

Summary
The filamentous fungus Fusarium graminearum possesses an RNA‐interference (RNAi) pathway that acts as a defence response against virus infections and exogenous double‐stranded (ds) RNA. Fusarium graminearum virus 1 (FgV1), which infects F. graminearum, confers hypovirulence‐associated traits such as reduced mycelial growth, increased pigmentation and reduced pathogenicity. In this study, we found that FgV1 can suppress RNA silencing by interfering with the induction of FgDICER2 and FgAGO1, which are involved in RNAi antiviral defence and the hairpin RNA/RNAi pathway in F. graminearum. In an FgAGO1‐ or FgDICER2‐promoter/GFP‐reporter expression assay the green fluorescent protein (GFP) transcript levels were reduced in FgV1‐infected transformed mutant strains. By comparing transcription levels of FgDICER2 and FgAGO1 in fungal transformed mutants expressing each open reading frame (ORF) of FgV1 with or without a hairpin RNA construct, we determined that reduction of FgDICER2 and FgAGO1 transcript levels requires only the FgV1 ORF2‐encoded protein (pORF2). Moreover, we confirmed that the pORF2 binds to the upstream region of FgDICERs and FgAGOs in vitro. These combined results indicate that the pORF2 of FgV1 counteracts the RNAi defence response of F. graminearum by interfering with the induction of FgDICER2 and FgAGO1 in a promoter‐dependent manner.
Keywords: Fusarium graminearum virus 1, mycovirus, ORF2, RNAi, silencing suppressor
The ORF2 protein of Fusarium graminearum virus 1 can directly repress the induction of FgDICER2 and FgAGO1 at the transcriptional level to counteract the RNAi defence response of Fusarium graminearum.

Introduction
Virus infection and propagation change the expression of many host genes. These complex changes result from general or specific interactions between viruses and hosts (Golem and Culver, 2003). Using a variety of newly developed techniques, researchers have increased our understanding of virus–host interactions and have determined how some host genes affect virus replication, symptom development and defence‐related responses (Alexander and Cilia, 2016; Zanardo et al., 2019).
Because RNA silencing has pivotal roles in host gene regulation, de novo DNA methylation and chromatin modification, this pathway has been considered essential for defence response against viruses and transposable elements in animals, plants and fungi (Dalakouras and Wassenegger, 2013). Gene silencing occurs through mRNA degradation, termed post‐transcriptional gene silencing (PTGS), or through repression of transcription, termed transcriptional gene silencing (TGS) (Vaucheret and Fagard, 2001). PTGS involves cleavage of target RNA, including viral RNA genomes and exogenous double‐stranded (ds) RNA. Once target RNAs are recognized, they are processed into the 21–24 nucleotides of small interfering (si) RNA by Dicer. They are loaded onto the RNA‐induced silencing complex, which includes the slicer endonuclease Argonaute for cleavage of cognate viral RNAs (Vaucheret and Fagard, 2001; Weinberg and Morris, 2016). TGS, which is also known as RNA‐directed DNA methylation (RdDM) in plants, siRNA precursors and long non‐coding (lnc) RNA scaffolds establish repressive chromatin modifications at target loci through interaction with Argonaute 4 (AGO4) to form an RdDM effector complex (Rowley et al., 2017).
TGS and PTGS pathways can be triggered by infection by both DNA and RNA viruses; this triggering depends on the presence of homologous sequence(s) in the host and the virus (Baltusnikas et al., 2018). In response to the host defence, many viruses encode viral suppressors of RNA silencing (VSRs) that inhibit distinct steps/components of host RNA silencing pathways. Such a counterdefence also enhances the replication of co‐infecting viruses (Fukuzawa et al., 2010; Pruss et al., 1997; Syller, 2012). One of the well‐characterized VSRs of plant viruses is the p19 protein of tombusviruses. The p19 homodimer proteins suppress RNA silencing by binding to the 21‐nt siRNA duplex and thus prevent the loading of siRNAs onto Argonaute protein (Incarbone and Dunoyer, 2013). Several VSRs of DNA viruses, such as the AC2 protein in tomato golden mosaic virus and beet curly top virus, inactivate the S‐adenosyl methionine pathway or the RdDM pathway and thereby affect methylation‐mediated gene silencing (Bisaro, 2006; Cui et al., 2005). Among mycoviruses, Cryphonectria parasitica hypovirus 1 (CHV1) encodes the papain‐like protease p29, which shares similarities with the helper component‐proteinase (HC‐Pro), a multifunctional protein encoded by plant RNA viruses. p29 can affect the antiviral RNA silencing pathway in Cryphonectria parasitica by interfering with the up‐regulation of two major genes encoding Dicer‐like (dcl‐2) and Argonaute‐like (agl‐2) proteins (Segers et al., 2006; Sun et al., 2009). Andika et al. (2017) recently demonstrated that the Spt–Ada–Gcn5 acetyltransferase (SAGA) complex is involved in the transcriptional activation of agl2 and dcl2 for RNA‐interference (RNAi) activation in C. parasitica. DCL2 might have critical roles for SAGA‐mediated host gene up‐regulation as well as for cleavage of virus‐derived dsRNA molecules into virus‐derived siRNA (Andika et al., 2019). Although the S10 protein encoded by Rosellinia necatrix mycoreovirus 3 (RnMyRV3) has the ability to suppress RNA silencing, it might not repress the up‐regulation of the RNA silencing‐related genes in Rosellinia necatrix but may instead repress another RNA‐silencing step (Yaegashi et al., 2013, 2016). Recent studies have reported that both RNA and DNA mycoviruses in Sclerotinia sclerotiorum trigger RNA silencing response. There is functional redundancy between two Dicer genes (dcl‐1 and dcl‐2) in the antiviral response, whereas only agl‐2 has a role in antiviral defence between two Argonaute genes (agl‐2 and agl‐4) in S. sclerotiorum (Mochama et al., 2018; Neupane et al., 2019). Unlike the products of plant virus genes, only a few mycovirus proteins are known to be suppressors of RNA silencing, and their mechanisms of suppression are mostly unclear.
Fusarium graminearum, a filamentous ascomycete fungus, causes head blight of wheat, barley, rice and oats, and stalk and ear rot of maize (Dweba et al., 2017). In addition to reducing yield, this fungal pathogen contaminates cereal grain with mycotoxins, such as trichothecene and zearalenone, that are harmful to animals and humans (Ferrigo et al., 2016). Several mycoviruses of F. graminearum have recently been isolated and characterized (Li et al., 2019). Our laboratory previously characterized four different mycoviruses of F. graminearum: Fusarium graminearum virus 1 (FgV1), FgV2, FgV3 and FgV4, phylogenetically related to members of the Fusariviridae, Chrysoviridae, Totiviridae and Partitiviridae, respectively (Cho et al., 2013, Lee et al., 2014). The FgV1 and FgV2 infections are associated with hypovirulence of the fungal host, i.e. these infections have been associated with reduced mycelial growth, increased pigmentation, reduced virulence, decreased mycotoxin production and impaired sexual development (Lee et al., 2014). In contrast, FgV3 and FgV4 have been found to infect fungal hosts asymptomatically (Yu et al., 2009).
Previous studies identified the functions of RNAi‐related genes in F. graminearum (Chen et al., 2015; Son et al., 2017; Yu et al., 2018). Fusarium graminearum contains two Dicer [F. graminearum DICER1 (FgDICER1) and FgDICER2], two Argonaute (FgAGO1 and FgAGO2) and five RdRP (FgRdRP1–5) genes. FgDICER2 and FgAGO1 have a pivotal role in hairpin RNA‐induced gene silencing (Chen et al., 2015). FgDICER1 and FgAGO2 have a primary role in the sex‐specific RNAi pathway (Son et al., 2017). Our laboratory recently reported that FgAGO1 functions in antiviral response on FgV1 infection, although the two Dicers and two Argonautes are partially redundant in F. graminearum (Yu et al., 2018). We observed the up‐regulation of transcript levels of FgDICER2 and FgAGO1 in F. graminearum PH‐1 following FgV2 or FgV3 infection but not following FgV1 infection (Yu et al., 2018). In addition, green fluorescent protein (GFP) hairpin RNA expression strains, which increase the transcript levels of FgDICER2 and FgAGO1, were also negatively affected by FgV1 infection (Yu et al., 2018). The latter study strongly suggested that FgV1 can interfere with one of the RNA silencing pathways in the fungal host at the transcriptional level.
Here, we describe how FgV1 suppresses the induction of FgDICER2 and FgAGO1 in the fungal host. We conducted FgDICER2‐ or FgAGO1‐promoter/GFP‐reporter assays to clarify how FgV1 represses transcription of FgDICER2 and FgAGO1. To identify which viral protein(s) of FgV1 function as transcriptional repressors of FgDICER2 and FgAGO1, we constructed fungal single‐ or double‐mutant strains that express each open reading frame (ORF) of FgV1 with or without the hairpin RNA construct, and we compared the transcript levels of FgDICER2 and FgAGO1 genes. These results suggest that the FgV1 protein pORF2 might function as a suppressor of the antiviral RNAi pathway in F. graminearum.
Results
Differential levels of GFP expression in RNAi‐induced transformed mutants following virus infection
A previous report showed that levels of FgDICER2 and FgAGO1 transcripts are lower following FgV1 infection than following FgV2 or FgV3 infection (Yu et al., 2018). Moreover, RNA levels of FgDICER2 and FgAGO1 induced by the GFP‐hairpin RNA construct were significantly reduced by FgV1 infection but not by FgV2 infection (Yu et al., 2018). In the current study, the transcript levels of FgDICER2 and FgAGO1 were lower in FgV1/FgV2 or FgV1/FgV3 double‐infected F. graminearum strains than in FgV2 or FgV3 single‐infected strains (Table 1). To confirm GFP silencing is affected by virus infection, we assessed whether a GFP‐hairpin RNA‐triggered silencing is altered by virus infection in mycelia of virus‐free and virus‐infected transformed mutants in which GFP silencing was triggered by a GFP‐hairpin RNA, which was named GFP + SA. GFP expression was not detected in GFP + SA with or without FgV2 infection. However, fluorescence was seen in the GFP + SA strain infected with FgV1 (Fig. 1), indicating that silencing was suppressed by FgV1 infection in the GFP + SA strain. These observations suggested that the RNAi pathway mediated by FgDICER2 and FgAGO1 in F. graminearum might be negatively affected by FgV1 infection in order to counter the antiviral defence response of the host.
Table 1.
Transcript levels of FgDICERs and FgAGOs in Fusarium graminearum following infection by Fusarium graminearum virus 1–3
| Relative mRNA expression* | ||||
|---|---|---|---|---|
| FgDICER1 | FgDICER2 | FgAGO1 | FgAGO2 | |
| PH‐1 | 0.9 ± 0.2a† | 0.8 ± 0.2a | 1.1 ± 0.2a | 1.0 ± 0.2a |
| PH‐1/FgV1 | 3.8 ± 1.3b | 0.1 ± 0.03a | 0.4 ± 0.1a | 2.0 ± 0.3i |
| PH‐1/FgV2 | 3.8 ± 0.8b | 18.1 ± 9.4d | 7.9 ± 1.6g | 2.3 ± 0.8i |
| PH‐1/FgV3 | 0.5 ± 0.3a | 10.9 ± 0.8e | 7.7 ± 2.0g | 0.2 ± 0.1j |
| PH‐1/FgV1 + FgV2 | 4.5 ± 2.0b | 0.2 ± 0.1a | 0.4 ± 0.2a | 2.1 ± 1.1i |
| PH‐1/FgV1 + FgV3 | 5.3 ± 1.3b | 1.1 ± 0.1a | 0.5 ± 0.1a | 2.0 ± 0.8i |
| PH‐1/FgV2 + FgV3 | 22.0 ± 3.5c | 6.0 ± 0.5f | 6.0 ± 1.7h | 28.4 ± 20.4k |
Relative mRNA levels of Dicer and Argonaute gene transcripts in virus‐free F. graminearum PH‐1 (PH‐1) and in single‐ or double virus‐infected PH‐1 after 120 h of incubation. Relative mRNA levels were measured by RT‐qPCR.
Values are means (±SD) of three independent experiments. Values in each row followed by the same letters are not significantly different from the mean for virus‐free PH‐1 according to Tukey’s test at P < 0.05.
Figure 1.

Green fluorescent protein (GFP) expression in hyphae of Fusarium graminearum strains (GFP or GFP + SA) that were not infected or were infected by FgV1 or FgV2. Fluorescence microscopic images showing GFP expression in virus‐free, FgV1‐infected and FgV2‐infected transformed mutant strains. After cultivation at 25 °C for 3 days in complete medium broth, mycelia were harvested and observed with a microscope. GFP, transformant with only the pSKGen vector; SA, transformant with pGFP‐SA construct; GFP + SA, GFP and pGFP‐SA were transformed in the wild type F. graminearum. DIC, differential interference contrast. Scale bar = 20 μm.
Suppression of promoter‐dependent transcriptional activity of FgDICER2 and FgAGO1 by FgV1 infection
As mentioned in the previous section, FgV1 infection down‐regulates the accumulation of FgDICER2 and FgAGO1 transcripts. The expression of RNAi‐related genes in the RNAi‐related gene overexpression (OE) mutant strains that contained the constitutively expressed elongation factor 1α promoter (PEF1α) region upstream of the putative coding sequence were significantly affected by FgV1 infection (Yu et al., 2018). Based on these results, we hypothesized that FgV1 infection might affect transcription of FgDICER2 and FgAGO1 in a promoter‐specific manner, i.e. FgV1 infection might reduce the transcriptional induction of FgDICER2 and FgAGO1 genes by reducing the promoter activity of FgDICER2 and FgAGO1. To test this hypothesis, we confirmed GFP expression was driven by the FgAGO1‐ or FgDICER2‐promoter. The plasmids named PAGO1/GFP and PDICER2/GFP were transformed into F. graminearum PH‐1. Those transformed mutants were then infected with FgV1 or FgV2 via hyphal anastomosis to compare the effect of viral infection on GFP expression under the control of the FgAGO1‐ or FgDICER2‐promoter in the fungal host. The colony morphologies and mycelial growth rates of individual virus‐free, FgV1‐infected and FgV2‐infected transformed mutants were similar to those of the corresponding virus‐free, FgV1‐infected and FgV2‐infected wild types (Fig. 2A). All transformed mutants were confirmed by Southern blot hybridization using a 32P‐labelled DNA fragment of GFP (Fig. 2B).
Figure 2.

Analysis of the FgDICER2‐ or FgAGO1‐promoter/green fluorescent protein (GFP)‐reporter expression mutants. (A) Colony morphologies of virus‐free (left), FgV1‐infected (middle) and FgV2‐infected (right) FgDICER2‐ or FgAGO1‐promoter/GFP‐reporter expression mutants. All cultures were photographed after 4 days on complete medium. (B) Southern blot hybridization of each transformed mutant. A 32P‐labelled GFP DNA fragment was used as a probe. Lane 1, wild‐type (WT) strain PH‐1; lanes 2 to 4, biological replicates of the indicated promoter/GFP‐reporter expression mutant. (C) Levels of target gene transcripts in virus‐free, FgV1‐infected and FgV2‐infected transformed mutant strains. The relative accumulation levels of GFP, FgDICER2 and FgAGO1 were analysed by RT‐qPCR after 5 days of incubation. Values are means (±SD) of three biological replicates and three independent experiments. Means with * or ** indicate a statistical difference (P < 0.05 or P < 0.01, respectively) relative to the mean for the noninfected fungus.
The GFP transcript levels were significantly elevated in the FgDICER2‐ and FgAGO1‐promoter/GFP mutant strains following FgV2 infection, and the RNA levels of GFP in those transformed mutants were similar or decreased in FgV1‐infected strains compared to those in virus‐free strains (Fig. 2C). The transcript levels of FgDICER2 and FgAGO1 genes were elevated only in FgV2‐infected transformed mutant strains compared to the virus‐free and FgV1‐infected transformed mutant strains. This result indicates that FgV1 has the ability to block the transcriptional induction of FgDICER2 or FgAGO1 in a promoter‐dependent manner.
Ability of FgV1 ORF2 to suppress the transcription of FgDICER2 and FgAGO1
In a previous study, our laboratory showed that FgV1 infection could down‐regulate FgDICER2 and FgAGO1 (Yu et al., 2018). To determine which FgV1 gene product(s) is involved in the repression of FgDICER2 or FgAGO1 transcription, we developed a single viral gene expression construct for the transformation of F. graminearum. Four FgV1‐encoded ORFs (ORF1, ‐2, ‐3 and ‐4) were expressed under the control of PEF1α as a constitutive expression promoter in these constructs, which were referred to as pEN1, pEN2, pEN3 and pEN4, respectively (Fig. 3A and Table S1). We transformed each of the four EN constructs into fungal cells, determined their insertion by Southern blot hybridization and referred to the transformed mutants as EN1 to EN4 (Figs S1 and S2). In addition, we generated mutants by introducing pEN ORFs and GFP‐hairpin RNA expression constructs into strain PH‐1 of F. graminearum. The colony morphologies of all single‐ and double‐transformed mutants were similar to those of the wild‐type PH‐1 strain (Fig. 3B).
Figure 3.
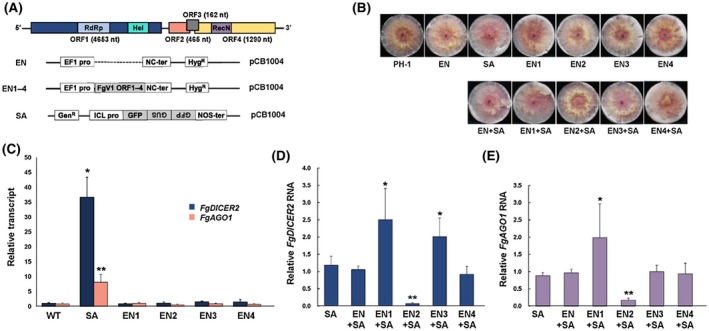
Suppression of FgDICER2 and FgAGO1 transcriptional induction by FgV1 ORF proteins. (A) Genome organization of FgV1 (top) and diagram of the plasmid used in this experiment (middle and bottom). EN, expression vector transformed mutant; EN1–4, FgV1 ORF1‐ to ORF4‐expressing mutant, respectively; SA, the green fluorescent protein (GFP) hairpin RNA‐expressing strain (GFP spacer and antisense strand). Four open reading frames (ORFs) were individually expressed under the control of the EF1α promoter and the SA construct was under the control of the isocitrate lyase (ICL) promoter. (B) Colony morphologies of fungal transformed mutants expressing FgV1‐encoded ORFs with (double) or without (single) the SA plasmid in Fusarium graminearum PH‐1. All cultures were photographed after 4 days on CM. EN1–4, FgV1 ORF1–4 expression mutant strains, respectively; EN1–4 + SA, each ORF expression construct and SA were transformed together into the wild‐type (WT) strain. (C)–(E) Levels of FgDICER2 and FgAGO1 gene transcripts in FgV1 ORF expression‐ and dsRNA‐producing transformed mutants with different combinations of SA and EN. All single‐mutant (C) and double‐mutant strains (D and E) of FgDICER2 or FgAGO1 transcript levels were analysed by RT‐qPCR at 5 days post‐inoculation. EF1α and UBH genes were used as the internal controls. Values are means (±SD) of at least three biological replicates and three independent experiments. Means with * or ** indicate a statistical difference (P < 0.05 or P < 0.01, respectively) relative to the means for the WT in (C) and for SA alone in (D) and (E).
We compared the transcript levels of FgDICER2 and FgAGO1 in all of these single mutants, double mutants and the wild type by quantitative reverse transcription polymerase chain reaction (RT‐qPCR) analysis (Fig. 3C). The transcript levels of FgDICER2 and FgAGO1 were approximately 38‐fold and 7‐fold higher, respectively, in the GFP hairpin dsRNA‐producing mutant (SA) than in the wild type, PH‐1. The accumulation of FgDICER2 and FgAGO1 transcripts in each FgV1 ORF expression mutant was similar to that in the wild type. FgDICER2 and FgAGO1 transcript levels in double‐mutant strains were similar to those in single SA‐mutant strains (SA strains) and in double SA‐mutant strains (EN + SA strains) that were transformed with the SA construct and the empty vector (EN) (Fig. 3). However, the transcript levels of those genes in the double‐mutant strains were different from those in the EN + SA transformed mutant. Although similar levels of FgDICER2 and FgAGO1 transcripts were accumulated in both EN + SA and EN4 + SA, we observed significantly higher accumulation of FgDICER2 and FgAGO1 transcripts in the EN1 + SA transformed mutant strain than in the EN + SA transformed mutant strain. The EN3 + SA double‐mutant strain contained higher levels of FgDICER2 transcripts than the EN + SA transformed mutant strain but similar levels of FgAGO1 transcripts. Interestingly, FgDICER2 and FgAGO1 transcript levels in the ORF2 overexpressing mutant (EN2 + SA) were significantly lower than those in other transformed mutants containing the hairpin RNA construct (Fig. 3D,E). Thus, FgV1‐encoded ORF2 can interfere with hairpin RNA‐induced transcriptional induction of FgDICER2 and FgAGO1.
Effect of interference of FgDICER2 and FgAGO1 expression by FgV1 ORF2 on unrelated viruses
Because the transcript levels of FgDICER2 and FgAGO1 were up‐regulated following FgV2 or FgV3 infection (Table 1), we hypothesized that the ORF2 of FgV1 negatively affects unrelated mycoviruses of F. graminearum and that the other FgV1 ORFs could not repress the transcriptional induction of FgDICER2 or FgAGO1 in response to infection by unrelated mycoviruses. To verify this possibility, we compared the transcript levels of FgDICER2 or FgAGO1 using semiquantitative RT‐PCR. The transcript levels of FgDICER2 or FgAGO1 were not changed in FgV2‐infected ORF1, ORF3 and ORF4 overexpression mutants (EN1 + FgV2, EN3 + FgV2 and EN4 + FgV2, respectively) compared to the FgV2‐infected wild‐type strain. However, transcript levels of FgDICER2 and FgAGO1 were lower in the FgV2‐infected ORF2 expression mutant (EN2 + FgV2) than in the FgV2‐infected wild‐type strain (Fig. 4A). To confirm the ORF2‐related suppression of FgDICER2 and FgAGO1 transcription, we assessed the viral dsRNA levels in the FgV2‐ or FgV3‐infected ORF2 overexpression mutant strain and found that levels of FgV2 and FgV3 dsRNA were higher in ORF2 overexpression strains than in the wild‐type strains (Fig. 4B). The relative transcript levels of two other genes, FgDICER1 and FgAGO2, were not significantly affected in any of the FgV1 ORF overexpression mutants (EN1–4 + FgV2) relative to those in the FgV2‐infected wild‐type PH‐1 strain. These results indicate that the FgV1‐encoded pORF2 interferes with the transcriptional induction only of FgDICER2 and FgAGO1.
Figure 4.

Effect of the FgV1 ORF2‐encoded protein (pORF2) on Fusarium graminearum PH‐1 infected by FgV2 and FgV3. (A) Semiquantitative RT‐PCR analysis to assess the effects of ORF1–4 expression on FgDICER and FgAGO transcript levels. The target PCR products of FgDICER2 and FgAGO1 (after 25 amplification cycles) and of FgDICER1 and FgAGO2 (after 33 amplification cycles) were visualized on a 1.5% agarose gel under UV light. Amplified fragment sizes are indicated to the right of the image. Three independent biological replicates were used for this experiment and the EF1α gene was used as a control. (B) Agarose gel (1%) analysis of the dsRNA levels for PH‐1 (WT)/FgV2, PH‐1/FgV3 and virus‐infected FgV1‐ORF2 or ‐ORF4 expression mutants. Five‐ and 30‐μg quantity of total RNAs of FgV2‐infected or FgV3‐ infected strains, respectively, were treated with DNase I and S1 nuclease. The lane marked M corresponds to lambda DNA digested with HindIII.
Binding of the FgV1 pORF2 to the upstream region of FgDICERs and FgAGOs
FgV1 ORF2 consists of 465 bp that encode 155 amino acid residues (Kwon et al., 2007). Based on computational prediction, there is no protein with a high degree of domain or sequence similarity to pORF2. To determine the possible function of pORF2, we predicted its protein structure and construction using the Phyre2 server (Protein Homology/analogY Recognition Engine V 2.0, http://www.sbg.bio.ic.ac.uk/phyre2/html/) (Kelley et al., 2015). Although we failed to obtain any structural model with a high degree of confidence for inferring protein function, some of the low‐confidence models were functionally annotated with the Swi3p, Rsc8p, and Moira (SWIRM) domain family at the C‐terminal region (34%) and with the zinc finger domain (24%) at the N‐terminal region (data not shown). Because zinc finger domains are among the most abundant domains in many DNA‐binding proteins, we speculated that the FgV1 pORF2 might transcriptionally regulate the expression of FgDICER2 and FgAGO1 genes by binding to their promoter regions.
To determine whether the pORF2 directly binds to the upstream region of FgDICER2 and FgAGO1, we carried out electrophoretic mobility shift assays (EMSAs). Considering the functional redundancy of FgDICER and FgAGO, we amplified approximately 350–500 bp upstream of the coding region in FgDICER1, FgDICER2, FgAGO1 and FgAGO2 (Fig. 5A). The complete ORF2 of FgV1 was cloned into a pET‐28a(+) vector and was expressed in Escherichia coli BL21(DE3). The empty pET28a(+) vector and ORF3 of FgV1 were used as controls in this experiment. Significant amounts of His‐tagged pORF2 and pORF3 fusion proteins were synthesized after induction with isopropyl β‐d‐thiogalactoside (IPTG). The His‐pORF2 fusion protein migrated in SDS‐PAGE gels to a position corresponding to an Mr of c. 18 kDa, and Coomassie blue staining indicated that both proteins were highly enriched after induction with IPTG. Purification of the fusion protein using HisPur Ni‐NTA magnetic beads resulted in the production of significant amounts of His‐pORF2 and His‐pORF3 fusion proteins (Figs 5B and S3B). Figure 5C shows a typical gel retardation assay that used the radiolabelled DNA fragments spanning 350–500 bp upstream of the coding region in FgDICER1, FgDICER2, FgAGO1 and FgAGO2 with His‐pORF2. The results show that distinct DNA–protein complexes with slower electrophoretic mobility were formed between the DNA fragments that form the upstream regions of FgDICER2 and FgAGO1 and the FgV1 pORF2; the degree of gel shift increased with the quantity of pORF2 added. Interestingly the FgV1 pORF2 also formed DNA–protein complexes with promoter regions of the other Dicer and Argonaute genes, i.e. with FgDICER1 and FgAGO2, the functions of which are partially redundant in terms of antiviral response. In the absence of ORF2 (Fig. 5C, lane 1 in all panels), the DNA fragment migrated as a discrete band at the bottom of the gel. The FgV1 pORF3 failed to form DNA–protein complexes (Fig. S3C). pORF2 formed DNA–protein complexes when we used excess amounts of a DNA fragment spanning the promoter region of the EF1α gene or an internal region of the actin DNA fragment was used for binding (Fig. 5D).
Figure 5.

Binding and competition assay between the upstream region of RNAi‐related genes in Fusarium graminearum and the FgV1 pORF2. (A) Schematic representation of the upstream regions of FgDICER1, FgDICER2, FgAGO1 and FgAGO2 that were used as probes in this assay. (B) SDS‐PAGE analysis of purified His‐tagged pORF2 from Escherichia coli. Samples were separated on 12% of SDS‐PAGE. CE, cell lysate; FT, flow through; E, elutions. (C) and (D) Effects of increasing amount of the pORF2 (6.7–20 nmol) with 32P‐labelled probes representing upstream region of FgDICER1, FgDICER2, FgAGO1, FgAGO2 and EF1α promoter from pSK2707, and partially amplified actin gene in F. graminearum, as indicated. (E)–(H) Competition experiment using homologous and non‐homologous DNA competitors. Electrophoreric mobility shift assay of DNA binding of 12 nM of the pORF2 and DNA at increasing concentrations of unlabelled DNA. All reaction mixtures contained 150 pM of 32P‐labelled specific probe and protein concentrations are indicated above each lane. Bovine serum albumen (BSA) alone was used as a protein control.
To determine the sequence specificity of the DNA–protein interactions, we conducted competition assays in the presence of increasing amounts (20, 100 and 200 molar excess) of the unlabelled promoter fragments of FgDICER1, FgDICER2, FgAGO1 and FgAGO2. When increasing amounts of the unlabelled homologous promoter fragments of FgDICER1, FgDICER2, FgAGO1 and FgAGO2 were incubated with pORF2 before the addition of the 32P‐labelled promoter fragments, DNA–protein complexes were decreased (Fig. 5E–H). In contrast, bovine serum albumin (BSA) or poly(dI‐dC) did not affect the binding between pORF2 and each promoter (Fig. 5E–H). These results show that the homologous competitor significantly reduced the formation of DNA–protein complexes and that the pORF2 binds less specifically to the other DNA fragments, including the promoter region of the EF1α gene.
Absence of FgV1 ORF‐specific suppressor activity in planta
Many plant viruses encode VSRs to counteract the antiviral RNA silencing response in plants. VSR activities of mycovirus gene products such as p29 of CHV1 or S10 of RnMyRV3 were also observed in Nicotiana benthamiana (Segers et al., 2006; Yaegashi et al., 2013). We therefore assessed the VSR activity of the FgV1‐encoded protein in a co‐agroinfiltration assay in N. benthamiana (Fig. 6A; (Park et al., 2009). First, we detected the expression of each FgV1 ORF‐encoded protein that contained the HA‐tag at the C‐terminal region by western blot analysis (Fig. S4). GFP expression was maintained at 4 days post‐inoculation (dpi) in leaves agroinfiltrated with the TBSV p19, but GFP intensity decreased over time in leaves that were co‐agroinfiltrated with each FgV1 ORF (Fig. 6B). In this agrobacterium co‐infiltration assay, the RNA silencing suppressor activity of FgV1‐encoded putative proteins was not detected. Although the FgV1 pORF2 might interfere with the RNA silencing pathway by blocking the induction of RNA silencing‐related genes in F. graminearum, including FgDICER2 and FgAGO1, FgV1‐encoded viral proteins seem to lack intracellular silencing suppression activity in N. benthamiana.
Figure 6.

Suppression of green fluorescent protein (GFP) silencing by transient overexpression of each FgV open reading frame (ORF) in Nicotiana benthamiana. (A) Diagram of the constructs used for co‐agroinfiltration. The PCR product of ORF1 containing from helicase domain or full‐length of ORFs was cloned into pPZP via StuI. All clones had three consecutive HA tags for immunodetection. (B) Images of N. benthamiana leaves that were infiltrated with a mixture of Agrobacterium tumefaciens carrying the indicated construct. The mixture of constructs in each infiltrated spot is indicated in the image on the left. GFP fluorescence was visualized under UV light at 2, 3 and 4 days post‐infiltration (dpi). The viral RNA‐silencing suppressor p19 was used as a positive control.
Discussion
Viruses can interact with and manipulate host cellular machinery in a variety of ways to optimize infection and cope with host immune responses. Suppression of RNA silencing by VSRs is one common way that viruses protect their genomes in host cells. VSRs also function in viral RNA accumulation, cell‐to‐cell movement, long‐distance movement, polyprotein processing and transcription regulation (Csorba et al., 2015). VSRs have evolved independently and exhibit substantial diversity in sequence and structure (Csorba et al., 2015). Therefore, VSR mechanisms have been studied in diverse contexts involving most RNA silencing processes, such as siRNA generation, effector complex assembly, targeting, secondary siRNA synthesis and destabilization of RNA‐silencing components (Wieczorek and Obrępalska‐Stęplowska, 2015). Here, we found a unique strategy of RNA‐silencing suppression by the mycovirus FgV1, through directly affecting the host’s RNAi‐related gene expression during transcription.
The FgV1 ORF2 encodes a 16.7 kDa protein. Neither putative conserved domain nor any sequence identity with proteins coded by other viruses belonging to the newly proposed Fusariviridae family, which includes FgV1, was detected with the pORF2 (Hrabáková et al., 2017; Zhong et al., 2016). Although heterologous expression of a protein from some mycoviruses caused morphological or cytological change in their host fungi (Suzuki et al., 1999; Urayama et al., 2014), none of the FgV1 ORF proteins resulted in significant morphological or cytological changes in F. graminearum (Fig. 4). We hypothesized that the FgV1 pORF2 might contribute to symptom development by repressing the transcription of RNA silencing‐related genes in the fungal host. Using the Phyre2 program to predict the protein structure, we were unable to identify conserved structural domains with a high degree of confidence. The presence of the zinc finger domain (24%) at the N‐terminal region and the SWIRM domain family at the C‐terminal region (34%) of pORF2 was predicted by low‐confidence models. Based on these models the FgV1 pORF2 bound to the promoter regions of FgDICER1, FgDICER2, FgAGO1 and FgAGO2. We also observed that pORF2 binds in vitro to the EF1α promoter and internal region of the actin gene. It is possible that pORF2 might nonspecifically associate to some other DNA regions and affect their gene expressions. SWIRM is a small α‐helical domain that occurs in many eukaryotic chromosomal proteins involved in the protein–protein interactions of chromatin remodelling and gene expression (Da et al., 2006). Because the presence of the SWIRM domain was estimated with low confidence, whether or not it has similar functions in F. graminearum during FgV1 infection remains to be determined.
As mentioned above, VSR genes occur in the genomes of several mycoviruses, but their mechanisms of action are mostly unknown. A previous study showed that the multifunctional protein CHV1 p29 serves as an RNA‐silencing suppressor, i.e. it significantly represses the transcriptional induction of dcl2 and agl2 transcripts in C. parasitica (Sun et al., 2009). Another study recently demonstrated that p29 might block the SAGA‐mediated up‐regulation of host genes (Andika et al., 2019). The mechanism of silencing suppression seems to be quite different for the FgV1 pORF2 in F. graminearum than for CHV1 p29 in C. parasitica. Among RNAi‐related genes in C. parasitica, only agl2 and dcl2 have role(s) in the antiviral RNA‐silencing response, and expression of agl2 regulates expression of dcl2 following CHV1 infection but not following infection by mycoreovirus 1 (Sun et al., 2009). In F. graminearum, there is functional redundancy in the response to viral infection, and a significant relationship between FgDICERs and FgAGOs was not observed at the transcriptional level (Yu et al., 2018). Although the two Dicer and two Argonaute genes have functional redundancy, the latter study also showed that FgAGO1 has a vital role in the antiviral response of F. graminearum against FgV1 infection. In the current study, we found that the RNA levels of FgDICER2 and FgAGO1 were suppressed when the same wild‐type strain was infected with either FgV2 or FgV3 together with FgV1, whereas RNA levels of FgDICER1 and FgAGO2 were significantly induced when the wild‐type strain was singly infected with FgV2 or FgV3 (Table 1). Although we confirmed that the pORF2 of FgV1 can bind to the upstream promoter regions of all FgDICER and FgAGO genes (Fig. 5), we observed suppression only of FgDICER2 and FgAGO1 following infection of the wild‐type PH‐1 by FgV1 alone or by FgV1 along with other viruses. Similar changes in expression were also observed in the hairpin dsRNA‐induced RNA‐silencing transformed mutant strain. However, FgV1 infection did not significantly suppress FgDICER1 and FgAGO2. Assuming that the FgV1 pORF2 is a gene silencing suppressor that binds to the promoter regions and down‐regulates the transcription of FgDICER2 and FgAGO1 genes, determining how and why transcription or expression of FgDICER1 and FgAGO2 genes are up‐regulated in response to FgV1 infection requires additional research, even though the FgV1 pORF2 can also bind to the promoter regions of these genes.
Among mycoviruses, CHV1‐encoded p29 and RnMyRV3‐encoded S10 contribute to silencing suppression in the fungal host. Because the S10 protein of RnMyRV3 showed VSR activity in an agroinfiltration transient assay in N. benthamiana (Yaegashi et al., 2013), we also tested the VSR activity of putative FgV1‐encoded proteins in planta. None of the putative FgV1‐encoded proteins, including pORF2, suppressed transgene‐induced local silencing in N. benthamiana (Fig. 6). As discussed earlier, pORF2 was probably able to enhance viral RNA replication by suppressing transcriptional induction of FgDICER2 and FgAGO1. It is possible that pORF2 failed to suppress transgene‐induced local silencing in N. benthamiana due to the lack of specific binding of pORF2 to promoter regions of plant homologue genes. Further study is needed to determine whether FgV1‐encoded viral proteins are involved in the PTGS pathway in fungi.
The regulation of transcription of FgDICER1 and FgAGO2 might depend on the other unknown pathway despite the fact that pORF2 binds to the promoter regions of these genes in vitro. Considering that (i) transcriptional induction of FgDICER1 and FgAGO2 was not detected in GFP hairpin RNA expression and that (ii) induction of these genes was observed in mixed infections with FgV1/FgV2 or FgV1/FgV3, transcriptional induction of Dicer and Argonaute genes in F. graminearum may be differently affected by the DNA‐binding activity of pORF2. Other factors might also be responsible for a particular protein conformation that enables specific DNA binding of pORF2. It is possible that binding affinity/specificity of pORF2 at promoter region in the fungal host is determined by the protein’s chemical alteration via phosphorylation or other post‐translational modification. Computational prediction suggested that the amino acid sequence of FgV1 ORF2 contains possible phosphorylation sites on serine and threonine residues. Moreover, we found that the FgV1 pORF2 could interact with itself in a yeast two‐hybrid assay (unpublished data). A previous report indicated that the tomato golden mosaic virus protein AL2 can function as a transcriptional activator or as a silencing suppressor and can have different subcellular localizations depending on its interactions with itself, i.e. depending on its oligomeric status (Bisaro, 2006). The activity of pORF2 as a transcriptional repressor/activator may require appropriate cellular localization, oligomeric status or phosphorylation. We did not determine the subcellular localization of pORF2 with or without FgV1 infection, and further study is needed to elucidate the biological/chemical properties of FgV1‐encoded viral proteins and the interactions among viral proteins.
Many mycoviruses, including FgV1, have narrow host ranges in nature and lack an extracellular route of infection. FgV1 presumably possesses a specific silencing‐suppression mechanism in order to exist as a dsRNA without capsid and in order to defend its genomic RNA from FgAGO1‐dependent antiviral host RNA silencing responses. The current study demonstrated that the FgV1 pORF2 can directly repress the induction of FgDICER2 and FgAGO1 at the transcriptional level in F. graminearum and that the FgV1 pORF2 thereby serves as an RNA‐silencing suppressor in the infected fungal host.
Experimental Procedures
Fungal strains and culture conditions
Virus‐free and virus‐infected F. graminearum PH‐1 isolates were stored in 20% (v/v) glycerol at –80 °C and were reactivated on potato dextrose agar (PDA) at 25 °C with a 12‐h light/12‐h dark cycle. Fusarium graminearum strains used for the extraction of total RNA or genomic DNA were grown in 50 mL of complete medium (CM) at 25 °C at 150 rpm for 5 days on an orbital shaker. Mycelia were collected on sterile 3MM paper, washed with distilled water and then stored at −80 °C after removal of the excess water with paper towels.
Promoter/GFP expression assay
To make a GFP expression construct controlled by the native promoter, we amplified the upstream region that included the predicted promoter regions of FgDICER2 (RefSeq accession number FGSG_04408, 1,085 bp) and FgAGO1 (RefSeq accession number FGSG_08752, 1571 bp) from the genomic DNA of F. graminearum PH‐1. The amplified promoter and NC‐terminator were inserted into the pCB1004 vector, which was carrying the hygromycin resistance‐gene cassette. Then the eGFP fragment was inserted between each promoter and NC‐terminator. Each of the final constructs was confirmed by sequence analysis. The expression of GFP driven by the FgDICER2 or FgAGO1 promoter was confirmed by fluorescence microscopy or by real‐time RT‐qPCR.
Construction of FgV1‐encoded ORF expression clones
Constructs expressing FgV1 ORF were generated in this study. The constitutive transcriptional promoter and terminator fragments were subcloned using the pSK2707 vector, which contains the elongation factor 1α promoter (PEF1α) from Fusarium verticillioides and the β‐tubulin gene terminator (NC‐ter) from Neurospora crassa (Lee et al., 2011). A BamHI restriction enzyme site was present between PEF1α and NC‐ter. Then, we inserted the PEF1α–BamHI–NC‐ter fragment into the pCB1004 vector and named the construct pEN. FgV1 cDNA was used for amplification of four FgV1 ORFs with gene‐specific F/R primer sets containing BamHI restriction enzyme sequences at the 5′ end. Each of four amplified fragments was cloned into the pGEM‐T Easy vector (Promega, Madison, USA) and was confirmed by sequence analysis. Each verified and selected clone was then digested with BamHI for insertion into pEN. The final clones FgV1 ORF1, 2, 3 and 4 in pEN were named pEN1–4, respectively. Clones and mutants in present study are listed in Table S1. A hygromycin phosphotransferase (Hyg) gene resistance cassette was used as a selectable marker in pEN. pSA‐Gen was modified from pGFP‐SA for this study (Yu et al., 2018). The Hyg cassette in pGFP‐SA was replaced with the geneticin‐resistance cassette that was isolated from the pII99 vector using the HpaI restriction enzyme. Three independent clones for each FgV ORF were selected for further study. All primer sets are listed in Table S2.
Generation of fungal transformed mutants, DNA extraction and Southern blot hybridization
Plasmid DNA carrying GFP or the viral gene was linearized with the appropriate restriction enzyme at a location downstream of the NC‐ter region and was purified with phenol/chloroform/isoamyl alcohol. The DNA was dissolved in 20 μL of distilled water after ethanol precipitation. The final DNA constructs were introduced into fungal protoplasts using the polyethylene glycol (PEG)‐uptake method as previously described (Son et al., 2013). Single or double transformants were obtained by growing on CM containing 50 μg/mL hygromycin (Invitrogen, Carlsbad, USA) and/or geneticin (Gibco by Life Technologies, New York, USA). All transformed mutant strains were determined by Southern blot hybridization with proper probes, as described previously (Yu et al., 2015). In brief, we extracted genomic DNA by using the standard cetyltrimethylammonium bromide (CTAB) method. For Southern blot hybridization, 10 μg of genomic DNA of wild‐type and transformed mutant strains were digested with the proper restriction enzyme. Digested genomic DNA was loaded on a 0.8% agarose gel, blotted onto a nitrocellulose membrane and hybridized with [α‐32P]dCTP‐labelled DNA probe at 65 °C. Selected virus‐free transformant strains were infected with FgV1 or FgV2 by hyphal anastomosis. Viral infection was detected by RT‐PCR using virus‐specific primer pairs.
Total RNA preparation and cDNA synthesis for RT‐PCR
Total RNA was extracted using RNAiso Plus (Takara, Shiga, Japan) following the manufacturer’s instructions and was then treated with RNase‐free DNase I (Takara Bio Inc., Kusatsu, Japan) to remove genomic DNA. Finally, the extracted RNA was dissolved in DEPC‐treated water and 5 µg of total RNA was then used for cDNA synthesis with 50 pmol oligo(dT)18 primer and GoScript Reverse Transcriptase (Promega) according to the manufacturer’s protocols. All synthesized cDNAs were diluted 1:10 with distilled water for RT‐qPCR and semiquantitative RT‐PCR analyses.
RT‐qPCR and semiquantitative RT‐PCR analyses
RT‐qPCR was performed as described previously with a Bio‐Rad CFX384 real‐time PCR system using gene‐specific primers (Yu et al., 2018). The 10 µL reaction mixture included 25 ng of total cDNA mix, 5 µL of 2 × iQ SYBR green supermix (Bio‐Rad, Hercules, CA, USA) and 10 pmol of forward and reverse primers. The thermal profile was as follows: 3 min at 95 °C, then 40 cycles of 10 s at 95 °C and 30 s at 58 °C, with melting curve data obtained by increasing the temperature from 55 to 95 °C. UBH (RefSeq, FGSG_01231) and EF1α (RefSeq, FGSG_08811) were used as internal controls. Relative expression levels of genes were analysed using Bio‐Rad CFX Manager software, v. 1.6.541.1028. The experiment included at least three independent biological replicates of each strain and three technical replicates. Semiquantitative RT‐PCR was performed using the specific primer pairs. After initial denaturation for 3 min at 94 °C, the thermal profile included 25 or 33 cycles of 30 s at 94 °C, 30 s at 56 °C and 20 s at 72 °C. After electrophoresis on 1.5% agarose gels, ethidium bromide‐stained gels were visualized under a UV light. The transcript of EF1α was used as an internal control to ensure a quantitative analysis.
Protein expression and purification from E. coli
The entire region of FgV1 ORF2 and ORF3 was amplified from full‐length viral cDNA, cloned into the pET‐28a(+) vector (Novagen, Madison, WI, USA) between NdeI and BamHI restriction enzyme sites, and expressed in E. coli BL21‐CodonPlus‐RIL. Cells were grown at 37 °C in Luria–Bertani (LB) medium to an optical density of 0.5 at 600 nm, and cultures were then induced with 0.3 mM IPTG at 30 °C for 3 h. The collected cells were lysed in phosphate‐buffered saline (PBS, pH 7.4) by sonication. After cell debris was removed by centrifugation, the supernatant from the extract was collected. Purified proteins were obtained with HisPur Ni‐NTA magnetic beads (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. The eluted proteins were separated on a 12% SDS‐PAGE and stained with Coomassie Brilliant Blue. Concentrations of protein were measured using the Bradford assay.
Electrophoretic mobility shift assays
EMSAs were performed according to the standard protocol (Hellman and Fried, 2007). Double‐stranded DNA probes were labelled as described previously (Yu et al., 2015). In brief, DNA fragments approximately 500 bp of the upstream sequence that included the putative promoter region of FgDICER1, FgDICER2, FgAGO1 and FgAGO2 were amplified by PCR. Purified DNA fragments were then end‐labelled with 30 μCi of [α‐32P]dCTP and used as probes for in vitro EMSA experiments. The end‐labelled DNA and various amounts (0–3 ng) of His‐tagged proteins were incubated in binding buffer [1 mM Tris–Cl (pH 7.5), 2.5% (v/v) glycerol, 0.1 mM EDTA, 12 mM KCl, 1mM MgCl2, 1 mM dithiothreitol and 1 μg sheared salmon sperm DNA, and 0.1 μg bovine serum albumin] at 30 °C for 30 min. Samples were separated on 4.2% or 4.5% nondenaturing polyacrylamide gel in 1 × Tris–borate–EDTA at 10 mA. Gels were scanned using a Typhoon 8000 scanner (Amersham, Uppsala, Sweden). A competitive binding experiment was conducted using specific and nonspecific DNA competitors. Unlabelled DNA fragments that were identical to each probe were used as specific competitors. Before incubation with the labelled DNA probe, protein and different concentrations of competitors were incubated for 15 min at 30 °C.
Agroinfiltration silencing suppression assay in planta
For a co‐agroinfiltration assay, each of four FgV1 ORFs was PCR amplified from cDNA of FgV1 using specific ORF primer pairs. Amplified products were cloned into the pPZP vector containing a 35S promoter via restriction enzyme sites of StuI. The three consecutive or single HA tags were amplified by PCR and cloned into the C‐terminal of pPZP for immunodetection of proteins. Three independent clones for each ORF were used for further study. Competent cells of Agrobacterium tumefaciens GV3101 were chemically transformed with about 1 μg of plasmid DNA. Clones were selected on plates containing LB medium supplemented with the following antibiotics: spectinomycin (150 μg/μL), kanamycin (50 μg/μL) and rifampicin (50 μg/μL). Transformed bacterial cells were grown on a shaker at 220 rpm at 30 °C in 5 mL of LB broth supplemented with appropriate antibiotics for 16 h. The overnight cultures were then harvested and resuspended in infiltration buffer (10 mM MgCl2, 10 mM MES, 150 μM acetosyringone, pH 5.5). Resuspended cultures were incubated at room temperature for 3 h before infiltration. Equal volumes of GFP construct and each ORF construct‐transformed culture were mixed for co‐infiltration into the leaves of 3‐week‐old N. benthamiana plants. Infiltrated N. benthamiana plants were grown at 25 °C with a 16‐h light and 8‐h dark cycle. The p19 protein of tomato bushy stunt virus (TBSV) was used as a positive control. The infiltrated leaves were visualized at 2, 3 and 4 dpi under UV light and were photographed with a camera (D700, Nikon, Tokyo, Japan) with a yellow filter.
Author Contributions
J.Y. and K.H.K. planned and designed the research; J.Y., J.Y.P. and J.I.H. performed the research; J.Y., J.Y.P., J.I.H. and K.H.K. analysed and interpreted data; J.Y. and K.H.K. wrote the manuscript.
Supporting information
Fig. S1 Determination of an FgV1 ORF‐expressing mutant and a dsRNA hairpin‐expressing mutant. (A) Schematic of plasmid constructs for fungi in this experiment. ORF‐expressing mutants (pEN1–4) and a dsRNA hairpin expressing mutant (SA) were generated and analysed. A hygromycin‐resistance cassette was replaced with a geneticin‐resistance cassette in SA. The EF1α promoter was amplified from the pSK2707 vector. Each ORF was inserted downstream of the EF1α promoter. The restriction enzymes used for Southern blot analyses are indicated above the plasmid constructs, and the 32P‐labelled DNA fragments used as probes are indicated by bars. Expected DNA size and detected regions are indicated by arrows under the construct. (B) Southern blot analysis of single‐gene expression mutants. Insertion of pEN1–4 and SA mutant strains was confirmed by Southern blot hybridization using appropriate 32P‐labelled DNA for each transformed mutant strain. The images of Southern blots used for detection of each gene insertion are in the left panel, and images confirming insertion copy numbers are in the right panel. Lane 1, wild type (WT) strain PH‐1; lane 2 to 5, different biological replicates of the indicated single ORF‐expressing mutants; n, ectopic.
Fig. S2 Determination of the double‐mutant of the FgV1 ORF‐expressing mutant and the dsRNA hairpin‐expressing mutant. (A) Schematic of plasmid constructs. The pEN1–4 SA restriction enzymes used for Southern blot analyses are indicated above the plasmid constructs, and the 32P‐labelled DNA fragments used as probes are indicated by bars. Expected DNA size and detected regions are indicated by arrows under the construct. (B) Results of Southern blot hybridization of the each transformed mutant. The restriction enzyme sites used are indicated under the blotting image. The images of Southern blots for detection of each gene insertion are in the left panel, and the images for confirmation of insertion copy numbers are in the right panel. For EN1+SA and EN4+SA, we used the ORF‐expressing mutant strains that were confirmed in Fig. S1 and that were individually transformed with an SA construct. EN2+SA and EN3+SA strains were generated by transformation of fungal protoplasts with both an ORF‐expressing construct and an SA construct. Lane 1, wild type (WT) strain PH‐1; lane 2 to 5, different biological replicates of the indicated single ORF‐expressing mutants.
Fig. S3 In vitro interaction between the upstream region of RNAi‐related genes in Fusarium graminearum and His‐tagged FgV1 ORF3 protein as indicated by electrophoretic mobility shift assay. (A) Schematic representation of the upstream regions of FgDICER1, FgDICER2, FgAGO1, and FgAGO2 that were used as probes in this experiment. (B) SDS‐PAGE analysis of purified His‐tagged ORF3 protein from Escherichia coli. Samples were separated on 15% of SDS‐PAGE. Cell lysate (CE), flow through (FT), elutions (E). (C) EMSA of different concentrations of ORF3 protein with 32P‐labelled probes representing the upstream region of FgDICER1, FgDICER2, FgAGO1, and FgAGO2 as indicated.
Fig. S4 Confirmation of expression of FgV1 ORF‐encoded protein in Nicotiana benthamiana by western blot. Nicotiana benthamiana plants were co‐agroinfiltrated with Agrobacterium tumefaciens GV3101 strains harbouring pPZP‐ORF1–4 tagged with HA. Expressed proteins were recognized by anti‐HA antibodies in western analysis. Samples were separated on 8%, 10%, 12%, or 15% SDS‐PAGE acrylamide gels. Coomassie Blue stained (CBB) RuBisCO proteins are shown as the loading control.
TABLE S1 Clones and mutants used in this study.
TABLE S2 Primers used in this study.
Acknowledgements
This research was supported in part by grants from the National Research Foundation (NRF‐2016R1D1A1B03936370) funded by the Ministry of Education, Science, and Technology (MEST) and the Vegetable Breeding Research Center (no. 710011‐03) from the Ministry of Agriculture, Food and Rural Affairs, Republic of Korea. J.Y.P. and J.I.H. were supported by research fellowships from the Brain Korea 21 Plus Project. The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
- Alexander, M.M. and Cilia, M. (2016) A molecular tug‐of‐war: global plant proteome changes during viral infection. Curr. Plant Biol. 5, 13–24. [Google Scholar]
- Andika, I.B. , Jamal, A. , Kondo, H. and Suzuki, N. (2017) SAGA complex mediates the transcriptional up‐regulation of antiviral RNA silencing. Proc. Natl Acad. Sci. USA, 114, E3499–E3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andika, I.B. , Kondo, H. and Suzuki, N. (2019) Dicer functions transcriptionally and posttranscriptionally in a multilayer antiviral defense. Proc. Natl Acad. Sci. USA, 116, 2274–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltusnikas, J. , Satkauskas, S. and Lundstrom, K. (2018) Long‐term transcriptional gene silencing by RNA viruses. Trends. Biochem. Sci. 43, 397–401. [DOI] [PubMed] [Google Scholar]
- Bisaro, D.M. (2006) Silencing suppression by geminivirus proteins. J. Virol. 344, 158–168. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Gao, Q. , Huang, M. , Liu, Y. , Liu, Z. , Liu, X. and Ma, Z. (2015) Characterization of RNA silencing components in the plant pathogenic fungus Fusarium graminearum . Sci. Rep. 5, 12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, W.K. , Lee, K.‐M. , Yu, J. , Son, M. and Kim, K.‐H. (2013). Insight into mycoviruses infecting Fusarium species. Adv. Virus Res. 86, 273–288. [DOI] [PubMed] [Google Scholar]
- Csorba, T. , Kontra, L. and Burgyán, J. (2015) Viral silencing suppressors: tools forged to fine‐tune host–pathogen coexistence. Virology, 479, 85–103. [DOI] [PubMed] [Google Scholar]
- Cui, X. , Li, G. , Wang, D. , Hu, D. and Zhou, X. (2005) A begomovirus DNAβ‐encoded protein binds DNA, functions as a suppressor of RNA silencing, and targets the cell nucleus. J. Virol. 79, 10764–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da, G. , Lenkart, J. , Zhao, K. , Shiekhattar, R. , Cairns, B.R. and Marmorstein, R. (2006) Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl Acad. Sci. USA, 103, 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakouras, A. and Wassenegger, M. (2013) Revisiting RNA‐directed DNA methylation. RNA Biol. 10, 453–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweba, C. , Figlan, S. , Shimelis, H. , Motaung, T. , Sydenham, S. , Mwadzingeni, L. and Tsilo, T. (2017) Fusarium head blight of wheat: pathogenesis and control strategies. J. Crop Prot. 91, 114–122. [Google Scholar]
- Ferrigo, D. , Raiola, A. and Causin, R. (2016) Fusarium toxins in cereals: occurrence, legislation, factors promoting the appearance and their management. Molecules, 21, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzawa, N. , Itchoda, N. , Ishihara, T. , Goto, K. , Masuta, C. and Matsumura, T. (2010) HC‐Pro, a potyvirus RNA silencing suppressor, cancels cycling of Cucumber mosaic virus in Nicotiana benthamiana plants. Virus Genes, 40, 440–446. [DOI] [PubMed] [Google Scholar]
- Golem, S. and Culver, J.N. (2003) Tobacco mosaic virus induced alterations in the gene expression profile of Arabidopsis thaliana . Mol. Plant‐Microbe Interact. 16, 681–688. [DOI] [PubMed] [Google Scholar]
- Hellman, L.M. and Fried, M.G. (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nat. Protoc. 2, 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabáková, L. , Grum‐Grzhimaylo, A.A. , Koloniuk, I. , Debets, A.J. , Sarkisova, T. and Petrzik, K. (2017) The alkalophilic fungus Sodiomyces alkalinus hosts beta‐and gammapartitiviruses together with a new fusarivirus. PLoS ONE, 12, e0187799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incarbone, M. and Dunoyer, P. (2013) RNA silencing and its suppression: novel insights from in planta analyses. Trends Plant Sci. 18, 382–392. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A. , Mezulis, S. , Yates, C.M. , Wass, M.N. and Sternberg, M.J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, S.‐J. , Lim, W.‐S. , Park, S.‐H. , Park, M.‐R. and Kim, K.‐H. (2007) Molecular characterization of a dsRNA mycovirus, Fusarium graminearum virus‐DK21, which is phylogenetically related to hypoviruses but has a genome organization and gene expression strategy resembling those of plant potex‐like viruses. Mol. Cells, 23, 304–315. [PubMed] [Google Scholar]
- Lee, S. , Son, H. , Lee, J. , Lee, Y.‐R. and Lee, Y.‐W. (2011) A putative ABC transporter gene, ZRA1, is required for zearalenone production in Gibberella zeae . Curr. Genet. 57, 343–351. [DOI] [PubMed] [Google Scholar]
- Lee, K.‐M. , Cho, W.K. , Yu, J. , Son, M. , Choi, H. , Min, K. , Lee, Y.‐W. and Kim, K.‐H. (2014) A comparison of transcriptional patterns and mycological phenotypes following infection of Fusarium graminearum by four mycoviruses. PLoS ONE, 9, e100989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Bhattacharjee, P. , Wang, S. , Zhang, L. , Ahmed, I. and Guo, L. (2019) Mycoviruses in Fusarium species: an updating review. Front. Cell. Infect. Microbiol. 9, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochama, P. , Jadhav, P. , Neupane, A. and Lee Marzano, S.‐Y. (2018) Mycoviruses as triggers and targets of RNA silencing in white mold fungus Sclerotinia sclerotiorum . Viruses, 10, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane, A. , Mochama, P. , Feng, C. , Saleem, H. and Lee Marzano, S.‐Y. (2019) Roles of argonautes and dicers on Sclerotinia sclerotiorum antiviral RNA silencing. Front. Plant Sci. 10, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, M.‐R. , Park, S.‐H. , Cho, S.‐Y. and Kim, K.‐H. (2009) Nicotiana benthamiana protein, NbPCIP1, interacting with Potato virus X coat protein plays a role as susceptible factor for viral infection. Virology, 386, 257–269. [DOI] [PubMed] [Google Scholar]
- Pruss, G. , Ge, X. , Shi, X.M. , Carrington, J.C. and Vance, V.B. (1997) Plant viral synergism: the potyviral genome encodes a broad‐range pathogenicity enhancer that transactivates replication of heterologous viruses. Plant Cell, 9, 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley, M.J. , Rothi, M.H. , Böhmdorfer, G. , Kuciński, J. and Wierzbicki, A.T. (2017) Long‐range control of gene expression via RNA‐directed DNA methylation. PLoS Genet. 13, e1006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segers, G.C. , Van Wezel, R. , Zhang, X. , Hong, Y. and Nuss, D.L. (2006) Hypovirus papain‐like protease p29 suppresses RNA silencing in the natural fungal host and in a heterologous plant system. Eukaryot. Cell, 5, 896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son, M. , Lee, K.‐M. , Yu, J. , Kang, M. , Park, J.M. , Kwon, S.‐J. and Kim, K.‐H. (2013) The HEX1 gene of Fusarium graminearum is required for fungal asexual reproduction and pathogenesis and for efficient viral RNA accumulation of Fusarium graminearum virus 1. J. Virol. 87, 10356–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son, H. , Park, A.R. , Lim, J.Y. , Shin, C. and Lee, Y.‐W. (2017) Genome‐wide exonic small interference RNA‐mediated gene silencing regulates sexual reproduction in the homothallic fungus Fusarium graminearum . PLoS Genet. 13, e1006595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Q. , Choi, G.H. and Nuss, D.L. (2009) A single Argonaute gene is required for induction of RNA silencing antiviral defense and promotes viral RNA recombination. Proc. Natl Acad. Sci. USA, 106, 17927–17932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, N. , Chen, B. and Nuss, D.L. (1999) Mapping of a hypovirus p29 protease symptom determinant domain with sequence similarity to potyvirus HC‐Pro protease. J. Virol. 73, 9478–9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syller, J. (2012) Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol. Plant Pathol. 13, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama, S.I. , Fukuhara, T. , Moriyama, H. , Toh‐E, A. and Kawamoto, S. (2014) Heterologous expression of a gene of Magnaporthe oryzae chrysovirus 1 strain A disrupts growth of the human pathogenic fungus Cryptococcus neoformans . Microbiol. Immunol. 58, 294–302. [DOI] [PubMed] [Google Scholar]
- Vaucheret, H. and Fagard, M. (2001) Transcriptional gene silencing in plants: targets, inducers and regulators. Trends Genet. 17, 29–35. [DOI] [PubMed] [Google Scholar]
- Weinberg, M.S. and Morris, K.V. (2016) Transcriptional gene silencing in humans. Nucleic Acids Res. 44, 6505–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek, P. and Obrępalska‐Stęplowska, A. (2015) Suppress to survive—implication of plant viruses in PTGS. Plant Mol. Biol. Rep. 33, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaegashi, H. , Yoshikawa, N. , Ito, T. and Kanematsu, S. (2013) A mycoreovirus suppresses RNA silencing in the white root rot fungus, Rosellinia necatrix . Virology, 444, 409–416. [DOI] [PubMed] [Google Scholar]
- Yaegashi, H. , Shimizu, T. , Ito, T. and Kanematsu, S. (2016) Differential inductions of RNA silencing among encapsidated double‐stranded RNA mycoviruses in the white root rot fungus Rosellinia necatrix . J. Virol. 90, 5677–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Kwon, S.‐J. , Lee, K.‐M. , Son, M. and Kim, K.‐H. (2009) Complete nucleotide sequence of double‐stranded RNA viruses from Fusarium graminearum strain DK3. Arch. Virol. 154, 1855–1858. [DOI] [PubMed] [Google Scholar]
- Yu, J. , Lee, K.M. , Son, M. and Kim, K.H. (2015) Effects of the deletion and over‐expression of Fusarium graminearum gene FgHal2 on host response to mycovirus Fusarium graminearum virus 1. Mol. Plant Pathol. 16, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Lee, K.‐M. , Cho, W.K. , Park, J.Y. and Kim, K.‐H. (2018) Differential contribution of RNA interference components in response to distinct Fusarium graminearum virus infections. J. Virol. 92, e01756–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanardo, L.G. , De Souza, G.B. and Alves, M.S. (2019) Transcriptomics of plant–virus interactions: a review. Theor. Exp. Plant Phys. 31, 103–125. [Google Scholar]
- Zhong, J. , Shang, H.H. , Zhu, C.X. , Zhu, J.Z. , Zhu, H.J. , Hu, Y. and Da Gao, B. (2016) Characterization of a novel single‐stranded RNA virus, closely related to fusariviruses, infecting the plant pathogenic fungus Alternaria brassicicola . Virus Res. 217, 1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Determination of an FgV1 ORF‐expressing mutant and a dsRNA hairpin‐expressing mutant. (A) Schematic of plasmid constructs for fungi in this experiment. ORF‐expressing mutants (pEN1–4) and a dsRNA hairpin expressing mutant (SA) were generated and analysed. A hygromycin‐resistance cassette was replaced with a geneticin‐resistance cassette in SA. The EF1α promoter was amplified from the pSK2707 vector. Each ORF was inserted downstream of the EF1α promoter. The restriction enzymes used for Southern blot analyses are indicated above the plasmid constructs, and the 32P‐labelled DNA fragments used as probes are indicated by bars. Expected DNA size and detected regions are indicated by arrows under the construct. (B) Southern blot analysis of single‐gene expression mutants. Insertion of pEN1–4 and SA mutant strains was confirmed by Southern blot hybridization using appropriate 32P‐labelled DNA for each transformed mutant strain. The images of Southern blots used for detection of each gene insertion are in the left panel, and images confirming insertion copy numbers are in the right panel. Lane 1, wild type (WT) strain PH‐1; lane 2 to 5, different biological replicates of the indicated single ORF‐expressing mutants; n, ectopic.
Fig. S2 Determination of the double‐mutant of the FgV1 ORF‐expressing mutant and the dsRNA hairpin‐expressing mutant. (A) Schematic of plasmid constructs. The pEN1–4 SA restriction enzymes used for Southern blot analyses are indicated above the plasmid constructs, and the 32P‐labelled DNA fragments used as probes are indicated by bars. Expected DNA size and detected regions are indicated by arrows under the construct. (B) Results of Southern blot hybridization of the each transformed mutant. The restriction enzyme sites used are indicated under the blotting image. The images of Southern blots for detection of each gene insertion are in the left panel, and the images for confirmation of insertion copy numbers are in the right panel. For EN1+SA and EN4+SA, we used the ORF‐expressing mutant strains that were confirmed in Fig. S1 and that were individually transformed with an SA construct. EN2+SA and EN3+SA strains were generated by transformation of fungal protoplasts with both an ORF‐expressing construct and an SA construct. Lane 1, wild type (WT) strain PH‐1; lane 2 to 5, different biological replicates of the indicated single ORF‐expressing mutants.
Fig. S3 In vitro interaction between the upstream region of RNAi‐related genes in Fusarium graminearum and His‐tagged FgV1 ORF3 protein as indicated by electrophoretic mobility shift assay. (A) Schematic representation of the upstream regions of FgDICER1, FgDICER2, FgAGO1, and FgAGO2 that were used as probes in this experiment. (B) SDS‐PAGE analysis of purified His‐tagged ORF3 protein from Escherichia coli. Samples were separated on 15% of SDS‐PAGE. Cell lysate (CE), flow through (FT), elutions (E). (C) EMSA of different concentrations of ORF3 protein with 32P‐labelled probes representing the upstream region of FgDICER1, FgDICER2, FgAGO1, and FgAGO2 as indicated.
Fig. S4 Confirmation of expression of FgV1 ORF‐encoded protein in Nicotiana benthamiana by western blot. Nicotiana benthamiana plants were co‐agroinfiltrated with Agrobacterium tumefaciens GV3101 strains harbouring pPZP‐ORF1–4 tagged with HA. Expressed proteins were recognized by anti‐HA antibodies in western analysis. Samples were separated on 8%, 10%, 12%, or 15% SDS‐PAGE acrylamide gels. Coomassie Blue stained (CBB) RuBisCO proteins are shown as the loading control.
TABLE S1 Clones and mutants used in this study.
TABLE S2 Primers used in this study.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.