018;123:57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohsin S, Khan M, Nguyen J, Alkatib M, Siddiqi S, Hariharan N, Wallach K, Monsanto M, Gude N, Dembitsky W, Sussman MA. Rejuvenation of human cardiac progenitor cells with pim‐1 kinase. Circ Res. 2013;113:1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samse K, Emathinger J, Hariharan N, Quijada P, Ilves K, Völkers M, Ormachea L, De La Torre A, Orogo AM, Alvarez R, Din S, Mohsin S, Monsanto M, Fischer KM, Dembitsky WP, Gustafsson ÅB, Sussman MA. Functional effect of Pim1 depends upon intracellular localization in human cardiac progenitor cells. J Biol Chem. 2015;290:13935–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Health. 2016;2016:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoenfelder KP, Fox DT. The expanding implications of polyploidy. J Cell Biol. 2015;209:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coward J, Harding A. Size does matter: why polyploid tumor cells are critical drug targets in the war on cancer. Front Oncol. 2014;4:123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Avitabile D, Ormachea L, Tsai EJ, Sussman MA, Mohsin S, Monsanto M, Samse K, Joyo A, Hariharan N, Quijada P, De La Torre A, McGregor MJ. Nucleostemin rejuvenates cardiac progenitor cells and antagonizes myocardial aging. J Am Coll Cardiol. 2015;65:133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quijada P, Toko H, Fischer KM, Bailey B, Reilly P, Hunt KD, Gude NA, Avitabile D, Sussman MA. Preservation of myocardial structure is enhanced by pim‐1 engineering of bone marrow cells. Circ Res. 2012;111:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jagsi R, Jiang J, Momoh AO, Alderman A, Giordano SH, Buchholz TA, Pierce LJ, Kronowitz SJ, Smith BD. HHS Public Access. 2017;263:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blackwood EA, Thuerauf DJ, Plate L, Glembotski CC, Paxman RJ, Wiseman RL, Azizi K, Kelly JW. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. 2019;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okabayashi FM, Okada Y, Tachibanat T. A series of hybrid cells containing different ratios of parental chromosomes formed by two steps of artificial fusion. Proc Natl Acad Sci. 1971;68:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suetsugu A, Matsumoto T, Hasegawa K, Nakamura M, Kunisada T, Shimizu M, Saji S, Moriwaki H, Bouvet M, Hoffman RM. Color‐coded live imaging of heterokaryon formation and nuclear fusion of hybridizing cancer cells. Anticancer Res. 2016;36:3827–3831. [PubMed] [Google Scholar]
- 25. Shirakata Y. Heparin‐binding EGF‐like growth factor accelerates keratinocyte migration and skin wound healing. J Cell Sci. 2005;118:2363–2370. [DOI] [PubMed] [Google Scholar]
- 26. Iwamoto R, Mekada E. ErbB and HB‐EGF signaling in heart development and function. Cell Struct Funct. 2006;31:1–14. [DOI] [PubMed] [Google Scholar]
- 27. Yi S, Chen JR, Viallet J, Schwall RH, Nakamura T, Tsao MS. Paracrine effects of hepatocyte growth factor/scatter factor on non‐small‐cell lung carcinoma cell lines. Br J Cancer. 1998;77:2162–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Islam MR, Yamagami K, Yoshii Y, Yamauchi N. Growth factor induced proliferation, migration, and lumen formation of rat endometrial epithelial cells in vitro . J Reprod Dev. 2016;62:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee JG, Jung E, Heur M. Fibroblast growth factor 2 induces proliferation and fibrosis via SNAI1‐mediated activation of CDK2 and ZEB1 in corneal endothelium. J Biol Chem. 2018;293:3758–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gude NA, Sussman MA. Chasing c‐Kit through the heart: taking a broader view. Pharmacol Res. 2018;127:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan M, Koch WJ. c‐kit + cardiac stem cells. Circ Res. 2016;118:783–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan J, Bu L, Zhang L, Moon A, Razzaque S, Cai W, Zhou B, Huang GY, Jeong D, Cai CL, Sultana N, Sheng W, Xu M, Hajjar RJ, Chen J. Resident c‐kit+ cells in the heart are not cardiac stem cells. Nat Commun. 2015;6:8701–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, Molkentin JD. C‐kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vajravelu BN, Hong KU, Al‐Maqtari T, Cao P, Keith MCL, Wysoczynski M, Zhao J, Moore JB, Bolli R. C‐Kit promotes growth and migration of human cardiac progenitor cells via the PI3KAKT and MEK‐ERK Pathways. PLoS One. 2015;10:e0140798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith AJ, Torella A, Shone V, Lewis FC, Cristiano F, Gritti G, Nadal‐Ginard B, Scalise M, Vicinanza C, Ellison‐Hughes GM, Terracciano CM, Torella D, Marotta P, Britti D, Indolfi C, Cianflone E, Veltri P, Aquila I, Mancuso T, Couch L, Marino F, Sacco W. Adult cardiac stem cells are multipotent and robustly myogenic: c‐kit expression is necessary but not sufficient for their identification. Cell Death Differ. 2017;24:2101–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfò M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal‐Ginard B. Adult c‐kitpos cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell. 2013;154:827–842. [DOI] [PubMed] [Google Scholar]
- 37. Sussman MA, Tuck S, Gude N, Cottage CT, Muraski J, Alvarez R, Bailey B, Avitabile D, Fischer KM, Quijada P, Collins B. Cardiac progenitor cell cycling stimulated by pim‐1 kinase. Circ Res. 2010;106:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monsanto M, White K, Fisher K, Wang JSM. CardioClusters: harnessing the power of multi‐lineage cardiac stem cells. Circ Res. 2018;121:A355(Abstract). [Google Scholar]
- 39. Carrera‐Quintanar L, Bernad A, Samper E, Torres Y, Benguría A, Pérez RA, Ramírez JC, Roche E, Dopazo A, Enríquez JA, Torres R, Estrada JC. Human mesenchymal stem cell‐replicative senescence and oxidative stress are closely linked to aneuploidy. Cell Death Dis. 2013;4:e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peterson SE, Westra JW, Rehen SK, Young H, Bushman DM, Paczkowski CM, Yung YC, Lynch CL, Tran HT, Nickey KS, Wang YC, Laurent LC, Loring JF, Carpenter MK, Chun J. Normal human pluripotent stem cell lines exhibit pervasive mosaic aneuploidy. PLoS One. 2011;6:e23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dong F, Harvey J, Finan A, Weber K, Agarwal U, Penn MS. Myocardial CXCR4 expression is required for mesenchymal stem cell mediated repair following acute myocardial infarction. Circulation. 2012;126:314–324. [DOI] [PubMed] [Google Scholar]
- 42. Herrmann JL, Wang Y, Meldrum DR, Abarbanell AM, Poynter JA, Manukyan MC, Weil BR, Brewster BD. Intracoronary mesenchymal stem cells promote postischemic myocardial functional recovery, decrease inflammation, and reduce apoptosis via a signal transducer and activator of transcription 3 mechanism. J Am Coll Surg. 2011;213:253–260. [DOI] [PubMed] [Google Scholar]
- 43. Shi B, Wang Y, Zhao R, Long X, Deng W, Wang Z. Bone marrow mesenchymal stem cell‐derived exosomal miR‐21 protects C‐kit+ cardiac stem cells from oxidative injury through the PTEN/PI3K/Akt axis. PLoS One. 2018;13:e0191616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Angelos MG, Noor M, Dougherty JA, Kumar N, Khan M, Chen CA, Khan M. Extracellular vesicles released by human induced‐pluripotent stem cell‐derived cardiomyocytes promote angiogenesis. Front Physiol. 2018;9:1794–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moore JB, Tang XL, Zhao J, Fischer AG, Wu WJ, Uchida S, Gumpert AM, Stowers H, Wysoczynski M, Bolli R. Epigenetically modified cardiac mesenchymal stromal cells limit myocardial fibrosis and promote functional recovery in a model of chronic ischemic cardiomyopathy. Basic Res Cardiol. 2018;114:3–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Balbi C, Bollini S. Fetal and perinatal stem cells in cardiac regeneration: moving forward to the paracrine era. Placenta. 2017;59:96–106. [DOI] [PubMed] [Google Scholar]
- 47. Cowan CA, Atienza J, Melton DA, Eggan K. Developmental biology: nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–1373. [DOI] [PubMed] [Google Scholar]
- 48. Davani S. Mesenchymal progenitor cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a rat cellular cardiomyoplasty model. Circulation. 2003;108:253–258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Media
Table S2. List of Antibodies
Table S3. qRT PCR Primer List
Figure S1. Flow cytometry plots of one representative fusion experiment including. A, Naive c‐kit+ cardiac interstitial cells (cCICs), (B) cCIC‐GFP (green fluorescent protein), (C) naive mesenchymal stem cells (MSCs), (D) MSC‐mCherry, (E) the combinatorial cCIC and MSC group as the negative control, and (F) Sendai virus–induced fused cells.
Figure S2. Live native fluorescent images of human CardioChimeras (hCCs) illustrating double positivity of the fused cells.
Figure S3. Immunocytochemistry images of (A) c‐kit+ cardiac interstitial cell (cCIC)‐GFP (green fluorescent protein), (B) MSC‐mCherry, and (C) nontransduced MSCs stained for GFP and mCherry. Sytox green was used as the nuclear stain.
Figure S4. Flow cytometry plots for propidium iodide/RNAse staining of human CardioChimeras (hCCs) D6 and F1.
Figure S5. Percentage of survival (live cells) of D2 clones in c‐kit+ cardiac interstitial cell (cCIC) media, cCICs in cCIC media, D2 clones in mesenchymal stem cell (MSC) media, and MSCs in MSC media (from left to right) after treatment with hydrogen peroxide (350 μmol/L). Error bars are ±SEM. ***P<0.001.
Figure S6. Percentage of apoptotic, necrotic, and live cells. A, Cultured in growth media in normoxic condition and (B) subjected to Krebs‐Heinsleit buffer in hypoxic condition. Error bars are ±SEM.
Figure S7. Cell doubling time of human CardioChimeras (hCCs) up to and after passage 10 in culture represented in hours. Error bars are ±SEM.