

Summary
Colorectal cancers (CRCs) initiate through distinct mutations, including in APC pathway components leading to tubular adenomas (TAs); in BRAF, with epigenetic silencing of CDX2, leading to serrated adenomas (SAs); and in the DNA mismatch repair machinery driving microsatellite instability (MSI). Transformation through the APC pathway involves loss of the hormone GUCA2A that silences the tumor-suppressing receptor GUCY2C. Indeed, oral hormone replacement is an emerging strategy to reactivate GUCY2C and prevent CRC initiation and progression. Moreover, retained expression by tumors arising from TAs has established GUCY2C as a diagnostic and therapeutic target to prevent and treat metastatic CRC. Here, we defined the potential role of the GUCA2A-GUCY2C axis and its suitability as a target in tumors arising through the SA and MSI pathways. GUCA2A hormone expression was eliminated in TAs, SAs, and MSI tumors compared to their corresponding normal adjacent tissues. In contrast to the hormone, the tumor-suppressing receptor GUCY2C was retained in TA and MSI tumors. Surprisingly, GUCY2C expression was nearly eliminated in SAs, reflecting loss of the transcription factor CDX2. Changes in the GUCA2A-GUCY2C axis in human SAs and MSI tumors were precisely recapitulated in genetic mouse models. These data reveal the possibility of GUCA2A loss silencing GUCY2C in the pathophysiology of, and oral hormone replacement to restore GUCY2C signaling to prevent, MSI tumors. Also, they highlight the potential for targeting GUCY2C to prevent and treat metastases arising from TA and MSI tumors. In contrast, loss of GUCY2C excludes patients with SAs as candidates for GUCY2C-based prevention and therapy.
Keywords: Serrated adenoma, Tubular adenoma, Colorectal cancer, Guanylin, Guanylate cyclase C, CDX2
1. Introduction
Colorectal cancer (CRC) is a heterogeneous disease, which can be classified into molecular subtypes originating through distinct biological pathways with some degree of overlap [1]. The conventional neoplasia pathway accounts for ~75% of CRCs and is frequently associated with chromosomal instability (CIN) [1]. This pathway is often linked to ligand-independent upregulation of canonical WNT signaling due to a loss-of-function mutation in the APC (Adenomatous Polyposis Coli) gene, facilitating nuclear translocation of β-catenin that promotes increased expression of proto-oncogenic targets such as c-MYC and CYCLIN-D1 [2]. The premalignant adenomatous polyps that arise from this pathway are often noted to have tubular, tubulovillous, or villous histology [3]. On the other hand, ~20% of all CRCs originate from serrated precursor lesions that exhibit serrated or sawtooth dysplastic crypts, and are subdivided into traditional serrated adenomas (TSA) or sessile serrated adenomas (SSAs) [4]. Adenomatous transformation and progression to carcinomas in the serrated pathway reflect molecular alterations rarely seen in the conventional pathway. Thus, mutations in the mitogen-activated protein kinase components, such as KRAS or BRAF, are early events in the serrated pathway [5,6]. Oncogenic BRAF (usually V600E) mutations are identified in only 10% of all CRCs, but 70%–80% of SSAs, and are associated with a worse prognosis [6].
Furthermore, serrated lesions are characterized by epigenetic silencing of various genes by methylation (CpG island methylator phenotype or CIMP) [5,7]. CIMP can silence tumor-suppressor genes such as p16, IGF2, MGMT, AXIN2, MCC, and CDX2 [5,7,8]. CDX2 is a caudal-type homeobox transcription factor that promotes intestinal homeostasis along the constantly renewing crypt villus axis, provides tissue identity to stem cells, and has emerged as a tumor suppressor [9,10]. The lack of CDX2 expression in CRC is associated with decreased 5-year disease-free survival and overall poor prognosis independent of tumor stage, grade, age, and sex [10]. Interestingly, BRAF mutations and loss of CDX2 expression occur together frequently in serrated tumors and are associated with worse prognosis [6,11].
In addition, a hypermutable phenotype known as microsatellite instability (MSI) pathway accounts for 12%–17% of all CRCs [8]. MSI results from a loss of genomic replicative fidelity due to deficiencies of the DNA mismatch repair (MMR) machinery. The hereditary subtype of MMR deficiency, also called Lynch syndrome or hereditary nonpolyposis CRC (HNPCC), accounts for 3% of MSI tumors and is characterized by germline mutations or epimutations in 1 or more of the 4 MMR genes: MSH2, MLH1, MSH6, and PMS2 [8,12]. In contrast, sporadic MSI tumors are characterized by biallelic methylation of the MLH1 promoter and functional loss of MLH1 and PMS2 proteins [8,12]. Interestingly, sporadic MSI tumors are associated with a background of CIMP and harbor the BRAF (V600E) mutation in half of cases, which is associated with a worse prognosis [8,12,13]. Thus, the serrated neoplasia phenotype can be associated with sporadic MSI; indeed, they are not mutually exclusive.
Guanylate cyclase C (GUCY2C) is a transmembrane receptor expressed on apical surfaces of enterocytes in small intestine, colon, and rectum, where it surveys the lumen for its peptide ligands [14]. The intestine expresses 2 peptide hormones that function as paracrine endogenous GUCY2C ligands: uroguanylin (GUCA2B) and guanylin (GUCA2A) in the small and large intestine, respectively [14]. In addition, GUCY2C is the intestinal target for enterotoxigenic bacterial heat-stable enterotoxins, which are structurally analogous to its endogenous ligands and the pathogenic basis for traveler’s diarrhea [14,15]. Once activated, GUCY2C produces the second messenger cyclic guanosine monophosphate (cGMP), activating downstream effectors such as cGMP-dependent protein kinase, which phosphorylates the cystic fibrosis transmembrane conductance regulator channel promoting salt and water secretion [15].
Beyond fluid homeostasis, GUCY2C has emerged as a tumor suppressor, and transformation is associated with the loss of guanylin expression which silences receptor signaling and disrupts intestinal homeostasis to promote CRC [16,17]. Indeed, GUCA2A expression is lost, whereas GUCY2C is retained, in human colorectal cancers, as well as in adenomas in ApcMin mice and patients with familial adenomatous polyposis, reflecting the conventional pathway of tumorigenesis [18–20]. In that context, GUCA2A loss coupled with retention of GUCY2C provides a unique opportunity for primary chemoprevention of the conventional pathway of transformation by reactivating receptor signaling through oral hormone replacement [19].
Furthermore, retention of GUCY2C by metastatic tumors arising by the conventional pathway of transformation from the colorectum also has emerged as a mucosally restricted tumor-associated antigen that is a diagnostic marker [20] and therapeutic target for cancer vaccines, adoptive cell therapies, and antibody-drug conjugates [21]. In that regard, luminal compartmentalization in apical membranes of normal epithelial cells, but systemic exposure in metastatic tumors, makes GUCY2C a unique target for systemic therapies, eliminating extraintestinal deposits of cancer without adverse effects on the normal intestinal mucosa [22].
The roles of silencing the GUCA2A-GUCY2C signaling axis in the pathogenesis of, and the potential utility of GUCY2C as a chemoprevention and therapeutic target in, tumors that arise from the conventional pathway involving mutations in APC or CTNNB1 genes are established. However, changes in expression of GUCA2A or GUCY2C, and the associated utility of the receptor in chemoprevention and targeted therapeutic paradigms, in tumors that arise by SA and MSI pathways remain unknown. Here, we define the expression of GUCY2C and GUCA2A in tubular adenomas (TAs), serrated adenomas (SAs), and MSI tumors and their normal adjacent tissues (NATs).
2. Materials and methods
2.1. Human tumor tissues and histological interpretation
The study was approved by the institutional review board of Thomas Jefferson University (IRB control #14D.376). To investigate GUCA2A and GUCY2C protein and mRNA expression, we obtained human tissues as formalin-fixed paraffin-embedded (FFPE) blocks from 47 patients from the archives of Department of Pathology at Thomas Jefferson University Hospital and Cooperative Human Tissue Network (CHTN: https://www.chtneast.org). These included normal colon (n = 7); TAs (n = 18); SAs (n = 15); MSI tumors (n = 7); and, where available, their matched NATs. The diagnosis of serrated lesions and MSI tumors was made based on established histopathologic criteria by a board-certified gastrointestinal pathologist (J. P.) [4,8]. Tumors and their matched normal epithelia were labeled on the hematoxylin- and eosin (H&E)-stained tissue sections by a pathologist (E. G.) blinded to immunofluorescence (IF) results. Clinicopathologic data included patient age, sex, location of tumor (proximal colon was classified as proximal to the splenic flexure, and the remaining region was defined as distal), and clinicopathologic subtypes and are summarized in the Table.
Table.
Patient characteristics
| TA (n = 118) | SA (n = 15) | Normal colon (n = 7) | MSI tumor (n = 7) | |
|---|---|---|---|---|
| Male/female ratio | 7:11 | 5:10 | 3:4 | 3:4 |
| Median age in years (IQR) | 54 (47–70) | 65 (55–69) | 50 (44–77) | 60 (41–61) |
| Proximal/distal colon a | 8:10 | 10:5 | 2:5 | 6:1 |
| NAT present | 55% | 80% | – | 86% |
| Subtype | Tubular 7 | SSA 13 | – | Sporadic 3 |
| Tubulovillous 8 | TSA 2 | Hereditary 4 | ||
| Villous 1 | ||||
| FAP2 |
Abbreviations: FAP, familial adenomatous polyposis; IQR, interquartile range.
Proximal colon: classified as proximal to the splenic flexure; the remaining region was defined as distal.
2.2. Mouse tissues
CDX2P-CreERT2 mice express Cre-ERT2 under the control of CDX2 promoter/enhancer regions, enabling tamoxifen-inducible targeting of loxP alleles from the ileal to rectal epithelium. Cdx2fl/fl mice exhibit biallelic Cdx2 loss (Cdxfl/fl) following Cre-mediated recombination. BrafCA mice express normal BRAF prior to Cre exposure and mutationally active BRAFV600E following Cre-mediated recombination [23]. Four-micrometer–thick tissue sections from tamoxifen-treated and -untreated mice harboring these transgenes were used for the work here [24]. These included 8 serrated cecal tumors with compound Cdx2 loss and BrafV600E expression. Selected tissues from wild-type and CreERT2-negative control mice also were used for comparison [24]. Also, Msh2fl/fl mice with a constitutively active vil-Cre transgene were acquired [25]. Tumors harvested from Msh2fl/fl mice, predominantly located in small intestine, were fixed in formalin and embedded in paraffin for histology and flash frozen in liquid nitrogen for protein and mRNA analysis. All mice were C57BL/6, and animal husbandry was compliant with local IACUC standards.
2.3. RNA isolation
Total RNA was isolated from animal tissues using RNeasy RNA extraction kits (Qiagen, Germantown, MD). Also, total RNA was extracted from human FFPE blocks. At least 20-μm slices of tissue (up to 4×) were cut using a microtome. Tissue slices were deparaffinized using 2 applications of Citra Solv detergent (Citra Solv LLC, Danbury, CT) overnight at room temperature. After removing Citra Solv, samples were washed with ethyl alcohol ×3. Air-dried samples were incubated in at least 90 μL of digestion buffer containing 0.1 mol/L NaCl (AM9760G, Life Technologies, Carlsbad, CA), 10 mmol/L Tris (AM9855G, Life technologies), 1 mmol/L EDTA (AM9260G, Life Technologies), 0.75% sodium dodecyl sulfate (AM9822, Life technologies), and 10 μL of proteinase K solution (AM2548, Life Technologies) at 56°C overnight. Proteinase K was then heat-inactivated by placing the solution at 90°C for 5 minutes, and samples were subjected to extraction with RNeasy kits (Qiagen). RNA was eluted from RNeasy column using molecular-grade water, concentration determined by spectrophotometry, and stored at −80°C.
2.4. Quantitative reverse-transcription polymerase chain reaction
Relative mRNA levels of GUCY2C, GUCA2A, and GUCA2B (relative to GAPDH levels) in both human and mouse tissue were determined by quantitative real-time reverse-transcription polymerase chain reaction (RT-PCR) by using commercially available primer-probe sets (for mouse: Gucy2c, Mm01267705_m1; Guca2a, Mm00433863_m1; Gapdh, Mm99999915_g1; Guca2b, Mm01192051_m1; for human: GUCY2C, Hs00990129_m1; GUCA2A, Hs00157859; GAPDH, Hs02758991_g1 [Life Technologies]). Target mRNA was amplified from total RNA using RNeasy mini kits (Qiagen) in a 25-μL reaction on an ABI 7000 Sequence Detection System (Applied Biosystems, Foster City, CA).
2.5. Immunoblot analysis
Protein was extracted from mouse tissues lysed in M-PER reagent (Pierce, Rockford, IL) containing phosphatase and protease inhibitor cocktails (Roche, Indianapolis, IN) and Ben-zonase nuclease (Sigma, St Louis, MO). Lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (NuPAGE 4%–12% Bis-Tris Gel; Novex Life Technologies) and transferred to a nitrocellulose membrane (Novex Life Technologies). Membranes were blocked in 10% milk (nonfat dry milk, M0841, LabScientific, Highlands, NJ) in 1× phosphate-buffered saline and 1% Tween 20 and probed overnight with antibodies to GUCY2C (1:500; Ms20 [26]), GAPDH (1:5000; Cell Signaling Technologies, Danvers, MA), GUCA2A (1:1000), and GUCA2B (1:1000) followed by incubation with goat anti-mouse horseradish peroxidase-conjugated and goat anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:30000; Jackson ImmunoResearch, West Grove, PA). Rabbit anti-GUCA2A (#2538) and anti-GUCA2B (#6910) antisera used for detecting mouse tissue guanylin and uroguanylin, respectively, were a gift from M. Goy (University of North Carolina) [27]. Blots were developed in SuperSignal West Dura enhanced chemilu-minescence substrate (Thermo Scientific, Waltham, MA). Relative intensity was quantified by densitometry using ImageJ software (v1.51 s, National Institute of Health, Bethesda, MD) and normalized to the intensity of GAPDH.
2.6. Immunofluorescence staining and signal quantification
Sections (4 μm) were obtained from human and mouse specimens in FFPE blocks. After tissue deparaffinization and rehydration, slides were subjected to heat-induced epitope retrieval by boiling in a pressure cooker for 15 minutes in pH 9 antigen retrieval buffer (Dako, Carpinteria, CA). Protein expression was determined by following primary antibodies for immunostaining: mouse anti-β-catenin IgG1 (1:100, sc-7963, Santa Cruz Biotechnology, Dallas, TX); rabbit anti-CDX2 conjugated with Alexa Fluor 647 (1:500, ab195008, Abcam, Cambridge, MA); rabbit anti-GUCA2A for human guanylin (1:500, HPA018215, Sigma-Aldrich, St Louis, MO), mouse anti-GUCY2C IgG2a (2 μg/mL; Ms20 [26]), rabbit anti-GUCA2A (1:1000), and rabbit anti-MSH2 (1:100, D24B5, Cell Signaling Technology, Danvers, MA). Slides were stained according to a modified Alexafluor protocol (Life Technologies, Carlsbad, CA) or tyramide signal amplification protocol [28]. Appropriate negative controls were used to improve specificity of IF staining. Thus, GUCA2A blocking peptide (APREST72418, Sigma, St Louis, MO) was preincubated with primary antibody at the manufacturer’s recommended concentration. Mouse IgG2a isotype control was used as a GUCY2C negative control (Biolegend, San Diego, CA). For all runs of IF, normal tissue served as a positive control. All images were captured using EVOS FL auto imaging system (Life Technologies) with a 20× objective. To quantify GUCA2A, GUCY2C, CDX2, and MSH2 protein expression, a single in-focus plane was acquired. Using ImageJ software, quantification in defined regions of interest was performed by calculating corrected total cell fluorescence (CTCF) using the following equation: CTCF = integrated density − (area of selected cell × mean fluorescence of background readings). CTCF for each tissue section was calculated relative to control region of interest.
2.7. Gene expression data
We obtained log2-transformed GUCY2C-GUCA2A gene expression data from colonic tissue in Cdx2fl/fl, BrafV600E, Wt control mice as well as in serrated tumors from compound Cdx2flfl and BrafV600E mice from Gene Expression Omnibus (accession number GSE85650) [24]. We obtained The Cancer Genome Atlas (TCGA) [29] gene expression data using UCSC genome browser (available at: http://xenabrowser.net) to compare GUCA2A-GUCY2C-CDX2 expression in human CRCs and normal colonic tissue. We classified human CRCs into MSI and microsatellite-stable (MSS) tumors to compare GUCA2A and GUCY2C gene expression (Supplementary Figs. 1 and 2). MSI was defined by reduced expression in 1 or more of following MMR genes: MLH1, MSH2, MSH6, and PMS2.
2.8. Statistical analysis
Statistical significance was analyzed using Student t test, 1e-way analysis of variance, and Pearson correlation coefficient. All Ps were 2-tailed with P<.05 considered statistically significant. Ps were corrected for multiple testing using Bonferroni method. Sample sizes of mouse and human tissues were based on experience with previous similar experiments. All statistical analyses were performed using GraphPad Prism version 7 (La Jolla, CA). Analyses represent mean ± SEM, unless otherwise indicated; *P<.05, **P<.01, ***P<.001, and ****P<.0001.
3. Results
3.1. Patient characteristics
Cohorts with SAs and TAs were balanced with respect to age and sex (P > .05; Table). Patients in the normal colon and MSI tumor cohorts also were balanced with respect to age and sex (P > .05; Table). SAs and MSI tumors were located predominantly in proximal colon, in agreement with previous observations, whereas TAs were distributed equally between proximal and distal colon. TAs included all major histologic subtypes including tubular, tubulovillous, and villous adenomas. In addition, adenomas from patients with familial adenomatous polyposis also were analyzed. SAs included both major pathogenic subtypes: SSA and TSA. The MSI tumor cohort included both sporadic and hereditary subtypes.
3.2. Tubular adenomas
In close agreement with earlier transcriptomic analyses [18,19], GUCA2A protein and mRNA expression was eliminated in TAs (Fig. 1A–E). As demonstrated previously, elimination of guanylin hormone expression was universal, and all TAs analyzed here exhibited complete loss of GUCA2A protein compared to NATs (P < .01; Fig. 1B–D). Similarly, GUCA2A mRNA expression was reduced in adenomas compared to normal colonic mucosa (Fig. 1E). In contrast to the hormone, the tumor-suppressing receptor GUCY2C mRNA and protein were retained in TAs compared to normal mucosa (Fig. 1F–I), in alignment with previous transcriptomic and immunohistochemical analyses of CRCs from patients [18,19]. Again, receptor expression was universal, and TAs analyzed here exhibited a 25-fold increase in GUCY2C protein compared to NATs (P < .05; Fig. 1G–I).
Fig. 1.
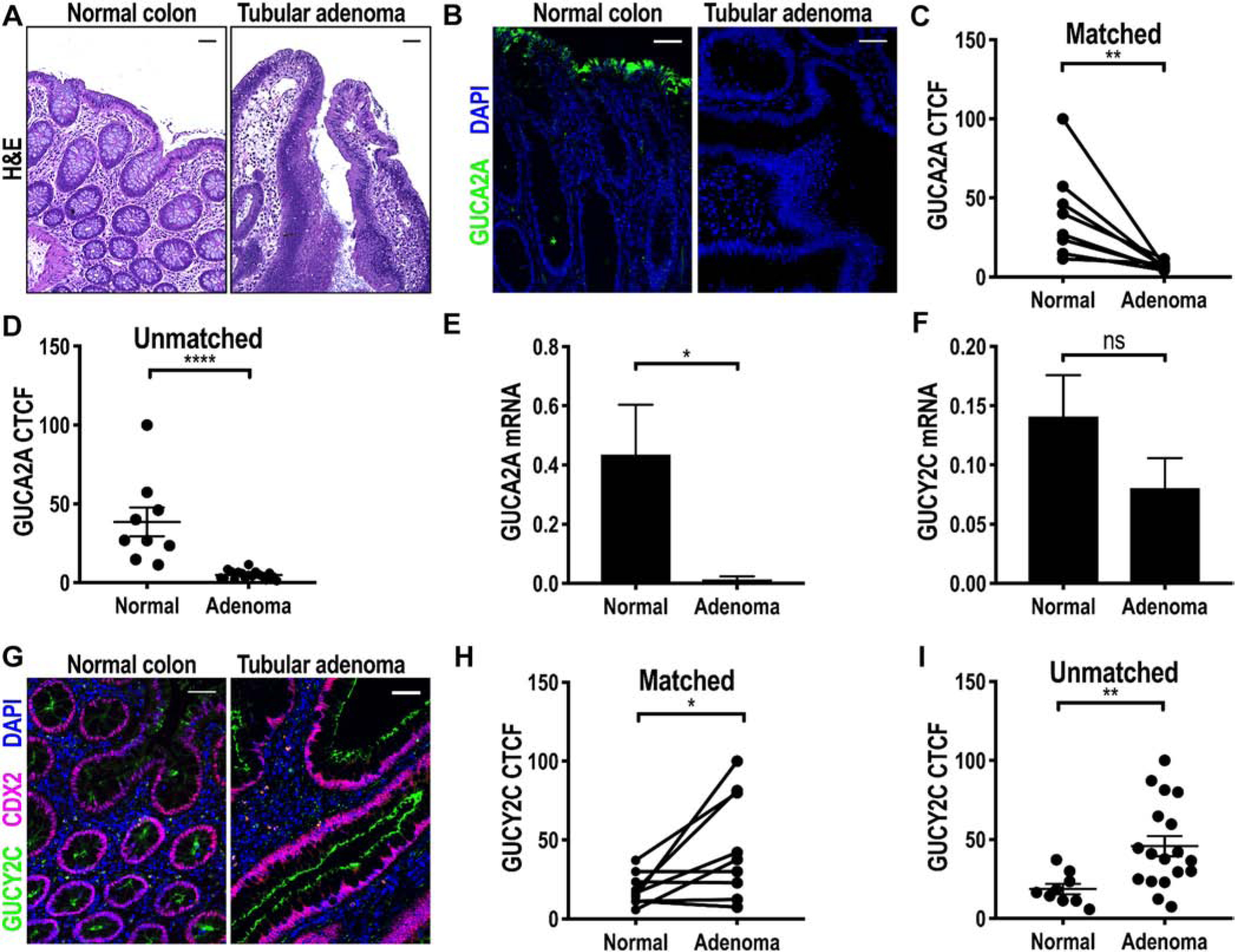
GUCA2A, but not GUCY2C, is lost in TAs. A, Representative H&E image of normal colon and TA. B, Representative IF image of human TAs and normal colonic mucosa with GUCA2A immunostain. C and D, GUCA2A (green) quantified by IF (B) in TAs and NAT in matched (n = 9; C) and unmatched (n = 18; D) specimens. E, GUCA2A mRNA quantified by RT-PCR in TAs (n = 9) compared to normal colonic mucosa (n = 8). F, GUCY2C mRNA quantified by RT-PCR in TAs (n = 9) compared to normal colonic mucosa (n = 8). G-I, GUCY2C (green; F) quantified by IF in TAs and NATs in matched (n = 9; H) and in unmatched (n = 18; I) specimens (purple, CDX2; blue, DAPI). *P < .05; **P < .01, ****P < .0001. Scale bar = 100 μm.
3.3. Serrated adenomas
As in TAs, hormone loss was universal, and SAs exhibited reduced expression (P < .001) of GUCA2A protein (Fig. 2A–C). Similarly, GUCA2A mRNA expression also was reduced in SAs compared to normal colonic mucosa (Fig. 2D). Unexpectedly, expression of GUCY2C mRNA and protein was eliminated in almost all SAs (Fig. 2E–H), in striking contrast to receptor retention observed in TAs (Fig. 1E–H). Again, receptor loss was universal, and all SAs exhibited reduced expression (P < .001) of GUCY2C protein compared to NAT (Fig. 2G and H). Co-staining CDX2 and GUCY2C in human SAs confirmed loss of GUCY2C receptor with absent nuclear CDX2 staining (Fig. 2F; Supplementary Fig. 3A and B). Indeed, retention of CDX2 expression within a crypt was associated with preservation of GUCY2C expression and normal morphology (arrowhead in Fig. 2F and I). Moreover, TCGA data revealed a direct relationship between CDX2 and GUCY2C in colonic adenocarcinomas (correlation coefficient: 0.74, Supplementary Fig. 4).
Fig. 2.
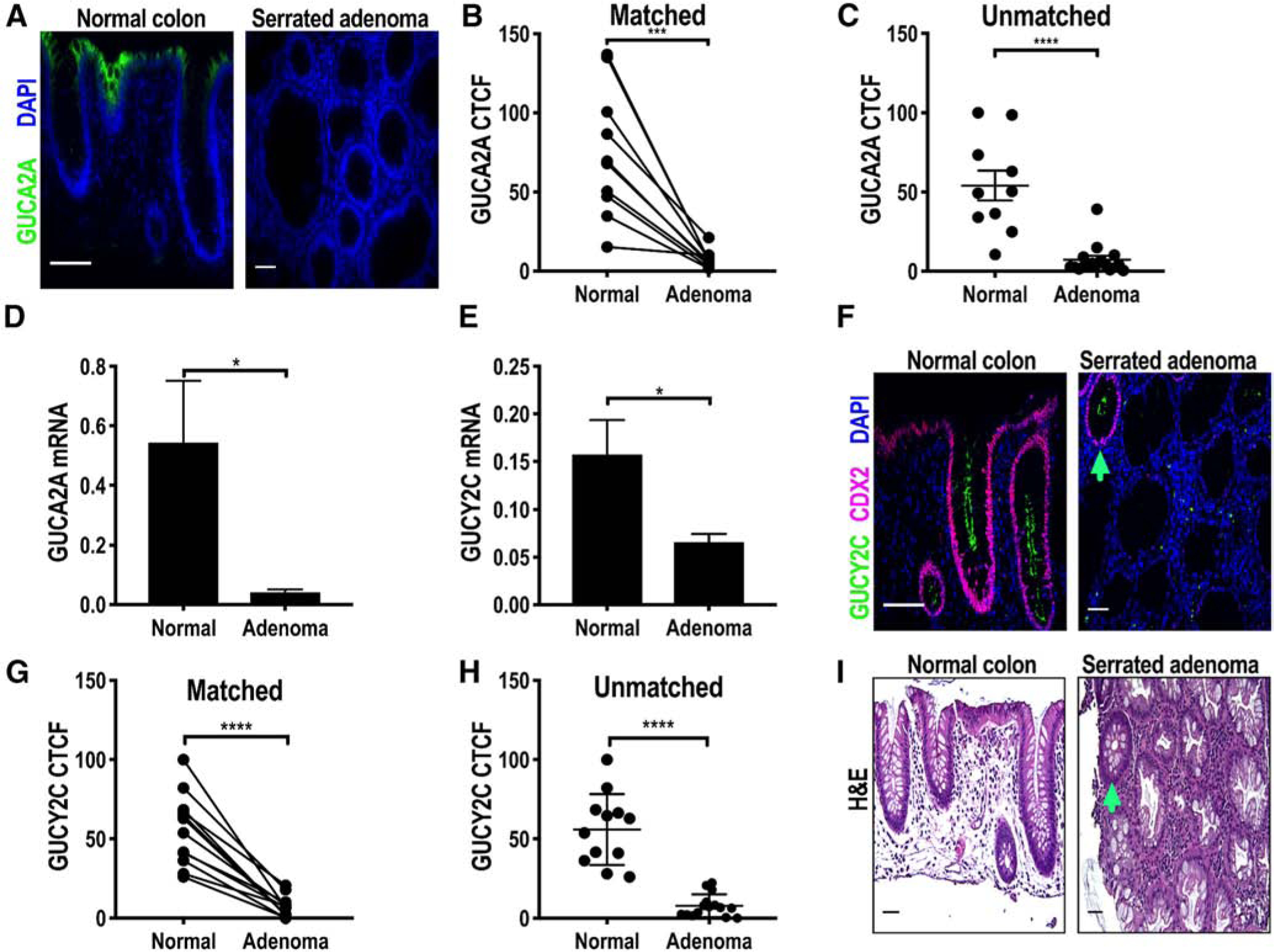
GUCY2C and GUCA2A are lost in SAs. A, Representative IF image of human SAs and normal colonic mucosa. A-C, GUCA2A (green) quantified by IF (A) in SAs compared to NAT in matched (n = 12; B) and unmatched (n = 15; C) specimens. D, GUCA2A and (E) GUCY2C mRNA quantified by RT-PCR in SAs (n = 6) compared to normal colonic mucosa (n = 6). F-H, GUCY2C (green) quantified by IF (F) in SAs and NATs in matched (n = 9; G) and unmatched (n = 18; H) specimens. I, Representative H&E image of normal colon and SA. Arrowhead on F and I corresponds to a normal-appearing crypt (I) with preservation of CDX2 and GUCY2C staining (F) (purple, CDX2; blue, DAPI). *P < .05; ***P < .001, ****P < .0001. Scale bar = 100 μm.
Serrated tumors from mouse cecum in CDX2P-CreERT2-Cdx2FL/FL-BrafV600E mice recapitulated GUCA2A loss observed in human SAs (Fig. 3A). Indeed, GUCA2A protein (Fig. 3B) and mRNA (Fig. 3C) were reduced in serrated tumors compared to Wt controls. In contrast, GUCA2A mRNA and protein were preserved in tissues from Cre recombinase-negative, CDX2P-CreERT2-Cdx2FL/FL, or mutant BrafV600E mice (Fig. 3A–C). Similarly, GUCY2C protein and mRNA were reduced in serrated tumors from CDX2P-CreERT2-Cdx2FL/FL-BrafV600E mice compared to Wt controls (Fig. 3D–F). GUCY2C protein and mRNA were unchanged in tissues from Cre recombinase-negative and mutant BrafV600E mice compared to Wt controls. In contrast, GUCY2C mRNA and protein were reduced in tissues from CDX2P-CreERT2-Cdx2FL/FL mice (Fig. 3D–F). Quantitative differences between changes in mRNA and protein expression reflect the mosaicism of gene loss following activation of Cre recombinase by tamoxifen. In that context, protein expression was analyzed in homogenous transformed tissues visualized by IF, compared to analyses of mRNA which quantify expression in heterogeneous samples of tumor admixed with NAT.
Fig. 3.
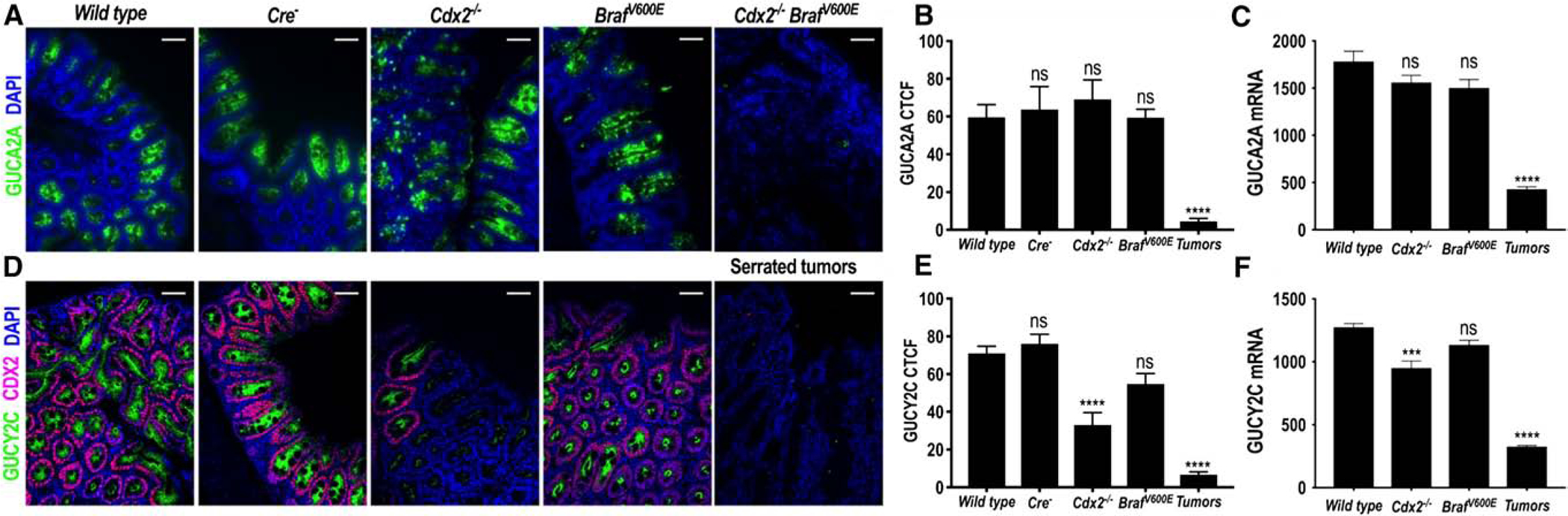
GUCA2A and GUCY2C expression following Cdx2 inactivation and/or BrafV600E mutation in mice. A, Representative IF images of expression of GUCA2A (green) from CDX2P-CreER T2-Cdx2fl/fl (Cdx2fl/fl), CDX2P-CreERT2-BrafCA (BrafV600E), CDX2P-CreERT2-Cdx2fl/fl -BrafCA (Cdx2fl/fl-BraJV600E), CreERT2 negative control (Cre−), and wild-type control mice. B, IF quantification revealed decreased GUCA2A expression in serrated tumors (n = 8) from compound Cdx2fl/fl -BrafV600E mice, but not Cre− control (n = 6), Cdx2fl/fl (n = 3), or BrafV600E (n = 3) mice, compared to wild-type controls (n = 6). C, GUCA2A mRNA was reduced in serrated tumors from compound Cdx2fl/fl BrafV600E mice, but not in Cdx2fl/fl or BrafV600E mice, compared to wild-type controls (n = 3). D, Representative IF images of expression of GUCY2C (green) and CDX2 (purple) from CDX2P-CreERT2-Cdx2fl/fl (Cdx2fl/fl), CDX2P-CreERT2-BrafCA (BrafV600E), CDX2P-CreERT2-Cdx2fl/fl-BrafCA (Cdx2fl/fl -BraJV600E), CreERT2 negative control (Cre−), and wild-type control mice. E, IF quantification revealed decreased GUCY2C expression in serrated tumors (n = 8) from compound Cdx2fl/fl BrafV600E mice compared to wild-type controls (n = 6). GUCY2C expression was also reduced in Cdx2fl/fl (n = 3) mice compared to wild-type controls, but not in Cre− control (n = 6), or BrafV600E (n = 3) mice. F, GUCY2C mRNA was reduced in both Cdx2fl/fl and serrated tumors in compound Cdx2fl/fl BrafV600E mice, but not in BrafV600E mice, compared to wild-type controls (n = 3). Blue, DAPI. **P < .01, ****P < .0001. Scale bar = 100, μm.
3.4. MSI tumors
The foregoing analyses reveal differences in expression of components of the GUCA2A-GUCY2C axis in tumors arising from the APC-β-catenin and CDX2-Braf pathways that, in turn, have implications for their utility as targets for cancer prevention and therapy. These differences underscore the necessity of defining their expression in CRC tumors initiating through divergent molecular pathways. In that context, inactivation of DNA mismatch repair genes through hypermethylation or heritable mutations creates MSI contributing to HNPCC and Lynch syndrome [8,12,13]. As in tumors arising through the APC-β-catenin and CDX2-Braf pathways, GUCA2A protein and mRNA expression was lost in human MSI tumors (Fig. 4A–E). However, in contrast to tumors arising through the CDX2-Braf pathway, GUCY2C mRNA and protein expression was retained in MSI tumors (Fig. 4F–I), which was comparable to tumors arising through the APC-β-catenin pathway. TCGA data confirmed loss of GUCA2A, but retention of GUCY2C, mRNA in MSI and MSS tumors (Supplementary Figs. 1 and 2).
Fig. 4.
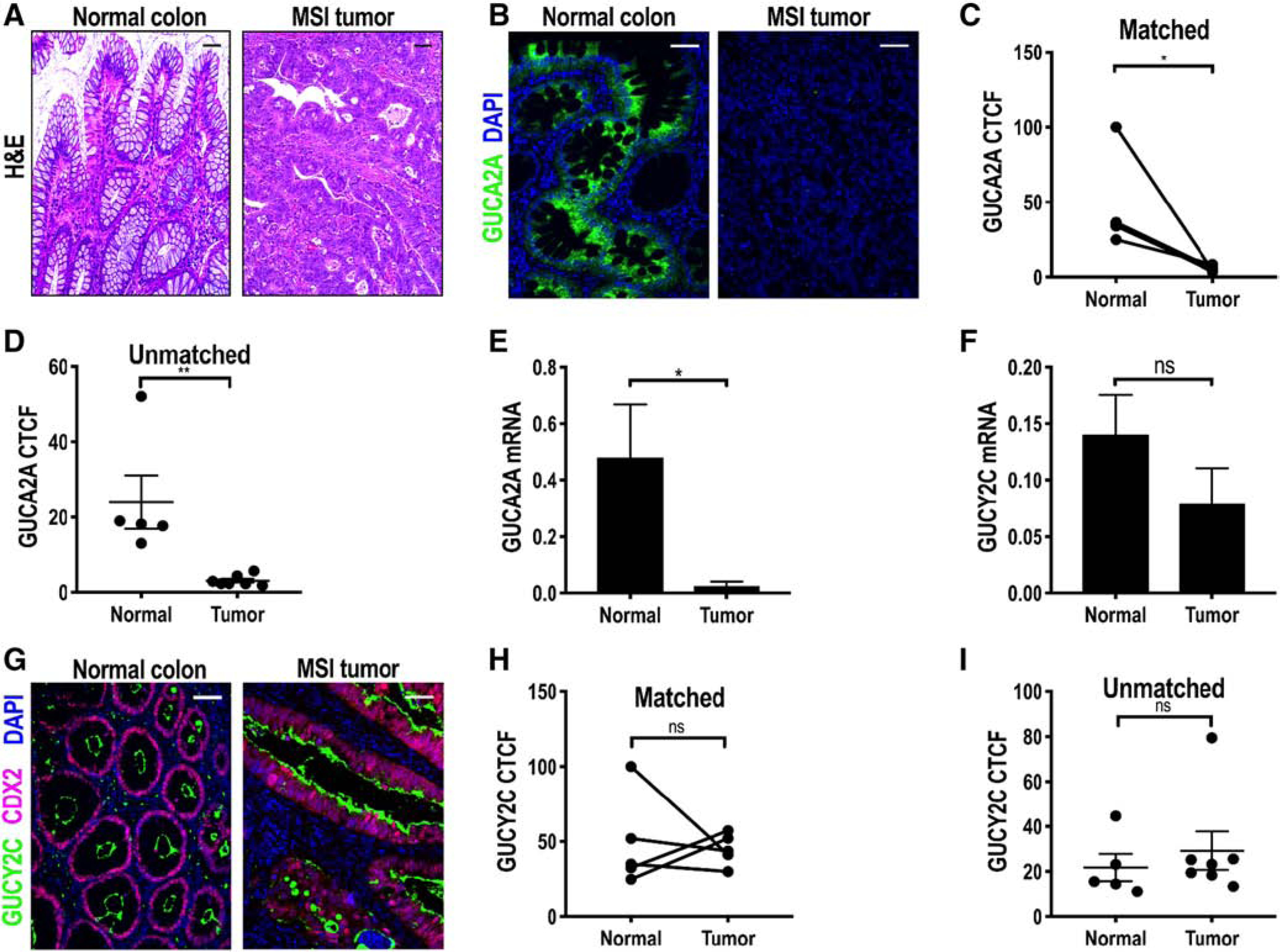
GUCA2A, but not GUCY2C, is lost in MSI tumors. A, Representative H&E image of normal colon and MSI tumor. B, Representative IF images of MSI tumors and normal colonic mucosa. C and D, GUCA2A (green) quantified by IF (B) in MSI tumors and NATs in matched (n = 5; C) and unmatched (n = 7; D) specimens. E, GUCA2A mRNA quantified by RT-PCR in MSI tumors (n = 6) compared to normal colonic mucosa (n = 7). F, GUCY2C mRNA quantified by RT-PCR in MSI tumors (n = 6) compared to normal colonic mucosa (n = 7). G-I, GUCY2C (green) quantified by IF (G) in MSI tumors and NATs in matched (n = 5; H) and unmatched (n = 7; I) specimens (purple, CDX2; blue, DAPI). *P < .05; **P < .01. Scale bar = 100, μm.
Msh2FL/FL mice develop jejunal tumors (Fig. 5A and B) with complete loss of the DNA mismatch repair protein MSH2 compared to Wt controls (Fig. 5C and D) [25]. Whereas GUCA2A is almost exclusively the GUCY2C ligand expressed in colon, GUCA2B is the primary ligand expressed in small intestine [14]. In agreement with human MSI tumors from colon, jejunal tumors from Msh2FL/FL mice exhibited loss of GUCA2A (Fig. 5E–G) and GUCA2B (Fig. 5H) protein. Interestingly, the mRNA for these ligands was not significantly different between wild-type and Msh2FL/FL tumors (Fig. 5I and J). Again, in agreement with human tumors, GUCY2C protein and mRNA were retained in Msh2FL/FL tumors (Fig. 5K–N).
Fig. 5.
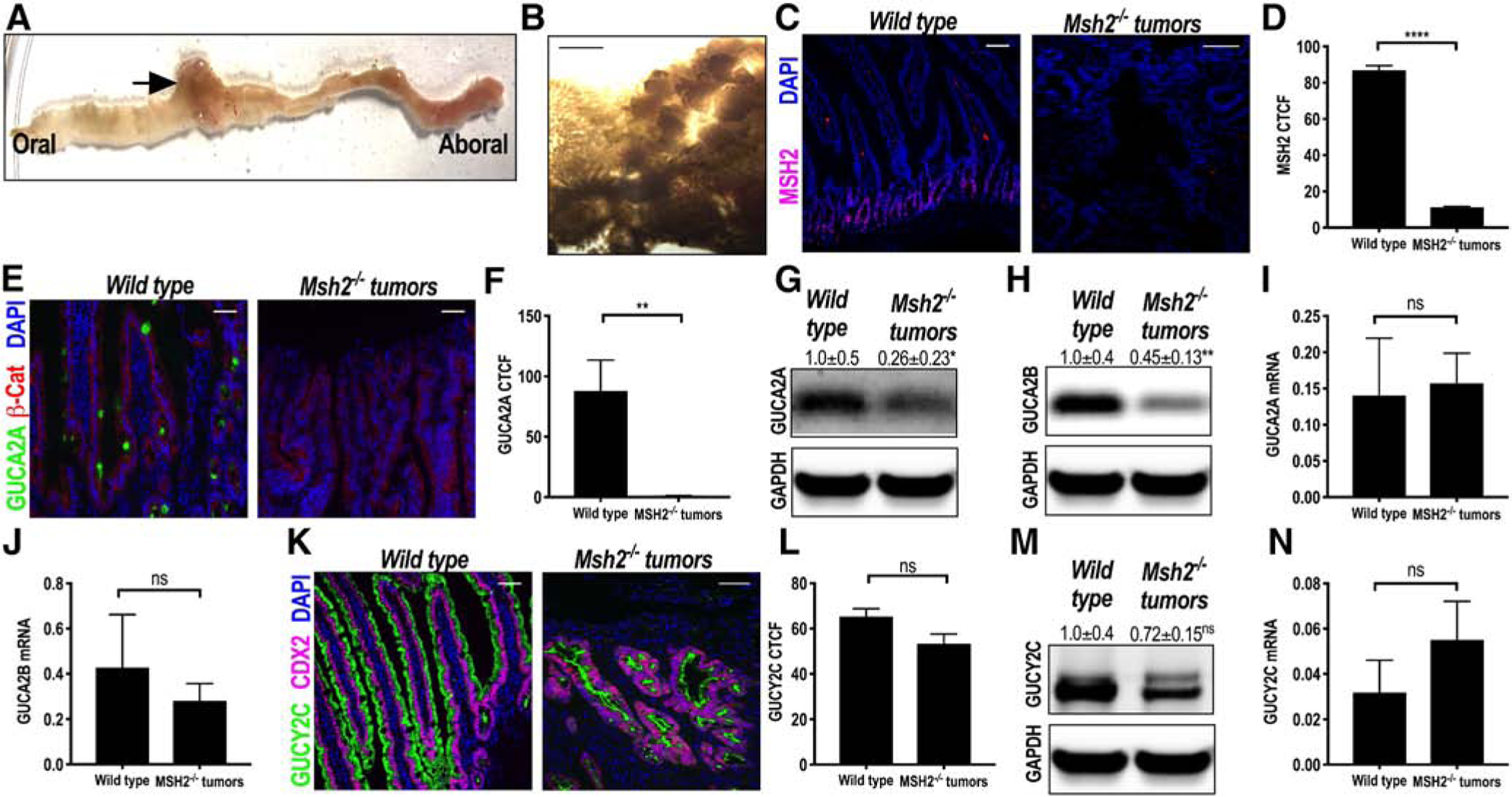
GUCA2A and GUCY2C in enterocytes following biallelic Msh2 inactivation in mice. Constitutively active vil-Cre-Msh2fl/fl (Msh2−/−) mice developed jejunal tumors (A, gross appearance; B, microscopic appearance). C and D, Representative IF image and quantification of MSH2 protein expression (purple) in Msh2−/− tumors (n = 3) compared to age-matched wild-type controls (n = 3). E and F, (GUCA2A, green; β-catenin, red) from Msh2−/− tumors (n = 5) compared to age-matched wild-type controls (n = 5) reveals loss of GUCA2A expression. G, GUCA2A protein expression was reduced by immunoblot analysis (n = 5). H, GUCA2B protein expression was reduced by immunoblot analysis (n = 5). GUCA2A (I) and GUCY2B (J) mRNA quantified by RT-PCR in Msh2−/− tumors (n = 5) was not reduced compared to age-matched wild-type controls (n = 6). K and L, Representative IF images (GUCY2C, green; CDX2, purple) from Msh2−/− tumors (n = 10) compared to age-matched wild-type controls (n = 6) reveal no change in GUCY2C protein expression. M and N, GUCY2C protein by immunoblot analysis (n = 5) and mRNA (n = 5) was unchanged in Msh2−/− tumors compared to Wt controls (n = 6). Blue, DAPI. *P < .05; **P < .01. ****P < .0001. Scale bar =100, μm.
4. Discussion
CRC continues to be the fourth leading cause of cancer and the second leading cause of cancer mortality, underscoring the significant unmet clinical need for effective prevention and therapy [30]. The well-established adenoma-carcinoma sequence, involving stepwise accumulation of genetic alterations resulting in sequential epithelial transformation over a 20-year horizon, offers unique opportunities to interrupt the formation of invasive and metastatic tumors by premalignant screening and removal, or chemoprevention [2,3]. However, surveillance has proven inadequate, and only 63% of eligible patients in the United States underwent regular screening in 2015 [31]. Moreover, chemoprevention is limited to low-dose aspirin only in patients 50–59 years old with increased risk of cardiovascular disease and without increased risk for bleeding [32]. Beyond prevention, >50% of patients who develop CRC die of metastatic disease [30]. Identifying patients who will develop metastases remains a diagnostic dilemma [20]. Furthermore, there are no therapeutic modalities that cure metastases. Thus, identifying and validating new targets for prevention and therapy remain a priority.
Although the evolution of CRC reflects a final common clinical pathway, there is recognition that these tumors arise through different genetic alterations. Tumors originating from the conventional pathway represent ~75% of sporadic CRCs and involve driver mutations in APC and CTNNB1 genes constitutively activating β-catenin-dependent WNT signaling [1–3]. These tumors are characterized by CIN resulting in the development of a median of >60 mutations, including critical driver mutations in TP53, SMAD4, and/or KRAS contributing to invasion and metastasis [3]. Alternatively, tumors originating from the serrated pathway represent ~20% of sporadic CRCs and frequently have driver mutations in mitogen-activated protein kinase pathway components—particularly BRAFV600E—that promote DNA hypermethylation and CIMP [4]. In turn, hypermethylation silences expression of CDX2, producing an epithelial stem-like niche permissive for mutant BRAF to drive transformation [33]. Finally, MSI tumors comprise ~15% of CRCs and reflect silencing of DNA mismatch repair genes—through mutations in inherited syndromes like HNPCC (Lynch syndrome) or through hypermethylation in sporadic tumors—that leads to a hypermutable phenotype [8,12,13]. This diversity in CRC initiation and evolution, characterized by different genetic drivers and dysregulated signaling pathways, highlights the importance of defining molecular targets in specific tumor types to individualize clinical utility.
The GUCY2C hormone axis has emerged as a marker and target for managing patients across the CRC disease continuum from prevention to metastatic therapy. GUYC2C is a tumor-suppressing receptor whose silencing drives transformation, including hyperproliferation, Warburg metabolism, genomic instability, and desmoplasia [16,17]. Furthermore, CRC is characterized by loss of GUCA2A expression, which silences GUCY2C contributing to transformation[18,19]. Additionally, GUCY2C ligands prevent transformation induced by mutations, carcinogens, or inflammation in preclinical models [17,19]. These observations support a role for oral hormone replacement to reconstitute GUCY2C signaling as chemoprevention for CRC [34]. Beyond prevention, tissue-selective expression on normal epithelial cells confined to intestine, but overexpression on cancer cells, makes GUCY2C a sensitive and specific marker of occult metastatic tumor cells in lymph nodes and blood that improves staging and postoperative surveillance of CRC patients [20]. Moreover, normally, GUCY2C is sequestered in apical membranes of epithelial cells in the luminal mucosa but represents a neoantigen in the systemic compartment in metastatic disease [35]. This anatomical and immunological compartmentalization makes GUCY2C uniquely suited as a therapeutic target to eradicate established metastases with antibody-drug conjugates [21] and adoptive immune cell therapies [36] and to prevent the development of secondary metastases with vaccines without intestinal toxicities [35]. However, this broad applicability in CRC prevention and therapy is contingent on differential expression of GUYC2C signaling components in tumors arising through different canonical genetic pathways.
Here, we defined the expression of GUCA2A and GUCY2C in tumors arising through conventional, serrated, and MSI pathways. This study revealed the loss of GUCA2A at the earliest stages in the development of precancerous lesions of conventional and serrated pathways as well as in hereditary and sporadic MSI tumors. These observations confirm previous studies exploring the expression of GUCA2A in intestinal tumors arising in mice and humans [18,19]. In that regard, they are consistent with the mechanistic hypothesis that paracrine hormone loss silencing the GUCY2C-cGMP signaling axis universally contributes to initiation and promotion of intestinal tumorigenesis arising through each of the canonical genetic mechanisms. Furthermore, GUCY2C expression was preserved in conventional adenomas and cancers and in tumors arising through the MSI pathway. Surprisingly, we observed complete loss of GUCY2C in tumors arising through the serrated pathway. These observations validate prior studies describing the general association of GUCY2C expression in most, but not all, colorectal tumors [20,37]. They reinforce the role of silencing the GUYC2C tumor suppressor, here defined by hormone and/or receptor loss, as an integral component of the process of transformation generally in mice and humans. Moreover, they underscore the importance of exploring the expression of individual markers by tumors arising through discreet molecular mechanisms to define their suitability for clinical application. Indeed, these studies reveal that, in contrast to tumors arising through the conventional and MSI pathways, those arising through the serrated pathway would not be amenable to prevention, diagnosis, or therapy targeting GUCY2C. In that context, the reconstitution of GUCY2C-cGMP signaling in premalignant lesions of the colorectum arising through the conventional and MSI pathways aimed at preventing malignant transformation remains an area of active investigation. Current work has direct translational relevance, as the paradigm of GUCY2C reactivation may not be applicable to the serrated neoplasia pathway due to the loss of GUCY2C receptors.
Although loss of GUCA2A is a universal feature characterizing tumorigenesis initiated through conventional, serrated, and MSI pathways, molecular mechanisms eliminating paracrine hormone expression remain incompletely defined. Indeed, regulation of GUCA2A transcription and translation has not been characterized previously. GUCA2A expression is suppressed in the colorectum by endoplasmic reticulum (ER) stress induced by consumption of excess calories in mice and humans [17]. In part, loss ofGUCA2A, associated with silencing of the GUCY2C tumor suppressor, by ER stress produced by excess calories is one molecular mechanism contributing to colorectal transformation in the context of obesity [17]. However, the precise contribution of ER stress to the pathophysiology of cancer generally, and to different pathways driving colorectal cancer specifically, remains to be elucidated. Beyond ER stress, GUCA2A expression is eliminated as an early event in intestinal mucosa of mice heterozygous for APC mutations [18,19]. These observations are compatible with a model in which constitutive activation of Wnt signaling, through accumulation of nuclear β-catenin, regulates transcriptional programs associated with silencing of GUCA2A expression [15]. This possibility is strengthened by the frequency of mutations in Wnt pathway components, resulting in constitutive activation, as part of the mechanism mediating transformation in serrated and MSI pathways [8,38]. In that context, we are exploring the role of transcriptional regulation of GUCA2A expression by constitutively activated Wnt signaling in tumors arising through different genomic pathways.
Although molecular processes underlying GUCA2A loss in CRC tumorigenesis remain to be defined, there are clear mechanistic insights into the differential loss of GUCY2C expression in tumors arising through the serrated, but not conventional or MSI, pathway. In that context, loss of CDX2 expression through hypermethylation is a key step in providing the context for mutations in BRAF to drive tumorigenesis [9,33]. Indeed, here, CDX2 expression was eliminated in tumors arising through the serrated, but not the conventional or MSI, pathway. Previous studies have demonstrated that intestine-specific expression of GUCY2C is driven by the canonical transcriptional activity of the CDX2 homeobox protein in mice and humans [39]. Furthermore, colorectal tumors which are deficient in CDX2 also lose GUCY2C expression [37]. Additionally, transcriptomic analyses revealed a direct relationship between CDX2 and GUCY2C expression (Supplementary Fig. 4). Finally, although isolated mutations in Braf had no effect, elimination of CDX2 expression alone reduced expression of GUCY2C in mouse colonic mucosa (Fig. 3). Taken together, these observations support the role of CDX2 in driving intestine-specific expression of GUCY2C and its loss in tumors arising through the serrated, but not conventional or MSI, pathway.
These studies establish loss of GUCA2A and silencing of GUCY2C as universal features of intestinal tumorigenesis produced by conventional, serrated, and MSI pathways [17–19]. They reinforce the utility of hormone loss as an actionable mechanism to develop strategies using oral GUCY2C ligand replacement to prevent the development of tumors arising through the conventional pathway [17,19]. In the context of the preservation of GUCY2C expression, these observations also suggest that oral hormone replacement might be a useful strategy to prevent tumors arising through the MSI pathway. This possibility is underscored by the role of the GUCY2C-cGMP signaling axis in amplifying DNA repair mechanisms and preventing injury and transformation by DNA-damaging agents [16,17,28,40]. The immediate translational application of these observations can be appreciated by considering that the oral GUCY2C ligands linaclotide (Linzess) and plecanatide (Trulance) are approved to treat chronic constipation syndromes. Importantly, these studies reveal that GUCY2C expression is preserved in tumors arising through conventional and MSI, but not serrated, pathways. In turn, they suggest the utility of GUCY2C-tageted therapies, including antibody-drug conjugates, adoptive cell therapies, and cancer vaccines, to prevent or treat metastatic disease originating from tumors arising through conventional and MSI, but not serrated, pathways [21,35,36].
Supplementary Material
Acknowledgments
S. A. W. is the Samuel MV Hamilton Professor of Medicine of Thomas Jefferson University. The authors would like to thank Dr Winfried Edelmann for providing Msh2FL/FL mice. Also, we would like to thank Dr Melanie Kucherlapati for providing mouse tumors from MSH2 knockout mice. We are grateful to Ms Elinor Leong for technical assistance with RNA isolation and quantitative RT-PCR assay. Results are, in part, based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Funding/Support: Supported by grants to S. A. W. from the National Institutes of Health (NIH) (R01 CA204881, R01 CA206026, P30 CA56036) and Targeted Diagnostics and Therapeutics, Inc, and to A. E. S. (PhRMA Foundations; Margaret Q. Landenberg Foundation). B. B. was supported by NIH institutional award T32 GM008562 for Postdoctoral Training in Clinical Pharmacology. D. M. was supported by a PhRMA Predoctoral Fellowship Award in Pharmacology/Toxicology and an NIH Ruth Kirschstein Individual Predoctoral MD/PhD Fellowship (F30DK 103492). J. R. was supported by a PhRMA Predoctoral Fellowship Award in Pharmacology/Toxicology and an NIH Ruth Kirschstein Individual Predoctoral MD/PhD Fellowship (F30CA 232469).
Footnotes
Competing interests: S. A. W. is a member of the Board and Chair of the Scientific Advisory Board (both uncompensated) of Targeted Diagnostics & Therapeutics, Inc., which provided research funding that, in part, supported this work and has a license to commercialize inventions related to this work. Also, he is the Chair of the Board of Directors of Feelux Company, Ltd. Furthermore, he receives research support from Synergy Pharmaceuticals, Inc.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.humpath.2018.11.032.
References
- [1].Marisa L, de Reyniès A, Duval A, et al. Gene expression classificationof colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10:e1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Morin PJ, Sparks AB, Korinek V, et al. Activation of (β-catenin-Tcf signaling in colon cancer by mutations in (β-catenin or APC. Science 1997; 275:1787–90. [DOI] [PubMed] [Google Scholar]
- [3].Strum WB. Colorectal adenomas. N Engl J Med 2016;374:1065–75. [DOI] [PubMed] [Google Scholar]
- [4].Langner C Serrated and non-serrated precursor lesions of colorectal cancer. Dig Dis 2015;33:28–37. [DOI] [PubMed] [Google Scholar]
- [5].Wynter CVA, Walsh MD, Higuchi T, Leggett BA, Young J, Jass JR. Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Gut 2004;53:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Clarke CN, Kopetz ES. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J Gastrointest Oncol 2015;6:660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang S, Farraye FA, Mack C, Posnik O, O’Brien MJ. BRAF and KRAS mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol 2004;28:1452–9. [DOI] [PubMed] [Google Scholar]
- [8].Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073–2087.e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Balbinot C, Armant O, Elarouci N, et al. The Cdx2 homeobox gene suppresses intestinal tumorigenesis through non-cell-autonomous mechanisms. J Exp Med 2018;215:911–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dalerba P, Sahoo D, Paik S, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374:211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Landau MS, Kuan S-F, Chiosea S, Pai RK. BRAF-mutatedmicrosatellite stable colorectal carcinoma: an aggressive adenocarcinoma with reduced CDX2 and increased cytokeratin 7 immunohistochemical expression. Hum Pathol 2014;45:1704–12. [DOI] [PubMed] [Google Scholar]
- [12].Sinicrope FA. Lynch syndrome-associated colorectal cancer. N Engl J Med 2018;379:764–73. [DOI] [PubMed] [Google Scholar]
- [13].Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009;58:90–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kuhn M Molecular physiology of membrane guanylyl cyclase receptors. Physiol Rev 2016;96:751–804. [DOI] [PubMed] [Google Scholar]
- [15].Pattison AM, Merlino DJ, Blomain ES, Waldman SA. Guanylyl cyclase C signaling axis and colon cancer prevention. World J Gastroenterol 2016;22:8070–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li P, Schulz S, Bombonati A, et al. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 2007;133:599–607. [DOI] [PubMed] [Google Scholar]
- [17].Lin JE, Colon-Gonzalez F, Blomain E, et al. Obesity-induced colorectal cancer is driven by caloric silencing of the guanylin-GUCY2C paracrine signaling axis. Cancer Res 2016;76:339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cohen MB, Hawkins JA, Witte DP. Guanylin mRNA expression in human intestine and colorectal adenocarcinoma. Lab Invest 1998;78:101–8. [PubMed] [Google Scholar]
- [19].Shailubhai K, Yu HH, Karunanandaa K, et al. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res 2000;60:5151–7. [PubMed] [Google Scholar]
- [20].Waldman SA, Hyslop T, Schulz S, et al. Association of GUCY2C expression in lymph nodes with time to recurrence and disease-free survival in pN0 colorectal cancer. JAMA 2009;301:745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Marszalowicz GP, Snook AE, Magee MS, Merlino D, Berman-Booty LD, Waldman SA. GUCY2C lysosomotropic endocytosis delivers immunotoxin therapy to metastatic colorectal cancer. Oncotarget 2014; 5:9460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Snook AE, Li P, Stafford BJ, et al. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res 2009;69:3537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev 2007;21:379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sakamoto N, Feng Y, Stolfi C, et al. BRAFV600E cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. Elife 2017;6:e20331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kucherlapati MH, Lee K, Nguyen AA, et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology 2010;138:993–1002.e1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Valentino MA, Lin JE, Snook AE, Li P, Kim GW, Marszalowicz G, Magee MS, Hyslop T, Schulz S, Waldman SA. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J Clin Invest 2011; 121:3578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Moss NG, Fellner RC, Qian X, et al. Uroguanylin, an intestinal natriuretic peptide, is delivered to the kidney as an unprocessed propeptide. Endocrinology 2008;149:4486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li P, Wuthrick E, Rappaport JA, et al. GUCY2C signaling opposes the acute radiation-induced GI syndrome. Cancer Res 2017;77:5095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- [31].Health, United States. 2015: with special feature on racial and ethnic health disparities. Hyattsville (MD): National Center for Health Statistics; 2016. [PubMed] [Google Scholar]
- [32].Bibbins-Domingo K, on behalf of the USPSTF. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2016;164:836–45. [DOI] [PubMed] [Google Scholar]
- [33].Tong K, Pellón-Cárdenas O, Sirihorachai VR, et al. Degree of tissue differentiation dictates susceptibility to BRAF-driven colorectal cancer. Cell Rep 2017;21:3833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Weinberg DS, Lin JE, Foster NR, et al. Bioactivity of oral linaclotide in human colorectum for cancer chemoprevention. Cancer Prev Res (Phila) 2017;10:345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Snook AE, Stafford BJ, Li P, et al. Guanylyl cyclase C–induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J Natl Cancer Inst 2008;100:950–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Magee MS, Abraham TS, Baybutt TR, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res 2018;6:509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Winn B, Tavares R, Matoso A, et al. Expression of the intestinal biomarkers guanylyl cyclase C and CDX2 in poorly differentiated colorectal carcinomas. Hum Pathol 2010;41:123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Borowsky J, Dumenil T, Bettington M, et al. The role of APC in WNT pathway activation in serrated neoplasia. Mod Pathol 2018;31: 495–504. [DOI] [PubMed] [Google Scholar]
- [39].Park J, Schulz S, Waldman SA. Intestine-specific activity ofthe human guanylyl cyclase C promoter is regulated by Cdx2. Gastroenterology 2000;119:89–96. [DOI] [PubMed] [Google Scholar]
- [40].Lin JE, Li P, Snook AE, et al. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology 2009;138:241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.