

The bacteria occupying the mammalian gut have evolved unique strategies to thrive in their environment. Bacteroides organisms, which often comprise 25 to 50% of the human gut microbiota, derive nutrients from structurally diverse complex polysaccharides, commonly called dietary fibers. This ability requires an expansive genetic repertoire that is coordinately regulated to achieve expression of those genes dedicated to utilizing only those dietary fibers present in the environment. Here we identify the global regulon of a transcriptional regulator necessary for dietary fiber utilization and gut colonization. We demonstrate that this transcription factor regulates hundreds of genes putatively involved in dietary fiber utilization as well as a putative translation factor dispensable for growth on such nutrients but necessary for survival in the gut. These findings suggest that gut bacteria coordinate cellular metabolism with protein synthesis via specialized translation factors to promote survival in the mammalian gut.
KEYWORDS: BT4338, EF-G2, fusA2, microbiota
ABSTRACT
Microbial colonization of the mammalian gut is largely ascribed to the ability to utilize nutrients available in that environment. To understand how beneficial microbes establish a relationship with their hosts, it is crucial to determine what other abilities promote gut colonization. We now report that colonization of the murine gut by the beneficial microbe Bacteroides thetaiotaomicron requires activation of a putative translation factor by the major transcriptional regulator of gut colonization and carbohydrate utilization. To ascertain how this regulator—called BT4338—promotes gut colonization, we identified BT4338-regulated genes and BT4338-bound DNA sequences. Unexpectedly, the gene whose expression was most reduced upon BT4338 inactivation was fusA2, specifying a putative translation factor. We determined that fusA2 activation by BT4338 is conserved in another Bacteroides species and essential for gut colonization in B. thetaiotaomicron because a mutant lacking the BT4338 binding site in the fusA2 promoter exhibited a colonization defect similar to that of a mutant lacking the fusA2 gene. Furthermore, we demonstrated that BT4338 promotes gut colonization independently of its role in carbohydrate utilization because the fusA2 gene was dispensable for utilization of carbohydrates that depend on BT4338. Our findings suggest that microbial gut colonization requires the use of alternative protein synthesis factors.
INTRODUCTION
The gut microbiota is a key contributor to human health and development. Symbiotic microbes in the gut benefit their hosts by supplying essential nutrients, facilitating energy extraction from food, hindering pathogen invasion, and shaping host immunity (1–4). The host’s diet controls the composition of the gut microbiota by providing nutrients utilized only by those microbiota members that can access them (5, 6). In addition, dietary components can serve as signaling molecules that regulate the production of specific colonization factors in certain gut commensal species (7). Therefore, the host diet governs the composition of the microbiota by promoting or inhibiting the expansion of specific microbial species in the gut.
Typically associated with healthy individuals, Bacteroides thetaiotaomicron is a gut microbe that devotes ∼18% of its genes to the acquisition and utilization of a wide variety of carbohydrates (8). One of these genes specifies a transcription factor—named BT4338—that is required both for the utilization of multiple polysaccharides and their monosaccharide constituents (9) and for murine gut colonization (10, 11). BT4338 is proposed to bind a DNA sequence present in the putative promoter regions of many genes mediating carbohydrate utilization (9, 12). While these results imply that BT4338 promotes gut colonization by enabling the utilization of specific dietary polysaccharides, a mutant lacking the BT4338 gene exhibited a similar gut colonization defect regardless of dietary polysaccharides present in the host diet (10). Therefore, BT4338 may control processes necessary for gut colonization but dispensable for carbohydrate utilization.
Protein synthesis requires ribosomes, amino acids, tRNAs, GTP, and several proteins that play distinct roles in the initiation, elongation, and termination of translation as well as the recycling of ribosomes. Elongation factor G (EF-G) is an indispensable GTPase that functions in two different phases of protein synthesis in Escherichia coli and other studied bacteria (13). During translation elongation, EF-G promotes translocation—the movement of the tRNA-peptidyl-tRNA-mRNA complex relative to the ribosome—after a peptide bond has been formed, making the A-site in the ribosome available for decoding the next codon. During ribosome recycling, EF-G works together with the ribosome recycling factor (14, 15) to catalyze the rapid dissociation of the ribosomal post-termination complex into subunits. Some bacterial species, such as E. coli, harbor a single EF-G-specifying gene (designated fusA), while others, such as B. thetaiotaomicron, harbor two EF-G-specifying genes (fusA and fusA2). The deduced amino acid sequences of the B. thetaiotaomicron fusA (BT2729) and fusA2 (BT2167) genes are 57% and 34% identical, respectively, to the E. coli EF-G protein. Whereas fusA is putatively essential, fusA2 is not (11). However, fusA2 inactivation compromises gut colonization in multiple Bacteroides species (10).
We now report that the master regulator of carbohydrate utilization in B. thetaiotaomicon—termed BT4338—controls gut colonization independently of its ability to govern carbohydrate utilization. We identify both the transcriptional profiles of isogenic wild-type and BT4338-deficient B. thetaiotaomicron and the sequences bound by the BT4338 protein in vivo genome-wide. We determine that BT4338 binds to promoters and regulates the transcription of genes involved in carbohydrate utilization, including one encoding a protein that promotes immunological tolerance in the murine gut. Surprisingly, the mRNA most highly activated by BT4338 corresponds to the fusA2 gene. We demonstrate that direct transcriptional activation of the fusA2 gene by BT4338 is essential for gut colonization despite fusA2 being dispensable for in vitro growth on all investigated BT4338-dependent nutrients. In addition, conditions that increase fusA2 mRNA abundance >200-fold result in a 10-fold decrease in fusA mRNA abundance. The transcriptional activation of fusA2 by BT4338 is conserved in other Bacteroides species, suggesting that gut colonization requires specific translation factors.
RESULTS
Carbon limitation promotes transcription of the BT4338-activated gene araM.
The BT4338 gene is required for expression of the arabinose utilization gene araM (BT0356) in B. thetaiotaomicron cultures supplied with arabinose and for growth on arabinose as the sole carbon source (9). Curiously, araM is also transcriptionally activated following a shift from growth on glucose to carbon limitation (9) (i.e., in the absence of arabinose), indicating that arabinose is not essential for araM transcription.
We have now determined that the mRNA abundance of the araM gene increases 18.1- and 5.6-fold following 10- and 60-min exposures of wild-type B. thetaiotaomicron to carbon limitation, respectively (Fig. 1). By contrast, the araM mRNA abundance in the BT4338 mutant increased <2-fold after 10 min and decreased after 60 min (Fig. 1). These results indicate that BT4338 is necessary for transient transcriptional activation of the araM gene in cells experiencing carbon limitation.
FIG 1.
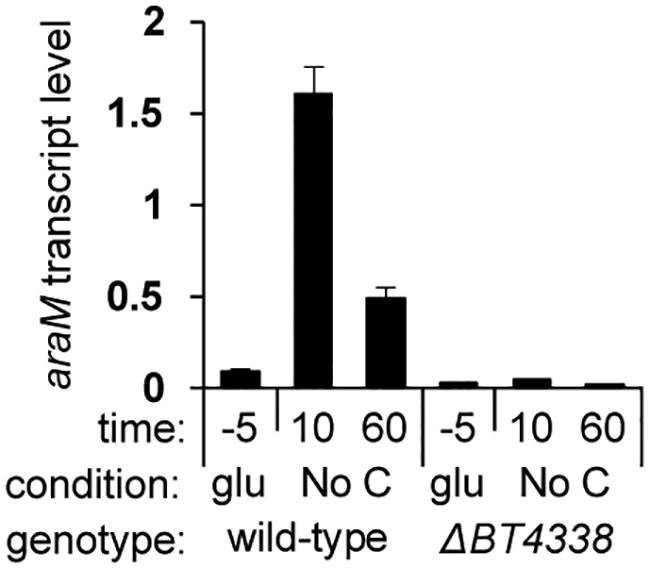
BT4338 transiently activates araM transcription during carbon limitation. Shown are araM mRNA abundances relative to those of 16S rRNA in wild-type (GT23) and BT4338 mutant (NS364) B. thetaiotaomicron during mid-log growth in minimal medium containing glucose as the sole carbon source (glu) and 10 and 60 min following a switch to carbon limitation conditions (No C). Measurements were carried out by qPCR and represent averages of 6 independent biological replicates; error bars represent SEM.
High-throughput RNA sequencing (RNA-seq) analysis identifies BT4338 as a global regulator.
We examined mRNA abundance genome-wide in wild-type and BT4338 mutant B. thetaiotaomicron at mid-logarithmic phase of growth on glucose (see Fig. S1A and Table S1 in the supplemental material) and after a 10-min exposure to carbon limitation (Fig. 2 and Table S2). During growth on glucose, the mRNA abundance of 11 genes was 5-fold lower, while that of 207 genes was 5-fold higher, in the BT4338 mutant than in the wild-type strain (Fig. 2A; adjusted P values [Padj] < 0.05; P values, determined by analysis of variance [ANOVA], were adjusted using the Benjamini-Hochberg procedure). The numbers of mRNAs whose abundances changed 2-fold are 225 and 832, respectively (Fig. S2B). For example, the mRNA abundance of BT2131 was 57-fold greater in the BT4338 mutant than in the wild-type strain and represents the greatest increase in transcription resulting from the loss of this transcription factor during growth in glucose (Fig. S1A and Table S1). Conversely, the BT4543 transcript was 20-fold lower in the BT4338 mutant than in the wild-type strain under this condition (Fig. S1A and Table S1). These results indicate that BT4338 is necessary for proper transcription of subsets of target genes during mid-exponential-phase growth in glucose, a condition in which the BT4338 mutant grows similarly to wild-type B. thetaiotaomicron (9).
FIG 2.

The BT4338 regulon in bacteria experiencing carbon limitation. Shown are the results of RNA-seq analysis of wild-type (GT23) and BT4338 mutant (NS364) B. thetaiotaomicron during growth in minimal medium containing glucose as the sole carbon source and at 10 min following a switch to carbon limitation conditions. (A) Venn diagrams representing genes whose corresponding mRNAs are >5-fold different between wild-type (GT23) and BT4338 mutant (NS364) B. thetaiotaomicron. (B) Volcano plot depicting the fold change versus the −log10 of the corresponding P value for all genes between wild-type (GT23) and BT4338 mutant (NS364) B. thetaiotaomicron switched from growth in glucose to carbon limitation for 10 min. Blue dots represent genes exhibiting a >2-fold decrease in expression in the BT4338 mutant compared to that in the wild type. Red dots represent genes exhibiting a >2-fold increase in expression in the BT4338 mutant compared to that in the wild type. Genes discussed in Results and Discussion are labeled. Data correspond to DESeq2 analysis of RNA-seq analysis of 3 biological replicates examined by featureCounts.
The BT4338 regulon during growth in glucose. (A) Volcano plot depicting the fold change versus the −log10 of the corresponding P value for all genes between wild-type (GT23) and BT4338-deficient (NS364) B. thetaiotaomicron strains during growth in glucose. Blue dots represent genes exhibiting a >2-fold decrease in expression in the BT4338 mutant compared to that in the wild type. Red dots represent genes exhibiting a >2-fold increase in expression in the BT4338 mutant compared to that in the wild type. Genes discussed in the Results and Discussion sections of the text are labeled. Data represent the averages of 3 biological replicates measured by RNA-seq. (B) Venn diagrams representing genes whose mRNAs are >2-fold different between wild-type (GT23) and BT4338-deficient (NS364) strains of B. thetaiotaomicron growing in mid-exponential phase in minimal medium containing glucose as a sole carbon source and at 10 min following a switch to carbon limitation conditions. Data represent the averages of 3 biological replicates measured by RNA-seq, with an adjusted P value of <0.05. Download FIG S1, PDF file, 0.2 MB (193.3KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Complementation of a BT4338-null mutant with a gene specifying an epitope-tagged BT4338 protein. (A to C) Growth (OD600) of wild-type B. thetaiotaomicron harboring an empty vector (GT1009; black lines) or a BT4338 mutant harboring either the empty vector (NS432; gray lines), a plasmid-encoded wild-type BT4338 (GT1498; blue lines), BT4338 with a C-terminal HA tag (NS433; orange lines), or BT4338 with a C-terminal HA epitope separated by a linker comprised of 4 glycine residues (GT1481; green lines). Bacteria were grown in minimal medium containing either arabinose (A), glucuronate (B), or xylose (C). (D) fusA2 mRNA abundances in strains GT1009, GT1498, GT1481, and NS432 during growth in glucose (glu, -5) and 10 or 60 min following a switch to carbon limitation conditions (No C). mRNA abundance was measured by qPCR. Data represent the averages of 4 biological replicates, and error bars represent SEM. Download FIG S2, PDF file, 0.3 MB (268.4KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes with significantly altered expression in a BT4338-deficient strain (NS364) relative to that in wild-type B. thetaiotaomicron (GT23) during mid-exponential-phase growth in minimal medium containing glucose. Download Table S1, XLSX file, 0.5 MB (558.1KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes with significantly altered expression in a BT4338-deficient strain (NS364) relative to that in wild-type B. thetaiotaomicron (GT23) following a 10-min exposure to carbon limitation. Download Table S2, XLSX file, 0.5 MB (553.8KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
After 10 min of carbon limitation, the mRNA abundance of 148 genes was 5-fold lower, while that of 322 genes was 5-fold higher, in the BT4338 mutant than in the wild-type strain (Fig. 2A; Padj values < 0.05; P values, determined by ANOVA, were adjusted using the Benjamini-Hochberg procedure). The numbers of mRNAs whose abundances changed 2-fold are 688 and 990, respectively (Fig. S1B). The mRNA abundance of certain genes changed >5-fold both during growth on glucose and upon carbon limitation: 8 genes were lower in the BT4338 mutant than in the wild-type strain, and 119 were higher in the BT4338 mutant than in the wild-type strain (Fig. 2A). Below we discuss some of the genes regulated by BT4338 during carbon limitation.
BT4338-regulated genes participating in carbohydrate utilization and central metabolism.
The BT4338 gene is required for growth on arabinose, fucose, glucuronate, and xylose (9). Moreover, the BT4338 protein was predicted to bind to the promoters of the genes involved in the utilization of these monosaccharides (9, 12). We have now established that when B. thetaiotaomicron experiences carbon limitation, the mRNA abundance of the genes in the following operons increase in a BT4338-dependent manner: the predicted BT0356-BT0350 operon, which encodes products corresponding to arabinose utilization genes; the BT1272-BT1277 operon, corresponding to fucose utilization genes; the BT1434-BT1432 operon, corresponding to glucuronate utilization genes; and the BT0791-BT0794 operon, corresponding to xylose utilization genes (Fig. 2B and Table S2). These increases took place despite the absence of the specific monosaccharides, the utilization of which requires the activated operons.
Expression of genes required for the utilization of particular polysaccharides is also BT4338 dependent during carbon limitation (Table S2). These genes include those in the BT2818-BT2825 locus (BT2818-BT2822 operon and BT2823, BT2824, and BT2825 genes), which is regulated by porcine mucin O-glycans (16); the BT0453-BT0449 and BT0439-BT0443 operons, which are induced in vivo by unknown substrates (16); and the BT4299-BT4295 operon, which is induced by N-acetyl-d-lactosamine (17). Several proteins are encoded in the BT4299-BT4295 operon, one of which is implicated in immune cell development (18).
Transcription of genes encoding products predicted to participate in central metabolism and bioenergetics requires BT4338 (Table S2). For example, the BT1450-BT1448 operon encodes a putative propionyl coenzyme A (propionyl-CoA) carboxylase beta-subunit, biotin carboxylase, and biotin carboxyl carrier protein. These proteins are predicted to form a multi-subunit complex that catalyzes the conversion of propionyl-CoA to d-methylmalonyl-CoA, which is necessary for multiple cellular functions, including acetyl-CoA assimilation in alphaproteobacteria (19), mycolic acid biosynthesis in Mycobacterium tuberculosis (20), and polyketide synthesis in Streptomyces species (21).
Additionally, the BT4338 mutant exhibited a 5-fold decrease in rnfABCDEG (BT0617-BT0622) mRNA abundance following carbon limitation (Fig. 2B and Table S2). The genes in this operon specify subunits of a protein complex that catalyzes the oxidation of reduced ferredoxin to NAD+, which generates an ion gradient across the inner membrane that is harnessed for cellular energy production (22, 23). This finding suggests that BT4338 controls the intracellular pools of NADH/NAD+, which are likely altered following a shift from rapid growth in glucose to carbon limitation conditions. In support of this notion, carbon limitation resulted in a 28-fold increase in the BT1554 mRNA amounts in a BT4338-dependent fashion (Table S2). The BT1554 gene encodes alanine dehydrogenase, an enzyme that couples the conversion of alanine into pyruvate with the reduction of NAD+ into NADH and serves to control the redox state in other bacterial species (24).
Cumulatively, these data indicate that B. thetaiotaomicron responds to carbon limitation by increasing transcription of determinants favoring energy production via the transcriptional regulator BT4338.
The BT4338 protein binds to the promoter regions of a subset of BT4338-activated genes.
To determine which of the genes differentially expressed in a BT4338-dependent manner following carbon limitation are direct targets of the BT4338 protein, we examined genome-wide occupancy by BT4338 using chromatin immunoprecipitation sequencing (ChIP-seq). Because antibodies directed against the BT4338 protein were not available, we engineered a strain that expressed a C-terminal epitope-tagged BT4338 protein from its normal promoter. This approach was successfully used with other DNA binding proteins (25, 26). The strain expressing the epitope-tagged BT4338 protein supported growth on three different BT4338-dependendent carbohydrates (Fig. S2A to C) and induced fusA2 expression upon carbon limitation (Fig. S2D). The engineered strain showed ChIP enrichment of the promoter regions of the araM and fusA2 genes during carbon limitation (Fig. 3A), which produced mRNAs in response to carbon limitation in a BT4338-dependent manner (Fig. 2B).
FIG 3.

Genome-wide DNA binding by the BT4338 protein. (A) Fold enrichment of the araM (BT0356) and fusA2 (BT2167) promoter regions during exponential-phase growth in minimal medium containing glucose and following a 10-min exposure to carbon limitation in a strain expressing an epitope-tagged BT4338 protein (GT1481). Measurements were carried out by ChIP-qPCR and represent averages of 4 independent biological replicates; error bars represent SEM. (B to H) BT4338 binding analyzed by high-throughput sequencing (ChIP-seq) upstream of the following genes: the arabinose utilization operon (BT0356 to BT0350) (B), fucose utilization genes (BT1272 to BT1277) (C), glucuronate utilization genes (BT1434 to BT1432) (D), xylose utilization genes (BT0791 to BT0794) (E), the alanine dehydrogenase gene (BT1554) (F), rnfABCDEF (G), and fusA2 (BT2167) (H). Black represents the input DNA, and red represents the immunoprecipitated DNA. Data represent the normalized abundances of reads mapped across the B. thetaiotaomicron genome and visualized in Artemis.
BT4338 bound to the promoter regions of the arabinose and fucose utilization genes and the alanine dehydrogenase gene (Fig. 3B to D). By contrast, BT4338 binding was not observed upstream of the glucuronate and xylose utilization genes (Fig. 3E and F), polysaccharide utilization genes, or the rnfABCDEG operon (Fig. 3G), even though they are all transcriptionally induced during carbon limitation in a BT4338-dependent fashion (Fig. 2B). These results suggest that BT4338 regulates the latter genes only indirectly, via another regulator. Alternatively, BT4338 binding to the latter genes may require additional signals or be compromised by the epitope at the C terminus of the BT4338 protein used in the ChIP-seq experiments, as this strain does not grow as well as the wild-type parent on xylose and glucuronate (Fig. S2B and C).
Our ChIP-seq analysis revealed that BT4338 binds to 834 locations in the B. thetaiotaomicron genome (Table S3A). These locations correspond to 8 intergenic peaks, 200 intragenic peaks, and 626 peaks that span both intergenic and intragenic regions. BT4338 binding is associated with changes in the mRNA abundances of 175 associated genes, corresponding to 35 BT4338-activated genes and 140 BT4338-repressed genes (Table S3B). Curiously, 184 of 200 intergenic sites, 5 of 8 intragenic sites, and 505 of 626 intra- and intergenic sites bound by BT4338 in vivo are not associated with changes in gene expression. (Note that one peak can be associated with multiple genes nearby.) This phenomenon has been reported for other DNA binding regulatory proteins in different bacterial species (27, 28).
Regions of the B. thetaiotaomicron genome bound by BT4338 following a 10-min exposure to carbon limitation. (A) All 834 regions bound by BT4338 in response to carbon limitation. (B) BT4338 binding corresponding to BT4338-dependent changes in gene expression during carbon limitation. Download Table S3, XLSX file, 0.4 MB (409.2KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
fusA2 is the BT4338-activated gene most highly induced upon carbon limitation.
The largest fold increase in mRNA abundance resulting from a switch from growth in glucose to carbon limitation in wild-type B. thetaiotaomicron (i.e., 238-fold) was in the fusA2 gene (BT2167) (Table S4 and Fig. S3A). fusA2 mRNA levels increased only 4.7-fold in the BT4338 mutant (Fig. 4A), while fusA mRNA amounts actually decreased 10-fold upon carbon limitation (Table S4 and Fig. S3A), and this decrease was independent of BT4338 (Fig. 4B). These results indicate that B. thetaiotaomicron coordinates the expression of genes specifying related translation elongation factors in response to carbon limitation.
FIG 4.

BT4338 directly controls fusA2 transcription in response to carbon limitation. Shown are fusA2 (A) and fusA (B) mRNAs in wild-type (GT23) and BT4338 mutant (NS364) B. thetaiotaomicron or a mutant lacking the putative BT4338 binding site in the fusA2 promoter (WH311) during growth in glucose and 10 min or 60 min following exposure to carbon limitation. mRNA abundance was determined by qPCR. Measurements are averages of 3 independent biological replicates; error bars represent SEM. A log10 scale is used for the y axis. (C) The fold enrichment versus the −log10 of the corresponding P value of all regions of the B. thetaiotaomicron chromosome bound by BT4338 following a switch to carbon limitation for 10 min. Enrichment was measured by ChIP-seq. Measurements were made using MACS2 callpeak from 3 independent biological replicates. (D) The DNA sequence upstream of the fusA2 coding region, with the putative BT4338 binding site highlighted in yellow, the putative −7 consensus sequence highlighted in green, and the start codon lettered in red. (E) Fold enrichment of the fusA2 and araM promoter regions with the BT4338 protein in B. thetaiotaomicron with a wild-type fusA2 promoter (wt; GT1481) or a strain lacking the 22-bp region (Δ22bp; WH335) highlighted in yellow in panel D. Measurements were carried out by qPCR of ChIP samples and represent averages of 4 independent biological replicates, and error bars represent SEM. (F) Western blot analysis of crude extracts prepared from the wild type (GT1301), a BT4338-deficient mutant (GT1308), and the Δ22bp mutant (WH389) during growth in glucose and 15 or 60 min following exposure to carbon limitation. The top blot was probed for the fusA2 gene product, EF-G2, and the bottom blot was probed for GroEL.
B. thetaiotaomicron genes regulated by carbon limitation. (A) Volcano plot depicting the fold change versus the −log10 of the corresponding P value for all genes in wild-type (GT23) B. thetaiotaomicron in carbon limitation conditions following growth in glucose (Table S4). Blue dots represent genes exhibiting a >2-fold decrease in expression during carbon limitation compared to growth in glucose. Red dots represent genes exhibiting a >2-fold increase in expression during carbon limitation compared to growth in glucose. fusA, fusA2, and araM are labeled. Data represent the averages of 3 biological replicates measured by RNA-seq. (B) fusA2 mRNA abundances in the wild type (GT23; gray bars) and a strain unable to synthesize (p)ppGpp0 (relA spoT; GT1181; purple bars) during growth in glucose (-5) and 10 or 60 min following a switch to carbon limitation conditions. mRNA abundance was measured by qPCR. Data represent the averages of 3 biological replicates, and error bars represent SEM. Download FIG S3, PDF file, 0.2 MB (191.8KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes with significantly altered expression in wild-type B. thetaiotaomicron (GT23) following a switch from exponential growth in glucose to carbon limitation conditions for 10 min. Download Table S4, XLSX file, 0.5 MB (567.2KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The BT4338 protein governs fusA2 transcription directly because of the following. First, the fusA2 promoter region was the most highly enriched DNA region in the ChIP-seq experiment (Table S3A and B, Fig. 3H, and Fig. 4C). Second, a 22-bp region upstream of fusA2 (CCTTATGTTACATAACATTGTT [Fig. 4D]) resembles the proposed consensus binding sequence for the BT4338 protein (wwwTATGTTnTAnAACATAwww), which is found upstream of many BT4338-activated genes (12). Third, removal of this sequence from the fusA2 promoter region prevented binding by the BT4338 protein to the fusA2 promoter, but not the araM promoter used as control, under carbon limitation conditions (Fig. 4E). And fourth, the mutant lacking the BT4338 binding site in the fusA2 promoter exhibited fusA2 mRNA amounts similar to those produced by the BT4338-null mutant under carbon limitation conditions (Fig. 4A). Thus, BT4338 activates fusA2 transcription by binding to the fusA2 promoter region.
To determine whether the BT4338 control of fusA2 mRNA abundance results in changes in EF-G2 protein amounts, we carried out Western blot analysis of crude extracts prepared from isogenic strains expressing an epitope-tagged version of the EF-G2 protein from the normal fusA2 promoter and chromosomal location. The EF-G2 protein abundance was very low during mid-exponential-phase growth in glucose but rapidly increased following carbon limitation (Fig. 4F). EF-G2 protein amounts did not increase in the BT4338-null mutant or in the mutant lacking the predicted BT4338 binding site in the fusA2 promoter (Fig. 4F).
Cumulatively, the results presented in this section demonstrate that the BT4338 protein directly activates transcription of the fusA2 gene when bacteria experience carbon limitation.
(p)ppGpp is dispensable for fusA2 transcriptional activation during carbon limitation.
We considered the possibility of the alarmone (p)ppGpp controlling fusA2 transcription because B. thetaiotaomicron produces (p)ppGpp when experiencing carbon limitation (29), and (p)ppGpp represses expression of components of the protein synthesis machinery under this condition (29). However, fusA2 transcription is (p)ppGpp independent because a mutant unable to make (p)ppGpp due to deletion of the BT0700 and BT3998 genes retained wild-type mRNA abundance of the fusA2 gene (Fig. S3B) (29). These experiments argue that the response to carbon limitation entails (p)ppGpp-dependent and -independent pathways.
BT4338-dependent transcription of the fusA2 gene is necessary for gut colonization.
The BT4338 and fusA2 genes are both highly conserved across the Bacteroidetes, required for gut colonization in three different Bacteroides species (10), and transcriptionally upregulated inside the murine gut relative to growth in laboratory media (29). To determine whether BT4338 promotes gut colonization by activating fusA2 transcription, we examined the abundance of barcoded wild-type B. thetaiotaomicron and mutants lacking either the BT4338 gene, the fusA2 gene, or the BT4338 binding site in the fusA2 promoter in germfree mice inoculated with nearly identical amounts of these strains.
Wild-type B. thetaiotaomicron outcompeted all three mutants and became predominant (over 90%) after a 1-week colonization (Fig. 5). These results argue that the colonization defects of the transposon-generated mutants of the BT4338 gene reported by others (10) are due to loss of function (rather than due to the truncated proteins encoded by the strain with a transposon insertion being toxic to the bacterium during gut colonization). The mutant lacking the BT4338 binding site in the fusA2 promoter was as defective for gut colonization as that lacking the fusA2 coding region (Fig. 5), indicating that BT4338 activation of fusA2 expression is essential for gut colonization. However, the latter two mutants were not as defective as the BT4338 mutant (Fig. 5), indicating that also BT4338 promotes gut colonization in a fusA2-independent manner.
FIG 5.
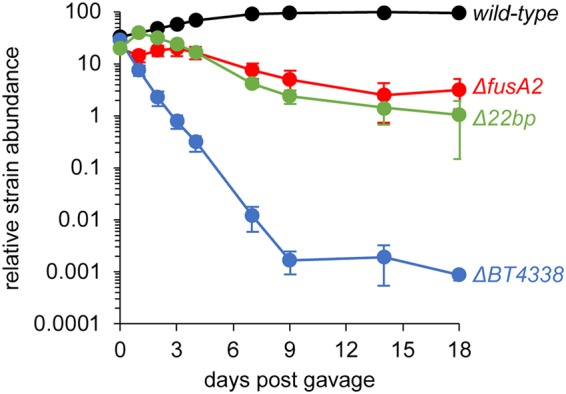
Transcriptional activation of the fusA2 gene by the BT4338 protein is required for colonization of the murine gut. Shown are the abundances of wild-type B. thetaiotaomicron (GT478), a BT4338-deficient strain (WH150), a fusA2-deficient strain (WH148), and a strain lacking the 22-bp BT4338 binding site in the fusA2 promoter (Δ22bp; WH324) at the indicated times after gavage of all four strains into germfree mice. The abundance of each strain was measured by qPCR of unique molecular barcodes. Data represent averages for 5 individual mice, and error bars represent SEM. A log10 scale is used for the y axis.
The fusA2 gene is dispensable for growth on BT4338-dependent carbon sources.
The B. thetaiotaomicron BT4338 gene is required for growth on multiple carbohydrates (9), likely due to its critical role in transcriptional activation of the corresponding genes (Tables S1 and S2). By contrast, the fusA2-null mutant retained a wild-type ability to grow on carbohydrates that require BT4338 for growth (Fig. S4). This is surprising because the BT4338 protein exhibited the strongest regulatory effect on the fusA2 gene (Table S2). These results argue that fusA2’s role in gut colonization is unrelated to utilization of the major carbohydrates that require BT4338 for growth in vitro.
fusA2 is dispensable for growth on BT4338-dependent carbon sources. (A to E) A wild-type B. thetaiotaomicron strain (GT23; black lines) and mutants lacking either BT4338 (NS364; blue lines) or fusA2 (GT1309; red lines) were grown in arabinose (A), xylose (B), fucose (C), glucuronate (D), or ribose (E) as a sole carbon source. Data represent the averages of 4 biological replicates and error bars represent SEM. Download FIG S4, PDF file, 0.2 MB (238.9KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
fusA2 regulation by BT4338 sequelogs is conserved among Bacteroides species.
The deduced amino acid sequences of the BT4338 and fusA2 genes are conserved in many Bacteroides species. For example, the BT4338 sequelog in Bacteroides ovatus (encoded by Bovatus_RS22425) is 96.1% identical to the BT4338 gene product and EF-G2 is 97.7% identical between these two species (BT2167 compared to Bovatus_RS10900). Moreover, the BT4338 and fusA2 genes are core fitness determinants necessary for gut colonization in three Bacteroides species (10). Thus, we wondered whether the transcriptional control of the fusA2 gene by BT4338 is conserved in other species, given that regulon compositions often differ across related bacterial species (30).
We determined that the abundance of the transcript corresponding to Bovatus_RS10900 increased 854-fold following carbon limitation in B. ovatus (Fig. 6). This increase is largely dependent on Bovatus_RS22425 because the Bovatus_RS10900 transcript increased only 5.2-fold in the Bovatus_RS22425-null mutant (Fig. 6). These results indicate that activation of the fusA2 gene by BT4338 in response to carbon limitation is conserved in B. ovatus.
FIG 6.
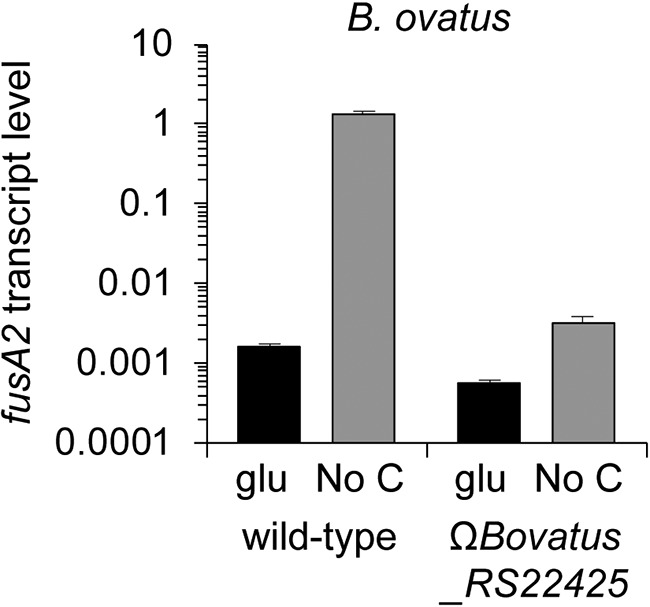
Transcriptional activation of the fusA2 gene by the BT4338 protein is conserved in Bacteroides ovatus. Shown are fusA2 (Bovatus_RS10900) mRNA abundances in wild-type B. ovatus (VR314) and a strain defective for the BT4338 sequelog (Bovatus_RS22425; WH275), during mid-log growth in minimal medium containing glucose as the sole carbon source and 10 min following a switch to carbon limitation conditions. mRNA abundance was determined by qPCR. Data represent the averages of 3 independent biological replicates, and error bars represent SEM. A log10 scale is used for the y axis.
DISCUSSION
We have now established that the master regulator of carbohydrate utilization in B. thetaiotaomicron controls gut colonization independently of its regulation of carbohydrate utilization genes. We identified the promoter region of the putative translation factor-encoding gene fusA2 as the DNA most highly bound by BT4338 (Fig. 4C) and the fusA2 mRNA as the transcript most highly activated by BT4338 (Fig. 2B and Table S2) and the most highly induced upon carbon limitation (Fig. S3A and Table S4). That fusA mRNA amounts decrease (Fig. 4B, Fig. S3A, and Table S4) concurrently with an increase in fusA2 mRNA amounts during carbon limitation (Fig. 4A, Fig. S3A, and Table S4) suggests that B. thetaiotaomicron switches components of its translation machinery during carbon limitation, a condition that bacteria may experience between meals taken by the host. BT4338 control of fusA2 transcription is conserved in other Bacteroides species (Fig. 6). In addition, fusA2 is dispensable for growth on all nutrients known to require BT4338 (Fig. S4). That transcriptional activation of fusA2 by BT4338 is critical for B. thetaiotaomicron to colonize the murine gut (Fig. 5) indicates that this master regulator governs gut colonization, in part, by controlling factors putatively responsible for protein synthesis.
Master regulators of carbohydrate utilization are active during carbon limitation.
Given BT4338’s role as an activator of carbohydrate utilization genes, it is paradoxical that this protein binds to its target DNA sequences during carbon limitation. This binding is physiologically relevant because it results in gene transcription (Fig. 4A), protein production (Fig. 4F), and gut colonization (Fig. 5).
The activity of the master regulator of carbohydrate utilization from E. coli—the cyclic AMP receptor protein (CRP)—is also stimulated by a brief exposure to carbon limitation. In the case of CRP, this is due to the transient synthesis of cyclic AMP (cAMP), the CRP-activating ligand, by adenylate cyclase following a sudden shift from growth in glucose to carbon limitation (31). However, BT4338 does not appear to operate by sensing cAMP produced under carbon limitation conditions because (i) the BT4338 protein exhibits low (i.e., <18%) amino acid identity with CRP, (ii) B. thetaiotaomicron lacks cya or crp sequelogs, (iii) addition of exogenous cAMP has no consistent effect on gene expression (32), and (iv) the closely related species Bacteroides fragilis encodes a BT4338 sequelog (BF9343_RS04500) but contains no detectable cAMP (33). Thus, BT4338 activates target gene transcription in response to an undefined signal, the availability of which changes in response to carbon limitation and is distinct from cAMP.
The targets of the master regulator BT4338.
We established that carbon limitation transcriptionally induces both mono- and polysaccharide utilization genes and that this induction is BT4338 dependent. These genes include BT0356 to BT0350 (arabinose utilization), BT1272 to BT1277 (fucose utilization), BT1434 to BT1432 (glucuronate utilization), and BT0791 to BT0794 (xylose utilization), as well as BT2818 to BT2825, BT0453 to BT0449, BT0439 to BT0443, and BT4299 to BT4295, which are predicted to mediate polysaccharide utilization (16). These results make sense given that the BT4338 mutant is defective for growth on various monosaccharides and polysaccharides (9).
Our findings also suggest the intriguing possibility of BT4338 controlling reactions in the tricarboxylic acid (TCA) cycle by regulating acetyl-CoA assimilation in response to various nutrients. This is because the operon encoding the propionyl-CoA carboxylase complex and the gene encoding alanine dehydrogenase are upregulated during carbon limitation in a BT4338-dependent manner.
Our direct experimental approach uncovered hundreds of DNA sequences bound by the BT4338 protein in vivo. These sequences are only partly in agreement with those predicted in a bioinformatic study (12) (Table S3A). That is, the bioinformatic study failed to identify many direct targets of BT4338, most conspicuously the fusA2 promoter, despite being the DNA region most strongly bound by BT4338 in vivo (Fig. 3H and Fig. 4C) and harboring sequences that strongly resemble the motif proposed by the bioinformatic analysis (12). The combination of ChIP-seq, RNA-seq, and genetic experiments indicates that the BT4338 protein operates as both a transcriptional activator and repressor.
Concluding remarks.
The gene most highly regulated by BT4338 during carbon limitation—fusA2—specifies a protein that exhibits 31% shared identity (52% similarity) with the deduced amino acid sequence of the B. thetaiotaomicron fusA gene, which encodes the canonical elongation factor EF-G. The EF-G and EF-G2 proteins appear to serve different functions in B. thetaiotaomicron because of the following. First, the fusA gene is essential under all conditions, whereas fusA2 is necessary for successful colonization of the murine gut but dispensable otherwise. Second, when fusA is highly expressed (i.e., during exponential growth) (Fig. 4B and Table S4), the abundance of fusA2 mRNA and its protein product, EF-G2, is very low (Fig. 4A and F and Table S4). By contrast, conditions that activate fusA2 expression, such as carbon limitation (this work) and stationary phase (34), are accompanied by a decrease in fusA expression (Fig. 4A and B and Table S4) (35). And third, the increase in fusA2 mRNA levels taking place during carbon limitation is dependent on BT4338, whereas the concomitant fusA mRNA decrease is BT4338 independent.
We suggest that EF-G2 plays two non-mutually exclusive functions in Bacteroides species. On the one hand, it provides additional molecules of a translation factor to compensate for the decrease in EF-G abundance. On the other hand, it enables translation of particular mRNAs, perhaps working with ribosomes having a composition distinct from those operating during rapid growth (36). That activation of fusA2 by BT4338 is required for gut colonization suggests that B. thetaiotaomicron utilizes a distinct protein synthesis machinery when experiencing nutrient fluctuations in its natural habitat.
MATERIALS AND METHODS
Bacterial culture conditions.
Bacterial strains and plasmids used in this study are listed in Table S5. E. coli organisms were cultured in Luria-Bertani broth (BD) containing 100 μg/ml of ampicillin (Sigma). B. thetaiotaomicron and B. ovatus strains were cultured in brain heart infusion medium containing 5% defibrinated horse blood, tryptone-yeast extract-glucose (TYG) medium, or Bacteroides minimal medium [100 mM KH2PO4 (pH 7.2), 15 mM NaCl, 8.5 mM (NH4)2SO4, 0.5 μg/ml of l-cysteine, 1.9 μM hematin, 200 μM l-histidine, 100 μM MgCl2, 1.4 μM FeSO4, 50 μM CaCl2, 1 μg/ml of vitamin K3, and 5 ng/ml of vitamin B12, plus individual carbon sources (0.5% wt/vol)] as described previously (9).
Strains used in this study. Download Table S5, PDF file, 0.1 MB (60.6KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Construction of strains.
All strains were constructed using oligonucleotides listed in Table S6. In-frame gene deletions and mutations were generated using counterselectable allelic exchange as described (37). Plasmids derived from pNBU2-tetQ were introduced into the B. thetaiotaomicron chromosome (NBU2 att-1 site) as described previously (37). Molecular barcodes encoded within pNBU2-tetQ vectors were introduced into the B. thetaiotaomicron genome (NBU2 att-1 site) as described (16). Epitope tagging of BT2167 (fusA2) was performed using the pKNOCK-tetQ vector by cloning the 750-bp sequence at the 3′ end of the fusA2, including additional nucleotide sequences at the 3′ end encoding the FLAG tag and a stop codon as described previously (38). Inactivation of the Bovatus_RS22425 gene was carried out by cloning the sequence from bp 157 to 556 of Bovatus_RS22425 into the pKNOCK-tetQ vector, followed by its introduction into the B. ovatus chromosome as described (38).
Oligonucleotides used in this study. Download Table S6, PDF file, 0.1 MB (75.3KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Carbon limitation experiments.
B. thetaiotaomicron and B. ovatus strains were grown in TYG medium anaerobically overnight before being subcultured into 2.0 ml of Bacteroides minimal medium containing 0.5% glucose as the sole carbon source. After growth to stationary phase, the resulting culture was diluted 1:50 into identical prereduced medium and allowed to grow to mid-exponential phase (optical density [OD] = 0.45 to 0.5), at which time an aliquot was collected by centrifugation for 1 min before decanting and immediate placement of the cell pellet on dry ice until the end of the experiment, which represents measurements designated “-5” in figures for all carbon limitation experiments. Subsequently, the remaining culture was centrifuged at 7,197 × g at room temperature for 3 min in sealed tubes and reintroduced into the anaerobic chamber where the tubes were unsealed, and the supernatants were decanted. Cell pellets were resuspended in an equivalent volume of prewarmed, prereduced minimal medium lacking a carbon source and incubated at 37°C anaerobically. At, 10, 15, or 60 min following incubation, aliquots were collected by centrifugation and the supernatant was decanted before the pellet was placed on dry ice prior to storage at –80°C.
Quantitative PCR (qPCR).
mRNA was prepared from 1.0 ml of B. thetaiotaomicron cell culture treated with RNAprotect (Qiagen) using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. cDNA was subsequently synthesized from 1.0 μg of RNA using Superscript VILO master mix (Thermo Fisher) according to the manufacturer’s directions. The mRNA levels of each gene were measured using a Fast ABI7500 machine (Applied Biosystems) by quantification of cDNA using Fast SYBR green PCR master mix (Applied Biosystems) and primers listed in Table S6. fusA and fusA2 transcript levels were measured from 10-fold-diluted cDNA using primers W3565 and W3566 or W4344 and W4345, respectively. Data were normalized to 16S rRNA from 1,000-fold-diluted cDNA using primers 10256 and 10257 as described previously (9).
Western blotting.
Frozen pellets from 12 ml of cells were thawed and resuspended in 400 μl of Tris-buffered saline (TBS; 50 mM Tris, 138 mM NaCl, 2.7 mM KCl [pH 8.0]) containing 1 mM EDTA and 0.5 mg/ml of chicken egg lysozyme (Sigma). Cell suspensions were transferred to a 2.0-ml tube containing lysing matrix B (MP Biomed) and subjected to disruption using a MiniBeadBeater for 5 cycles of 40 s, with 5-min incubations on ice between each cycle. Samples were centrifuged for 2 min at 10,000 × g at 4°C to remove cell debris. Protein concentrations were estimated by measuring absorbance at 280 nm using a NanoDrop 8000 (Thermo Fisher). A volume corresponding to 100 μg of protein from each sample was combined with 5 μl of 4× LDS buffer (Thermo Fisher) containing 100 mM dithiothreitol and subjected to heating at 95°C for 5 min. Samples were loaded onto a NuPAGE 4 to 12% bis-Tris protein gel (Thermo Fisher) and fractionated for 60 min at 180 V in 1× morpholinepropanesulfonic acid (MOPS) running buffer (Thermo Fisher). Fractionated proteins were transferred to a nitrocellulose membrane using an iBlot device (Invitrogen), and the resulting membrane was cut below the 65-kDa marker and both portions were blocked for 1 h in TBS containing 3% skim milk. FLAG-tagged EF-G2 were detected on the top portion of the membrane using a 1:5,000 dilution of a mouse anti-FLAG antibody (Sigma) followed by a 1:5,000 dilution of a horseradish peroxidase (HRP)-conjugated anti-mouse antibody (Promega). GroEL was detected on the bottom portion of the membrane using a 1:5,000 dilution of anti-GroEL (Sigma) followed by a 1:5,000 dilution of an HRP-conjugated anti-rabbit antibody (GE). Membranes were washed before and after addition of secondary antibody with TBS containing 0.05% Tween 20 (Sigma) and finally rinsed with TBS prior to detection with Amersham ECL Western blotting detection reagent (GE) using a LAS-4000 (GE).
RNA-seq.
Ten milliliters of bacterial culture was collected from triplicate cultures growing exponentially in minimal medium containing glucose (OD at 600 nm [OD600] = 0.45 to 0.5) or 10 min after centrifugation and resuspension in minimal medium lacking a carbon source. Cell pellets were immediately frozen on dry ice and stored at −80°C. The mRNA was stabilized by treatment with 10 ml of RNAprotect (Qiagen) (diluted 2 parts RNAprotect to 1 part water) prior to extraction using the RNeasy minikit (Qiagen) with on-column DNase I treatment. The eluates were treated with Turbo DNase (Invitrogen) for 30 min before a subsequent round of purification using the RNeasy minikit. RNA-seq was performed at the Yale Center for Genome Analysis, where the RNA quality was examined using a BioAnalyzer (Agilent) before rRNA was depleted using the RiboZero bacterial kit (Illumina). Approximately 50 million 75-bp paired-end reads per sample were collected using a HiSeq 4000 (Illumina).
ChIP.
A strain expressing a C-terminal hemagglutinin (HA)-tagged BT4338 (GT1481) was grown anaerobically overnight in TYG medium before being subcultured into 2.0 ml of minimal medium containing 0.5% glucose as the sole carbon source. After growth to stationary phase, 2 ml of the resulting culture was inoculated into 100 ml of identical medium and allowed to grow to mid-exponential phase (OD = 0.45 to 0.5). Forty milliliters of cells was combined with 1.08 ml of 37% formaldehyde and gently mixed at room temperature by nutation for 15 min and represented the “-5” time point. Concomitantly, 50 ml of the remaining culture was centrifuged at 7,197 × g at room temperature for 3 min. The pelleted cells were resuspended in 50 ml of prewarmed, prereduced minimal medium and incubated at 37°C anaerobically for 10 min before 40 ml of cell culture was combined with 1.08 ml of 37% formaldehyde and gently mixed by nutation for 15 min at room temperature. All cross-linking reactions were halted by the addition of 4 ml of 2.5 M glycine (Sigma), gently mixed by nutation for 5 min at room temperature, and placed on ice until the end of the experiment. Cells were pelleted by centrifugation at 5,000 × g for 8 min and washed with 40 ml of ice-cold phosphate-buffered saline (PBS). This wash step was repeated once, and the resulting pellet was resuspended in 1.0 ml of ice-cold PBS, transferred to a 1.5-ml microcentrifuge tube, and pelleted by centrifugation at 5,000 × g for 8 min. Cells pellets were immediately frozen on dry ice and stored at −80°C.
The cell pellets were resuspended in 0.5 ml of lysis buffer (10 mM Tris [pH 8.0], 50 mM NaCl, 10 mM EDTA, 20% sucrose) containing 20 mg/ml of lysozyme (Sigma) and protease inhibitor cocktail (Roche). The resuspended cells were combined with 25 μl of RNase A (Qiagen) before incubation at 37°C for 35 min with agitation at 600 rpm in a ThermoMixer (Eppendorf). Following the addition of 80 μl of 10× radioimmunoprecipitation (RIPA) buffer (Millipore) containing protease inhibitor cocktail (Roche), samples were further incubated for 10 min with agitation at 600 rpm in a ThermoMixer (Eppendorf). The samples were cooled on ice prior to DNA fragmentation by sonication at 4°C in a cup horn sonicator (Misonix) with 12- to 15-s pulses at an amplitude of 80, with 30-s rests between each pulse. Debris was pelleted by centrifugation at 18,000 × g for 15 min at 4°C, and the supernatant was reserved and stored at −80°C. Upon thawing, 25 μl was reserved as input DNA and the remainder was combined with 10 μl of MagnaChIP protein G beads (Millipore) and incubated at room temperature for 30 min with nutation. The supernatant was recovered and split in half, with one half combined with MagnaChIP protein G beads (Millipore) preincubated with 2 μl of an HA tag-specific antibody (Sigma; IP samples) and the other half combined with MagnaChIP protein G without antibody (control samples). After 2 h of incubation at room temperature with nutation, the beads were recovered using a magnetic stand for 60 s and the bead pellet was washed twice by adding 1 ml of 1× RIPA buffer and gently agitated for 5 min on a nutator. The beads were then washed twice by adding 1 ml of LiCl solution (10 mM Tris [pH 8.0], 250 mM LiCl, 1 mM EDTA, 0.5% Igepal CA-630, 0.5% sodium deoxycholate) and gently agitated for 5 min on a nutator before recovering on a magnetic stand. The beads were then washed twice by adding 1 ml of TE Solution, pH 8.0 (10 mM Tris-HCl, 1 mM EDTA; Millipore) and gently agitated for 5 min on a nutator before recovering on a magnetic stand. Bound DNA was eluted from the beads by the addition of 100 μl of elution buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 1% SDS) and incubation at 65°C for 15 min with mixing at 600 rpm in a ThermoMixer. The beads were collected on a magnetic stand and the supernatant was recovered. The input DNA was brought to 100 μl by addition of 75 μl of elution buffer, and all samples were combined with 100 μl of proteinase K solution and incubated at 37°C for 3 h, followed by 9 h of incubation at 67°C. The DNA was recovered from all input, IP, and control samples using a Qiagen PCR purification kit and eluted in 50 μl of TE buffer. The abundance of the BT1311 (rpoD, for normalization), araM, fusA2, and BT3348 promoters were measured in 50-fold-diluted input DNA and 2-fold-diluted IP or control samples using qPCR. The fold enrichment was calculated as described previously (30).
ChIP-seq.
Triplicate ChIP samples prepared as described above were analyzed by the Yale Center for Genome Analysis, which collected approximately 50 million 75-bp paired-end reads per sampled using a HiSeq 4000 (Illumina).
High-throughput sequencing data analysis.
The RNA-seq and ChIP-seq alignments and analysis were performed on the Galaxy web server (https://usegalaxy.org). The reads were aligned to the B. thetaiotaomicron genome (strain VPI-5482; GenBank accession number NC_004663) using Bowtie2 (v2.3.4). For RNA-seq samples, the mapped reads were quantified using featureCounts (v1.6.3) and differential gene expression was measured using DEseq2 (v1.18.1). To identify ChIP-seq peaks associated with differential gene expression, the significant ChIP peaks called by MACS2 were assigned as intragenic peaks if they were within annotated genes according to genomic reference NC_004663.1 (GenBank accession number). Conversely, intergenic peaks were defined as those positioned outside annotated coding region.
For analysis of the BT4338 regulon and BT4338-associated regions, genes with a 4-fold (2-log) difference of expression between GT23 and NS364 were selected and assigned with intergenic and/or intragenic peaks that were within a 500-bp distance, if any. For each of the selected genes, the information regarding gene annotation, RNA expression, associated peak location, and associated peak sequences was extracted and reported (Table S3B). The pipeline was written in Python3 with packages pandas, numpy, Biopython, pybedtools, pyBigWig, and pysam. Source code will be made available upon request.
Examining bacterial abundance in the murine gut.
All experiments using mice were performed using protocols approved by the Yale University Institutional Animal Care and Use Committee. Germfree Swiss Webster mice were maintained in flexible plastic gnotobiotic isolators with a 12-h light/dark cycle and provided a standard, autoclaved mouse chow (5K67 LabDie; Purina) ad libitum. Mice were gavaged with around 108 CFU of each strain suspended in 200 μl of phosphate-buffered saline. Input (day 0) abundance of each strain was determined by CFU plating. Fecal pellets were collected at the desired times and genomic DNA was extracted as described previously (16). The abundance of each strain was measured by qPCR, using barcode-specific primers (wild-type strain, primers W1701 and W1713; ΔBT4338 strain, primers W1704 and W1713; ΔBT2167 strain, primers W1702 and W1713; and Δ22 strain, primers W1711 and W1713) as described previously (16).
Data availability.
The RNAseq data set is available as GEO submission GSE134115. For ChIPseq samples, peaks were called from pooled triplicate input and IP samples using MACS2 (v 2.1.1.20160309), and the corresponding data set is available as GEO submission GSE134116.
ACKNOWLEDGMENTS
We thank Jennifer Aronson for comments on the manuscript.
This research was supported by grants GM123798 (to E.A.G.) and GM118159 (to A.L.G.) from the National Institutes of Health of the United States.
G.E.T., W.H., N.D.S., and E.A.G. designed the research, G.E.T., W.H., N.D.S., X.H., and N.A.B.-B. performed the research, G.T., W.H., N.D.S., X.H., A.L.G., and E.A.G. analyzed the data, and G.E.T., W.H., and E.A.G. wrote the paper. All authors approved the final version of the manuscript.
We declare no conflict of interest.
Footnotes
Citation Townsend GE, II, Han W, Schwalm ND, III, Hong X, Bencivenga-Barry NA, Goodman AL, Groisman EA. 2020. A master regulator of Bacteroides thetaiotaomicron gut colonization controls carbohydrate utilization and an alternative protein synthesis factor. mBio 11:e03221-19. https://doi.org/10.1128/mBio.03221-19.
REFERENCES
- 1.Cani PD, Van Hul M, Lefort C, Depommier C, Rastelli M, Everard A. 2019. Microbial regulation of organismal energy homeostasis. Nat Metab 1:34–46. doi: 10.1038/s42255-018-0017-4. [DOI] [PubMed] [Google Scholar]
- 2.Flint HJ, Scott KP, Louis P, Duncan SH. 2012. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 9:577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 3.Round JL, Mazmanian SK. 2009. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skelly AN, Sato Y, Kearney S, Honda K. 2019. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat Rev Immunol 19:305–323. doi: 10.1038/s41577-019-0144-5. [DOI] [PubMed] [Google Scholar]
- 5.Zmora N, Suez J, Elinav E. 2019. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol 16:35–56. doi: 10.1038/s41575-018-0061-2. [DOI] [PubMed] [Google Scholar]
- 6.Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, Sonnenburg JL. 2018. An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557:434–438. doi: 10.1038/s41586-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Townsend GE II, Han W, Schwalm ND III, Raghavan V, Barry NA, Goodman AL, Groisman EA. 2019. Dietary sugar silences a colonization factor in a mammalian gut symbiont. Proc Natl Acad Sci U S A 116:233–238. doi: 10.1073/pnas.1813780115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 9.Schwalm ND III, Townsend GE II, Groisman EA. 2016. Multiple signals govern utilization of a polysaccharide in the gut bacterium Bacteroides thetaiotaomicron. mBio 7:e01342-16. doi: 10.1128/mBio.01342-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, Osterman AL, Gordon JI. 2015. Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350:aac5992. doi: 10.1126/science.aac5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. 2009. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravcheev DA, Godzik A, Osterman AL, Rodionov DA. 2013. Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics 14:873. doi: 10.1186/1471-2164-14-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodnina MV, Savelsbergh A, Katunin VI, Wintermeyer W. 1997. Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature 385:37–41. doi: 10.1038/385037a0. [DOI] [PubMed] [Google Scholar]
- 14.Janosi L, Hara H, Zhang S, Kaji A. 1996. Ribosome recycling by ribosome recycling factor (RRF)—an important but overlooked step of protein biosynthesis. Adv Biophys 32:121–201. doi: 10.1016/0065-227x(96)84743-5. [DOI] [PubMed] [Google Scholar]
- 15.Ito K, Fujiwara T, Toyoda T, Nakamura Y. 2002. Elongation factor G participates in ribosome disassembly by interacting with ribosome recycling factor at their tRNA-mimicry domains. Mol Cell 9:1263–1272. doi: 10.1016/s1097-2765(02)00547-6. [DOI] [PubMed] [Google Scholar]
- 16.Martens EC, Chiang HC, Gordon JI. 2008. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynch JB, Sonnenburg JL. 2012. Prioritization of a plant polysaccharide over a mucus carbohydrate is enforced by a Bacteroides hybrid two-component system. Mol Microbiol 85:478–491. doi: 10.1111/j.1365-2958.2012.08123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wegorzewska MM, Glowacki RWP, Hsieh SA, Donermeyer DL, Hickey CA, Horvath SC, Martens EC, Stappenbeck TS, Allen PM. 2019. Diet modulates colonic T cell responses by regulating the expression of a Bacteroides thetaiotaomicron antigen. Sci Immunol 4:eaau9079. doi: 10.1126/sciimmunol.aau9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alber BE. 2011. Biotechnological potential of the ethylmalonyl-CoA pathway. Appl Microbiol Biotechnol 89:17–25. doi: 10.1007/s00253-010-2873-z. [DOI] [PubMed] [Google Scholar]
- 20.Gago G, Kurth D, Diacovich L, Tsai SC, Gramajo H. 2006. Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J Bacteriol 188:477–486. doi: 10.1128/JB.188.2.477-486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Boghigian BA, Pfeifer BA. 2010. Investigating the role of native propionyl-CoA and methylmalonyl-CoA metabolism on heterologous polyketide production in Escherichia coli. Biotechnol Bioeng 105:567–573. doi: 10.1002/bit.22560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller V, Imkamp F, Biegel E, Schmidt S, Dilling S. 2008. Discovery of a ferredoxin:NAD+-oxidoreductase (Rnf) in Acetobacterium woodii: a novel potential coupling site in acetogens. Ann N Y Acad Sci 1125:137–146. doi: 10.1196/annals.1419.011. [DOI] [PubMed] [Google Scholar]
- 23.Hess V, Gallegos R, Jones JA, Barquera B, Malamy MH, Muller V. 2016. Occurrence of ferredoxin:NAD(+) oxidoreductase activity and its ion specificity in several Gram-positive and Gram-negative bacteria. PeerJ 4:e1515. doi: 10.7717/peerj.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong J-A, Park SW, Yoon D, Kim S, Kang H-Y, Oh J-I, Jeong J-A, Park SW, Yoon D, Kim S, Kang H-Y, Oh J-I. 2018. Roles of alanine dehydrogenase and induction of its gene in Mycobacterium smegmatis under respiration-inhibitory conditions. J Bacteriol 200:e00152-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin D, Lee EJ, Huang H, Groisman EA. 2006. A positive feedback loop promotes transcription surge that jump-starts Salmonella virulence circuit. Science 314:1607–1609. doi: 10.1126/science.1134930. [DOI] [PubMed] [Google Scholar]
- 26.Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, Fang FC. 2006. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313:236–238. doi: 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- 27.Grainger DC, Aiba H, Hurd D, Browning DF, Busby SJ. 2007. Transcription factor distribution in Escherichia coli: studies with FNR protein. Nucleic Acids Res 35:269–278. doi: 10.1093/nar/gkl1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitzgerald DM, Smith C, Lapierre P, Wade JT. 2018. The evolutionary impact of intragenic FliA promoters in proteobacteria. Mol Microbiol 108:361–378. doi: 10.1111/mmi.13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schofield WB, Zimmermann-Kogadeeva M, Zimmermann M, Barry NA, Goodman AL. 2018. The stringent response determines the ability of a commensal bacterium to survive starvation and to persist in the gut. Cell Host Microbe 24:120–132.e126. doi: 10.1016/j.chom.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez JC, Shin D, Zwir I, Latifi T, Hadley TJ, Groisman EA. 2009. Evolution of a bacterial regulon controlling virulence and Mg(2+) homeostasis. PLoS Genet 5:e1000428. doi: 10.1371/journal.pgen.1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gstrein-Reider E, Schweiger M. 1982. Regulation of adenylate cyclase in E. coli. EMBO J 1:333–337. doi: 10.1002/j.1460-2075.1982.tb01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pudlo NA, Urs K, Kumar SS, German JB, Mills DA, Martens EC. 2015. Symbiotic human gut bacteria with variable metabolic priorities for host mucosal glycans. mBio 6:e01282-15. doi: 10.1128/mBio.01282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegel LS, Hylemon PB, Phibbs PV Jr.. 1977. Cyclic adenosine 3′,5′-monophosphate levels and activities of adenylate cyclase and cyclic adenosine 3′,5′-monophosphate phosphodiesterase in Pseudomonas and Bacteroides. J Bacteriol 129:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonnenburg ED, Sonnenburg JL, Manchester JK, Hansen EE, Chiang HC, Gordon JI. 2006. A hybrid two-component system protein of a prominent human gut symbiont couples glycan sensing in vivo to carbohydrate metabolism. Proc Natl Acad Sci U S A 103:8834–8839. doi: 10.1073/pnas.0603249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI. 2005. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 36.Ferretti MB, Karbstein K. 2019. Does functional specialization of ribosomes really exist? RNA 25:521–538. doi: 10.1261/rna.069823.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koropatkin NM, Martens EC, Gordon JI, Smith TJ. 2008. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16:1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghavan V, Lowe EC, Townsend GE II, Bolam DN, Groisman EA. 2014. Tuning transcription of nutrient utilization genes to catabolic rate promotes growth in a gut bacterium. Mol Microbiol 93:1010–1025. doi: 10.1111/mmi.12714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The BT4338 regulon during growth in glucose. (A) Volcano plot depicting the fold change versus the −log10 of the corresponding P value for all genes between wild-type (GT23) and BT4338-deficient (NS364) B. thetaiotaomicron strains during growth in glucose. Blue dots represent genes exhibiting a >2-fold decrease in expression in the BT4338 mutant compared to that in the wild type. Red dots represent genes exhibiting a >2-fold increase in expression in the BT4338 mutant compared to that in the wild type. Genes discussed in the Results and Discussion sections of the text are labeled. Data represent the averages of 3 biological replicates measured by RNA-seq. (B) Venn diagrams representing genes whose mRNAs are >2-fold different between wild-type (GT23) and BT4338-deficient (NS364) strains of B. thetaiotaomicron growing in mid-exponential phase in minimal medium containing glucose as a sole carbon source and at 10 min following a switch to carbon limitation conditions. Data represent the averages of 3 biological replicates measured by RNA-seq, with an adjusted P value of <0.05. Download FIG S1, PDF file, 0.2 MB (193.3KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Complementation of a BT4338-null mutant with a gene specifying an epitope-tagged BT4338 protein. (A to C) Growth (OD600) of wild-type B. thetaiotaomicron harboring an empty vector (GT1009; black lines) or a BT4338 mutant harboring either the empty vector (NS432; gray lines), a plasmid-encoded wild-type BT4338 (GT1498; blue lines), BT4338 with a C-terminal HA tag (NS433; orange lines), or BT4338 with a C-terminal HA epitope separated by a linker comprised of 4 glycine residues (GT1481; green lines). Bacteria were grown in minimal medium containing either arabinose (A), glucuronate (B), or xylose (C). (D) fusA2 mRNA abundances in strains GT1009, GT1498, GT1481, and NS432 during growth in glucose (glu, -5) and 10 or 60 min following a switch to carbon limitation conditions (No C). mRNA abundance was measured by qPCR. Data represent the averages of 4 biological replicates, and error bars represent SEM. Download FIG S2, PDF file, 0.3 MB (268.4KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes with significantly altered expression in a BT4338-deficient strain (NS364) relative to that in wild-type B. thetaiotaomicron (GT23) during mid-exponential-phase growth in minimal medium containing glucose. Download Table S1, XLSX file, 0.5 MB (558.1KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes with significantly altered expression in a BT4338-deficient strain (NS364) relative to that in wild-type B. thetaiotaomicron (GT23) following a 10-min exposure to carbon limitation. Download Table S2, XLSX file, 0.5 MB (553.8KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Regions of the B. thetaiotaomicron genome bound by BT4338 following a 10-min exposure to carbon limitation. (A) All 834 regions bound by BT4338 in response to carbon limitation. (B) BT4338 binding corresponding to BT4338-dependent changes in gene expression during carbon limitation. Download Table S3, XLSX file, 0.4 MB (409.2KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
B. thetaiotaomicron genes regulated by carbon limitation. (A) Volcano plot depicting the fold change versus the −log10 of the corresponding P value for all genes in wild-type (GT23) B. thetaiotaomicron in carbon limitation conditions following growth in glucose (Table S4). Blue dots represent genes exhibiting a >2-fold decrease in expression during carbon limitation compared to growth in glucose. Red dots represent genes exhibiting a >2-fold increase in expression during carbon limitation compared to growth in glucose. fusA, fusA2, and araM are labeled. Data represent the averages of 3 biological replicates measured by RNA-seq. (B) fusA2 mRNA abundances in the wild type (GT23; gray bars) and a strain unable to synthesize (p)ppGpp0 (relA spoT; GT1181; purple bars) during growth in glucose (-5) and 10 or 60 min following a switch to carbon limitation conditions. mRNA abundance was measured by qPCR. Data represent the averages of 3 biological replicates, and error bars represent SEM. Download FIG S3, PDF file, 0.2 MB (191.8KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes with significantly altered expression in wild-type B. thetaiotaomicron (GT23) following a switch from exponential growth in glucose to carbon limitation conditions for 10 min. Download Table S4, XLSX file, 0.5 MB (567.2KB, xlsx) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
fusA2 is dispensable for growth on BT4338-dependent carbon sources. (A to E) A wild-type B. thetaiotaomicron strain (GT23; black lines) and mutants lacking either BT4338 (NS364; blue lines) or fusA2 (GT1309; red lines) were grown in arabinose (A), xylose (B), fucose (C), glucuronate (D), or ribose (E) as a sole carbon source. Data represent the averages of 4 biological replicates and error bars represent SEM. Download FIG S4, PDF file, 0.2 MB (238.9KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains used in this study. Download Table S5, PDF file, 0.1 MB (60.6KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotides used in this study. Download Table S6, PDF file, 0.1 MB (75.3KB, pdf) .
Copyright © 2020 Townsend et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The RNAseq data set is available as GEO submission GSE134115. For ChIPseq samples, peaks were called from pooled triplicate input and IP samples using MACS2 (v 2.1.1.20160309), and the corresponding data set is available as GEO submission GSE134116.