

This study offers an effective strategy to enhance immunotherapy by promoting CCR9+CD8+ T cell tumor infiltration.
Abstract
CCR9+ T cells have an increased potential to be activated and therefore may mediate strong antitumor responses. Here, we found, however, that CCL25, the only chemokine for CCR9+ cells, is not expressed in human or murine triple-negative breast cancers (TNBCs), raising a hypothesis that intratumoral delivery of CCL25 may enhance antitumor immunotherapy in TNBCs. We first determined whether this approach can enhance CD47-targeted immunotherapy using a tumor acidity–responsive nanoparticle delivery system (NP-siCD47/CCL25) to sequentially release CCL25 protein and CD47 small interfering RNA in tumor. NP-siCD47/CCL25 significantly increased infiltration of CCR9+CD8+ T cells and down-regulated CD47 expression in tumor, resulting in inhibition of tumor growth and metastasis through a T cell–dependent immunity. Furthermore, the antitumor effect of NP-siCD47/CCL25 was synergistically enhanced when used in combination with programmed cell death protein–1/programmed death ligand-1 blockades. This study offers a strategy to enhance immunotherapy by promoting CCR9+CD8+ T cell tumor infiltration.
INTRODUCTION
Triple-negative breast cancer (TNBC), characterized by the lack of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2), accounts for approximately 15 to 20% of all invasive breast cancers (1) but has a poor prognosis with a high mortality and early relapse (2). Because of the lack of therapeutic targets, specific therapies for TNBC have been lacking (3) and effective therapeutic strategies are urgently needed. Although immunotherapy with programmed cell death protein–1 (PD-1)/programmed death ligand-1 (PD-L1) blocking antibodies appears to be a potentially promising therapeutic option for TNBC, the objective response rate was low in these patients (4, 5), even in those receiving PD-1/PD-L1 blockade and nab-paclitaxel (6). Even so, tumor-infiltrating lymphocytes (TILs) are associated with favorable outcomes in patients with TNBC, including disease-free survival and overall survival (7, 8), and cancer prognosis and immunotherapy outcome are also largely attributed to TILs.
Chemokines, through interaction with the corresponding receptors, play critical roles in controlling immune trafficking and tissue infiltration (9–11). The chemokine CCL25 (also known as thymus-expressed chemokine) is the only ligand for CCR9 (12) and is selectively expressed by medullary dendritic cells and cortical epithelial cells in the thymus and the epithelium of the small intestine under normal conditions (13–15). The CCL25/CCR9 interaction was found to be essential for homing of CCR9+ T cell progenitors into the thymus (16) and for the development and maintenance of the unique immune cell composition of the gut, with most of the lymphocytes expressing CCR9 (13, 17, 18). In line with these findings, the CCL25/CCR9 interaction plays an important role in the pathogenesis of inflammatory bowel diseases (19). It has been reported that CCR9+ T cells have an increased potential to be activated (20, 21) and to produce proinflammatory cytokines (19, 22, 23) and that CCR9+ T helper cells can promote expansion and survival of CD8+ T cells (24). Furthermore, CCL25/CCR9 signaling inhibits CD4+ T cell differentiation into regulatory T (Treg) cells (23). The correlation between increased circulating CCR9+CD8+ naïve T cells and prolonged survival in patients with melanoma (25) implicates that these T cells may possibly mediate stronger antitumor responses than other T cells. In support of this possibility, treatment with anti–CCL25-neutralizing antibodies accelerated tumor growth in a CCL25-expressing mouse tumor model (25).
In this study, we found that neither human nor mouse TNBCs express CCL25. By establishing a tumor acidity–responsive nanoparticle (NP-siCD47/CCL25) delivery system that can sequentially release CCL25 in tumor stroma and CD47 small interfering RNA (siRNA) into tumor cells, we demonstrated that intratumoral CCL25 delivery promotes CCR9+ T cell infiltration and enhances CD47-targeted immunotherapy in a murine TNBC model. The antitumor effect induced by NP-siCD47/CCL25 is primarily T cell dependent and can be synergistically enhanced by combining this approach with PD-1/PD-L1 blockades. This study provides insights into the antitumor effect of CCR9+CD8+ T cells and offers an effective strategy to enhance immunotherapy by promoting CCR9+CD8+ T cell tumor infiltration.
RESULTS
CCR9+CD8+ T cells exhibit an enhanced activation phenotype but limited tumor infiltration in TNBCs
We first evaluated the frequency of CCR9+ T cells in peripheral blood and CCL25 expression in tumors from patients with TNBC. Both healthy volunteers and patients with TNBC had comparably low levels of CCR9+ T cells (CD3+CD45+) and T cell subsets (CD3+CD4+, CD3+CD8+, CD3+CD45RA+, CD3+CD45RO+, and CD3+CD71+) in peripheral blood mononuclear cells (PBMCs) (fig. S1, A to C). Immunohistochemistry (IHC) revealed that none of the TNBC tumor samples showed detectable CCL25 (fig. S2A). CCL25 was also undetectable in a mouse TNBC tumor expression (fig. S2B). These data suggest that the CCL25/CCR9 interaction is unlikely to be directly involved in immune cell infiltration in TNBC tumors. Furthermore, IHC revealed that CCR9+ cells, which were clearly detected in human CCL25+ colon cancer samples (fig. S2C), were barely detected or sporadically found in both human TNBC tumors (fig. S2C) and mouse 4T1 tumors (fig. S3). Given that TNBC tumor tissues consisted of predominantly tumor cells that are CCR9− (fig. S4, left), we further analyzed 4T1 tumor–infiltrating CD45+ cells for CCR9 expression by flow cytometry. We found that CD4+ T cells, CD8+ T cells, CD49b+ natural killer (NK) cells, and CD19+ B cells were clearly detected in tumors (Fig. 1, A and B). Although CCR9+ cells were detected in all TIL (Fig. 1C) and myeloid cell (fig. S1D) populations at increased levels, compared to the levels in the spleens from tumor-bearing and normal mice, the ratios of CCR9+ cells remained low (approximately 15%) in both CD4+ and CD8+ T cells (Fig. 1, C and D). However, CCR9+ T cells displayed a more activated phenotype defined by increased proportions of CD62L−CD44hi cells in the spleens of normal mice (Fig. 1, E and F), which was consistent with previous studies (20). In line with this and with a previous study, we found that T cells showed CCR9 up-regulation upon T cell receptor stimulation (24) by anti-CD3/CD28 antibodies (Fig. 1, G and H). Even in 4T1 tumor–bearing mice, CCR9+CD8+ T cells appeared to reach a more activated state in the spleens and tumors (Fig. 1, I and J). Nonetheless, CCR9+CD8+ T cells also displayed an increased expression of PD-1 compared to CCR9−CD8+ T cells in the tumors and spleens of tumor-bearing mice and normal mice (Fig. 1, K and L). The frequency of PD-1+ cells among tumor-infiltrating CCR9+CD8+ T cells was also higher than in the spleens of normal mice or tumor-bearing mice (Fig. 1L). Since CCL25 was not detected in the tumors (fig. S2, A and B) and lymphocytes can express multiple chemokine receptors (26), we proposed that the low level of CCR9+ lymphocyte tumor infiltration was CCL25/CCR9 independent and mediated by other chemotactic signals.
Fig. 1. Activated characteristics of CCR9+ T cells in spleen and 4T1 tumors.
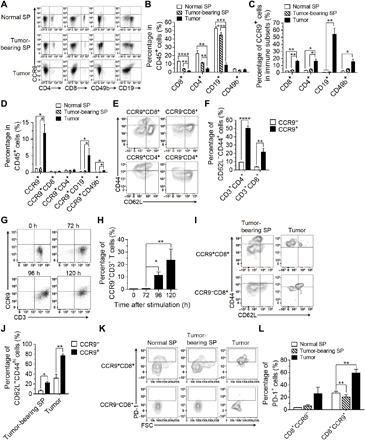
Spleens and tumors were harvested from 4T1 tumor–bearing mice when tumor volume was about 500 mm3 and analyzed for surface CCR9 expression in splenic lymphocytes and TILs. Spleen cells from normal BALB/c mice were used as controls. (A) Representative flow cytometry plots demonstrating the CCR9+ cells in CD4+, CD8+, CD49b+, and CD19+ cells in CD45+ cells in the spleens and tumors. (B) Frequencies of CD4+ T cells, CD8+ T cells, B cells, and NK cells in CD45+ cells in spleens and 4T1 tumors. (C) Frequencies of CCR9+ cells in CD4+ T cells, CD8+ T cells, B cells, and NK cells in the spleens and 4T1 tumors. (D) Frequencies of CCR9+ cells, CCR9+CD4+ T cells, CCR9+CD8+ T cells, CCR9+ B cells, and CCR9+ NK cells in CD45+ cells in the spleens and 4T1 tumors. (E and F) Representative flow cytometry plots (E) for gating strategy to define frequencies (F) of CD62L−CD44hi cells in CCR9+CD8+, CCR9+CD4+ (CCR9+CD3+CD8−) and CCR9−CD8+, and CCR9−CD4+ (CCR9−CD3+CD8−) T cells isolated from the spleens harvested from normal BALB/c mice. (G and H) CCR9−CD3+ T cells were purified from the spleen of normal BALB/c mice and activated for 5 days by plating 2 × 105 cells in plates coated with CD3 and CD28 antibodies. Representative flow cytometry plots showing CCR9 expression on CD3+ T cells (G) and frequencies of CCR9+ cells in CD3+ T cells (n = 4 per group) (H) at 0, 72, 96, and 120 hours after activation. (I and J) Representative flow cytometry plots (I) and frequencies (J) CD62L−CD44hi cells in CCR9+CD8+ and CCR9−CD8+ T cells in the spleens and 4T1 tumors when tumor volumes were about 500 mm3 (n = 3 to 4 per group). (K and L) CCR9+CD8+ and CCR9−CD8+ T cells were prepared from the spleens (SP) of normal BALB/C mice and the spleens and tumors of 4T1 tumor–bearing BALB/c mice (tumor volumes were about 500 mm3) and analyzed for PD-1 expression by flow cytometry. (K) Representative flow cytometry profiles showing PD-1 expression in gated CCR9+CD8+ and CCR9−CD8+ T cells. (L) Frequencies of PD-1+ cells in CCR9+CD8+ and CCR9−CD8+ T cells (n = 3 to 4 per group). Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P< 0.001; ****P < 0.0001.
NP-siCD47/CCL25 significantly increases tumor infiltration of CCR9+CD8+ T cells and down-regulates the CD47 expression in TNBC tumors in vivo
We investigated whether intratumoral delivery of CCL25 can increase CCR9+ T cell infiltration and enhance the antitumor responses of CD47-targeting immunotherapy. As shown in Fig. 2A, the positively charged and CD47 siRNA-loaded micellar nanoparticles (⊕NP/siCD47) were used as a core (fig. S5A). Then, we added the tumor acidity–responsive negatively charged polyethylene glycol (PEG)–ylated deblock copolymer PPC-DA [PPC, PEG-b-poly(allyl ethylene phosphate) modified with cysteamine; DA, 2,3-dimethylmaleic anhydride], synthesized as previously described (27), for the rational preparation of core-shell-corona polyion complex nanoparticles (NP-siCD47). The chemokine CCL25 proteins were then adsorbed onto the nanoparticles to generate nanoparticles carrying both CD47 siRNA and CCL25 (NP-siCD47/CCL25). The encapsulation efficiency of CCL25 on NP-siCD47/CCL25 was 68.4 ± 8.79%. Cryo–transmission electron microscopic analysis of NP-siCD47/CCL25 revealed nanoparticles with spherical morphology and similar diameters (fig. S5B). The hydrodynamic diameters were 217.47 ± 6.57 nm for ⊕NP/siCD47, 239.43 ± 13.91 nm for NP-siCD47, and 257.10 ± 25.83 nm for NP-siCD47/CCL25 (fig. S5C). The zeta potentials for ⊕NP/siCD47, NP-siCD47, and NP-siCD47/CCL25 were 6.79 ± 0.77 mV, −2.90 ± 0.53 mV, and − 3.38 ± 0.50 mV, respectively (fig. S5D). To evaluate the drug release ability of NP-siCD47/CCL25 after exposed to the tumor extracellular microenvironment with pH 6.5 to 6.8 (28), we measured the changes in size and CCL25 release behavior of NP-siCD47/CCL25 after incubation in slightly acidic buffer (pH 6.8) in vitro. The diameter of NP-siCD47/CCL25 was decreased to 216.25 ± 17.75 nm and 2.45 ± 1.06 nm, which was similar to ⊕NP/siCD47 and CCL25, indicating an efficient detachment of the PPC-DA shell from the nanoparticle and release of CCL25 (fig. S5E). The lightly acidic tumor microenvironment (pH 6.8) significantly increased both the release rate and the release amount of CCL25 from NP-siCD47/CCL25 (fig. S5F). We then determined whether NP-siCD47 can be effectively endocytosed, leading to down-regulation of CD47 in mouse TNBC cell line 4T1 cells that express a high level of CD47 (fig. S4, middle). We determined the delivery efficacy into 4T1 cells in vitro using fluorescein amidite–labeled CD47 siRNA (FAM-siCD47) and fluorescent dye Cy3-labeled CCL25 (Cy3-CCL25 and NP-FAM-siCD47/Cy3-CCL25). We incubated 4T1 cells with NP-FAM-siCD47/Cy3-CCL25 in culture medium at pH 6.8 or 7.4 for 30 min. Flow cytometry revealed that 4T1 cells incubated at pH 6.8 showed a markedly enhanced signal of FAM-siCD47 and reduced signal of Cy3-CCL25 compared to those incubated at pH 7.4 (Fig. 2, B and C, and fig. S6A). Confocal laser scanning microscopy (CLSM) images showed that Cy5-siCD47 mainly distributed intracellularly (and was enriched in nuclei), with little detectable Cy3-CCL25 localized to cell membranes in 4T1 cells incubated at pH 6.8 (Fig. 2D). However, only limited amounts of Cy5-siCD47 and Cy3-CCL25 were colocalized and distributed on the cell membrane when 4T1 cells were incubated in neutral conditions (Fig. 2D). Furthermore, 4T1 cells incubated with NP-siCD47/CCL25 at pH 6.8, but not at pH 7.4, showed a significantly reduced CD47 expression at mRNA and protein levels as revealed by quantitative polymerase chain reaction (qPCR), Western blotting, and flow cytometry (Fig. 2, E to G, and fig. S6B). Moreover, NP-siCD47/CCL25 was found as effective as Lipofectamine 2000 in delivering siRNA for inhibiting target gene expression (Fig. 2, E to G, and fig. S6B). These results suggest that, upon exposure to an acidic tumor environment, NP-siCD47/CCL25 can rapidly release CCL25 in the extracellular environment and the remaining NP-siCD47 core particles can be effectively endocytosed by tumor cells.
Fig. 2. Tumor acidic environment facilitates CCL25 and CD47 siRNA progressive delivery and enhances the target gene silencing efficiency.
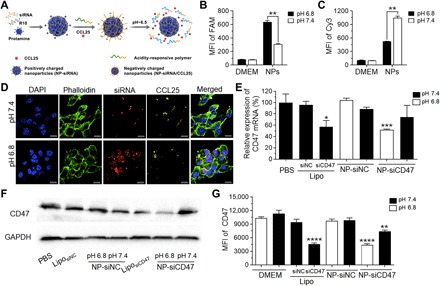
(A) Schematic illustration of CCL25 and CD47 siRNA coloaded tumor acidity–responsive nanoparticle (NP-siCD47/CCL25). NP-siCD47/CCL25 sequentially release CCL25 in tumor stroma and CD47 siRNA into tumor cells in the acidic environment of tumor. (B and C) Quantitative evaluation of cellular uptake of CD47 siRNA (B) and CCL25 (C) by 4T1 cells after incubation with NP-siCD47/CCL25 at pH 7.4 or 6.8 for 30 min using flow cytometry (n = 4). The CD47 siRNA and CCL25 were labeled with FAM and Cy3, respectively. MFI, mean fluorescence intensity; DMEM, Dulbecco’s modified Eagle’s medium. (D) Confocal laser scanning microscopy (CLSM) images of the 4T1 cells after incubation with NP-siCD47/CCL25 at pH 7.4 or 6.8 for 30 min. The CD47 siRNA and CCL25 were labeled with Cy5 (red) and Cy3 (yellow), respectively. The cell membrane and nuclei were stained with phalloidin–FITC (green) and 4′, 6-diamidino-2-phenylindole (DAPI) (blue), respectively. Scale bar, 10 μm. (E) Relative mRNA levels of CD47 in 4T1 cells upon treatment with NP-siCD47/CCL25 and other controls at pH 7.4 or 6.8 for 24 hours were assayed by quantitative real-time PCR. The siRNA concentration was 100 nM. The data were averaged from two independent experiments ± SEM. (F) CD47 protein levels were analyzed by Western blotting using anti-CD47 antibody. The 4T1 cells were treated with NP-siCD47/CCL25 and other controls at pH 7.4 or 6.8 for 48 hours. The siRNA concentration was 100 nM. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (G) The CD47 levels on cell surface of 4T1 cells continuously incubated with NP-siCD47/CCL25 and other controls at pH 7.4 or 6.8 for 4 days were determined by flow cytometry (n = 4). Data present means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0005.
We further assessed in vivo distribution of NP-siCD47/CCL25 in a mouse orthotopic 4T1 breast cancer model. Compared to free Cy5-siCD47 and Cy3-CCL25, NP-Cy5-siCD47/Cy3-CCL25 showed a significantly increased accumulation in tumors 24 hours after intravenous injection (Fig. 3A and fig. S7A). A similar Cy5-siCD47 distribution over the tumor was observed between NP-Cy5-siCD47– and NP-Cy5-siCD47/Cy3-CCL25–treated mice (Fig. 3, A and B), indicating that loading CCL25 onto the nanoparticles had no significant impact on the ability of PPC-DA to respond to tumor acidity, due to the ability of passive tumor-targeted drug delivery of nanoparticles through the enhanced permeability and retention effect (29). Among the other organs evaluated, Cy5-siCD47 was found predominantly distributed in kidneys in NP-Cy5-siCD47– and NP-Cy5-siCD47/Cy3-CCL25–treated mice, whereas Cy3-CCL25 was preferentially accumulated in the liver and kidneys in mice treated with NP-Cy5-siCD47/Cy3-CCL25 (fig. S7B). Furthermore, CLSM images revealed a markedly enhanced cellular uptake of Cy5-siCD47 and extracellular distribution of Cy3-CCL25 in NP-Cy5-siCD47/Cy3-CCL25–treated tumors compared to tumors treated with free Cy5-siCD47 and Cy3-CCL25 (Fig. 3C). Tumors from mice treated with free Cy5-siCD47 and Cy3-CCL25 showed undetectable Cy5 or Cy3 fluorescence signals, and NP-Cy5-siCD47 treated tumors showed obvious intracellular Cy5 fluorescence. In contrast, strong Cy5 and Cy3 signals were detected in tumors from mice treated with NP-Cy5-siCD47/Cy3-CCL25, with Cy5-siRNA distributed in the cytoplasm and Cy3-CCL25 colocalized with cell membrane (Fig. 3C and fig. S7C). Lack of colocalization of Cy5-siCD47 and Cy3-CCL25 (Fig. 3, A and C) indicates that Cy5-CCL25 was released from NP-Cy5-siCD47/Cy3-CCL25 before engulfment by tumor cells. IHC analysis of tumor tissues from mice receiving multiple injections of nanoparticles (Fig. 3D) confirmed the presence of CCL25 proteins in the tumors (mainly in the extracellular matrix) from mice treated with NP-siCD47/CCL25 and NP-siNC/CCL25, but not those injected with NP-siNC, NP-siCD47, or phosphate-buffered saline (PBS) (Fig. 3E). Furthermore, IHC and flow cytometry confirmed the reduction of CD47 expression in tumors from mice treated with NP-siCD47/CCL25 or NP-siCD47 but not NP-siNC or NP-siNC/CCL25, compared to PBS-injected controls (Fig. 3, F and G). In line with the intratumoral CCL25 release, IHC revealed a notable increase in tumor-infiltrating CCR9+ cells in mice treated with NP-siNC/CCL25 compared to those treated with NP-siNC (fig. S3). Furthermore, flow cytometry revealed a significant increase in tumor-infiltrating CD8+ T cells (Fig. 3H), including tumor-infiltrating CCR9+CD8+ T cells (Fig. 3I), and a significant decrease in tumor-infiltrating myeloid (CD4−CD8−CD19−CD49b−) cells (Fig. 3H) in NP-siNC/CCL25–treated mice, compared to that in NP-siNC–treated mice. Intratumoral delivery of CCL25 did not promote the infiltration of CD4+ T cells, NK cells, or B cells into 4T1 tumors (Fig. 3, H and I). These data indicate that codelivery of siCD47 and CCL25 by NP-siCD47/CCL25 increases the frequency of CCR9+CD8+ T cells in tumors while reducing the expression of CD47 on tumor cells.
Fig. 3. NP-siCD47/CCL25 significantly down-regulates the expression of CD47 in 4T1 tumor cells and enhances the infiltration of CCR9+ cytotoxic T cells into tumor tissues.
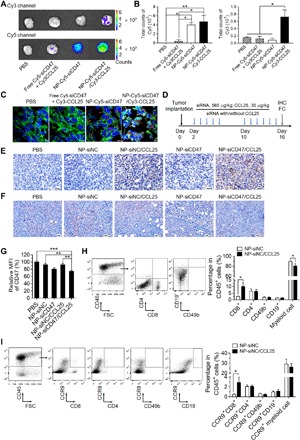
(A) Fluorescent images of 4T1 tumors harvested at 24 hours after intravenous injection of PBS, free Cy5-siCD47 + Cy3-CCL25, NP-Cy5-siCD47, and NP-Cy5-siCD47/Cy3-CCL25. The CD47 siRNA and CCL25 were labeled with Cy5 and Cy3, respectively. (B) Corresponding quantification of Cy5 (left) and Cy3 (right) signals in tumor tissues in (A). (C) CLSM images showed the distribution and cellular uptake of Cy5-labeled CD47 siRNA (red arrowheads) and Cy3-labeled CCL25 (yellow arrowheads) in tumor tissues in (A). The cell membrane and nuclei were stained with phalloidin-FITC (green) and DAPI (blue), respectively. Scale bar, 5 μm. (D) Schedule of treatment of the tumor-bearing mice with drug-loaded nanoparticles. CD47 siRNA or negative control siRNA-loaded nanoparticle (NP-siCD47 or NP-siNC), siRNA and CCL25 coloaded nanoparticle (NP-siCD47/CCL25 or NP-siNC/CCL25), or PBS was introduced via tail vein injection from the second day after 4T1 tumor implantation (3 × 105 cells per mouse). The doses of siRNA and CCL25 were 565 and 35 μg/kg in each injection, respectively. (E and F) IHC analysis of CCL25 (E) and CD47 expression (F) in tumor tissues treated by PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 as scheduled in (D). (G) MFI of CD47 in CD45− cells in tumors harvested from tumor-bearing mice after injection of PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 as schedule in (D) (n = 7 to 9 per group). (H) Representative flow cytometry plots demonstrating the CD8+, CD4+, CD19+, and CD49b+ cells in CD45+cells in tumors harvested from tumor-bearing mice after NP-siNC or NP-siNC/CCL25 injection as scheduled in (D) [(H), left]. Frequencies of CCR9+ cells, CD8+ T cells, CD4+ T cells, CD49b+ NK cells, CD19+ B cells, and myeloid cells in tumor-infiltrating CD45+ cells harvested from tumor-bearing mice after NP-siNC or NP-siNC/CCL25 injection as scheduled in (D) (n = 8 to 9 per group) [(H), right]. FSC, forward scatter. (I) Representative flow cytometry plots exhibiting CCR9+ immune subsets in CD45+ cells in tumors treated by NP-siNC or NP-siNC/CCL25 as scheduled in (D) (n = 8 to 9 per group) [(I), left)]. Frequencies of tumor-infiltrating CCR9+CD8+ T cells, CCR9+CD4+ T cells, CCR9+CD49b+ NK cells, CCR9+CD19+ B cells, and CCR9+ myeloid cells in CD45+ cells in tumors treated by NP-siNC or NP-siNC/CCL25 as scheduled in (D) (n = 8 to 9 per group) [(I), right]. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
NP-siCD47/CCL25 significantly inhibits TNBC tumor growth and metastasis through a T cell–dependent antitumor immunity
We then determined whether down-regulated CD47 expression and increased CCR9+CD8+ T cell infiltration in tumors may act synergistically to improve antitumor responses in mice treated with NP-siCD47/CCL25. Briefly, BALB/c mice were inoculated with 4T1 tumor cells and treated with intravenous injection of PBS, NP-siNC, NP-siCD47, NP-siNC/CCL25, or NP-siCD47/CCL25 for a total of 11 times starting from the second day after tumor inoculation (Fig. 4, A and C to M) or 7 times, starting from tumor volumes reaching an average of about 50 mm3 (Fig. 4B). Compared to PBS, administration of NP-siCD47/CCL25 significantly delayed tumor formulation (Fig. 4A) and even reduced the tumor growth rate in mice with established tumors (Fig. 4B). However, NP-siNC/CCL25 and NP-siNC did not affect the 4T1 tumor growth, while NP-siCD47 slightly inhibited the tumor growth in the case of early and long-term treatment (Fig. 4A). All the mice survived throughout the experimental period without body weight loss (fig. S8). Tumor metastasis was further evaluated in a stable luciferase-expressing 4T1 (4T1-luc) tumor model. Treatment with NP-siCD47/CCL25 not only significantly inhibited local tumor growth (fig. S9A) but also substantially suppressed tumor metastasis (Fig. 4C). In vivo imaging at day 30 posttumor challenge showed remarkable reduction in distant metastases in mice receiving NP-siCD47/CCL25 (Fig. 4C). Histological examination was performed to visualize the microscopic tumor lesions in the lung. Both the number and size of the lung metastases were remarkably reduced in NP-siCD47/CCL25–treated mice compared to other groups (Fig. 4D). Autopsy revealed that the number of metastatic tumor nodules in the lung was significantly reduced in NP-siCD47/CCL25–injected mice compared to those treated with PBS, NP-siNC, NP-siCD47, or NP-siNC/CCL25 (Fig. 4E and fig. S9B).
Fig. 4. NP-siCD47/CCL25 significantly inhibits 4T1 tumor growth and metastasis through enhanced antitumor immune responses.
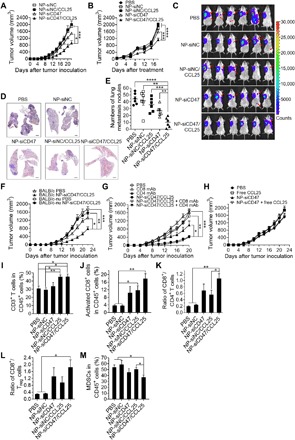
(A) Tumor growth curves. NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 were given by intravenous injection 11 times according to the schedule in Fig. 3D (n = 6 per group). (B) BALB/c mice were inoculated with 4T1 tumor cells; and PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, or NP-siCD47/CCL25 were given by intravenous injection every day seven times when the tumor volume reached about 50 mm3 (n = 7 per group). The doses of siRNA and CCL25 for each injection were 565 and 35 μg/kg, respectively. Tumor volumes at the indicated times after the first treatment are shown. (C) Tumor metastasis were monitored by noninvasive bioluminescence imaging at the 30th day after mammary fat pad injection of 3 × 105 4T1-luc cells. The mice were treated with NP-siCD47/CCL25 and other controls as the same as in (A). Red arrows, metastatic lesions of 4T1-luc tumors in the lung and abdomen. (D and E) The representative H&E staining of lung sections (D) and number of metastatic nodules in the lungs (E) harvested from the mice at the end of the tumor metastasis experiment. Scale bar, 1 mm. (F) Tumor growth was measured in BALB/c and BALB/c-nu mice from the day after tumor inoculation. NP-siCD47/CCL25 and PBS control were given by intravenous injection as the same as in (A). (G) BALB/c mice were inoculated with 4T1 tumor, followed by treatment with PBS, NP-siCD47/CCL25 (as detailed in Fig. 3D), anti-mouse CD8α (53-6.7, 12.5 mg/kg), anti-mouse CD4 (GK1.5, 12.5 mg/kg), or with NP-siCD47/CCL25 and anti-mouse CD8 or anti-mouse CD4 (anti-mouse CD8 or CD4 antibodies were given by intraperitoneal injection on days −2, 0, and 10 with respect to tumor cell inoculation). Tumor size was measured every other day. (H) Tumor growth was measured in BALB/c mice from the day after tumor inoculation. PBS, free CCL25, NP-siCD47, and NP-siCD47 and free CCL25 were given by intravenous injection as the same as in (A). (I to M) Frequencies of CD3+ T cells (I), activated (CD44hiCD62L−) CD8+ T cells (J), MDSCs (CD11b+Gr-1+) (M) in tumor-infiltrating CD45+ cells, and the ratio of CD8+/CD4+ (K) and CD8+/Treg cells (L) in tumors were measured at the end of the tumor inhibition experiment (n = 4 to 5 per group). The tumor-bearing mice were treated as the same as in (A). Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To determine the roles of T cells in the antitumor responses observed in NP-siCD47/CCL25–treated mice, we compared 4T1 tumor growth in BALB/c mice and T cell–deficient nude mice (on the BALB/c background) following administration of NP-siCD47/CCL25. Treatment with NP-siCD47/CCL25 significantly inhibited tumor growth in BALB/c mice, but not in nude mice, compared to PBS controls (Fig. 4F). Furthermore, the antitumor effect of NP-siCD47/CCL25 was completely inhibited in CD8+ T cell–depleted 4T1 tumor–bearing mice but slightly enhanced in CD4+ T cell–depleted tumor-bearing mice (Fig. 4G and fig. S10A). Meanwhile, NP-siCD47 combined with CCL25 treatment did not lead to tumor growth inhibition (Fig. 4H). These results demonstrate that the antitumor responses induced by NP-siCD47/CCL25 were CD8+ T cell dependent. Thus, we further assessed the effect of NP-siCD47/CCL25 on tumor-infiltrating immune cells. Significant increases in CD3+ TILs and activated (CD44hiCD62L−) CD8+ TILs were detected in mice treated with NP-siNC/CCL25 or NP-siCD47/CCL25 but not in mice treated with NP-siCD47, compared to PBS- and NP-siNC–injected controls (Fig. 4, I and J). Significant increases in CD8+/CD4+ T cell ratios (Fig. 4K) and CD8+/Treg cell ratios (Fig. 4L) were also detected in mice treated with NP-siCD47/CCL25 compared to those injected with PBS or NP-siNC. Treatment with NP-siCD47/CCL25 significantly increased the CD8+/CD4+ T cell ratios in tumors more than NP-siNC/CCL25 did (Fig. 4K). There was a tendency toward increases in the levels of activated (CD44hiCD62L−) CD8+ T cells (Fig. 4J), CD8+/CD4+ T cell ratios (Fig. 4K), and CD8+/Treg cell ratios (Fig. 4L) in NP-siCD47–injected tumors and in the levels of CD8+/CD4+ T cell ratios (Fig. 4K), and CD8+/Treg cell ratios (Fig. 4L) in NP-siNC/CCL25 injected tumors, compared to those injected with PBS or NP-siNC. In addition, the immunosuppressive microenvironment in tumor was modulated by administration of NP-siCD47/CCL25. The percentage of myeloid-derived suppressor cells (MDSCs, CD11b+Gr1+), an important cellular component of the immunosuppressive tumor microenvironment, was also significantly decreased after NP-siCD47/CCL25 treatment (Fig. 4M). Therefore, a strong synergistic antitumor effect can be achieved by simultaneously delivering CCL25 and siCD47 into the tumor.
NP-siCD47/CCL25 significantly improves the potency of TNBC immunotherapy through combination with PD-1/PD-L1 blockade
Although the 4T1 tumor cells are PD-L1+ (fig. S4, right), like the patient TNBCs (30, 31), blockade of PD-1/PD-L1 was poorly effective in a 4T1 tumor model (fig. S10B) (32, 33), so this model represents a class of patients that is extremely insensitive to immunotherapy. Since the effect of PD-1/PD-L1 blockades is highly dependent on TIL numbers and tumor-associated myeloid cell profiles (33–35), we sought to improve antitumor responses by combined treatment with anti–PD-L1 antibodies and NP-siCD47/CCL25. Flow cytometric analysis revealed that 4T1 tumor cells from all nanoparticle-treated groups expressed similar levels of PD-L1 comparable to that of PBS controls (Fig. 5A). As expected, significant antitumor responses were detected in mice treated with NP-siCD47/CCL25 alone, but not in those treated with NP-siNC/CCL25 or anti–PD-L1 antibodies alone compared to PBS controls (Fig. 5B). Combination of anti–PD-L1 antibodies with NP-siNC/CCL25 significantly inhibited 4T1 tumor growth (Fig. 5B) and prolonged the overall survival (Fig. 5C) to a level comparable to mice treated with NP-siCD47/CCL25. Further improvement in antitumor responses was detected in mice treated with anti–PD-L1 antibodies and NP-siCD47/CCL25, as shown by more extensive delay of tumor formation (Fig. 5B), inhibition of tumor growth (Fig. 5B), and prolongation of the overall survival (Fig. 5C). These results indicate that the antitumor effect of anti–PD-L1 antibodies can be enhanced by promoting tumor infiltration of CCR9+CD8+ T cells and resetting the profiles or function of tumor-infiltrating myeloid cells via targeting CD47–signal regulatory protein-α (SIRPα) signaling. The data also demonstrate that the antitumor activity of CCR9+ cytotoxic TILs is highly susceptible to inhibition by the tumor microenvironment, which is in agreement with the increased PD-1 expression on CCR9+CD8+ T cells in the 4T1 tumor (Fig. 1, K and L). Similar results were observed in mice treated with anti–PD-1 antibodies. Treatment with anti–PD-1 antibodies did not induce detectable antitumor responses in 4T1 tumor–bearing mice compared to PBS-treated controls, as shown by comparable tumor formation time (Fig. 5D), tumor growth (Fig. 5D), and overall survival (Fig. 5E) between the two groups of mice. However, anti–PD-1 antibodies induced significant antitumor responses in mice that were simultaneously treated with NP-siNC/CCL25 and did so to a significantly greater extent in those that received NP-siCD47/CCL25 simultaneously (Fig. 5, D and E). Notably, the primary tumor volumes in NP-siCD47/CCL25 + PD-L1/PD-1 monoclonal antibody (mAb)–treated mice at death were, in general, smaller than those in other groups (Fig. 5, B and D), suggesting that their death is predominantly associated with the metastatic burden. Together, these results demonstrate the great potential of NP-siCD47/CCL25 to improve the potency of TNBC immunotherapy using PD-1/PD-L1 blocking antibodies.
Fig. 5. NP-siCD47/CCL25 significantly improves the antitumor efficiency of PD-L1 and PD-1 antibodies on 4T1 tumors.
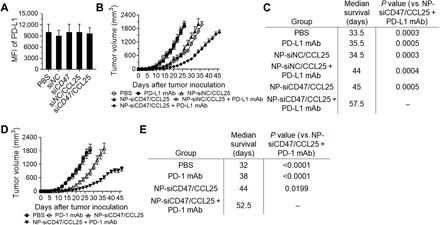
(A) Surface PD-L1 expression on CD45− tumor cell was measured in the tumor from mice treated by PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 according to the schedule in Fig. 3C (n = 6 to 8 per group). The doses of siRNA and CCL25 for each injection were 565 and 35 μg/kg, respectively. (B) Tumor growth curves measured form the day of tumor inoculation up to the day when death was found in each group (n = 6 to 8 per group). PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 were given by intravenous injection on days 2 to 8 and 12 to 15; and PD-L1 antibodies were given by intraperitoneal injection on days 2, 4, 6, 8, and 12. The doses of siRNA, CCL25, and PD-L1 antibody for each injection were 565 and 35 μg/kg and 10 mg/kg per mouse, respectively. (C) Median survival time values of mice treated in (B) are shown in the table. (D) Tumor growth curves measured form the day of tumor inoculation up to the day when death was found in each group (n = 7 or 8 per group). PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 were given by intravenous injection on days 2 to 8 and 12 to 15; and PD-1 antibodies were given by intraperitoneal injection on days 2, 4, 6, 8, 12, 14, 16, and 18. The doses of siRNA, CCL25, and PD-1 antibody for each injection were 565 and 35 μg/kg and 12.5 mg/kg per mouse, respectively. (E) Median survival time values of mice treated in (D) are shown in the table. Data are presented as means ± SEMs.
DISCUSSION
Treatment of TNBC remains challenging, because the tumors are highly aggressive and lack therapeutic targets (2, 3). Among the therapeutic options for TNBC, immunotherapy is emerging as a promising approach, but clinical studies in patients with TNBC revealed unsatisfactory outcomes, which is largely attributed to the lack of TILs (36).
The interactions between chemokines and chemokine receptors are of major importance in guiding the recruitment of different lymphocytes into the tumor microenvironment (37). Recent studies indicate that CCL25/CCR9 interactions are critical components of an anti-inflammatory tissue response, including the small intestine and liver (19, 38), through regulating the migration, infiltration, and functions of CCR9+ T cells. Compared to CCR9− T cells, we found that CCR9+ T cells displayed a more activated phenotype both in vitro and in vivo, indicating that these T cells may possibly mediate stronger antitumor responses than other T cells when they infiltrate into tumor microenvironment. However, the role of CCL25/CCR9 axis in antitumor immune response is still unclear, because CCL25 is hardly expressed by most tumor cells (39, 40). In this study, we found that CCL25 is undetectable in both human TNBC samples and a mouse TNBC 4T1 tumor. We also found that, although tumor infiltration by CCR9+ T cells could be detected in CCL25− tumors, likely mediated by other chemotactic signals (e.g., CCL5/CXCR2 and CXCL10/CXCR3) (41, 42), TILs in 4T1 tumors consisted of a limited number of CCR9+ T cells. These results support the possibility that intratumoral delivery of CCL25 proteins may promote tumor infiltration by CCR9+ T cells and hence enhance cancer immunotherapy in TNBC.
The advent of nanomedicine marks an unparalleled opportunity to improve pharmacotherapeutic outcomes through specifically enhanced accumulation and controlled release of various drugs in the sites of the body where they are needed (43, 44). In our study, we successfully delivered CCL25 proteins into the extracellular environment of CCL25− mouse TNBC tumor using NP-siNC/CCL25, a tumor acidity–responsive nanoparticle delivery system. Treatment of 4T1 tumor–bearing mice with NP-siNC/CCL25 significantly increased tumor infiltration by CCR9+CD8+ T cells, indicating a critical role of CCR9/CCL25 signaling in CCR9+ T cell tumor infiltration. These findings also provide direct evidence for the feasibility and effectiveness of targeted delivery of protein drugs (such as chemokines) to the tumor stroma by acidity-responsive nanoparticles.
CD47, via interaction with myeloid inhibitory receptor SIRPα, mediates potent inhibition of macrophage phagocytosis (45) and dendritic cell activation (46). CD47 is overexpressed on a broad range of solid tumors, including breast cancer, contributing to their escape from immune surveillance and destruction by phagocytes (47). Blockade of the CD47/SIRPα interaction has been shown to augment macrophage phagocytosis of tumor cells (48) and promote T cell–dependent antitumor immunity (49). Because CD47 is ubiquitously expressed on normal cells, raising concern for the phagocytosis of red blood cells (RBCs) and platelets following systemic administration of anti-CD47 antibodies (11, 47, 50), intratumoral delivery of anti-CD47 antibodies (49) or siRNA (51) were attempted with encouraging results. Thus, we sought to improve CD47-targeted immunotherapy by promoting CCR9+ T cell tumor infiltration using a “smart” delivery system that can sequentially release CCL25 proteins and CD47 siRNA in tumor tissues. Given that the stably transduced luciferase gene may increase the immunogenicity of 4T1 cells (52), all in vivo experiments, except the metastasis studies, were carried out with the unmodified 4T1 tumor model. Intratumoral delivery of CCL25 or CD47 siRNA alone failed to induce significant antitumor responses, suggesting that both the number of CCR9+ TILs and the tumor microenvironment are crucial in antitumor immunity. Treatment with NP-siCD47/CCL25, but not monotherapy with NP-siNC/CCL25, resulted in a significant increase in activated CD8+ TILs, and the ratios of CD8/CD4 TILs and CD8/Treg cells, and decrease in tumor-infiltrating MDSCs, indicating that intratumoral delivery of CCL25 is important for recruiting T cells into the tumor, but siCD47 plays an important role in altering T cell profiles and/or activation, and the immunosuppressive microenvironment. Accordingly, simultaneously delivering CD47 siRNA and CCL25 into the tumor significantly inhibited the tumor growth and metastasis in mice with established tumors, reflecting a synergistic effect between CCL25-mediated CD8 T cell tumor infiltration and CD47 siRNA-induced resetting of the tumor immune microenvironment.
As one of the most critical immune checkpoint treatments, PD-1/PD-L1 blockade has emerged as a highly effective therapeutic strategy for a subset of patients with solid tumors and hematologic malignancies (53–55). However, PD-1/PD-L1 blockades are usually ineffective for patients with TNBC (4, 5), assumably due to the lack of sufficient TILs. In addition, a recent study indicates that increasing the recruitment of CCR9+CXCR3+CD4+ T lymphocytes into mouse tumor beds can restore the antitumor efficacy of PD-1 blockade (56). Thus, we intended to achieve further improved outcome by combined treatment with NP-siCD47/CCL25 and anti–PD-1/anti–PD-L1 antibodies. We show that treatment with anti–PD-L1 or anti–PD-1 antibodies did not induce detectable antitumor responses when used alone but markedly enhanced the antitumor responses in NP-siCD47/CCL25–treated mice, leading to extensively delayed tumor onset, inhibited tumor growth, and prolonged overall survival. Our results indicate that the antitumor effect of anti–PD-L1 or anti–PD-1 antibodies can be enhanced by promoting tumor infiltration of CCR9+CD8+ T cells and resetting the profiles or function of tumor-infiltrating myeloid cells via targeting CD47-SIRPα signaling.
In summary, we have established a smart nanoparticle delivery system that can sequentially release CCL25 proteins and CD47 siRNA in tumor tissue in a mouse TNBC model. Using this system, we demonstrate that intratumoral delivery of CCL25 can significantly enhance immunotherapy, by means of blockades of the CD47/SIRPα and PD-1/PD-L1 signaling pathways, by promoting CCR9+CD8+ T cell tumor infiltration. Our results highlight the great potential of CCR9+ T cells in mediating antitumor immunity. Since CCL25, the only chemokine that interacts with CCR9, is not expressed in TNBC tumors, intratumoral delivery of CCL25 offers an effective strategy for selectively promoting CCR9+ T cell tumor infiltration and hence enhancing antitumor responses.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM), PBS, penicillin/streptomycin, l-glutamine, fetal bovine serum (FBS), Aqua Dead Cell Stain Kit, Lipofectamine 2000, TRIzol, and collagenase type IV were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Protamine and Histopaque-1083 were purchased from Sigma-Aldrich (St. Louis, MO, USA). R10 peptide (Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg) was purchased from ChinaPeptides Co. Ltd. (Hangzhou, China). RBC lysis buffer was purchased from Solarbio (Beijing, China). Recombinant mouse CCL25 was obtained from BioLegend (San Diego, CA, USA). The anti–PD-L1 mAb (clone no. 10F.9G2) and anti–PD-1 mAb (clone no. RMP1-14) used for in vivo blockade experiments were purchased from Bio X Cell (West Lebanon, NH, USA). siRNA-targeting mouse CD47 mRNA (antisense strand, 5′-UGGUGAAAGAGGUCAUUCCdTdT-3′), negative control siRNA with a scrambled sequence (antisense strand, 5′-ACGUGACACGUUCGGAGAAdTdT-3′), and fluorescently labeled negative control siRNA (FAM-siRNA and Cy5-siRNA) were synthesized by Suzhou Biosyntech Co. Ltd. (Suzhou, China).
Cell culture
The murine breast cancer 4T1 cells were obtained from The American Type Culture Collection and were maintained in DMEM (Carlsbad, CA, USA) supplemented with 10% FBS (Waltham, MA, USA) and 1% penicillin with streptomycin (complete DMEM) at 37°C with 5% CO2 humidified atmosphere. The cells were stably transduced with luciferase gene with a retroviral vector.
Animals and tumor model
Female BALB/c mice and BALB/c nude mice (6 to 7 weeks old) were purchased from Charles River Laboratories (Beijing, China) and raised in a specific pathogen–free environment with free access to food and water. All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Jilin University Animal Care and Use Committee. To set up the tumor-bearing mouse model, 4T1 cells (3 × 105) were suspended in 100 μl of PBS and were administered by subcutaneous injection into the armpit of the mice. Tumor volume (cubic millimeter) was determined by measuring length (l) and width (w) and calculated as V = lw2/2 (49).
Clinical samples
For experiments examining expression patterns of CCR9 in human peripheral blood T cells, we used samples of peripheral blood from patients with TNBC (n = 10) and disease-free female volunteers (n = 11). To examine the expression patterns of CCL25 and CCR9 in TNBC tumors, we used paraffin-embedded primary site specimens of TNBC (n = 9), a colon cancer and a fetal thymus obtained from the specimen library of the First Hospital of Jilin University. Eligible patients were women aged 50 to 72 years old and histologically documented TNBC (lack of estrogen and progesterone receptor expression and no overexpression of HER2, according to American Society of Clinical Oncology/College of American Pathologists guidelines) (57). For all the experiments using clinical samples, we had ensured blinded outcome assessment. All participants were given written informed consent, and all procedures in this study were approved by the Ethics Committee of the First Hospital of Jilin University.
Preparation and characterization of CD47 siRNA- and CCL25-loaded nanoparticles
The PPC-DA was synthesized as described in a previous study (58). First, 300 pmol CD47 siRNA was added into the PBS solution containing 144 μg of protamine and 48 μg of R10 peptide and incubated for 20 min at room temperature (RT) to form a positively charged core. Then, 90 μl of PPC-DA (5 mg/ml in PBS) was added into the protamine/R10/siRNA solution to form the sheddable nanoparticle (NP-siRNA). Last, 300-ng mouse CCL25 was added into the sheddable nanoparticle and incubated at RT for 20 min to form the nanoparticle carrying CCL25 and CD47 siRNA (NP-siCD47/CCL25). Size and ζ potential of the prepared nanoparticles were monitored by a dynamic light scattering (Zetasizer Nano ZS90, Malvern Instruments, Southborough, UK) at 25°C. The morphology of nanoparticles was observed using an FEI Tecnai G2-F20 Electron Microscope (FEI Company, Hillsboro, OR, USA) operated at 120 kV.
Loading efficiency of CCL25 and in vitro release
The unencapsulated Cy3-CCL25 was removed by dialyzing the NP-siCD47/Cy3-CCL25 solution against PBS through a dialysis tubing (molecular weight cutoff = 100 kDa). The amounts of Cy3-CCL25 in the dialysate and total Cy3-CCL25 added were calculated by measuring the fluorescence intensity of Cy3 using a Xenogen IVIS Lumina system (Caliper Life Sciences, USA). Encapsulation efficiency is calculated by (total drug added − free nonentrapped drug) divided by the total drug added.
To evaluate the release of CCL25 from NP-siCD47/CCL25, 1 ml of NP-siCD47/Cy3-CCL25 solution was dialyzed against 13 ml of PBS under pH 7.4 or 6.8. The dialysate was completely changed to fresh PBS with pH 7.4 or 6.8 at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 hours. The amount of Cy3-CCL25 in the dialysate was calculated by measuring the fluorescence intensity of Cy3 using a Xenogen IVIS Lumina system (Caliper Life Sciences, USA).
Cellular uptake study
4T1 cells were seeded in 24-well plates at a density of 1 × 105 cells per well with 500 μl of complete DMEM and incubated overnight at 37°C incubator with 5% CO2. Nanoparticles loaded with FAM-siRNA (150 pmol) and Cy3-CCL25 (150 ng) in 500 μl of DMEM (pH 7.4 or 6.8) were added into each well. After incubation at 37°C for 30 min, the cells were analyzed by flow cytometry.
For fluorescence microscopy observation, 1 × 105 4T1 cells were cultured in 24-well plates containing a coverslip and 500 μl of complete DMEM overnight. Then, the cells were incubated with NP-Cy5-siCD47/Cy3-CCL25 suspended in complete DMEM (pH 7.4 or 6.8) at a concentration of 300 nM Cy5-siCD47 and 300 ng/ml Cy3-CCL25 at 37°C for 30 min. The medium was removed, and the cells were rinsed twice with cold PBS, followed by fixation with paraformaldehyde for 20 min at 37°C and perforation with 0.1% Triton X-100 for 10 min at RT. The cells were further stained with phalloidin–fluorescein isothiocyanate (FITC) (Sigma-Aldrich, St. Louis, MO, USA) and cytoskeleton and 4′, 6-diamidino-2-phenylindole (DAPI, Invitrogen, Waltham, MA, USA) according to the standard protocols provided by the suppliers. The cellular uptake of nanoparticles was visualized by a laser scanning confocal microscopy (LSM 880, Zeiss, Jena, Germany).
In vitro CD47 gene silencing with NP-siCD47/CCL25
4T1 cells were seeded in 24-well plates at a density of 3 × 104 cells per well and cultured at 37°C overnight. Cells were transfected with NP-siCD47/CCL25 and other controls in DMEM (pH 7.4 or 6.8) at an siRNA dose of 100 nM once a day four times. DMEM- and Lipofectamine 2000–transfected CD47 siRNAs were used as negative control and positive control, respectively. After 6 hours of incubation, the medium in each well was replaced with complete DMEM. Twenty-four hours after the last transfection, the cells were harvested and the expression of CD47 was determined by flow cytometry, quantitative real-time PCR, and Western blots.
Quantitative real-time PCR analysis
Total RNA from NP-siCD47/CCL25 and other control transfected cells was extracted using TRIzol. cDNA was synthesized using TransScript One-Step genomic DNA Removal and cDNA Synthesis SuperMix Kit (TransGen Biotech, Beijing, China). To measure the expression level of CD47 mRNA, cDNA was subjected to qPCR analysis targeting CD47 and β-actin using SYBR Premix Ex Taq II kit (Takara, Dalian, China). The quantitative real-time PCR was performed using the StepOnePlus Real-Time PCR system (Applied Biosystems), and cycle threshold (Ct) values were calculated with the StepOne Software (Applied Biosystems). The primers used for mouse CD47 and β-actin were as follows: CD47, 5′-CAAAACTACCACATTCCCTACCC-3′ (forward) and 5′-ACATACAAACACGCCCACAAAC-3′ (reverse); and β-actin, 5′-TTCAACACCCCAGCCATG-3′ (forward) and 5′-CCTCGTAGATGGGCACAGT-3′ (reverse).
Western blotting analysis
Whole-cell lysates were prepared using radioimmunoprecipitation assay buffer with 1% phenylmethylsulfonyl fluoride (Beyotime, Shanghai, China). After incubation on ice for 30 min, the lysates were collected after centrifuging at 12,000 rpm for 20 min at 4°C. The protein concentration of each sample was determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA). Proteins were separated by electrophoresis on 10% SDS–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane. Total CD47 was detected by blotting with polyclonal rabbit anti-CD47 antibody (1:2000) and was visualized with peroxidase-conjugated goat anti-rabbit secondary and enhanced chemiluminescence substrate (Pierce, Rockford, IL, USA).
Tumor accumulation and tissue distribution of NP-siCD47/CCL25
When 4T1 tumors reached to about 300 mm3, the tumor-bearing mice were administrated with NP-Cy5-siCD47/Cy3-CCL25, NP-Cy5-siCD47, free Cy5-siRNA + Cy3-CCL25, or PBS by intravenous injection. The doses of siRNA and CCL25 were 4.7 mg/kg and 294 μg/kg, respectively. The fluorescent image acquisition was performed at 24 hours after injection using the Xenogen IVIS Lumina system (Caliper Life Sciences, USA). Data were analyzed by the Living Image software (Caliper Life Sciences).
The distributions of Cy5-siRNA and Cy3-CCL25 in tumor tissues and tumor cells were analyzed using CLSM. The 4T1 tumors treated with NP-Cy5-siCD47/Cy3-CCL25, NP-Cy5-siCD47, free Cy5-siRNA + Cy3-CCL25, or PBS were fixed in 4% paraformaldehyde and immersed using 30% sucrose solution. Tumor tissues were sectioned (6 μm thick), counterstained with DAPI and phalloidin-FITC, and imaged using a laser scanning confocal microscopy (LSM 880).
Treatment of tumor-bearing mice with NP-siCD47/CCL25
Tumor-bearing BALB/c or BALB/c nude mice were prepared by injection of 3 × 105 4T1 cells into the mammary fat pad and were given NP-siNC, NP-siNC/CCL25, NP-siCD47, NP-siCD47/CCL25, or combined with free CCL25 by intravenous injection 11 times. Although significant body weight lost was not observed following daily injection of NP-siCD47/CCL25, the animals developed shortness of breath immediately following injection, if have been treated consecutively for several days. Thus, the administration of NP-siCD47/CCL25 and other controls was performed on days 2 to 6 and 10 to 15. The doses of siRNA and CCL25 for each injection were 565 and 35 μg/kg, respectively. To combinate with PD-L1/PD-1 antibodies therapy, tumor-bearing mice were further given PD-L1 antibodies on days 2, 4, 6, 8, and 12 or PD-1 antibodies on days 2, 4, 6, 8, 12, 14, 16, and 18 by intraperitoneal injection. The doses of PD-L1 and PD-1 antibody for each injection were 10 and 12.5 mg/kg per mouse, respectively. To deplete CD4+ and CD8+ T cells, mice were given anti-mouse CD8 (clone no. 53-6.7, Bio X Cell) or anti-mouse CD4 (clone no. GK1.5, Bio X Cell) antibodies both at a dose of 12.5 mg/kg by intraperitoneal injection on days −2, 0, and 10 with respect to tumor cell inoculation. The depletion of CD4+ or CD8+ T cells in the peripheral blood and spleen was confirmed by flow cytometry on days 2 and 10 with respect to tumor cell inoculation. Tumor volumes and the mouse weights were measured every other day. In the established tumor growth inhibition experiment, 4T1 tumor–bearing mice were administrated with NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 every day seven times, starting from the tumor volumes that reached about 50 mm3. The doses of siRNA and CCl25 were the same as above. Tumor volumes were measured every 3 days. The mice were euthanized 24 hours after the last injection, and the immune cells in tumor tissues were measured by flow cytometry.
In the 4T1 metastasis experiment, BALB/c mice were orthotopically implanted with 3 × 105 4T1-luc cells and treated with NP-siCD47/CCL25 and other controls on days 2 to 6 and 10 to 15. At selected time points, mice were given an intravenous injection of d-luciferin firefly potassium salt at a dose of 150 mg/kg and imaged using the Xenogen IVIS system.
In vivo immune response analysis
4T1 tumor–bearing mice were given PBS, NP-siNC, NP-siNC/CCL25, NP-siCD47, and NP-siCD47/CCL25 11 times by intravenous injection. At the end of treatment, tumors were harvested to analyze the phenotypes of immune cells by flow cytometry.
Detection of CCR9 expression on lymphocytes
Normal and 4T1 tumor–bearing mice were euthanized for the collection of tumors and spleens. Spleens were gently grinded into single-cell suspension with PBS, and RBCs were removed by RBC lysis buffer (33). CCR9−CD3+ splenic cells harvested from normal BALB/c mice were plated into a 96-well plate (2 × 105 cells per well) and stimulated with plate-bound anti-mouse CD3ε mAb (2 μg/ml; 145-2C11, BioLegend) and soluble anti-mouse CD28 mAb (1 μg/ml; 37.51, BioLegend), as described in a previous study (59). The expression of CCR9 on these T cells was analyzed by flow cytometry after incubation for 72, 96, or 120 hours.
Flow cytometric analysis
Flow cytometry was used to determine the phenotypes of human lymphocytes and mouse immune cells using various combinations of the following fluorochrome-conjugated mAbs: anti-human CCR9 (L053E8), CD4 (OKT4), CD8 (HIT8a), CD45RO (UCLH1), CD45RA (HI100), and CD71 (CY1G4) from BioLegend; CD3 (SK7) from BD Pharmingen; anti-mouse CCR9 (CW-1.2), CD47 (miap301), PD-1 (29F.1A12), PD-L1 (10F.9G2), F4/80 (BM8), CD49b (DX5), CD19 (6D5), CD3 (17A2), CD4 (GK1.5), CD8 (53-6.7), CD44 (IM7), CD62L (MEL-14), CD45 (30-F11), CD11b (M1/70), CD11c (N418), GR-1 (RB6-8C5), I-A/I-E (M5/114.15.2), CD25 (3C7), Foxp3 (MF-14), and CD206 (C068C2) from BioLegend. Isotype controls used include rat immunoglobulin G2B (IgG2b) (RTK4530), rat IgG2a (RTK2758), and mouse IgG2a (MOPC-173) from BioLegend. For cell isolation from tumor tissues, the tumors were diced into small pieces (~1 mm3) and incubated with digestion solution [0.1% (w/v) type IV collagenase and 0.1% (w/v) deoxyribonuclease I] at 37°C for 30 min in a water bath shaker. Spleens harvested from normal mice or 4T1 tumor–bearing mice were triturated and incubated with RBC lysing buffer to remove RBC. PBMCs of healthy volunteers and patients with TNBC were isolated by incubating with RBC lysing buffer. All the samples were filtered through a nylon mesh filter (40 μm) to provide a single-cell suspension and stained with fluorochrome-conjugated mAb against respective surface antigens. For intracellular staining, cells were first stained for surface antigens, fixed, and permeabilized with intracellular fixation and permeabilization buffer (eBioscience), followed by staining with fluorochrome-conjugated mAb against respective intracellular proteins. All samples were collected on a fluorescence-activated cell sorter (LSRFortessa, Becton Dickinson) and analyzed by FlowJo software (TreeStar).
Histopathology and IHC
The lungs were collected from 4T1-luc tumor-bearing mice at the end of the tumor metastasis experiment, fixed in 4% paraformaldehyde, and sliced into 10-μm-thick sections. Lung sections were stained with hematoxylin and eosin (H&E). IHC was performed on paraffin-embedded sections of human or mouse thymus and tumor tissues to stain for CCL25, CCR9, and CD47 expression. Briefly, tissue sections were stained with anti-human CCL25 and CCR9 from Abcam (Cambridge, UK) or anti-mouse CCL25 (Leinco Technologies, MS, USA), CCR9 (Thermo Fisher Scientific), and CD47 (Abcam). All the slides were observed and photographed with a laser scanning confocal microscopy (LSM 880).
Statistical analysis
Statistics were performed on GraphPad Prism 6, and unpaired Student’s t tests or one-way analysis of variance (ANOVA) were used to compare the paired and unpaired analyses. The statistical evaluation of mouse survival was performed using the Kaplan-Meier test. P values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from Chinese MOST (2015CB964400), National Key Research and Development Program of China (2017YFA0208100), NSFC (81422026, 81571798, 91642208, and 81871478), Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R24), Special Fund Project of Jilin Province Provincial Industrial Innovation (2018C052-1), and Jilin Scientific and Technological Development Program (20160101028JC). Author contributions: H.C. designed, performed, and analyzed data from most of the studies, as well as helped write the manuscript with input from all authors. X.Y. and J.L. conducted chemical synthesis of PPC-DA. X.C. assisted in orthotopic tumor inoculation. C.W., X.Wu., K.M., X.M., X.S., X.Wa., S.L., and S.Z. helped in the performance of flow cytometry studies. J.W. aided in immunofluorescence staining experiments. G.Z. and F.L. helped with human sample preparation and in vitro T cell stimulation study. J.S. provided collecting clinical samples. Y.S., T.S., and Y.-G.Y. conceived, designed, and supervised all studies and participated in the drafting and editing of the manuscript. All authors contributed to the critical review of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax4690/DC1
Fig. S1. CCR9 expression in human peripheral blood T cells and mouse CD11b+ cells.
Fig. S2. CCL25 expression in thymus and TNBCs and CCR9 expression in human colon cancer and TNBC.
Fig. S3. Distribution of CCR9+ cells in tumor tissue.
Fig. S4. Expression of CCR9, CD47, and PD-L1 in 4T1 cells in vitro and in vivo.
Fig. S5. Characterization of NP-siCD47/CCL25 under neutral and tumoral acidic pH environments.
Fig. S6. Cellular uptake of CD47 siRNA and CCL25 by 4T1 cells in vitro and down-regulation of the surface CD47 expression in 4T1 cells after treated with NP-siCD47/CCL25.
Fig. S7. Tissue distribution of NP-siCD47/CCL25 in vivo.
Fig. S8. Body weights of 4T1 tumor–bearing mice.
Fig. S9. NP-siCD47/CCL25 significantly inhibits tumor growth and metastasis in 4T1-luc tumor-bearing mice.
Fig. S10. Flow cytometric analysis of T cell depletion in anti-CD8 or anti-CD4 antibody–treated mice and the antitumor effects of anti–PD-1 antibodies in the 4T1 tumor model.
REFERENCES AND NOTES
- 1.Foulkes W. D., Smith I. E., Reis-Filho J. S., Triple-negative breast cancer. N. Engl. J. Med. 363, 1938–1948 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Dent R., Trudeau M., Pritchard K. I., Hanna W. M., Kahn H. K., Sawka C. A., Lickley L. A., Rawlinson E., Sun P., Narod S. A., Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 13, 4429–4434 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Cardoso F., Senkus E., Costa A., Papadopoulos E., Aapro M., André F., Harbeck N., Aguilar Lopez B., Barrios C. H., Bergh J., Biganzoli L., Boers-Doets C. B., Cardoso M. J., Carey L. A., Cortés J., Curigliano G., Diéras V., El Saghir N. S., Eniu A., Fallowfield L., Francis P. A., Gelmon K., Johnston S. R. D., Kaufman B., Koppikar S., Krop I. E., Mayer M., Nakigudde G., Offersen B. V., Ohno S., Pagani O., Paluch-Shimon S., Penault-Llorca F., Prat A., Rugo H. S., Sledge G. W., Spence D., Thomssen C., Vorobiof D. A., Xu B., Norton L., Winer E. P., 4th ESO–ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)†. Ann. Oncol. 29, 1634–1657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dirix L. Y., Takacs I., Jerusalem G., Nikolinakos P., Arkenau H. T., Forero-Torres A., Boccia R., Lippman M. E., Somer R., Smakal M., Emens L. A., Hrinczenko B., Edenfield W., Gurtler J., von Heydebreck A., Grote H. J., Chin K., Hamilton E. P., Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 167, 671–686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nanda R., Chow L. Q., Dees E. C., Berger R., Gupta S., Geva R., Pusztai L., Pathiraja K., Aktan G., Cheng J. D., Karantza V., Buisseret L., Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 34, 2460–2467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmid P., Adams S., Rugo H. S., Schneeweiss A., Barrios C. H., Iwata H., Diéras V., Hegg R., Im S. A., Shaw Wright G., Henschel V., Molinero L., Chui S. Y., Funke R., Husain A., Winer E. P., Loi S., Emens L. A.; IMpassion130 Trial Investigators , Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Adams S., Gray R. J., Demaria S., Goldstein L., Perez E. A., Shulman L. N., Martino S., Wang M., Jones V. E., Saphner T. J., Wolff A. C., Wood W. C., Davidson N. E., Sledge G. W., Sparano J. A., Badve S. S., Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 32, 2959–2966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loi S., Michiels S., Salgado R., Sirtaine N., Jose V., Fumagalli D., Kellokumpu-Lehtinen P. L., Bono P., Kataja V., Desmedt C., Piccart M. J., Loibl S., Denkert C., Smyth M. J., Joensuu H., Sotiriou C., Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 25, 1544–1550 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Homey B., Müller A., Zlotnik A., Chemokines: Agents for the immunotherapy of cancer? Nat. Rev. Immunol. 2, 175–184 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Peske J. D., Woods A. B., Engelhard V. H., Control of CD8 T-cell infiltration into tumors by vasculature and microenvironment. Adv. Cancer Res. 128, 263–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang H., Qiao J., Fu Y. X., Immunotherapy and tumor microenvironment. Cancer Lett. 370, 85–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaballos Á., Gutiérrez J., Varona R., Ardavin C., Márquez G., Cutting edge: Identification of the orphan chemokine receptor GPR-9-6 as CCR9, the receptor for the chemokine TECK. J. Immunol. 162, 5671–5675 (1999). [PubMed] [Google Scholar]
- 13.Papadakis K. A., Prehn J., Nelson V., Cheng L., Binder S. W., Ponath P. D., Andrew D. P., Targan S. R., The role of thymus-expressed chemokine and its receptor CCR9 on lymphocytes in the regional specialization of the mucosal immune system. J. Immunol. 165, 5069–5076 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Vicari A. P., Figueroa D. J., Hedrick J. A., Foster J. S., Singh K. P., Menon S., Copeland N. G., Gilbert D. J., Jenkins N. A., Bacon K. B., Zlotnik A., TECK: A novel CC chemokine specifically expressed by thymic dendritic cells and potentially involved in T cell development. Immunity 7, 291–301 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Wurbel M. A., Philippe J. M., Nguyen C., Victorero G., Freeman T., Wooding P., Miazek A., Mattei M. G., Malissen M., Jordan B. R., Malissen B., Carrier A., Naquet P., The chemokine TECK is expressed by thymic and intestinal epithelial cells and attracts double- and single-positive thymocytes expressing the TECK receptor CCR9. Eur. J. Immunol. 30, 262–271 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Uehara S., Grinberg A., Farber J. M., Love P. E., A role for CCR9 in T lymphocyte development and migration. J. Immunol. 168, 2811–2819 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Norment A. M., Bogatzki L. Y., Gantner B. N., Bevan M. J., Murine CCR9, a Chemokine Receptor for Thymus-Expressed Chemokine That Is Up-Regulated Following Pre-TCR Signaling. J. Immunol. 164, 639–648 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Wurbel M.-A., Malissen B., Campbell J. J., Complex regulation of CCR9 at multiple discrete stages of T cell development. Eur. J. Immunol. 36, 73–81 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Papadakis K. A., Prehn J., Moreno S. T., Cheng L., Kouroumalis E. A., Deem R., Breaverman T., Ponath P. D., Andrew D. P., Green P. H., Hodge M. R., Binder S. W., Targan S. R., CCR9–positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology 121, 246–254 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Papadakis K. A., Landers C., Prehn J., Kouroumalis E. A., Moreno S. T., Gutierrez-Ramos J. C., Hodge M. R., Targan S. R., CC chemokine receptor 9 expression defines a subset of peripheral blood lymphocytes with mucosal T cell phenotype and Th1 or T-regulatory 1 cytokine profile. J. Immunol. 171, 159–165 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Kim C. H., Rott L., Kunkel E. J., Genovese M. C., Andrew D. P., Wu L., Butcher E. C., Rules of chemokine receptor association with T cell polarization in vivo. J. Clin. Invest. 108, 1331–1339 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kathuria N., Kraynyak K. A., Carnathan D., Betts M., Weiner D. B., Kutzler M. A., Generation of antigen-specific immunity following systemic immunization with DNA vaccine encoding CCL25 chemokine immunoadjuvant. Hum. Vaccin. Immunother. 8, 1607–1619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun J., Evans-Marin H. L., Cao A. T., Yao S., Chen F., He C., Liu H., Wu W., Gonzalez M. G., Dann S. M., Cong Y., Unexpected regulatory role of CCR9 in regulatory T cell development. PLOS ONE 10, e0134100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGuire H. M., Vogelzang A., Ma C. S., Hughes W. E., Silveira P. A., Tangye S. G., Christ D., Fulcher D., Falcone M., King C., A subset of interleukin-21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 34, 602–615 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Jacquelot N., Enot D. P., Flament C., Vimond N., Blattner C., Pitt J. M., Yamazaki T., Roberti M. P., Daillère R., Vétizou M., Poirier-Colame V., Semeraro M., Caignard A., Slingluff C. L. Jr., Sallusto F., Rusakiewicz S., Weide B., Marabelle A., Kohrt H., Dalle S., Cavalcanti A., Kroemer G., Di Giacomo A. M., Maio M., Wong P., Yuan J., Wolchok J., Umansky V., Eggermont A., Zitvogel L., Chemokine receptor patterns in lymphocytes mirror metastatic spreading in melanoma. J. Clin. Invest. 126, 921–937 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffith J. W., Sokol C. L., Luster A. D., Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 32, 659–702 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Yang X.-Z., Du X.-J., Liu Y., Zhu Y.-H., Liu Y.-Z., Li Y.-P., Wang J., Rational design of polyion complex nanoparticles to overcome cisplatin resistance in cancer therapy. Adv. Mater. 26, 931–936 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Zhou K., Huang G., Hensley C., Huang X., Ma X., Zhao T., Sumer B. D., DeBerardinis R. J., Gao J., A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat. Mater. 13, 204–212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi J., Kantoff P. W., Wooster R., Farokhzad O. C., Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buisseret L., Garaud S., de Wind A., Van den Eynden G., Boisson A., Solinas C., Gu-Trantien C., Naveaux C., Lodewyckx J.-N., Duvillier H., Craciun L., Veys I., Larsimont D., Piccart-Gebhart M., Stagg J., Sotiriou C., Willard-Gallo K., Tumor-infiltrating lymphocyte composition, organization and PD-1/ PD-L1 expression are linked in breast cancer. Oncoimmunology 6, e1257452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali H. R., Glont S. E., Blows F. M., Provenzano E., Dawson S. J., Liu B., Hiller L., Dunn J., Poole C. J., Bowden S., Earl H. M., Pharoah P. D., Caldas C., PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann. Oncol. 26, 1488–1493 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Hirano F., Kaneko K., Tamura H., Dong H., Wang S., Ichikawa M., Rietz C., Flies D. B., Lau J. S., Zhu G., Tamada K., Chen L., Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096 (2005). [PubMed] [Google Scholar]
- 33.De Henau O., Rausch M., Winkler D., Campesato L. F., Liu C., Cymerman D. H., Budhu S., Ghosh A., Pink M., Tchaicha J., Douglas M., Tibbitts T., Sharma S., Proctor J., Kosmider N., White K., Stern H., Soglia J., Adams J., Palombella V. J., McGovern K., Kutok J. L., Wolchok J. D., Merghoub T., Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 539, 443–447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herbst R. S., Soria J.-C., Kowanetz M., Fine G. D., Hamid O., Gordon M. S., Sosman J. A., McDermott D. F., Powderly J. D., Gettinger S. N., Kohrt H. E. K., Horn L., Lawrence D. P., Rost S., Leabman M., Xiao Y., Mokatrin A., Koeppen H., Hegde P. S., Mellman I., Chen D. S., Hodi F. S., Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tumeh P. C., Harview C. L., Yearley J. H., Shintaku I. P., Taylor E. J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., West A. N., Carmona M., Kivork C., Seja E., Cherry G., Gutierrez A. J., Grogan T. R., Mateus C., Tomasic G., Glaspy J. A., Emerson R. O., Robins H., Pierce R. H., Elashoff D. A., Robert C., Ribas A., PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solinas C., Gombos A., Latifyan S., Piccart-Gebhart M., Kok M., Buisseret L., Targeting immune checkpoints in breast cancer: An update of early results. ESMO Open 2, e000255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagarsheth N., Wicha M. S., Zou W., Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 17, 559–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eksteen B., Grant A. J., Miles A., Curbishley S. M., Lalor P. F., Hübscher S. G., Briskin M., Salmon M., Adams D. H., Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J. Exp. Med. 200, 1511–1517 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svensson M., Agace W. W., Role of CCL25/CCR9 in immune homeostasis and disease. Expert Rev. Clin. Immunol. 2, 759–773 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Chen H. J., Edwards R., Tucci S., Bu P., Milsom J., Lee S., Edelmann W., Gümüs Z. H., Shen X., Lipkin S., Chemokine 25-induced signaling suppresses colon cancer invasion and metastasis. J. Clin. Invest. 122, 3184–3196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adler E. P., Lemken C. A., Katchen N. S., Kurt R. A., A dual role for tumor-derived chemokine RANTES (CCL5). Immunol. Lett. 90, 187–194 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Jin W. J., Kim B., Kim D., Park Choo H.-Y., Kim H.-H., Ha H., Lee Z. H., NF-κB signaling regulates cell-autonomous regulation of CXCL10 in breast cancer 4T1 cells. Exp. Mol. Med. 49, e295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu X., Ho W., Zhang X., Bertrand N., Farokhzad O., Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 21, 223–232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blanco E., Shen H., Ferrari M., Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oldenborg P. A., Zheleznyak A., Fang Y.-F., Lagenaur C. F., Gresham H. D., Lindberg F. P., Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Wang H., Wu X., Wang Y., Oldenborg P.-A., Yang Y.-G., CD47 is required for suppression of allograft rejection by donor-specific transfusion. J. Immunol. 184, 3401–3407 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willingham S. B., Volkmer J.-P., Gentles A. J., Sahoo D., Dalerba P., Mitra S. S., Wang J., Contreras-Trujillo H., Martin R., Cohen J. D., Lovelace P., Scheeren F. A., Chao M. P., Weiskopf K., Tang C., Volkmer A. K., Naik T. J., Storm T. A., Mosley A. R., Edris B., Schmid S. M., Sun C. K., Chua M.-S., Murillo O., Rajendran P., Cha A. C., Chin R. K., Kim D., Adorno M., Raveh T., Tseng D., Jaiswal S., Enger P. Ø., Steinberg G. K., Li G., So S. K., Majeti R., Harsh G. R., van de Rijn M., Teng N. N. H., Sunwoo J. B., Alizadeh A. A., Clarke M. F., Weissman I. L., The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 6662–6667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiskopf K., Jahchan N. S., Schnorr P. J., Cristea S., Ring A. M., Maute R. L., Volkmer A. K., Volkmer J.-P., Liu J., Lim J. S., Yang D., Seitz G., Nguyen T., Wu D., Jude K., Guerston H., Barkal A., Trapani F., George J., Poirier J. T., Gardner E. E., Miles L. A., de Stanchina E., Lofgren S. M., Vogel H., Winslow M. M., Dive C., Thomas R. K., Rudin C. M., van de Rijn M., Majeti R., Garcia K. C., Weissman I. L., Sage J., CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Invest. 126, 2610–2620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X., Pu Y., Cron K., Deng L., Kline J., Frazier W. A., Xu H., Peng H., Fu Y.-X., Xu M. M., CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 21, 1209–1215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Majeti R., Chao M. P., Alizadeh A. A., Pang W. W., Jaiswal S., Gibbs K. D. Jr., van Rooijen N., Weissman I. L., CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y., Xu Z., Guo S., Zhang L., Sharma A., Robertson G. P., Huang L., Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol. Ther. 21, 1919–1929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baklaushev V. P., Kilpeläinen A., Petkov S., Abakumov M. A., Grinenko N. F., Yusubalieva G. M., Latanova A. A., Gubskiy I. L., Zabozlaev F. G., Starodubova E. S., Abakumova T. O., Isaguliants M. G., Chekhonin V. P., Luciferase expression allows bioluminescence imaging but imposes limitations on the orthotopic mouse (4T1) model of breast cancer. Sci. Rep. 7, 7715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan S., Zhang C. W.-H., Gao G. F., Seeing is believing: Anti-PD-1/PD-L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Signal Transduct. Target. Ther. 1, 16029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Y., Pang Z., Ding N., Dong W., Ma W., Li Y., Du J., Liu Q., The efficacy and potential predictive factors of PD-1/PD-L1 blockades in epithelial carcinoma patients: A systematic review and meta analysis. Oncotarget 7, 74350–74361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenblatt J., Avigan D., Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 129, 275–279 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Routy B., Le Chatelier E., Derosa L., Duong C. P. M., Alou M. T., Daillere R., Fluckiger A., Messaoudene M., Rauber C., Roberti M. P., Fidelle M., Flament C., Poirier-Colame V., Opolon P., Klein C., Iribarren K., Mondragon L., Jacquelot N., Qu B., Ferrere G., Clemenson C., Mezquita L., Masip J. R., Naltet C., Brosseau S., Kaderbhai C., Richard C., Rizvi H., Levenez F., Galleron N., Quinquis B., Pons N., Ryffel B., Minard-Colin V., Gonin P., Soria J. C., Deutsch E., Loriot Y., Ghiringhelli F., Zalcman G., Goldwasser F., Escudier B., Hellmann M. D., Eggermont A., Raoult D., Albiges L., Kroemer G., Zitvogel L., Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Hammond M. E. H., Hayes D. F., Dowsett M., Allred D. C., Hagerty K. L., Badve S., Fitzgibbons P. L., Francis G., Goldstein N. S., Hayes M., Hicks D. G., Lester S., Love R., Mangu P. B., McShane L., Miller K., Osborne C. K., Paik S., Perlmutter J., Rhodes A., Sasano H., Schwartz J. N., Sweep F. C. G., Taube S., Torlakovic E. E., Valenstein P., Viale G., Visscher D., Wheeler T., Williams R. B., Wittliff J. L., Wolff A. C.; American Society of Clinical Oncology; College of American Pathologists , American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 134, e48–e72 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Yang X.-Z., Du J.-Z., Dou S., Mao C.-Q., Long H.-Y., Wang J., Sheddable ternary nanoparticles for tumor acidity-targeted siRNA delivery. ACS Nano 6, 771–781 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Li P., Spolski R., Liao W., Wang L., Murphy T. L., Murphy K. M., Leonard W. J., BATF–JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax4690/DC1
Fig. S1. CCR9 expression in human peripheral blood T cells and mouse CD11b+ cells.
Fig. S2. CCL25 expression in thymus and TNBCs and CCR9 expression in human colon cancer and TNBC.
Fig. S3. Distribution of CCR9+ cells in tumor tissue.
Fig. S4. Expression of CCR9, CD47, and PD-L1 in 4T1 cells in vitro and in vivo.
Fig. S5. Characterization of NP-siCD47/CCL25 under neutral and tumoral acidic pH environments.
Fig. S6. Cellular uptake of CD47 siRNA and CCL25 by 4T1 cells in vitro and down-regulation of the surface CD47 expression in 4T1 cells after treated with NP-siCD47/CCL25.
Fig. S7. Tissue distribution of NP-siCD47/CCL25 in vivo.
Fig. S8. Body weights of 4T1 tumor–bearing mice.
Fig. S9. NP-siCD47/CCL25 significantly inhibits tumor growth and metastasis in 4T1-luc tumor-bearing mice.
Fig. S10. Flow cytometric analysis of T cell depletion in anti-CD8 or anti-CD4 antibody–treated mice and the antitumor effects of anti–PD-1 antibodies in the 4T1 tumor model.