

Abstract
Genetic variation across the HLA is known to influence renal-transplant outcome. However, the impact of genetic variation beyond the HLA is less clear. We tested the association of common genetic variation and clinical characteristics, from both the donor and recipient, with post-transplant eGFR at different time-points, out to 5-years post-transplantation.
We conducted GWAS meta-analyses across 10,844 donors and recipients from five European ancestry cohorts. We also analysed the impact of polygenic risk scores (PRS), calculated using genetic variants associated with non-transplant eGFR, on post-transplant eGFR.
PRS calculated using the recipient genotype alone, as well as combined donor and recipient genotypes were significantly associated with eGFR at 1-year post-transplant. 32% of the variability in eGFR at 1-year post-transplant was explained by our model containing clinical covariates (including weights for death/graft-failure), principal components and combined donor-recipient PRS, with 0.3% contributed by the PRS. No individual genetic variant was significantly associated with eGFR post-transplant in the GWAS.
This is the first study to examine PRS, composed of variants that impact kidney function in the general population, in a post-transplant context. Despite PRS being a significant predictor of eGFR post-transplant, the effect size of common genetic factors is limited compared to clinical variables.
1. Introduction
Since the discovery of the HLA in the 1950s, it has been clear that genetic factors are important in kidney transplant outcomes1. A number of studies have examined candidate genes or variants beyond the HLA and their impact on graft function2–4, however, few have been replicated5. To date, only a small number of genome-wide association studies (GWAS) have analysed medium/long-term allograft outcome6–8. A study from 2013 identified recipient genotype loci on chromosomes 14 and 18 which significantly associated with 5 years serum creatinine and long-term graft survival6. However, this study was limited by its sample size (n=326) and an independent effort failed to replicate the findings9. This highlights the need for larger, robust GWAS of allograft outcome to determine the extent to which common variation affects the outcome of kidney transplantation.
Kidney transplants are a relatively rare phenotype and collaborative efforts such as the UK and Ireland Renal Transplant Consortium (UKIRTC7) and International Genetics & Translational Research in Transplantation Network (iGeneTRAiN10) facilitate well-powered studies of transplant related phenotypes. Recently the UKIRTC published a GWAS of kidney transplant outcomes which examined graft survival and acute rejection in the first twelve months post-transplant, in a deceased donor cohort of 2,094 transplant pairs7. Although much better powered than earlier efforts, no significant associations were found beyond the previously known effect of the HLA. A GWAS involving pooled DNA from 4,127 renal transplant recipients identified signals of association with T-cell mediated acute rejection at the PTPRO and CCDC67 loci. The finding was replicated, but not as part of an independent study8. Neither the PTPRO or CCDC67 signal replicated in the UKIRTC GWAS of acute rejection, however the definition of acute rejection in the UKIRTC study11 was not specific to T-cell mediated rejection which may explain the discordance. Here, we hypothesise that a continuous variable of outcome (i.e. eGFR) would provide additional power to detect the impact of genetic variation on markers of graft outcome.
In non-transplant populations, there has been a number of large well-powered GWAS that have identified genetic variants that associate robustly with kidney function12–15, the largest of which (n= 133,814) was carried out by Pattaro et al 13. However, despite the identification of a number of risk loci, univariate effect sizes were small (OR of 0.93 to 1.06 for the 23 novel loci identified).
Common genetic variants typically have small effects on complex human traits, and thus are usually not of clinical relevance. Polygenic risk scores (PRS) quantify the cumulative effects of a number of loci, which may individually have a small predictive ability. By doing so, PRS’s may be more clinically relevant than looking at common genetic variants independently. One study, by Gorski et al12, used summary statistics from their GWAS of estimated glomerular filtration rate (eGFR) to create a polygenic risk score (PRS) which was then tested against eGFR in an independent cohort of 1,017 individuals. They found the PRS to explain 2.2% of the trait variance compared to 1.3% when just considering genome-wide significant loci.
In this study, we set out to test the hypothesis that common variation from donor or recipient genotype is associated with short and medium term allograft kidney function, using 1-year, 5-year and ∆ (change between 1 and 5 year) eGFR as measures of kidney function. Involving 10,844 transplant donors and recipients, and delivered through the iGeneTRAiN consortium, this study is the largest GWAS of allograft function to date. In addition to studying the association of individual SNPs with eGFR, we also set out to test the hypothesis that higher polygenic load for increased eGFR in the donor, recipient and combined (donor and recipient) genomes is associated with increased graft function. To do this, we tested if PRS estimated using alleles from a GWAS of kidney function (using eGFR as a proxy) in a non-transplant population13 is predictive of kidney function in a number of renal transplant cohorts.
2. Materials and Methods
2.1. Cohorts
We assembled five cohorts via the iGeneTRAiN consortium10. They are 1) ‘Transplant Lines’ (the Netherlands), 2) the Vienna/Prague cohort (Austria), 3) the Deterioration of Kidney Allograft Function Genomics (DeKAF) cohort (United States, NCT00270712), 4) the Genomics of Kidney Transplantation (GEN03) cohort (United States, NCT01714440) and 5) the United Kingdom and Ireland Renal Transplant Consortium (UKIRTC) cohort. See Table 1 for cohort descriptions and supplementary methods for genotyping and imputation information. The appropriate ethics committees at each site approved the protocol for this study.
Table 1.
Descriptive statistics for each cohort of significant clinical predictors and eGFR measures
| UKIRTC | TxLines | GEN03 | DeKAF | Vienna/ Prague | ||
|---|---|---|---|---|---|---|
| Recipients | N | 2233 | 983 | 673 | 1864 | 616 |
| Range of year of transplant | 1981−2007 | 1993−2008 | 2012−2016 | 2005−2011 | 2005−2015 | |
| % male | 64% | 57% | 62% | 63% | 63% | |
| Average length of follow-up post-transplant - years | 8.47 (4) | 9.77 (5) | 1.90 (0.8) | 2.25 (1) | 4.75(3) | |
| Average age at transplant | 46.32 (13) | 48.26 (13) | 50.25 (15) | 50.46 (14) | 52 (14) | |
| % with failure by year 1 | 1% | 4% | 0% | 0% | 5% | |
| % with death by year 1 | 0.3% | 2% | 0% | 1% | 3% | |
| % with failure by year 5 | 8% | 10% | 1% | 3% | 10% | |
| % with death by year 5 | 4% | 8% | 1% | 3% | 8% | |
| % on mycophenolate mofetil | 37%* | 72% | 100% | 95% | 99% | |
| % with delayed graft function | NA | 30% | 6% | 7% | 30% | |
| % Acute rejection episode by year 1 | 20.57%* | 30% | 15% | 14% | 36% | |
| % Acute rejection episode by year 5 | NA | 33% | 16% | 18% | 40% | |
| Donors | N | 1875 | 823 | 366 | 802 | 609 |
| % male | 57% | 52% | 43% | 41% | 56% | |
| Average age | 43.1 (16) | 43.02 (15) | 45.50 (12) | 44.31 (11) | 50 (14) | |
| % Living | 0% | 17% | 100% | 100% | 15% | |
| eGFR at 1 year | N observations | 1905 | 937 | 673 | 1864 | 556 |
| Mean | 48.1 | 45.6 | 61.9 | 58.1 | 52.5 | |
| Median | 47.1 | 44.6 | 59.2 | 55.9 | 50.8 | |
| Max | 120.5 | 108.7 | 201.2 | 217.6 | 109.8 | |
| Min | 4.5 | 2.5 | 15.0 | 4.0 | 2.0 | |
| Stdev | 17.8 | 18.8 | 21.8 | 20.8 | 18.8 | |
| eGFR at 5 year | N observations | 1700 | 906 | NA | NA | 315 |
| Mean | 46.4 | 46.2 | NA | NA | 51.5 | |
| Median | 44.9 | 45.9 | NA | NA | 50.8 | |
| Max | 122 | 181 | NA | NA | 105 | |
| Min | 4.1 | 2.5 | NA | NA | 9.4 | |
| Stdev | 19.4 | 22.5 | NA | NA | 19.9 | |
| ∆ eGFR | N observations | 1283 | 719 | NA | NA | 296 |
| Mean | -1.8 | 2.0 | NA | NA | -2.5 | |
| Median | -1.8 | 2.7 | NA | NA | -2.9 | |
| Max | 65.6 | 73.3 | NA | NA | 45.9 | |
| Min | -57.3 | -60.4 | NA | NA | -45.4 | |
| Stdev | 13.4 | 16.1 | NA | NA | 12.3 | |
The above table provides descriptive statistics for the UK and Ireland Renal Transplant consortium cohort (UKIRTC), TransplantLines cohort (TxLines), GEN03, DeKAF and Vienna/Prague cohort. N = number of individuals, % = Percentage, % with failure by year 1/5 = Percentage with a failure event within 1/5 years post-transplant, % with death by year 1/5= Percentage who died within 1/5 years post-transplant, % on mycophenolate mofetil = percentage of patients who received mycophenolate mofetil at the start of their transplant (intention to treat), % living = percentage of living donors, stdev = standard deviation, min = minimum value observed, max = maximum value observed. Delayed graft function status was unavailable for the UKIRTC cohort. Acute rejection status for the UKIRTC cohort was only available for the first twelve months post-transplant. ∆ eGFR and 5-year eGFR were unavailable for the GEN03 and DeKAF cohorts.
A large number of the UKIRTC patients had missing information for MMF and AR status and so these percentages were calculated from those who had yes/no status for MMF and individuals with missingness were excluded. MMF % was calculated as follows: 1045 N – not exposed to MMF, 604 – were exposed, 584 = missing. (604/ (1045 + 604))*100/1 = 36.62%. For acute rejection at 1 year, 1093 – did not experience an AR episode in the first twelve months, 283 did have an AR episode and 857 were missing. AR % was calculated as follows: (283 / (1093 + 283)) * 100/1= 20.57%.
Notably, GEN03 and DeKAF Genomics cohorts were entirely living donor transplants whereas UKIRTC, TransplantLines and Vienna/Prague cohort was predominantly deceased donor transplants. In TransplantLines, the year of transplant ranged from 1993–2008. In the UKIRTC, the year of transplant ranged from 1981–2007. In DEKAF Genomics, transplants were performed between 2005–2011 and in the GEN03 study from 2012–2016. In the Vienna/Prague cohort, the year of transplant ranged from 2005 to 2015.
2.2. Phenotype
We set out to analyse three phenotypes in each of these cohorts: eGFR at one-year post-renal transplant, eGFR at five years post-transplant and the change in eGFR from one to five years (Δ) post-transplant (see supplementary methods for details on calculating eGFR).
The following inclusion/exclusion criteria were applied: Participants must 1) be of European ancestry, 2) have donated/received a renal transplant and 3) be unrelated to level of 3rd degree relative.
To identify individuals of European ancestry, all patient cohorts were merged with samples from the Human Genome Diversity project (representing seven different global populations) or the 1000 Genomes Project and analysed using principal components analysis (PCA). Principal component (PC) 1 was plotted against PC2 and individuals in the patient cohorts that did not overlap with the European individuals in the HGDP were removed. To identify individuals unrelated beyond 3rd degree, we used PLINK’s --genome function to calculate identity-by-descent (PIHAT) scores16. One individual from each pair of related individuals (i.e. 3rd degree, or closer relatives) was excluded from the analysis16.
2.3. Clinical analyses
We first assessed a number of available clinical predictors of allograft function (see supplementary table 1 for list and definitions of clinical variables tested). These clinical variables were tested on a dataset of over 1,400 renal transplant recipients from TransplantLines and Dublin (subset of UKIRTC cohort). Significant clinical variables identified in the univariate linear regression analysis (see Table 2) were then tested in a stepwise regression model (see Supplementary Tables 2–7). Collinear variables were removed and the remaining clinical variables were included as covariates in our genetic analyses. See supplementary methods for further details.
Table 2.
Linear regression results for clinical variables associated with eGFR at 1 year, 5 years and ∆ eGFR from 1 to 5 years
| Clinical Variable |
Log 1-year eGFR |
Log 5-year eGFR |
∆ eGFR |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coef. | SE | Pbon | N | Coef. | SE | Pbon | N | Coef. | SE | Pbon | N | |
| Acute Rejection | −0.07 | 0.01 | 1.53×10−8 | 1407 | −0.09 | 0.01 | 3.48×10−11 | 1399 | −4.57 | 0.99 | 8.08×10−5 | 1108 |
| Azathioprine | 0.02 | 0.01 | 1 | 1406 | −0.01 | 0.01 | 1 | 1399 | −3.61 | 1.09 | 0.02 | 1108 |
| Cold Ischemic Time | −0.00004 | 0.00001 | 0.001 | 1400 | −0.00004 | 0.00001 | 0.0009 | 1392 | −0.001 | 0.001 | 1 | 1108 |
| CMV | −0.04 | 0.01 | 0.0004 | 1407 | -0.04 | 0.01 | 0.06 | 1340 | −1.54 | 0.97 | 1 | 1108 |
| Corticosteroids | 0.09 | 0.03 | 0.02 | 1402 | 0.08 | 0.03 | 0.24 | 1399 | −3.89 | 2.65 | 1 | 1108 |
| Cyclosporin | 0.04 | 0.02 | 0.48 | 1355 | 0.03 | 0.02 | 1 | 1399 | 1.34 | 1.48 | 1 | 1108 |
| Delayed graft function | −0.09 | 0.01 | 2.42×10−13 | 1407 | −0.09 | 0.01 | 1.39×10−8 | 1398 | 0.86 | 1.15 | 1 | 1103 |
| Donor age | −0.004 | 0.0003 | 3.84×10−30 | 1407 | −0.004 | 0.0004 | 1.41×10−20 | 1399 | −0.02 | 0.03 | 1 | 1063 |
| Donor sex | −0.04 | 0.01 | 0.001 | 1407 | −0.03 | 0.01 | 0.08 | 1399 | 0.47 | 0.93 | 1 | 1108 |
| Donor Type | −0.07 | 0.01 | 3.54×10−7 | 1407 | −0.05 | 0.02 | 0.006 | 1399 | −0.39 | 1.16 | 1 | 937 |
| HLA-mismatch A | 0.00 | 0.01 | 1 | 1407 | −0.01 | 0.01 | 1 | 1196 | −1.11 | 0.78 | 1 | 1108 |
| HLA-mismatch B | −0.01 | 0.01 | 1 | 1407 | 0.00 | 0.01 | 1 | 1196 | 0.40 | 0.74 | 1 | 936 |
| HLA-mismatch DR | 0.01 | 0.01 | 1 | 1407 | 0.003 | 0.01 | 1 | 1195 | −0.72 | 0.84 | 1 | 937 |
| Mycophenolate | 0.02 | 0.01 | 1 | 1196 | 0.05 | 0.01 | 0.003 | 1399 | 3.52 | 0.94 | 0.004 | 1107 |
| Recipient age | −0.003 | 0.0004 | 1.3×10−9 | 1195 | −0.0005 | 0.0004 | 1 | 1399 | 0.17 | 0.04 | 2.68×10−5 | 1108 |
| Recipient sex | −0.01 | 0.01 | 1 | 1196 | −0.03 | 0.01 | 0.44 | 1399 | −0.20 | 0.94 | 1 | 937 |
| Sirolimus | −0.09 | 0.03 | 0.14 | 1407 | −0.03 | 0.04 | 1 | 1399 | 5.64 | 2.86 | 0.96 | 1108 |
| Site | 0.01 | 0.01 | 1 | 1407 | 0.001 | 0.02 | 1 | 1399 | −2.92 | 1.15 | 0.22 | 1108 |
| Tacrolimus | −0.01 | 0.02 | 1 | 1196 | −0.02 | 0.02 | 1 | 1399 | −2.25 | 1.82 | 1 | 1108 |
| Total MM | 0.00 | 0.00 | 1 | 1407 | −0.003 | 0.005 | 1 | 1196 | -0.27 | 0.36 | 1 | 1108 |
Coef. = regression coefficient; SE=standard error; Pbon = Bonferroni corrected p value (unadjusted p value*20); N = number of individuals tested (i.e. who had the measure available); Site = cohort associated with given individual where Dublin =one and TransplantLines =zero. Graft failure and death with a functioning graft were adjusted for in the log10 1-year and 5 year eGFR analyses. Acute Rejection = acute rejection within 1 year post-transplant for log10 1-year eGFR analysis and within 5 years for ∆ eGFR and log10 5-year eGFR analysis. Azathioprine = azathioprine - intention to treat at start of transplant. Cold Ischemic Time = cold ischemic time in minutes. CMV = cytomegalovirus infection in recipient post-transplant. Corticosteroids = intention to treat at start of transplant. Cyclosporin = intention to treat at start of transplant. Delayed graft function = yes/no. Donor age = donor age at time of transplant. Donor sex = male = 0, female = 1. Donor Type = Living/deceased = living = 1, deceased = 0. HLA-mismatch A = Number of HLA mismatches between donor and recipient at A locus. HLA-mismatch B = Number of HLA mismatches between donor and recipient at B locus. HLA-mismatch DR = Number of HLA mismatches between donor and recipient at DR locus. Mycophenolate = Mycophenolate - intention to treat at start of transplant. Recipient age = recipient age at time of transplant. Recipient sex = recipient sex - male=0, female=1. Sirolimus = Sirolimus - intention to treat at start of transplant. Site = Dublin/TransplantLines, Dublin = 1, TransplantLines = 0. Tacrolimus = Tacrolimus - intention to treat at start of transplant. Total MM = Total number of HLA mismatches across A, B and DR locus.
2.4. Genome-wide association study
Genome-wide association studies were carried out at each site independently for each available outcome variable (log10 1-year eGFR, log10 5-year eGFR and ∆ eGFR) with PCs (to correct for population-specific allelic differences), and available clinical variables with weights for death and failure in the log10 1-year eGFR and log10 5-year eGFR GWAS included as covariates (see supplementary methods section). GWAS were conducted separately for donor and recipient genotypes. See supplementary methods section 4 for further details.
The results from each site were then combined using a meta-analysis approach (see supplementary methods for further details). The meta-analysis approach utilised in this study has previously been applied in a number of other genome-wide analyses of common genetic variation including analyses of kidney function13,17–19. Genome-wide level of significance was set at 5×10−8 for the GWAS meta-analysis.
Retrospective power calculations were carried out using a combination of R’s pchisq and qchisq functions. In the recipient full model (significant clinical covariates (including weights for death/failure for the log10 1-year eGFR and log10 5-year eGFR analyses – see supplementary methods), principal components and given genetic variant) we had 80% power to detect a variant which explains 0.75%, 1.87% and 2.20% of outcome variance in the one-year, five-year and Δ recipient analysis respectively. In the donor full model GWAS, we had 80% power to detect a variant which explains 1.12%, 2.13% and 2.49% of outcome variance in the one-year, five-year and Δ donor analysis respectively.
In the recipient baseline model (just PCs, failure and death as covariates), we had 80% power to detect a genetic variant which explains 0.67%, 1.39% and 1.78% of outcome variance in the one-year, five-year and Δ recipient analysis respectively. In the donor baseline GWAS, we had 80% power to detect a variant which explains 0.98%, 1.58% and 2.01% of outcome variance in the one-year, five-year and Δ donor analysis respectively.
2.5. Polygenic risk analysis
For the PRS analysis, we employed the same clinical covariates and outcome measures as the GWAS analysis. We defined PRS as the sum of the alleles associated with a given trait weighted by the effect size of that allele as determined by a previous GWAS20. PRSs were calculated using results of a previous GWAS of eGFR in a non-transplant population13. We calculated the PRS at multiple p-value thresholds to enable us to examine at different sets of SNPs (for example, is the top 1,000 or top 10,000 most significant SNPs associated with eGFR in the general population a better predictor of eGFR post-transplant). See supplementary methods for further details on how we calculated these scores. PRSs were then tested as predictors of each outcome variable correcting for available significant clinical covariates (as discussed previously) and PCs using in a linear regression model (see supplementary methods for further details). Linear regressions were carried out separately at each site and then combined using a meta-analysis approach in line with previously published meta-analyses of PRS 21. See supplementary methods for further details.
3. Results
This study involved 10,844 transplant donors and recipients recruited from UKIRTC (n=4,108), TransplantLines (n=1,806), GEN03 (n=1,039), DeKAF (n=2,666) and Vienna/Prague (n=1,225). The mean age of recipients and other clinical variables are reported in Table 1. Subjects had a mean eGFR of 52.43ml/min at 1 year and 51.39ml/min at 5 years post-transplant. The mean Δ eGFR was −1.01ml/min. Distributions of eGFR in each of the five cohorts can be seen in Supplementary Figures 1 to 4.
3.1. Clinical analysis
Clinical covariates were tested in a combined cohort comprising over 1,400 renal transplant recipients from TransplantLines and the Dublin subset of the UKIRTC (see supplementary methods for further details). After correcting for multiple testing and collinearity (see Supplementary Table 2–7), we identified donor age, donor type, donor sex, recipient age, delayed graft function and acute rejection as significant predictors of log10eGFR at 1-year. These variables explained 22% of the outcome variance.
Donor age, mycophenolate mofetil exposure, delayed graft function, acute rejection and donor type were identified as significant predictors of log10eGFR at 5-years. These variables explained 21% of the variance in eGFR at 5 years post-transplant.
Recipient age at time of transplantation, mycophenolate mofetil exposure and acute rejection were identified as significant predictors of ∆ eGFR. These variables explained 4% of the variance in delta eGFR. These clinical predictors of eGFR were brought forward for the following GWAS and polygenic risk score analyses.
3.2. Genome-wide association study
To test the hypothesis that, in a univariate model, common donor or recipient genotype is associated with graft function, imputed genotype data was tested against log10 eGFR at 1-year and 5-years post-transplant and ∆ eGFR between 1 and 5 years taking a GWAS approach.
No genome-wide significant signals were detected in the donor or recipient GWAS in either the baseline or full model for log10 eGFR at 1-year or 5-year post-transplant or ∆ eGFR (see Figure 1 and Supplementary Figures 5–16). The top ten most significant variants for each GWAS are described in Supplementary Tables 8–13. The genomic inflation for each of the GWASs was minimal and the GWASs appeared to behave normally when the expected versus observed p-values were plotted (see Supplementary Figures 5–16).
Figure 1. Manhattan plots of recipient and donor log10 1-year, log10 5-year and Δ eGFR – full model.

The red line indicates the genome-wide significance threshold (5×10−8), the blue line indicates suggestive significance threshold (1×10−5). A = recipient log10 1-year eGFR – full model (λ (genomic inflation factor) = 1.01). The top SNP was an intergenic variant on chr17p13.3. N SNPs = 3,673,881. B= donor log10 1-year eGFR – full model (λ = 1.01). N SNPs = 3,641,041. The most significant SNP was found in an intergenic region on chr12p13.1. C= recipient log10 5-year eGFR – full model (λ = 0.99). The top SNP was an intergenic variant on chr17q22. N SNPs = 3,924,633. D= donor log10 5-year eGFR – full model (λ = 1.01). N SNPs = 3,938,549. The most significant SNP was found on chromosome 8 in an intron of CSMD1. E= recipient Δ eGFR – full model (λ = 1.01). The top SNP was found in the gene ZNF551 on chromosome 19. N SNPs = 3,915,961. F= donor delta eGFR – full model (λ = 1.01). N SNPs = 3,927,634. The most significant SNP was found on chromosome 22 in an intron of OSBP2.
3.3. Polygenic risk analysis
Having assessed the donor and recipient genotype in a univariate model, we next examined common genetic variation in a multivariate polygenic risk model. Using linear regression models, we tested the hypothesis that a polygenic load for increased eGFR (calculated using alleles from non-transplant populations, see methods section 2.5) in the kidney donor, recipient or combined (donor and recipient) genotype, is associated with increased post-transplant eGFR (see Table 3). The PRS at pT 0.0001 calculated using the recipient genotype and the combined donor-recipient genotype significantly predicted log10 eGFR at 1-year post-transplant (see Figure 2 and 3). These figures illustrate that the effect size was consistent across sites with an increased PRS leading to a higher eGFR post-transplant - i.e. increased number of alleles that predict higher eGFR in non-transplant populations correlates with higher eGFR post-transplant.
Table 3.
Most significantly associated polygenic risk scores with eGFR post-transplant
| D/R | pT | eGFR | Estimate | SE | Pun | Padj | I2 | N | N SNPs |
|---|---|---|---|---|---|---|---|---|---|
| combined | 0.0001 | 1year | 0.011 | 0.003 | 4.35×10−5 | 0.001 | 0.0% | 3234 | 6229 |
| recipient | 0.0001 | 1year | 0.008 | 0.002 | 8.68×10−5 | 0.005 | 6.2% | 5295 | 6229 |
| donor | 0.0001 | 1year | 0.006 | 0.003 | 0.01 | 0.755 | 2.7% | 3564 | 6229 |
| donor | 0.1 | 5year | 0.011 | 0.005 | 0.05 | 1 | 35.0% | 2152 | 287016 |
| recipient | 0.0001 | 5year | 0.008 | 0.004 | 0.06 | 1 | 0.0% | 2494 | 6229 |
| combined | 0.0001 | 5year | 0.008 | 0.005 | 0.11 | 1 | 0.0% | 1930 | 6229 |
| recipient | 0.0001 | ∆ | 0.668 | 0.367 | 0.07 | 1 | 0.0% | 2191 | 6229 |
| donor | 0.001 | ∆ | -0.348 | 0.358 | 0.33 | 1 | 0.0% | 1904 | 14097 |
| combined | 0.001 | ∆ | -0.513 | 0.413 | 0.21 | 1 | 0.0% | 1722 | 14097 |
Meta-analysis results for linear regression of polygenic risk scores vs. eGFR measures. 1/5 year = log10 eGFR at 1/5 year post-transplant. ∆ = change in eGFR between 1 and 5 years post-transplant. Model was adjusted for all available significant clinical covariates and the first eight principal components at each site (see section 2.5 and 3.1). D/R = indicates whether the test was carried out on the recipient, donor or combined donor-recipient genotype. pT = p-value threshold of the calculated polygenic risk score. Estimate = estimated effect size. SE = standard error of the estimate. Pun = uncorrected p-value. Padj=approximate adjusted p-value. I² = the proportion of total variation in study estimates that is due to heterogeneity. N = number of individuals tested. N SNPs = number of SNPs at the given p-value threshold (prior to pruning for linkage disequilibrium).
Figure 2. Recipient PRS at pT 0.0001 as a predictor of log10 eGFR at 1-year post-transplant.

Site = study cohort. TxLines= TransplantLines. Vienna = Vienna/Prague cohort. Weight= proportion of data the given site contributed to overall model; coef [95% CI] = effect size with lower and upper 95% confidence intervals. PRS_pT_0.0001 = normalized recipient PRS of eGFR at p-value threshold 0.0001, RE model = Random-effects model. Model was adjusted for significant clinical covariates and the first eight principal components at each site (see section 3.1).
Figure 3. Combined PRS at pT 0.0001 as a predictor of log10 eGFR at 1-year post-transplant.
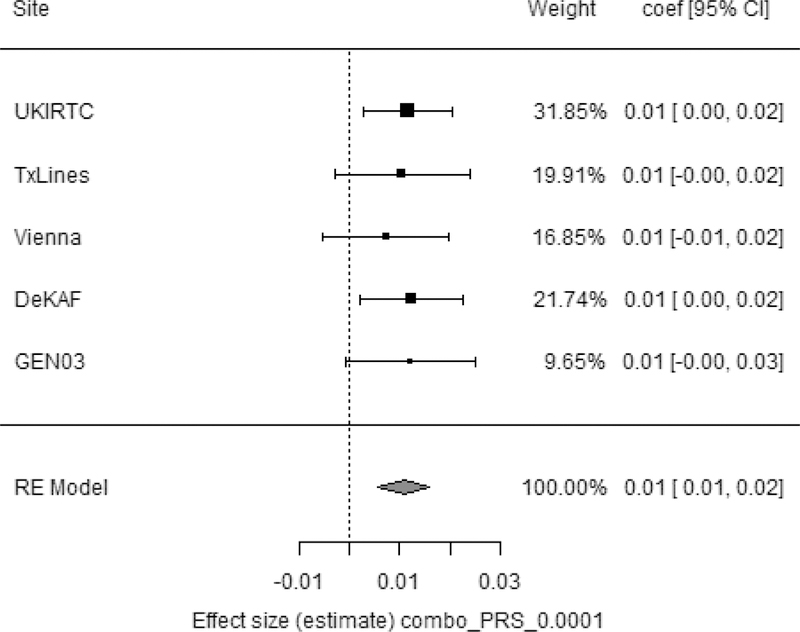
Site = study cohort. TxLines= TransplantLines. Vienna = Vienna/Prague cohort. Weight= proportion of data the given site contributed to overall model; coef [95% CI] = effect size with lower and upper 95% confidence intervals. Combo_PRS_pT_0.0001 = normalized combined PRS of eGFR at p-value threshold 0.0001. RE model = Random-effects model. The model was adjusted for significant clinical covariates and the first eight principal components at each site (see section 3.1).
The amount of variance explained by the full model (significant clinical covariates including weights for death/failure, principal components and polygenic risk score at pT 0.0001) for the combined donor-recipient PRS was 32% with 0.3% contributed by the PRS. The amount of variance explained by the full model for the recipient PRS was 30% with 0.2% contributed by the PRS (see Supplementary Table 14). Notably, approximately 9% of the variance explained by clinical variables is attributed to the death/failure weights (see Supplementary Table 14).
None of the PRS (at any pT) significantly predicted log10 eGFR 5 years or ∆ eGFR when calculated using the donor, recipient, or combined (sum of the donor and recipient alleles) genotype. Also, none of the PRS calculated using solely the donor alleles were significant predictors of any of the outcomes tested. Low heterogeneity was found across sites (see Table 3).
4. Discussion
In this study, we set out to test the impact of clinical variables and common genetic variation from both the donor and recipient on eGFR post-transplant using both univariate and polygenic methods. Although both clinical variables and polygenic risk scores were found to be predictive of eGFR post-transplant, clinical variables explained several orders of magnitude more of trait variance.
We identified a number of significant clinical predictors of eGFR at 1 and 5 years including donor age, donor type (living/deceased), donor sex, recipient age, delayed graft function, acute rejection and mycophenolate mofetil exposure. Collectively, these variables explained over 20% of the variance in eGFR at 1 and 5 years post-transplant. We also found that recipient age at time of transplantation, mycophenolate mofetil exposure and acute rejection predicted change in renal function over the first 5 years post-transplant. These findings and the direction of effect of these variables are in line with the literature and have all been previously implicated in eGFR post-transplant22–24. Notably, mismatches at HLA A, B and DR and the total number of mismatches across these three loci (calculated using serological testing) were not found to be significant in our clinical analysis (see Table 2). This likely due to advancements in immunosuppression as well as most donors and recipients being matched based on preferential HLA typing, which in combination are masking the effects of the HLA.
We did not find any donor or recipient SNP that associated (post-correction for multiple testing) with eGFR at 1 year, 5 years or ∆ eGFR post-renal transplantation. However, in the top SNPs from a number of the GWAS there were some interesting and potentially biologically relevant signals – although we stress these were not significant and so their role in graft function remains uncertain. For example, in our donor log10 5-year eGFR full model GWAS, the most significant SNP was found in the fifth intron of the Cub and Sushi Multiple domains 1 gene (CSMD1). CSMD1 has been implicated in a variety of diseases including schizophrenia and colorectal cancer25,26. CSMD1 has also been proposed as a regulator of the complement pathway – a pathway essential for inflammation and immune regulation27. Over the past decade, evidence has emerged which implicates the complement pathway in allograft ischemia-reperfusion as well as alloimmunity that results in graft injury thereby affecting the life-span of the graft 28. Further work is required to decipher if common variants in this gene play a role in allograft function.
For our donor GWAS of delta eGFR, in both the baseline and full models, the most significantly associated variant with delta eGFR was rs136237 (full model p= 7.89×10−7, baseline model p = 7.04×10−7), a SNP in the third intron of the oxysterol-binding protein 2 gene (OSBP2). This gene encodes a protein that binds some oxygenated forms of cholesterol called oxysterols and inhibits their functions 29. Oxysterols are involved in a vast range of important biological processes including apoptosis and platelet aggregation 30. OSBP2 is expressed at low levels in the kidney31,32. Further work is required to validate this finding.
Despite some interesting signals towards the tail of the distribution, none reached statistical significance and so our findings indicate that no single common genetic variant, in either the donor or recipient genome, explains a clinically relevant proportion (>2%) of the variation in eGFR post-kidney transplantation. It is probable that there are SNPs explaining a smaller proportion of eGFR post-transplant, but we were underpowered to detect these under a univariate model. This study focused specifically on common genetic variation, it is possible that rare variation in the donor and/or recipient genotype is influencing allograft function, but further work is required to clarify this.
We demonstrated that both recipient PRS and our combined PRS model significantly associated with log10 eGFR at 1-year post-transplant. We found that the recipient and combined PRS at pT 0.0001 was significantly associated with log10 1-year eGFR, indicating that common sub-genome-wide significance threshold (5×10−8) genetic variants influence graft outcome. This PRS was not found to associate with ∆ eGFR or log10 5-year GFR. This may indicate that the genetic variation that influences graft outcome is different for short-term outcome than that for medium-term or long-term graft function. This is consistent with our clinical findings, where concordance between the clinical variables that predict 1-year eGFR and 5-year eGFR is incomplete, indicating that different factors affect early stage vs medium stage graft function. However, we had more samples in our 1-year eGFR, than in our 5-year eGFR analysis and so potentially with larger numbers in the 5-year analysis this PRS may become significant as the same effect size and direction was seen.
Approximately 30% of the variance in log10 1-year eGFR was explained by our full model (clinical covariates including weights for death/graft-failure, PC and PRS) in both the recipient PRS and combined PRS analysis. The majority of this variation was explained by the clinical covariates and less than ~ 0.3% of the variance is explained by either PRS. Interestingly, Gorski et al12 found that a PRS based on eGFR associated variants calculated at the same p-value threshold (pT 0.0001) explained 1.7% of the outcome variance in non-transplant populations indicating that although the genetic basis for GFR post-transplant overlaps with that in non-transplant individuals, there are differences. However, our PRS was based off a different set of GWAS results13 than that in Gorski et al and their paper used a different method for calculating the PRS which may account for differences between the variance explained. The PRS, albeit not clinically relevant, does highlight that the recipient genotype, as well as the donor genotype, is associated with early-stage graft function. Notably, the donor genotype is only significant in the context of the recipient genotype (i.e. in our combined model).
Our study had a number of limitations. Firstly, although we had relatively comprehensive phenotypic data and corrected for clinical covariates, we likely did not have sufficient data to account for all clinical heterogeneity within and between our cohorts. In particular, our cohorts had different eras of transplantations and therefore different immunosuppression protocols which may have led to additional heterogeneity between cohorts. Potentially, through analysis of more similar cohorts we may have had more significant findings. However, in the PRS analyses, we did test for heterogeneity between our cohorts and found it to be minimal and any SNPs with high levels of heterogeneity in our GWAS were removed.
Secondly, these meta-analyses were carried out on European ancestry populations and therefore further work is needed to investigate single variant and polygenic effects in other non-European populations.
In conclusion, we found that polygenic effects of common genetics variants influence short-term allograft function but did not find any significant associations in our univariate model. This study is the first of its kind to look at the impact of polygenic effects of variants that impact kidney function in the general population in a post-transplant context. Our finding suggests that although common genetic variation does impact graft outcome, the effect size is limited compared to clinical variables.
Supplementary Material
Acknowledgments
We would like to thank the patients who contributed their DNA and phenotype data, without whom these analyses would not have been possible. C.P.S is supported by the Irish Research Council and Punchestown Kidney Research Fund (grant number EPSPG2015). P.J.P. is supported by an NRS Career Research Fellowship. Further thanks to funding support from Northern Ireland Kidney Research Fund and SFI-DfE (15/IA/3152). TransplantLines is registered at ClinicalTrials.gov with Identifier NCT03272841. The DEKAF and GEN03 studies are supported by 5U19-AI070119 & 5U01-AI058013 from NIAID.
UK and Ireland Renal Transplant Consortium investigators:
Maria P. Hernandez‐Fuentes, King’s College London, MRC Centre for Transplantation, London, UK, UCB Celltech, Slough, UK; Christopher Franklin, Welcome Trust Sanger Institute, Human Genetics, Cambridge, UK Irene Rebollo‐Mesa, King’s College London, MRC Centre for Transplantation, London, UK, Jennifer Mollon, King’s College London, MRC Centre for Transplantation, London, UK, Department of Haematology, University of Cambridge, Cambridge, UK; Florence Delaney, King’s College London, MRC Centre for Transplantation, London, UK, NIHR Biomedical Research Centre at Guy’s and St Thomas’, NHS Foundation Trust and King’s College London, London, UK; Esperanza Perucha, King’s College London, MRC Centre for Transplantation, London, UK, Caragh Stapleton, Royal College of Surgeons in Ireland, Dublin, Ireland; Richard Borrows, Renal Institute of Birmingham, Department of Nephrology and Transplantation, Birmingham, UK; Catherine Byrne, Nottingham Renal and Transplant Unit, Nottingham University Hospitals NHS Trust, Nottingham, UK; Gianpiero Cavalleri, Royal College of Surgeons in Ireland, Dublin, Ireland; Brendan Clarke, Transplant and Cellular Immunology, Leeds Teaching Hospitals NHS Trust, Leeds, UK; Menna Clatworthy, Department of Medicine, University of Cambridge, Cambridge, UK; John Feehally, Leicester General Hospital, Leicester, UK; Susan Fuggle, Transplant Immunology & Immunogenetics, Churchill Hospital, Oxford, UK; Sarah A. Gagliano, Center for Statistical Genetics, Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA; Sian Griffin, Cardiff & Vale University Health Board, Cardiff University, Cardiff, UK; Abdul Hammad, The Royal Liverpool and Broadgreen University Hospitals, Liverpool, UK; Robert Higgins, University Hospitals Coventry and Warwickshire NHS Trust, Coventry, UK; Alan Jardine, School of Medicine, Dentistry and Nursing, University of Glasgow, Glasgow, UK; Mary Keogan, Beaumont Hospital, Dublin, Ireland; Timothy Leach, Queen Alexandra Hospital, Portsmouth, UK; Iain MacPhee, St Georges’ Hospital NHS Trust, London, UK; Patrick B. Mark, School of Medicine, Dentistry and Nursing, University of Glasgow, Glasgow, UK; James Marsh, Epsom and St Helier University Hospitals Trust, Carshalton, UK; Peter Maxwell, School of Medicine, Dentistry and Biomedical Sciences, Queens University Belfast, Belfast, UK; William McKane, Sheffield Kidney Institute, Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, UK; Adam McLean, Kidney and Transplant, Imperial College Healthcare NHS Trust, London, UK; Charles Newstead, Leeds Teaching Hospitals NHS Trust, Leeds, UK; Titus Augustine, Central Manchester University Hospitals NHS Trust, Manchester, UK; Paul Phelan, NHS Lothian, Edinburgh, UK; Steve Powis, Division of Medicine, University College London, London, UK; Peter Rowe, Plymouth Hospitals NHS Trust, Plymouth, UK; Neil Sheerin, The Medical School, Newcastle University Newcastle, Newcastle upon Tyne, UK; Ellen Solomon, Division of Genetics& Molecular Medicine, King’s College London, London, UK; Henry Stephens, NHS Lothian, Edinburgh, UK; Raj Thuraisingham, Barts Health NHS Trust, London, UK; Richard Trembath, Division of Genetics& Molecular Medicine, King’s College London, London, UK; Peter Topham, Leicester General Hospital, Leicester, UK; Robert Vaughan, Clinical Transplantation Laboratory at Guy’s Hospital, Guy’s and St Thomas’ NHS Trust, London, UK; Steven H. Sacks, King’s College London, MRC Centre for Transplantation, London, UK, NIHR Biomedical Research Centre at Guy’s and St Thomas’, NHS Foundation Trust and King’s College London, London, UK; Peter Conlon, Royal College of Surgeons in Ireland, Dublin, Ireland, Beaumont Hospital, Dublin, Ireland; Gerhard Opelz, University of Heidelberg, Transplantation Immunology, Heidelberg, Germany; Nicole Soranzo, Welcome Trust Sanger Institute, Human Genetics, Cambridge, UK, Department of Haematology, University of Cambridge, Cambridge, UK; Michael E. Weale, Division of Genetics& Molecular Medicine, King’s College London, London, UK, Genomics plc, Oxford, UK; Graham M. Lord, King’s College London, MRC Centre for Transplantation, London, UK, NIHR Biomedical Research Centre at Guy’s and St Thomas’, NHS Foundation Trust and King’s College London, London, UK.
Deterioration of Kidney Allograft Function Genomics and Genomics of Kidney Transplantation investigators:
Arthur Matas, MD; J. Michael Cecka, MD; John Connett, PhD, Division of Biostatistics, University of Minnesota, Minneapolis, MN; Fernando G. Cosio, MD, Division of Nephrology, Mayo Clinic, Rochester, MN; Robert Gaston, MD, Division of Nephrology, University of Alabama, Division of Nephrology, Birmingham, AL; Rosalyn Mannon, MD, Division of Nephrology, University of Alabama, Division of Nephrology, Birmingham, AL; Sita Gourishankar,MD, Division of Nephrology and Immunology, University of Alberta, Edmonton, Alberta, Canada; Joseph P. Grande, MD, PhD, Mayo Clinic College of Medicine, Rochester, MN; Lawrence Hunsicker, MD, Nephrology Division, Iowa City, IA; Bertram Kasiske, MD, Division of Nephrology, Hennepin County Medical Center, Minneapolis, MN; and David Rush, MD, Health Sciences Center, Winnipeg MB.
Abbreviations
- CSMD1
Cub and Sushi Multiple domains 1 gene
- DeKAF
Deterioration of Kidney Allograft Function Genomics
- eGFR
Estimated glomerular filtration rate
- iGeneTRAiN
Genetics & Translational Research in Transplantation Network
- GWAS
Genome-wide association studies
- GEN03
Genomics of Kidney Transplantation
- HGDP
Human Genome Diversity project
- OSBP2
Oxysterol-binding protein 2 gene
- PRS
Polygenic risk scores
- PC
Principal component
- PCA
Principal components analysis
- UKIRTC
UK and Ireland Renal Transplant Consortium
Footnotes
Consortium members listed in the acknowledgements section the paper along with their affiliations
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of this article.
References
- 1.Thorsby E A short history of HLA. Tissue antigens 2009;74(2):101–116. [DOI] [PubMed] [Google Scholar]
- 2.Israni AK, Li N, Sidhwani S, et al. Association of hypertension genotypes and decline in renal function after kidney transplantation. Transplantation 2007;84(10):1240–1247. [DOI] [PubMed] [Google Scholar]
- 3.Gunesacar R, Opelz G, Erken E, et al. VEGF 936 C/T gene polymorphism in renal transplant recipients: association of the T allele with good graft outcome. Human immunology 2007;68(7):599–602. [DOI] [PubMed] [Google Scholar]
- 4.Lavin PJ, Laing ME, O’Kelly P, et al. Improved renal allograft survival with vitamin D receptor polymorphism. Renal failure 2007;29(7):785–789. [DOI] [PubMed] [Google Scholar]
- 5.Stapleton CP, Conlon PJ, Phelan PJ. Using omics to explore complications of kidney transplantation. Transpl Int 2018;31(3):251–262. [DOI] [PubMed] [Google Scholar]
- 6.O’Brien RP, Phelan PJ, Conroy J, et al. A genome-wide association study of recipient genotype and medium-term kidney allograft function. Clinical transplantation 2013;27(3):379–387. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez-Fuentes MP, Franklin C, Rebollo-Mesa I, et al. Long- and short-term outcomes in renal allografts with deceased donors: A large recipient and donor genome-wide association study. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2018. [DOI] [PMC free article] [PubMed]
- 8.Ghisdal L, Baron C, Lebranchu Y, et al. Genome-Wide Association Study of Acute Renal Graft Rejection. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2017;17(1):201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pihlstrom HK, Mjoen G, Mucha S, et al. Single Nucleotide Polymorphisms and Long-Term Clinical Outcome in Renal Transplant Patients: A Validation Study. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2017;17(2):528–533. [DOI] [PubMed] [Google Scholar]
- 10.International G, Translational Research in Transplantation N. Design and Implementation of the International Genetics and Translational Research in Transplantation Network. Transplantation 2015;99(11):2401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez-Fuentes M, Stapleton CP, Cavalleri GL, et al. The genetic determinants of renal allograft rejection. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2018;18(8):2100–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorski M, van der Most PJ, Teumer A, et al. 1000 Genomes-based meta-analysis identifies 10 novel loci for kidney function. Scientific reports 2017;7:45040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pattaro C, Teumer A, Gorski M, et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nature communications 2016;7:10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kottgen A, Pattaro C, Boger CA, et al. New loci associated with kidney function and chronic kidney disease. Nature genetics 2010;42(5):376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, Li Y, Weeks O, et al. SOS2 and ACP1 Loci Identified through Large-Scale Exome Chip Analysis Regulate Kidney Development and Function. Journal of the American Society of Nephrology : JASN 2017;28(3):981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature genetics 2013;45(12):1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518(7538):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boger CA, Gorski M, Li M, et al. Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD. PLoS genetics 2011;7(9):e1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Schizophrenia C, Purcell SM, Wray NR, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460(7256):748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward J, Graham N, Strawbridge RJ, et al. Polygenic risk scores for major depressive disorder and neuroticism as predictors of antidepressant response: Meta-analysis of three treatment cohorts. PloS one 2018;13(9):e0203896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.First MR. Renal function as a predictor of long-term graft survival in renal transplant patients. Nephrol Dial Transplant 2003;18 Suppl 1:i3–6. [DOI] [PubMed] [Google Scholar]
- 23.Salvadori M, Rosati A, Bock A, et al. Estimated one-year glomerular filtration rate is the best predictor of long-term graft function following renal transplant. Transplantation 2006;81(2):202–206. [DOI] [PubMed] [Google Scholar]
- 24.Siddiqi N, McBride MA, Hariharan S. Similar risk profiles for post-transplant renal dysfunction and long-term graft failure: UNOS/OPTN database analysis. Kidney Int 2004;65(5):1906–1913. [DOI] [PubMed] [Google Scholar]
- 25.Havik B, Le Hellard S, Rietschel M, et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol Psychiatry 2011;70(1):35–42. [DOI] [PubMed] [Google Scholar]
- 26.Zhang R, Song C. Loss of CSMD1 or 2 may contribute to the poor prognosis of colorectal cancer patients. Tumour Biol 2014;35(5):4419–4423. [DOI] [PubMed] [Google Scholar]
- 27.Kraus DM, Elliott GS, Chute H, et al. CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J Immunol 2006;176(7):4419–4430. [DOI] [PubMed] [Google Scholar]
- 28.Cravedi P, Heeger PS. Complement as a multifaceted modulator of kidney transplant injury. J Clin Invest 2014;124(6):2348–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreira EF, Jaworski C, Li A, Rodriguez IR. Molecular and biochemical characterization of a novel oxysterol-binding protein (OSBP2) highly expressed in retina. J Biol Chem 2001;276(21):18570–18578. [DOI] [PubMed] [Google Scholar]
- 30.Schroepfer GJ Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev 2000;80(1):361–554. [DOI] [PubMed] [Google Scholar]
- 31.Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 32.The Human Protein Atlas. www.proteinatlas.org. Accessed 12-07-18.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.