

Psoriasis is a chronic inflammatory disease of the skin, nails, and joints. In the last two decades, there has been enormous progress in our understanding of the genetics, immunology, and associated comorbidities (Figure 1). This has been accompanied by a marked improvement in the number of therapeutic agents and their effectiveness. Much of this progress has been outlined in numerous articles, commentaries and reviews in the Journal of Investigative Dermatology (JID) over the years (Figure 2). This commentary, which is part of the JID Collections, serves to outline how we have arrived at our present state of knowledge on psoriasis and provides an overview of some exciting future directions.
Figure 1. Simplified overview of interrelation between psoriasis predisposing factors, inflammatory mechanisms, clinical manifestations, and consequences.

Crosstalk between the innate and adaptive immune systems involves secretion of IL-12 and IL-23, which are important for priming and maintaining Th1 and Th17 responses. Inflammatory responses prime keratinocytes that in turn amplify the inflammatory response and “feed forward,” creating a sustaining cycle of inflammation. These inflammatory responses shape the clinical manifestations of psoriasis, which exist between pustular forms (dominated by higher IL-36 responses) and plaque psoriasis (characterized by high IL-17A).
Figure 2.

Number of psoriasis publications in the Journal of Investigative Dermatology (JID) per year since 1945.
Historical perspective
For many decades, psoriasis was thought to be a disease characterized by keratinocyte hyperplasia (Figure 3). This was the main motivation for use of the anti-proliferative chemotherapy agent methotrexate for treatment of psoriasis (Weinstein and Frost, 1968; Weinstein and Velasco, 1972) (Figure 4). Its therapeutic benefit was attributed primarily to direct effects of methotrexate on the skin (Weinstein and McCullough, 1976).
Figure 3.

Timeline of discoveries in genetics and (top) genomics of psoriasis and (bottom) pathogenesis of psoriasis.
Figure 4. Timeline of discoveries of psoriasis (top) comorbidities and (bottom) treatment.

MI, myocardial infarction.
Related to the focus on hyperproliferation of the psoriatic epidermis was research on cyclic AMP (Voorhees et al., 1972) and inhibitors of phosphodiesterases (PDE) as potential therapeutic agents in psoriasis (Rusin et al., 1978; Stawiski et al., 1975; Stawiski et al., 1979) – work that predated development and use of the PDE4 antagonist apremilast as a psoriasis therapy (Nast et al., 2015) by several decades.
As the discrete nature of psoriatic plaques suggested a local phenomenon, there was a hunt for a hormone or similar messenger directing localized genesis of lesions. While cyclic AMP garnered much attention, the arachidonic acid derivative leukotriene B4 (LTB4) also emerged as a likely candidate, as it was increased in psoriatic plaques (Brain et al., 1984; Brain et al., 1982; Grabbe et al., 1982) and had mitogenic effects on keratinocytes (Bauer et al., 1986; Kragballe et al., 1985). Cyclosporine, an inhibitor of phospholipase A2 (Fan and Lewis, 1985) and therefore potential suppressor of LTB4 production, thus surfaced as a potential anti-psoriatic agent.
In 1979, a small case series of four patients treated with cyclosporine was published in the New England Journal of Medicine (Mueller and Herrmann, 1979), followed 5 years later by a single case report in the Lancet in 1984 (Harper et al., 1984). In 1986, two clinical trials motivated in part by the LTB4 findings demonstrated conclusively the clinical efficacy of cyclosporine for chronic plaque psoriasis (Ellis et al., 1986; Griffiths et al., 1986). While lesional LTB4 was found to decrease with treatment (Ellis et al., 1986), the dramatic response prompted consideration of an alternative hypothesis – that the primary defect in psoriasis was not keratinocyte hyperproliferation but rather immunologic in nature. This represented a watershed moment in psoriasis research, rapidly shifting the focus toward the immune system, and particularly T cells, as a critical pathogenic driver (Gottlieb et al., 1992). Further work demonstrating disease improvement with selective blockade of activated T cells by interleukin (IL)-2 conjugated to diphtheria toxin fragments definitively implicated T cells in psoriasis pathogenesis (Gottlieb et al., 1995).
In 1991 Brian Nickoloff proposed the idea of the “cytokine network” in psoriasis (Nickoloff, 1991; Uyemura et al., 1993). In the “cytokine network” hypothesis, inflammatory cells and keratinocytes interact to drive the inflammatory process through secretion of various pro-inflammatory cytokines. Initial work focused primarily on the cytokines interferon (IFN)-γ and tumor necrosis factor (TNF)-α (Uyemura et al., 1993).
However, in 1998, IL-17A involvement was first recognized (Teunissen et al., 1998). Subsequently, psoriasis was shown to contain discrete populations of T helper (Th)1 and Th17 lymphocytes (Lowes et al., 2008). Whereas IL-12 is primarily responsible for driving Th1 responses, IL-23 has a key role in maintaining Th17 responses (Stritesky et al., 2008). IL-12 and IL-23 are closely related heterodimers (consisting of two subunits), each including a common p40 subunit, with IL-12 having a p35 subunit and IL-23 a p19 subunit (Lee et al., 2004). In 2004, it was noted that while the p40 and p19 subunits were prominently expressed in psoriatic skin, the p35 subunit was decreased (Lee et al., 2004), suggesting that IL-23 with its p19 subunit plays a more dominant role than IL-12, and therefore Th1 responses, in psoriasis.
Genetic studies align with these observations, showing association between psoriasis and variants close to the p40 gene (IL12B) and within the IL-23 receptor (IL23R) (Nair et al., 2008), as well as NF-κB (Nair et al., 2009) and the IL-17 signaling pathway (Ellinghaus et al., 2010).
This has consolidated the view of the cytokine network in psoriasis anchored by IL-23/IL-17/TNF responses (Martin et al., 2013), not only in chronic plaque psoriasis but also in other psoriasis subtypes (Xing et al., 2016). The foundational role of the “cytokine network” in psoriasis has been conclusively demonstrated by the remarkable efficacy of drugs targeting various cytokines in psoriasis including TNF, IL-17A and more recently IL-23 (Chaudhari et al., 2001; Gordon et al., 2015; Gordon et al., 2018; Langley et al., 2014; Leonardi et al., 2012; Papp et al., 2012; Reich et al., 2017; Reich et al., 2019).
Recently, a “feed forward” mechanism as a driver of psoriasis pathogenesis has been proposed by James Krueger and colleagues (Hawkes et al., 2017). This proposed mechanism links together the “cytokine network” with the epidermal responses and hyperplasia, as follows: activation and increased expression of IL-17 in pre-psoriatic skin produces a feed forward inflammatory response in keratinocytes that is self-amplifying and drives the development of psoriatic plaques through induction of epidermal cell proliferation, hyperplasia, and further recruitment of inflammatory cells into the skin (Hawkes et al., 2017). This model more readily enables incorporation of other inflammatory mediators and processes such as for the epidermal derived cytokines such as IL-17C (Fritz et al., 2017; Guttman-Yassky and Krueger, 2018), the IL-36 family of cytokines (Carrier et al., 2011; Johnston et al., 2011), and signaling pathways such as TNFAIP3 (Devos et al., 2019), CARD14 (Mellett et al., 2018), and MCPIP1, a regulator of IL-17A and IL-17C responses in psoriatic epidermis (Monin et al., 2017; Ruiz-Romeu et al., 2016), into the pathogenesis of psoriasis.
Thus, the pathogenesis of psoriasis continues to evolve after decades of intense study, and pathogenesis remains the most highly represented area of investigation in the JID’s body of recent psoriasis literature (Figure 5).
Figure 5. Areas of research focus for psoriasis publications in the Journal of Investigative Dermatology (JID) in the preceding 2 years.
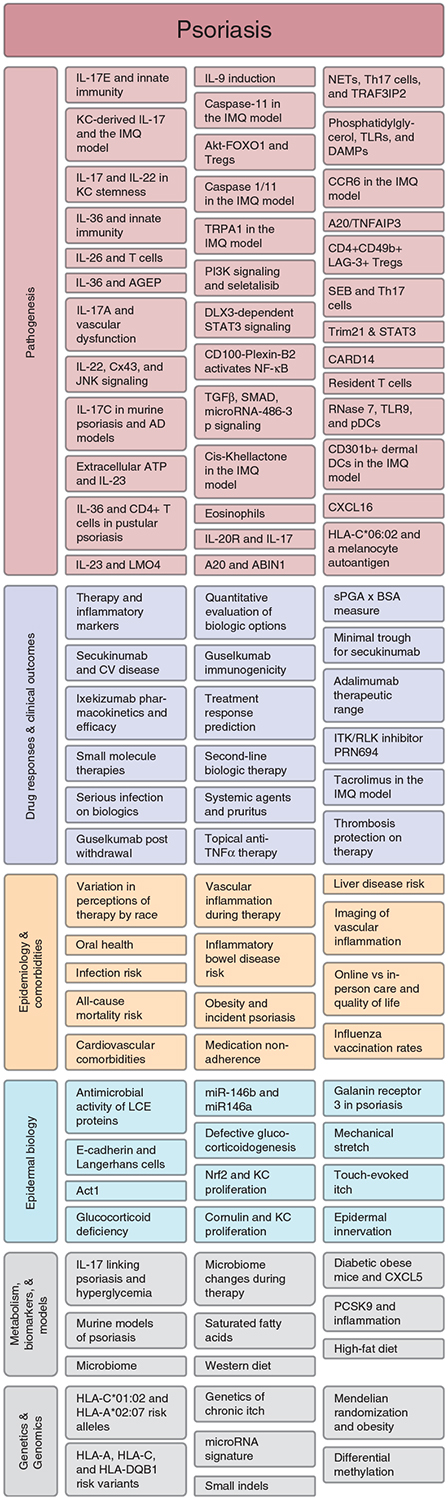
Subcategorization reveals predominant focus on pathogenesis, drug responses, and clinical outcomes but also a strong emphasis on epidemiology and comorbidities. Literature search includes JID publications from July 2017 through June 2019 containing the search term “psoria*” with direct relevance to psoriasis. Some topics are common to multiple publications but represented only once in the figure. Many publications could be appropriately classified in multiple ways; the figure is intended to provide an overall impression of distribution of scientific focus. AD, atopic dermatitis; AGEP, acute generalized exanthematous pustulosis; BSA, body surface area; CV, cardiovascular; DAMPs, damage-associated molecular patterns; DCs, dendritic cells; IMQ, imiquimod; KC, keratinocyte; LCE, late cornified envelope; NETs, neutrophil extracellular traps; sPGA, static physician global assessment; TLRs, toll-like receptors.
Comorbidities of psoriasis
Due to strong associations with a multitude of comorbidities, psoriasis is increasingly conceptualized as a systemic inflammatory disease. While psoriasis is classically associated with psoriatic arthritis, there is mounting evidence for associations with cardiometabolic disease, immune-mediated inflammatory disease, malignancy, and infection. In many cases, whether these associations are evidence of spill-over of cutaneous inflammation, shared susceptibility, or pervasive immune dysregulation remains to be determined.
Cardiometabolic disease has been reported in association with psoriasis for over a century (Strauss, 1897). Since 2000, there has been a marked proliferation in publications describing these associations (Gelfand et al., 2006; Neimann et al., 2006; Sommer et al., 2006), with severe psoriasis conferring higher risk. With regard to cardiovascular disease, these studies generally promoted the hypothesis that chronic skin inflammation and the concomitant increase in circulating proinflammatory cytokines favor development of atherosclerosis and that systemic anti-psoriatic therapy may protect against this process and consequent adverse cardiovascular outcomes. Indeed, studies have demonstrated reductions in major adverse cardiovascular events (MACE) in psoriasis patients on TNF-α antagonists (Ahlehoff et al., 2015; Wu et al., 2012).
However, the mechanism may not be so straightforward: circulating inflammatory markers decrease in as little as 4 weeks in psoriasis patients treated with anti-TNF-α therapy (Kim et al., 2018), yet 52 weeks of anti-TNF-α therapy had no effect on vascular inflammation in the ascending aorta and in fact slightly increased vascular inflammation in the carotids (Bissonnette et al., 2017).
More recently, the question of whether obesity promotes psoriasis was tackled using a powerful approach called Mendelian randomization (Budu-Aggrey and Paternoster, 2019) that overcomes challenges of observational epidemiological studies in which reverse causation or confounding factors may obscure causality. The technique uses genetic variants as proxies – termed instruments – for relevant exposures. Allotted randomly at conception, genetic variants are independent from confounders and the outcome itself and thus can be used to estimate the effect of the exposure on the outcome. As psoriasis has long represented a thriving forefront for genome-wide association studies (Elder et al., 2010), a nearly unparalleled amount of genetic data for psoriasis patients has been amassed. Psoriasis is thus particularly well positioned for approaches such as Mendelian randomization. Analyses of these large datasets using obesity-associated genetic variants as a proxy for actual measured body mass index (BMI) strongly suggested that higher BMI causally increases risk of psoriasis; the converse analyses showed little to no causal effect of psoriasis genetic risk on BMI (Budu-Aggrey et al., 2019; Ogawa et al., 2019). Additional comorbid conditions will likely benefit from similar analysis to help deconvolute the complex relationships between psoriasis and cardiometabolic diseases.
For some psoriasis comorbidities, however, there is direct evidence that the association is due not to unidirectional causality but rather to shared susceptibility. This is best exemplified by the overlap of genetic risk variants – many of which affect immune regulatory genes – between psoriasis and certain associated conditions. Chief among these is Crohn’s disease, but shared risk loci have also been identified for diseases such as type II diabetes mellitus that are not classically considered to be autoinflammatory (Capon et al., 2007; Cargill et al., 2007; Wolf et al., 2008). As might be anticipated for conditions with shared susceptibility, incident Crohn’s disease is more common among psoriasis patients (Li et al., 2013), and incident psoriasis is more common among patients with Crohn’s disease (Egeberg et al., 2019). Also not unexpectedly, these conditions share many therapies, and investigations into their immunopatho-genesis have revealed considerable similarities.
For psoriatic arthritis (PsA), the best-known psoriasis comorbidity, both shared genetic susceptibility and spill-over of cutaneous inflammation likely contribute to the association. PsA and psoriasis susceptibility genes are largely overlapping, yet variants have been identified that are more strongly associated with PsA than psoriasis (Ellinghaus et al., 2012; Stuart et al., 2015), suggesting the possibility of independent genetic drivers for PsA. However, PsA is generally diagnosed after the appearance of cutaneous psoriasis and seldom occurs in the absence of cutaneous psoriasis, and data indicate that cutaneous inflammation of psoriasis may exacerbate or even directly drive PsA: development of joint disease in a genetic mouse model of autoimmune arthritis is dramatically accelerated in the presence of psoriasis-like skin inflammation due to hyperactivation of Stat3 in the epidermis (Yamamoto et al., 2015). Furthermore, epidermally restricted hyperactivation (Winge et al., 2016) or deletion (Zenz et al., 2005) of genes implicated in human psoriasis can promote spontaneous development of PsA-like joint disease. Thus, controlling cutaneous psoriasis may be of benefit in preventing or limiting joint disease, lending credence to the concept of PsA as a ‘disease within a disease’ (Eder et al., 2011).
While not entirely distinct from the above mechanisms, a third possible explanation for psoriasis comorbidities is pervasive immune dysregulation. This mechanism is often invoked for associations of psoriasis with malignancy and infection. In one nationwide study, malignancy carried the highest absolute and excess risks of death in psoriasis (Lee et al., 2017). In larger cohort and meta-analysis studies, patients with psoriasis show higher risks for multiple cancers, particularly lymphohematopoietic malignancy, that persist even when controlling for confounders such as increased smoking and alcohol consumption among psoriasis patients and use of potentially malignancy-promoting anti-psoriatic therapies (Brauchli et al., 2009; Lee et al., 2017; Pouplard et al., 2013).
In the case of lymphoma, many have posited that the chronically dysregulated immune state of psoriasis drives increased risk. Now in the era of immunotherapy, as our understanding of the critical role of the immune system in limiting malignancy increases, it is tempting to speculate that immune dysregulation and associated impairment of antineoplastic immune surveillance underlies all excess malignancy risk among psoriasis patients. However, this will be challenging to ever prove. Chronic immune dysfunction also likely contributes to the increased risk of serious infections that is observed in patients with psoriasis, even when excluding those on immunosuppressive therapies (Takeshita et al., 2018).
While the true natures of the relationships between psoriasis and associated conditions have proven difficult to define, investigations of their intersection continue to advance understanding of psoriasis immunopatho-genesis and guide therapeutic pursuits. Accordingly, publications addressing epidemiology and comorbidities are now well represented research in the JID psoriasis literature (Figure 5).
Modeling of psoriasis – mouse models and beyond
With two reported exceptions, a rhesus monkey and a cynomolgus monkey, psoriasis is not observed in animals other than humans (Gudjonsson et al., 2007). However, over the past three decades, numerous mouse models of psoriasis have been described, created through genetic modifications, topical application, or intradermal cytokine injection. Furthermore, various non-animal in vitro models such as the epidermal raft system (Barker et al., 2004; Gordon et al., 2013) have been developed; these have become more frequently used in recent years, and many of these landmark advances in psoriasis have been published in the JID.
The first mouse model of psoriasis described was a xenograft model in which lesional psoriatic skin was grafted onto congenitally athymic (nude) mice (Krueger et al., 1975). These mice lack a thymus and are therefore unable to mount an immune response against the grafted skin, enabling maintenance of the skin graft for up to 11 weeks.
A major step forward was made in 2004 when spontaneous development of psoriasis was described in a novel xenograft model where non-lesional psoriatic skin spontaneously developed histologic hallmarks of psoriasis after grafting (Boyman et al., 2004). This model is uniquely dependent upon the recipient mouse, which is deficient in type I and type II interferon receptors along with being deficient in the recombination activating gene 2 (Rag2), as this spontaneous development of psoriasis-like disease is not observed with skin grafting onto other immunodeficient mouse strains. This model has been used to show the importance of plasmacytoid dendritic cells in initiation of psoriasis (Nestle et al., 2005), role of epidermal T cells (Conrad et al., 2007), and role of CD8+ T cells (Di Meglio et al., 2016).
The majority of the spontaneous and transgenic mouse models of psoriasis were developed and described in the mid-1990s to mid-to-late 2000s (Gudjonsson et al., 2007; Hawkes et al., 2018). Over 40 unique mouse models have been described (Hawkes et al., 2018). With the initial description of the imiquimod (IMQ) inducible model of psoriasis (van der Fits et al., 2009) and cytokine injection models (Zheng et al., 2007), acute or inducible models have rapidly become one of the most widely used systems for studying human psoriasis (Hawkes et al., 2018). Recent examples of the utilization of this model include the demonstration of IL-1β/IL-1R signaling pathway in skin inflammation (Cai et al., 2019); therapeutic intervention to determine the response to topical tacrolimus (Pischon et al., 2018); link between skin inflammation and hyperglycemia (Ikumi et al., 2019); role of IL-20 receptor signaling (Ha et al., 2019) and IL-17E (Senra et al., 2019) in psoriasis pathogenesis; and to elucidate the function of Trim32 in psoriasis and atopic dermatitis (Liu et al., 2017). However, some concerns have been raised about the overutilization of this model and its variability depending on the mouse strain used (Swindell et al., 2017). Nonetheless, since the first report of this model in 2009, the IMQ psoriasis-like model has been used in over 200 publications (Hawkes et al., 2017).
Other models entail intradermal injection of pro-inflammatory cytokines, with the first instance of this reported in 2003 with IL-23 (Kopp et al., 2003), and later with injections of IL-22 (Zheng et al., 2007), IL-21 (Caruso et al., 2009), and most recently IL-36A (Campbell et al., 2019). Other publications in the JID exploiting these models have explored the role of IL-6 (Lindroos et al., 2011), superoxide dismutase (Lee et al., 2013), and the toll-like-receptors (TLRs) 7, 8, and 9 in psoriasis (Jiang et al., 2013). The raft models have also been used to explore specific mechanisms in psoriasis including glucocorticoid deficiency in lesional psoriatic skin (Sarkar et al., 2017) and biology of Ephrin-A signaling (Gordon et al., 2013).
Complex diseases such as psoriasis create a formidable challenge for attempts to model pathogenesis. However, while no model can capture the entirety of psoriasis pathogenesis, these models are indispensable and help us understand the roles of specific mediators and signaling pathways.
Future directions in psoriasis
Despite dramatic advances in understanding of psoriasis immunopatho-genesis and treatment over the last several decades, psoriasis remains a vibrant field of investigation. This is perhaps because psoriasis serves as a readily accessible – although deeply complex – immune-mediated inflammatory disease paradigm for exploration of forefronts such as the role of tissue-resident memory T cells and the interplay between metabolism and autoinflammation. These and several other emerging entities are reviewed in brief below.
Tissue-resident memory T cells (TRMs) and the psoriasis autoantigen.
Psoriasis patients are frequently frustrated by the propensity for their psoriasis to recur at sites of previous lesions upon cessation of therapy. Early investigation of this phenomenon revealed a ‘residual disease genomic profile’ including expression of cytokines and T cell-specific transcripts in skin previously affected by psoriasis, suggesting the presence of persistent and likely disease-perpetuating memory T cells residing in resolved lesions between flares (Suarez-Farinas et al., 2011). Shortly thereafter, these TRMs were found to include IL-22-producing CD4+ T cells and IL-17-producing CD8+ T cells (Cheuk et al., 2014), providing a cytokine source for the instigation of recurrent lesions.
In 2017, however, high-throughput T cell receptor (TCR) screening revealed that resolved psoriasis lesions contain oligoclonal IL-17A-producing T cell populations; some of the expressed TCR sequences were found to be identical across multiple patients but absent in skin from healthy controls and patients with other cutaneous inflammatory diseases (Matos et al., 2017). Interestingly, while γδ T cells represent the primary source of IL-17 in the IMQ psoriasis-like mouse model (Laggner et al., 2011; Pantelyushin et al., 2012), all of the most frequent putatively pathogenic T cell clones were αβ T cells. This is concordant with the findings of another high-throughput examination of T cell repertoires in psoriasis that reported that γδ T cells constitute a very small population of cutaneous psoriatic T cells and do not correlate with IL-17A expression in humans (Merleev et al., 2018).
The presence of oligoclonal TRM populations in psoriasis has also breathed new life into a longstanding question: What is the psoriasis autoantigen? The most strongly associated psoriasis susceptibility loci correspond to specific human leukocyte antigen (HLA) alleles, particularly HLA-Cw6 (Nair et al., 2006), consistent with the existence of autoantigen presentation to pathogenic CD8+ T cells. Multiple studies have identified candidate epidermal autoantigens, including keratinocyte proteins with similarity to streptococcal antigens (Besgen et al., 2010; Valdimarsson et al., 2009), the antimicrobial peptide LL37 (Lande et al., 2014), neolipid antigens (Cheung et al., 2016; Kim et al., 2016), and an HLA-C*06:02–presented melanocytic protein, ADAMTSL5, recognized by IL-17A-producing CD8+ T cells of the Va3S1/Vb13S1 TCR (Arakawa et al., 2015; Prinz, 2017). The autoantigen presentation by HLA-Cw*06:02 in psoriasis has been characterized, and compared to other HLA-C alleles, HLA-Cw*06:02 has the greatest accessible contact area for the bound antigen, which might promote binding to greater number of T-cell receptors (Wei et al., 2017). Notably, LL-37 and M-protein antigens of streptococci were all predicted to be able to be presented by HLA*06:02 (Wei et al., 2017). Intriguingly, Vb13 is among the putative pathogenic T cell clones identified by the high-throughput TCR screening approach above (Matos et al., 2017), suggesting that deeper investigation into the antigen recognition of TRMs in psoriasis may serve to definitively establish psoriasis as a primary autoimmune condition.
Influence of metabolism on psoriasis and inflammation.
A shift in focus toward metabolism and inflammation has recently occurred. As mentioned above, obesity and the metabolic syndrome are closely linked with psoriasis, and obesity was recently shown to have a causal link to psoriasis (Budu-Aggrey et al., 2019; Ogawa et al., 2019). High-fat diet has been shown to exacerbate psoriatic skin inflammation in a mouse model of psoriasis (Herbert et al., 2018; Shimoura et al., 2018). Some of this effect may be mediated by fatty acids, which have been shown to shift immune responses in dendritic cells towards IL-23 and exacerbate psoriasis-like skin inflammation (Mogilenko et al., 2019).
Psoriasis subtypes and related insights.
While chronic plaque psoriasis, or psoriasis vulgaris, is the most common subtype of psoriasis, representing approximately 90% of all cases, research on other clinical subtypes of cutaneous psoriasis, such as inverse, erythrodermic, guttate, and pustular forms, have greatly increased in recent years. The common thread among all subtypes of psoriasis is the prominence of IL-17 responses (Xing et al., 2016), although these appear to be present on a gradient and are less dominant in pustular forms of psoriasis, in which IL-36 activity has greater prominence (Johnston et al., 2017). In contrast, IL-36 activity is less prominent in chronic plaque psoriasis (Johnston et al., 2011). Mutation in IL36RN, encoding the IL-36 receptor antagonist, has been associated with a spectrum of psoriasis-associated pustular phenotypes (Setta-Kaffetzi et al., 2013), and in particular generalized pustular psoriasis without psoriasis vulgaris (Sugiura et al., 2013), reiterating the role of the IL-36 inflammatory axis in pustular subtypes of psoriasis. Interestingly, IL-36 may contribute to and promote IL-17/Th17 responses (Arakawa et al., 2018), providing a link between these two inflammatory responses beyond the role of IL-17A as one of the major inducers of IL-36 expression (Carrier et al., 2011).
Furthermore, other presentations of psoriasis may be related to imbalances in the cytokine network. One example of this is the balance between TNF and type I interferon responses. A recent publication has shown that TNF blockade induces a dysregulated type I interferon response without autoimmunity in “paradoxical” psoriasis (Conrad et al., 2018). Furthermore, IL-6 blockade can induce new-onset psoriasis-like disease (Blauvelt, 2017), a phenomenon that has been explored in mouse models of psoriasis (Fritz et al., 2017) and clinically can present like chronic plaque psoriasis with guttate-like lesions (Laurent et al., 2010). Taken together, these data suggest that variation in the cytokine network may be a key factor in shaping different clinical manifestations of psoriasis.
Conclusion
The historical perspective of the field of psoriasis provides many intriguing connections between past and present. Recurring characters such as the PDE inhibitors highlight how a long-lived and thriving field can shed new light on old players. Nevertheless, our understanding of psoriasis pathogenesis continues to grow deeper and our therapeutics ever more effective, and the rise of psoriasis as an archetypal systemic immune-mediated disease assures a vibrant future for psoriasis research.
ACKNOWLEDGMENTS
ACB was supported by the Dermatology Foundation and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) T32 postdoctoral research training grant (2T32AR007197-41).
Abbreviations:
- HLA
human leukocyte antigen
- Psa
psoriatic arthritis
- TCR
T cell receptor
Footnotes
CONFLICT OF INTEREST
JEG serves on the Advisory Boards for Novartis, AbbVie, Almirall, and MiRagen. Research support was received from AbbVie, Novartis, AnaptysBio, and Celgene. ACB and JJV state no conflict of interest.
REFERENCES
- Ahlehoff O, Skov L, Gislason G, Gniadecki R, Iversen L, Bryld LE, et al. Cardiovascular outcomes and systemic anti-inflammatory drugs in patients with severe psoriasis: 5-year follow-up of a Danish nationwide cohort. J Eur Acad Dermatol Venereol 2015;29:1128–34. [DOI] [PubMed] [Google Scholar]
- Arakawa A, Siewert K, Stohr J, Besgen P, Kim SM, Ruhl G, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 2015;212:2203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa A, Vollmer S, Besgen P, Galinski A, Summer B, Kawakami Y, et al. Unopposed IL-36 Activity Promotes Clonal CD4(+) T-Cell Responses with IL-17A Production inþGeneralized Pustular Psoriasis. J Invest Dermatol 2018;138:1338–47. [DOI] [PubMed] [Google Scholar]
- Barker CL, McHale MT, Gillies AK, Waller J, Pearce DM, Osborne J, et al. The development and characterization of an in vitro model of psoriasis. J Invest Dermatol 2004;123:892–901. [DOI] [PubMed] [Google Scholar]
- Bauer FW, van de Kerkhof PC, Maassen-de Grood RM. Epidermal hyperproliferation following the induction of microabscesses by leukotriene B4. Br J Dermatol 1986;114: 409–12. [DOI] [PubMed] [Google Scholar]
- Besgen P, Trommler P, Vollmer S, Prinz JC. Ezrin, maspin, peroxiredoxin 2, and heat shock protein 27: potential targets of a streptococcal-induced autoimmune response in psoriasis. J Immunol 2010;184:5392–402. [DOI] [PubMed] [Google Scholar]
- Bissonnette R, Harel F, Krueger JG, Guertin MC, Chabot-Blanchet M, Gonzalez J, et al. TNF-alpha Antagonist and Vascular Inflammation in Patients with Psoriasis Vulgaris: A Randomized Placebo-Controlled Study. J Invest Dermatol 2017;137:1638–45. [DOI] [PubMed] [Google Scholar]
- Blauvelt A IL-6 Differs from TNF-alpha: unpredicted clinical effects caused by IL-6 blockade in psoriasis. J Invest Dermatol 2017;137: 541–2. [DOI] [PubMed] [Google Scholar]
- Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 2004;199: 731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain S, Camp R, Dowd P, Black AK, Greaves M. The release of leukotriene B4-like material in biologically active amounts from the lesional skin of patients with psoriasis. J Invest Dermatol 1984;83:70–3. [DOI] [PubMed] [Google Scholar]
- Brain SD, Camp RD, Dowd PM, Black AK, Woollard PM, Mallet AI, et al. Psoriasis and leukotriene B4. Lancet 1982;2:762–3. [DOI] [PubMed] [Google Scholar]
- Brauchli YB, Jick SS, Miret M, Meier CR. Psoriasis and risk of incident cancer: an inception cohort study with a nested case-control analysis. J Invest Dermatol 2009;129:2604–12. [DOI] [PubMed] [Google Scholar]
- Budu-Aggrey A, Brumpton B, Tyrrell J, Watkins S, Modalsli EH, Celis-Morales C, et al. Evidence of a causal relationship between body mass index and psoriasis: A mendelian randomization study. PLoS Med 2019;16:e1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budu-Aggrey A, Paternoster L. Research Techniques Made Simple: Using Genetic Variants for Randomization. J Invest Dermatol 2019;139:1416–14121.e1. [DOI] [PubMed] [Google Scholar]
- Cai Y, Xue F, Quan C, Qu M, Liu N, Zhang Y, et al. A Critical Role of the IL-1beta-IL-1R Signaling Pathway in Skin Inflammation and Psoriasis Pathogenesis. J Invest Dermatol 2019;139: 146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JJ, Ebsworth K, Ertl LS, McMahon JP, Wang Y, Yau S, et al. Efficacy of Chemokine Receptor Inhibition in Treating IL-36alpha-Induced Psoriasiform Inflammation. J Immunol 2019;202:1687–92. [DOI] [PubMed] [Google Scholar]
- Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet 2007;122:201–6. [DOI] [PubMed] [Google Scholar]
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet 2007;80:273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol 2011;131:2428–37. [DOI] [PubMed] [Google Scholar]
- Caruso R, Botti E, Sarra M, Esposito M, Stolfi C, Diluvio L, et al. Involvement of interleukin-21 in the epidermal hyperplasia of psoriasis. Nat Med 2009;15:1013–5. [DOI] [PubMed] [Google Scholar]
- Chaudhari U, Romano P, Mulcahy LD, Dooley LT, Baker DG, Gottlieb AB. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: a randomised trial. Lancet 2001;357: 1842–7. [DOI] [PubMed] [Google Scholar]
- Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 2014;192:3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med 2016;213: 2399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A, et al. Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med 2007;13:836–42. [DOI] [PubMed] [Google Scholar]
- Conrad C, Di Domizio J, Mylonas A, Belkhodja C, Demaria O, Navarini AA, et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun 2018;9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos M, Mogilenko DA, Fleury S, Gilbert B, Becquart C, Quemener S, et al. Keratinocyte Expression of A20/TNFAIP3 Controls Skin Inflammation Associated with Atopic Dermatitis and Psoriasis. J Invest Dermatol 2019;139: 135–45. [DOI] [PubMed] [Google Scholar]
- Di Meglio P, Villanova F, Navarini AA, Mylonas A, Tosi I, Nestle FO, et al. Targeting CD8(+) T cells prevents psoriasis development. J Allergy Clin Immunol 2016;138:274–6.e6. [DOI] [PubMed] [Google Scholar]
- Eder L, Chandran V, Pellett F, Pollock R, Shanmugaragjah S, Rosen CF, et al. IL13 gene polymorphism is a marker for psoriatic arthritis among psoriasis patients. Ann Rhuem Dis 2011;70:1594–8. [DOI] [PubMed] [Google Scholar]
- Egeberg A, Thyssen JP, Burisch J, Colombel JF. Incidence and Risk of Inflammatory Bowel Disease in Patients with Psoriasis-A Nationwide 20-Year Cohort Study. J Invest Dermatol 2019;139:316–23. [DOI] [PubMed] [Google Scholar]
- Elder JT, Bruce AT, Gudjonsson JE, Johnston A, Stuart PE, Tejasvi T, et al. Molecular dissection of psoriasis: integrating genetics and biology. J Invest Dermatol 2010;130:1213–26. [DOI] [PubMed] [Google Scholar]
- Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet 2010;42: 991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus E, Stuart PE, Ellinghaus D, Nair RP, Debrus S, Raelson JV, et al. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J Invest Dermatol 2012;132:1133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis CN, Gorsulowsky DC, Hamilton TA, Billings JK, Brown MD, Headington JT, et al. Cyclosporine improves psoriasis in a double-blind study. JAMA 1986;256:3110–6. [PubMed] [Google Scholar]
- Fan TP, Lewis GP. Mechanism of cyclosporin A-induced inhibition of prostacyclin synthesis by macrophages. Prostaglandins 1985;30:735–47. [DOI] [PubMed] [Google Scholar]
- Fritz Y, Klenotic PA, Swindell WR, Yin ZQ, Groft SG, Zhang L, et al. Induction of Alternative Proinflammatory Cytokines Accounts for Sustained Psoriasiform Skin Inflammation in IL-17CþIL-6KO Mice. J Invest Dermatol 2017;137:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006;296:1735–41. [DOI] [PubMed] [Google Scholar]
- Gordon K, Kochkodan JJ, Blatt H, Lin SY, Kaplan N, Johnston A, et al. Alteration of the EphA2/Ephrin-A signaling axis in psoriatic epidermis. J Invest Dermatol 2013;133: 712–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon KB, Duffin KC, Bissonnette R, Prinz JC, Wasfi Y, Li S, et al. A Phase 2 Trial of Guselkumab versus Adalimumab for Plaque Psoriasis. N Engl J Med 2015;373:136–44. [DOI] [PubMed] [Google Scholar]
- Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 2018;392:650–61. [DOI] [PubMed] [Google Scholar]
- Gottlieb AB, Grossman RM, Khandke L, Carter DM, Sehgal PB, Fu SM, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol 1992;98:302–9. [DOI] [PubMed] [Google Scholar]
- Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, Gottlieb AB, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med 1995;1:442–7. [DOI] [PubMed] [Google Scholar]
- Grabbe J, Czarnetzki BM, Mardin M. Chemotactic leukotrienes in psoriasis. Lancet 1982;2:1464. [DOI] [PubMed] [Google Scholar]
- Griffiths CE, Powles AV, Leonard JN, Fry L, Baker BS, Valdimarsson H. Clearance of psoriasis with low dose cyclosporin. Br Med J (Clin Res Ed) 1986;293:731–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, Elder JT. Mouse models of psoriasis. J Invest Dermatol 2007;127: 1292–308. [DOI] [PubMed] [Google Scholar]
- Guttman-Yassky E, Krueger JG. IL-17C: A unique epithelial cytokine with potential for targeting across the spectrum of atopic dermatitis and psoriasis. J Invest Dermatol 2018;138:1467–9. [DOI] [PubMed] [Google Scholar]
- Ha HL, Wang H, Claudio E, Tang W, Siebenlist U. IL-20-receptor signaling delimits IL-17 production in psoriatic inflammation. J Invest Dermatol 2019. 10.1016/j.jid.2019.06.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JI, Keat AC, Staughton RC. Cyclosporin for psoriasis. Lancet 1984;2:981–2. [DOI] [PubMed] [Google Scholar]
- Hawkes JE, Adalsteinsson JA, Gudjonsson JE, Ward NL. Research Techniques Made Simple: Murine Models of Human Psoriasis. J Invest Dermatol 2018;138:e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol 2017;140:645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert D, Franz S, Popkova Y, Anderegg U, Schiller J, Schwede K, et al. High-fat diet exacerbates early psoriatic skin inflammation independent of obesity: saturated fatty acids as key players. J Invest Dermatol 2018;138: 1999–2009. [DOI] [PubMed] [Google Scholar]
- Ikumi K, Odanaka M, Shime H, Imai M, Osaga S, Taguchi O, et al. Hyperglycemia Is Associated with Psoriatic Inflammation in Both Humans and Mice. J Invest Dermatol 2019;139: 1329–38.e27. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhu FG, Bhagat L, Yu D, Tang JX, Kandimalla ER, et al. A Toll-like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL-23-induced psoriasis. J Invest Dermatol 2013;133:1777–84. [DOI] [PubMed] [Google Scholar]
- Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol 2011;186:2613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol 2017;140:109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Tomalin L, Lee J, Fitz LJ, Berstein G, Correada Rosa J, et al. Reduction of Inflammatory and Cardiovascular Proteins in the Blood of Patients with Psoriasis: Differential Responses between Tofacitinib and Etanercept after 4 Weeks of Treatment. J Invest Dermatol 2018;138: 273–81. [DOI] [PubMed] [Google Scholar]
- Kim JH, Hu Y, Yongqing T, Kim J, Hughes VA, Le Nours J, et al. CD1a on Langerhans cells controls inflammatory skin disease. Nat Immunol 2016;17:1159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp T, Lenz P, Bello-Fernandez C, Kastelein RA, Kupper TS, Stingl G. IL-23 production by cosecretion of endogenous p19 and transgenic p40 in keratin 14/p40 transgenic mice: evidence for enhanced cutaneous immunity. J Immunol 2003;170:5438–44. [DOI] [PubMed] [Google Scholar]
- Kragballe K, Desjarlais L, Voorhees JJ. Leukotrienes B4, C4 and D4 stimulate DNA synthesis in cultured human epidermal keratinocytes. Br J Dermatol 1985;113:43–52. [DOI] [PubMed] [Google Scholar]
- Krueger GG, Manning DD, Malouf J, Ogden B. Long-term maintenance of psoriatic human skin on congenitally athymic (nude) mice. J Invest Dermatol 1975;64:307–12. [DOI] [PubMed] [Google Scholar]
- Laggner U, Di Meglio P, Perera GK, Hundhausen C, Lacy KE, Ali N, et al. Identification of a novel proinflammatory human skin-homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. J Immunol 2011;187:2783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun 2014;5:5621. [DOI] [PubMed] [Google Scholar]
- Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 2014;371:326–38. [DOI] [PubMed] [Google Scholar]
- Laurent S, Le Parc JM, Clerici T, Breban M, Mahe E. Onset of psoriasis following treatment with tocilizumab. Br J Dermatol 2010;163: 1364–5. [DOI] [PubMed] [Google Scholar]
- Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 2004;199:125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Yeh YC, Chang YT, Lai MS. All-cause and cause-specific mortality in patients with psoriasis in Taiwan: a nationwide population-based study. J Invest Dermatol 2017;137:1468–73. [DOI] [PubMed] [Google Scholar]
- Lee YS, Cheon IS, Kim BH, Kwon MJ, Lee HW, Kim TY. Loss of extracellular superoxide dismutase induces severe IL-23-mediated skin inflammation in mice. J Invest Dermatol 2013;133:732–41. [DOI] [PubMed] [Google Scholar]
- Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012;366:1190–9. [DOI] [PubMed] [Google Scholar]
- Li WQ, Han JL, Chan AT, Qureshi AA. Psoriasis, psoriatic arthritis and increased risk of incident Crohn’s disease in US women. Ann Rheum Dis 2013;72:1200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroos J, Svensson L, Norsgaard H, Lovato P, Moller K, Hagedorn PH, et al. IL-23-mediated epidermal hyperplasia is dependent on IL-6. J Invest Dermatol 2011;131:1110–8. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang Z, De La Torre R, Barling A, Tsujikawa T, Hornick N, et al. Trim32 deficiency enhances Th2 immunity and predisposes to features of atopic dermatitis. J Invest Dermatol 2017;137:359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 2008;128:1207–11. [DOI] [PubMed] [Google Scholar]
- Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, et al. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol 2013;133:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos TR, O’Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest 2017;127:4031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellett M, Meier B, Mohanan D, Schairer R, Cheng P, Satoh TK, et al. CARD14 Gain-of-Function Mutation Alone Is Sufficient to Drive IL-23/IL-17-Mediated Psoriasiform Skin Inflammation In Vivo. J Invest Dermatol 2018;138: 2010–23. [DOI] [PubMed] [Google Scholar]
- Merleev AA, Marusina AI, Ma C, Elder JT, Tsoi LC, Raychaudhuri SP, et al. Meta-analysis of RNA sequencing datasets reveals an association between TRAJ23, psoriasis, and IL-17A. JCI Insight 2018;3:120682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilenko DA, Haas JT, L’Homme L, Fleury S, Quemener S, Levavasseur M, et al. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 2019;178:263. [DOI] [PubMed] [Google Scholar]
- Monin L, Gudjonsson JE, Childs EE, Amatya N, Xing X, Verma AH, et al. MCPIP1/Regnase-1 Restricts IL-17A- and IL-17C-Dependent Skin Inflammation. J Immunol 2017;198:767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller W, Herrmann B. Cyclosporin A for psoriasis. N Engl J Med 1979;301:555. [DOI] [PubMed] [Google Scholar]
- Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 2009;41: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RP, Ruether A, Stuart PE, Jenisch S, Tejasvi T, Hiremagalore R, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol 2008;128:1653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S, et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet 2006;78:827–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nast A, Jacobs A, Rosumeck S, Werner RN. Efficacy and safety of systemic long-term treatments for moderate-to-severe psoriasis: a systematic review and meta-analysis. J Invest Dermatol 2015;135:2641–8. [DOI] [PubMed] [Google Scholar]
- Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol 2006;55:829–35. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med 2005;202:135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff BJ. The cytokine network in psoriasis.Arch Dermatol 1991;127:871–84. [PubMed] [Google Scholar]
- Ogawa K, Stuart PE, Tsoi LC, Suzuki K, Nair RP, Mochizuki H, et al. A Transethnic Mendelian Randomization Study Identifies Causality of Obesity on Risk of Psoriasis. J Invest Dermatol 2019;139:1397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest 2012;122:2252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012;366:1181–9. [DOI] [PubMed] [Google Scholar]
- Pischon H, Radbruch M, Ostrowski A, Schumacher F, Honzke S, Kleuser B, et al. How Effective Is Tacrolimus in the Imiquimod-Induced Mouse Model of Psoriasis? J Invest Dermatol 2018;138:455–8. [DOI] [PubMed] [Google Scholar]
- Pouplard C, Brenaut E, Horreau C, Barnetche T, Misery L, Richard MA, et al. Risk of cancer in psoriasis: a systematic review and meta-analysis of epidemiological studies. J Eur Acad Dermatol Venereol 2013;27(Suppl 3):36–46. [DOI] [PubMed] [Google Scholar]
- Prinz JC. Melanocytes: Target Cells of an HLAC*06:02-Restricted Autoimmune Response in Psoriasis. J Invest Dermatol 2017;137:2053–8. [DOI] [PubMed] [Google Scholar]
- Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet 2017;390:276–88. [DOI] [PubMed] [Google Scholar]
- Reich K, Rich P, Maari C, Bissonnette R, Leonardi C, Menter A, et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate-to-severe plaque psoriasis: results from a randomized phase II study. Br J Dermatol 2019;181:88–95. [DOI] [PubMed] [Google Scholar]
- Ruiz-Romeu E, Ferran M, Gimenez-Arnau A, Bugara B, Lipert B, Jura J, et al. MCPIP1 RNase is aberrantly distributed in psoriatic epidermis and rapidly induced by IL-17A. J Invest Dermatol 2016;136:1599–607. [DOI] [PubMed] [Google Scholar]
- Rusin LJ, Duell EA, Voorhees JJ. Papaverine and Ro 20–1724 inhibit cyclic nucleotide phosphodiesterase activity and increase cyclic AMP levels in psoriatic epidermis in vitro. J Invest Dermatol 1978;71:154–6. [DOI] [PubMed] [Google Scholar]
- Sarkar MK, Kaplan N, Tsoi LC, Xing X, Liang Y, Swindell WR, et al. Endogenous glucocorticoid deficiency in psoriasis promotes inflammation and abnormal differentiation. J Invest Dermatol 2017;137:1474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senra L, Mylonas A, Kavanagh RD, Fallon PG, Conrad C, Borowczyk-Michalowska J, et al. IL-17E (IL-25) Enhances Innate Immune Responses during Skin Inflammation. J Invest Dermatol 2019;139:1732–42.e17. [DOI] [PubMed] [Google Scholar]
- Setta-Kaffetzi N, Navarini AA, Patel VM, Pullabhatla V, Pink AE, Choon SE, et al. Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol 2013;133:1366–9. [DOI] [PubMed] [Google Scholar]
- Shimoura N, Nagai H, Fujiwara S, Jimbo H, Nishigori C. Exacerbation and prolongation of psoriasiform inflammation in diabetic obese mice: a synergistic role of CXCL5 and endoplasmic reticulum stress. J Invest Dermatol 2018;138:854–63. [DOI] [PubMed] [Google Scholar]
- Sommer DM, Jenisch S, Suchan M, Christophers E, Weichenthal M. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res 2006;298:321–8. [DOI] [PubMed] [Google Scholar]
- Stawiski MA, Powell JA, Lang PG, Schork A, Duell EA, Voorhees JJ. Papaverine: its effects on cyclic AMP in vitro and psoriasis in vivo. J Invest Dermatol 1975;64:124–7. [DOI] [PubMed] [Google Scholar]
- Stawiski MA, Rusin LJ, Burns TL, Weinstein GD, Voorhees JJ. Ro 20–1724: an agent that significantly improves psoriatic lesions in double-blind clinical trials. J Invest Dermatol 1979;73:261–3. [DOI] [PubMed] [Google Scholar]
- Strauss H Zur Lehre von der neurogenen und der thyreogenen Glykosurie. Dtsch med Wochenschr 1897;23:275–8. [Google Scholar]
- Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol 2008;181:5948–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart PE, Nair RP, Tsoi LC, Tejasvi T, Das S, Kang HM, et al. Genome-wide association analysis of psoriatic arthritis and cutaneous psoriasis reveals differences in their genetic architecture. Am J Hum Genet 2015;97:816–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Farinas M, Fuentes-Duculan J, Lowes MA, Krueger JG. Resolved psoriasis lesions retain expression of a subset of disease-related genes. J Invest Dermatol 2011;131:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura K, Takemoto A, Yamaguchi M, Takahashi H, Shoda Y, Mitsuma T, et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J Invest Dermatol 2013;133:2514–21. [DOI] [PubMed] [Google Scholar]
- Swindell WR, Michaels KA, Sutter AJ, Diaconu D, Fritz Y, Xing X, et al. Imiquimod has strain-dependent effects in mice and does not uniquely model human psoriasis. Genome Med 2017;9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita J, Shin DB, Ogdie A, Gelfand JM. Risk of serious infection, opportunistic infection, and herpes zoster among patients with psoriasis in the United Kingdom. J Invest Dermatol 2018;138:1726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol 1998;111:645–9. [DOI] [PubMed] [Google Scholar]
- Uyemura K, Yamamura M, Fivenson DF, Modlin RL, Nickoloff BJ. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol 1993;101:701–5. [DOI] [PubMed] [Google Scholar]
- Valdimarsson H, Thorleifsdottir RH, Sigurdardottir SL, Gudjonsson JE, Johnston A. Psoriasis–as an autoimmune disease caused by molecular mimicry. Trends Immunol 2009;30: 494–501. [DOI] [PubMed] [Google Scholar]
- van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009;182:5836–45. [DOI] [PubMed] [Google Scholar]
- Voorhees JJ, Duell EA, Bass LJ, Powell JA, Harrell ER. The cyclic AMP system in normal and psoriatic epidermis. J Invest Dermatol 1972;59:114–20. [DOI] [PubMed] [Google Scholar]
- Wei P, Yang Y, Liu Z, Luo Z, Tu W, Han J, et al. Characterization of autoantigen presentation by HLA-C*06:02 in psoriasis. J Invest Dermatol 2017;137:2238–41. [DOI] [PubMed] [Google Scholar]
- Weinstein GD, Frost P. Abnormal cell proliferation in psoriasis. J Invest Dermatol 1968;50:254–9. [PubMed] [Google Scholar]
- Weinstein GD, McCullough JL. Cytokinetics and chemotherapy of psoriasis. J Invest Dermatol 1976;67:26–30. [DOI] [PubMed] [Google Scholar]
- Weinstein GD, Velasco J. Selective action of methotrexate on psoriatic epidermal cells. J Invest Dermatol 1972;59:121–7. [DOI] [PubMed] [Google Scholar]
- Winge MC, Ohyama B, Dey CN, Boxer LM, Li W, Ehsani-Chimeh N, et al. RAC1 activation drives pathologic interactions between the epidermis and immune cells. J Clin Invest 2016;126: 2661–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf N, Quaranta M, Prescott NJ, Allen M, Smith R, Burden AD, et al. Psoriasis is associated with pleiotropic susceptibility loci identified in type II diabetes and Crohn disease. J Med Genet 2008;45:114–6. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Poon KY, Channual JC, Shen AY. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol 2012;148: 1244–50. [DOI] [PubMed] [Google Scholar]
- Xing X, Liang Y, Sarkar MK, Wolterink L, Swindell WR, Voorhees JJ, et al. IL-17 responses are the dominant inflammatory signal linking inverse, erythrodermic, and chronic plaque psoriasis. J Invest Dermatol 2016;136: 2498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Nakajima K, Takaishi M, Kitaba S, Magata Y, Kataoka S, et al. Psoriatic inflammation facilitates the onset of arthritis in a mouse model. J Invest Dermatol 2015;135: 445–53. [DOI] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 2005;437:369–75. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007;445:648–51. [DOI] [PubMed] [Google Scholar]