

Normal breast epithelia from BRCA2 mutation carriers display DNA damage and attenuated checkpoint responses.
Abstract
Women harboring heterozygous germline mutations of BRCA2 have a 50 to 80% risk of developing breast cancer, yet the pathogenesis of these cancers is poorly understood. To reveal early steps in BRCA2-associated carcinogenesis, we analyzed sorted cell populations from freshly-isolated, non-cancerous breast tissues of BRCA2 mutation carriers and matched controls. Single-cell whole-genome sequencing demonstrates that >25% of BRCA2 carrier (BRCA2mut/+) luminal progenitor (LP) cells exhibit sub-chromosomal copy number variations, which are rarely observed in non-carriers. Correspondingly, primary BRCA2mut/+ breast epithelia exhibit DNA damage together with attenuated replication checkpoint and apoptotic responses, and an age-associated expansion of the LP compartment. We provide evidence that these phenotypes do not require loss of the wild-type BRCA2 allele. Collectively, our findings suggest that BRCA2 haploinsufficiency and associated DNA damage precede histologic abnormalities in vivo. Using these hallmarks of cancer predisposition will yield unanticipated opportunities for improved risk assessment and prevention strategies in high-risk patients.
INTRODUCTION
Breast cancers arising in women who inherit heterozygous mutations in BRCA2 are associated with a high prevalence of genomic alterations and aggressive clinical behavior (1, 2). Because of the high risk of these cancers in BRCA2 mutation carriers, many such women elect to undergo bilateral mastectomy for breast cancer prevention. However, despite the unmet need for more effective breast cancer prevention approaches in this setting, the stepwise evolution from an otherwise normal BRCA2 heterozygous mutant (BRCA2mut/+) cell to an invasive malignancy has not been defined. Homozygous loss of BRCA2 is embryonically lethal (3–5), and acute loss in cultured cells rapidly leads to DNA damage and growth arrest or cell death (6–8). These observations suggest a multistep pathogenesis in which homozygous BRCA2 loss is not the earliest genetic event but rather that the wild-type BRCA2 allele may remain intact as early genetic changes accumulate. Critically, however, this scenario leaves unresolved the nature and enabling mechanism for early cancer evolution. Haploinsufficiency for BRCA2 has been proposed as a possible driver of early pathogenesis, but direct evidence for such an effect in the normal human mammary gland is inconsistent. Furthermore, heterozygous genetically engineered mouse models (GEMMs) of BRCA2 are not tumor prone and therefore represent a poor model of precancerous evolution in this setting (3–5, 8, 9). While the BRCA1 tumor suppressor shares many of these features (9, 10), the pathogenesis of BRCA1- versus BRCA2-associated breast cancers may differ in important ways, as the former are primarily hormone receptor (HR) and HER2-negative tumors, while the latter are primarily HR positive (11).
We sought to unveil the earliest steps in the pathogenesis of BRCA2-associated breast tumors through detailed analysis of histologically normal glands from women harboring germline deleterious mutations who elected to undergo bilateral prophylactic mastectomy. Genomic analysis of individual cells revealed frequent polyclonal chromosomal damage, which was most prevalent among the subset of epithelial cells that are the suspected cells of origin of these cancers. Corresponding defects in replication stress and DNA damage checkpoint responses in these same cells collectively define a previously unappreciated phenotype for BRCA2 that precedes histologic abnormalities in the human breast. The discovery of these precancerous hallmarks paves the way for improving clinical risk prediction and cancer prevention in this population.
RESULTS
Single-cell whole-genome analysis reveals subchromosomal aneuploidy in BRCA2mut/+ human primary breast epithelial cells
We carried out detailed analysis of noncancerous glands from BRCA2 carriers who elected to undergo bilateral prophylactic mastectomy, using as control tissues from women matched for age, menopausal status, and hormonal exposure electing cosmetic breast surgery (Fig. 1A and table S1). None of these women had a previous breast cancer diagnosis or chemotherapy exposure, and no occult cancers were detected upon histologic analysis of the tissues we analyzed (table S1). We used established markers to carry out flow cytometry–based isolation and sorting of the three major epithelial cell subpopulations: mature luminal (ML), luminal progenitor (LP), and basal epithelial cells (Fig. 1A). Notably, data from GEMMs and gene expression analyses of human tumors have suggested that the cell of origin of BRCA1-associated breast cancer is the LP cell (12, 13), while BRCA2-associated tumors may arise from an LP-related cell or a more ML cell (14).
Fig. 1. Single-cell whole-genome analysis of BRCA2mut/+ human primary breast epithelial cells.
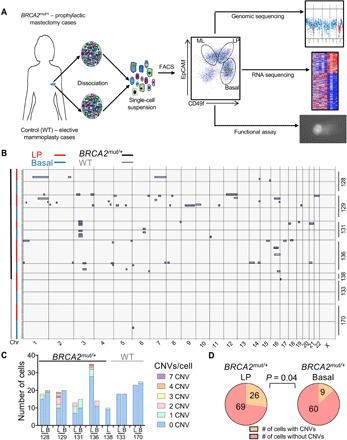
(A) Workflow depicts dissociation and isolation of human breast epithelial cells from BRCA2 carrier (BRCA2mut/+) prophylactic mastectomy and control [wild-type (WT)] elective mammoplasty cases for subsequent analyses, as indicated. Dot plot at center shows representative flow cytometry sorting via CD49f and EpCAM of ML, LP, and basal epithelial cells. FACS, fluorescence-activated cell sorting. (B) Summary of single-cell whole-genome sequencing (WGS) analysis of flow-sorted, primary uncultured breast epithelial cells. Copy number variation (CNV) calls for individual cells (rows) across the genome (x axis; Chr, chromosome) are shown, with gains and losses boxed. Cell types and genotypes are indicated at the top left, and individual patient ID numbers are indicated at the right. In total, 252 sequenced breast epithelial cells from BRCA2mut/+ (n = 5) and control (n = 2) tissue specimens are depicted. (C) Bar chart depicting the prevalence of CNVs in LP (L) and basal (B) cells of BRCA2 carrier and control (WT) patients. Color code depicts the number of CNVs identified per cell. (D) LP cells from BRCA2 carriers are significantly more likely to harbor CNVs than basal cells. P value is determined by χ2 test.
Among the earliest events in cancer evolution are thought to be polyclonal somatic genomic alterations. Accordingly, we looked for the presence of somatic copy number variations (CNVs) at high resolution through single-cell whole-genome sequencing (WGS) of uncultured, flow-sorted primary LP and basal epithelial cells from BRCA2 carriers and controls. Low-coverage WGS provides sufficiently high resolution to identify subchromosomal CNVs as small as 10 Mb, and our methodology for single-cell whole-genome amplification and analysis has been previously validated (15, 16). We carried out WGS to an average depth between 0.1× to 0.05× and then used two independent algorithms (HMMcopy and DNAcopy) to assign and confirm copy number changes across the genome (15, 16). Previous studies using this methodology have demonstrated that in unselected individuals, the proportion of cells with any such CNVs is very low (<5% of cells) in normal epithelial and brain tissues (16). In contrast, among nearly 100 individual LP cells from a cohort of BRCA2 carriers analyzed by WGS, we observed that 27% demonstrated one or more CNVs of >10 Mb (Fig. 1, B to D). Applying this methodology to an equal number of basal breast epithelial cells from the same individuals also revealed a substantial excess of cells harboring CNVs (13%), although significantly less than the proportion of CNV-positive LP cells (P = 0.04) (Fig. 1, B to D). By comparison, a parallel WGS analysis of sorted LP and basal cells from noncarriers revealed a single CNV in 90 cells (Fig. 1, B and C). As further validation of our sequencing and analysis pipelines, we reanalyzed existing data from normal skin and brain cells sequenced on the same platform. The overall sequence quality was comparable between these cells and the breast epithelial cells, and we confirmed the low prevalence of CNV-positive cells in 142 skin and brain cells sequenced (fig. S1). Thus, breast epithelia, and particularly, LP cells from noncancerous breast tissue of BRCA2 carriers harbor frequent subchromosomal aneuploid events (Fig. 1D and fig. S2A).
One notable CNV we observed was duplication of the entire chromosome 1q arm, which is a common genomic abnormality in breast cancer (Fig. 2A) (17). Most of the identified CNVs were subchromosomal haploid losses, consistent with the widespread pattern of losses observed in BRCA2-associated breast cancer (Fig. 2A and fig. S2B) (1). In some cases, identical losses were shared between multiple cells of the same patient, a finding that could conceivably correspond to early clonal evolution (Fig. 2B). None of the losses in any cell involved the BRCA2 locus on chromosome 13 (Fig. 1B and fig. S2, B to E). Previous analyses of germline BRCA2-associated breast cancers have demonstrated that most genetic loss-of-heterozygosity (LOH) events for BRCA2 itself are >10 Mb and therefore would have been detected by our analysis (18). This observation suggests that the wild-type BRCA2 allele is intact in our cases, implying that accumulation of subchromosomal aneuploidy may be a haploinsufficient phenotype. To confirm the integrity of the wild-type allele, we performed targeted polymerase chain reaction (PCR) amplification of the locus surrounding the patient-specific BRCA2 mutation from individual cells. Although efficiency for detection of either allele was low, we did not observe a bias toward detection of the mutant allele alone in the cells analyzed (Fig. 2C and table S2). Together, these findings imply that subchromosomal aneuploidy is an early and potentially haploinsufficient phenotype in BRCA2mut/+ breast epithelia.
Fig. 2. Polyclonal, subchromosomal aneuploidy is a hallmark of BRCA2mut/+ breast epithelial cells.

(A) Representative segmentation plots of individual LP (n = 4) and basal (n = 2) cells harboring CNVs from four BRCA2 mutation carriers. Y axis depicts normalized WGS read counts across the genome (x axis). Red dots indicate region of gain, whereas blue dots indicate losses. Patient ID numbers are indicated at the right. (B) Segmentation plots of three LP cells that share a clonal loss (red box) in a BRCA2 carrier (patient 131). Zoomed-in images of the clonal loss are shown at the right. (C) Representative chromatograms from single-cell PCR-based Sanger sequencing of genomic DNA in a BRCA2mut/+ LP cell. The presence of a heterozygous single-nucleotide polymorphism (SNP) and the superimposition of sequences adjacent to the frameshift mutation suggest that LOH has not occurred.
BRCA2mut/+ primary cells exhibit DNA damage and a deregulated replication stress response
The presence of viable aneuploid cells in BRCA2mut/+ tissues suggested ongoing DNA damage and/or a deregulated stress/damage response. Thus, we next used an independent method to directly assess DNA damage in single cells, the comet assay. This assay uses cells embedded in agarose that are lysed and then subjected to electrophoresis, causing broken DNA structures to migrate toward the anode, thus forming a comet tail (19). We briefly cultured freshly collected cells from BRCA2 carriers or controls under ultralow attachment conditions (48 to 72 hours) to select for epithelial progenitor cells before plating (20). Consistently, cells from BRCA2 carriers demonstrated increased DNA breaks at baseline compared to controls (Fig. 3A). In addition, inducing replication stress by treatment with hydroxyurea (HU) led to further increases in DNA damage in BRCA2mut/+ cells, potentially reflecting the established role of BRCA2 in protection of stalled replication forks (Fig. 3A) (21).
Fig. 3. BRCA2mut/+ breast epithelial cells exhibit DNA damage and an impaired replication stress checkpoint response.
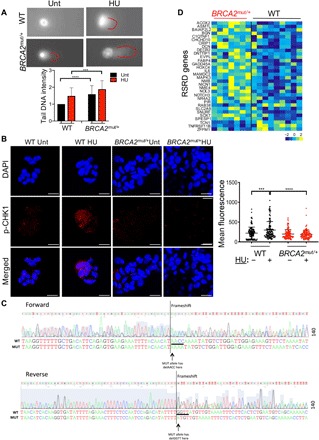
(A) Representative images of comet assays performed on primary human breast epithelial cells isolated from control (WT) and BRCA2mut/+ tissues. Red lines highlight “tail” of broken DNA. Graph below summarizes data from n = 3 patients per genotype (50 cells per patient). Cells were either untreated (Unt) or treated with HU for 4 hours. Data are depicted as fold change in tail DNA intensity. P values are determined by unpaired t test. ***P < 0.001 and ****P < 0.0001. Error bars indicate SD. (B) Representative confocal immunofluorescence staining of primary breast epithelial cells for p-CHEK1 (Ser317) shows increased nuclear staining following HU treatment only in control (WT) but not in BRCA2mut/+ cells. Graph at the right summarizes nuclear fluorescence of individual cells (dots) (n = 4 patients for control and n = 3 patients for BRCA2mut/+; four fields counted per condition per patient). P values are determined by unpaired t test. ***P < 0.001 and ****P < 0.0001. Horizontal lines indicate means and SDs. Scale bars, 20 μm. (C) Chromatograms depicting Sanger DNA sequencing of a cytospin of primary breast cells assayed in (B) from a BRCA2mut/+ patient harboring BRCA2 5799_5802delCCAA (p.Asn1933Lysfs). The superimposition of sequences adjacent to the frameshift mutation suggests that LOH has not occurred. (D) Heat map of RNA sequencing (RNA-seq) data from freshly sorted cells shows differential expression of RSRD (replication stress response deficiency) genes (24) in BRCA2mut/+ LP cells (n = 7 patients) compared to control (WT) LP cells (n = 9 patients). Columns correspond to individual patients.
We then examined the response to this genomic stress by analyzing phosphorylation of CHEK1, a central coordinator of the response to replication stress and DNA damage (22, 23). Cytospins of primary epithelial progenitor cultures prepared as above were stained for phosphorylated CHEK1 at baseline or following 4 hours of exposure to HU. As anticipated, control primary epithelia exhibited increased CHEK1 phosphorylation within 4 hours of HU treatment (Fig. 3B). In contrast, however, cells from BRCA2 carriers exhibited a failure to activate CHEK1 in response to HU, despite normal levels of total CHEK1 protein (Fig. 3B and fig. S3A). DNA sequencing of these cells revealed the presence of both wild-type and mutant BRCA2 alleles (Fig. 3C). These findings provide further support for a haploinsufficient phenotype of BRCA2 in the response to genomic stress.
Because we observed a deregulated genomic stress response in vitro, we wanted to know whether this also occurs in vivo. Thus, we carried out RNA sequencing (RNA-seq) analysis of freshly sorted LP and basal epithelial cell populations from BRCA2 carrier tissues or controls (Fig. 1A). Analysis of these data revealed enrichment in BRCA2mut/+ LP cells of an established signature reflecting a failure of the ATR (Ataxia telangiectasia mutated and Rad3-related)/CHEK1 (checkpoint kinase 1)–mediated replication stress checkpoint in nontransformed mammary epithelial cells (Fig. 3D and fig. S3B) (24). This replication stress response deficiency signature is known to predict future cancer risk (24), and it contains some of the top most differentially expressed genes between BRCA2mut/+ and control LP cells (Fig. 3D). Among these are genes of potential relevance to HR-positive breast cancer (which comprise 80% of BRCA2-associated breast cancers), including the estrogen receptor target gene HOXC4 and the GATA transcription factor–binding partner gene ZFPM1 (Fig. 3D) (25, 26). Furthermore, evaluation of differentially expressed programs through gene set enrichment analysis (GSEA) revealed the highly significant deregulation of a radiation response signature in BRCA2mut/+ LP cells (fig. S3C) (27). Notably, the differential expression of this signature between BRCA2mut/+ and control cells was far more significant within the LP compared to the basal population, in keeping with the more frequent occurrence of CNVs among LP cells (fig. S3C). Again, consistent with haploinsufficiency for BRCA2, the RNA-seq data showed no evidence for exclusive expression of the mutant allele in BRCA2mut/+ LP cells (fig. S3D). Thus, BRCA2mut/+ LP cells exhibit evidence of aberrant replication stress and DNA damage responses in vivo.
BRCA2mut/+ LP cells show increased TP53 activity and decreased NF-κB/SASP pathway expression
We then turned to examine the downstream consequences of the DNA damage detected in LP cells of BRCA2 carriers. A hallmark genetic event that cooperates with BRCA2 deficiency in cancer pathogenesis is loss of TP53, suggesting that activation of TP53 may be an early barrier to malignant progression in this setting (28). We therefore hypothesized that the failed CHEK1-dependent replication stress response we observed might ultimately lead to DNA double-strand breaks and thereby trigger TP53 activation through a CHEK1-independent pathway (29). Recent studies suggest that CHEK1 is not required for TP53 activation in primary breast epithelial cells following DNA damage (30). RNA-seq analysis did suggest activation of TP53 in BRCA2mut/+ LP cells, evidenced by the increased expression of multiple direct TP53 target genes (Fig. 4A) (31, 32). This in vivo effect was associated with a strong transcriptional profile indicating suppression of nuclear factor κB (NF-κB) signaling, including numerous cytokine and inflammatory factors associated with the senescence-associated secretory phenotype (SASP) (Fig. 4, B to D). TP53 is known to suppress the NF-κB/SASP response (33, 34), and this effect is emerging as a relevant component of TP53-dependent tumor suppression given that accumulation of SASP-expressing cells is an established driver of tumorigenesis (35). We independently validated the corresponding alterations in NF-κB protein expression, demonstrating that the NFKB1 (p50) and NFKB2 (p52) subunits were expressed at lower levels in BRCA2 carrier tissues compared to controls (Fig. 4C). Furthermore, knockdown of BRCA2 in nontransformed mammary epithelial cells via lentiviral short hairpin RNA attenuated expression of the same cytokine and NF-κB target genes that were down-regulated in BRCA2mut/+ progenitor cells in vivo (fig. S4A). Similar to the damage response signature (fig. S3C), deregulation of the SASP program was selective for LP cells in BRCA2 carriers, as no significant suppression of SASP was observed in the corresponding basal epithelial cells of these same patients (fig. S4B). These results suggest that DNA damage and TP53 activation in BRCA2mut/+ LP cells are associated with suppression of the NF-κB/SASP response.
Fig. 4. BRCA2mut/+ LP cells display increased TP53 activity and suppressed NF-κB/SASP pathway expression.
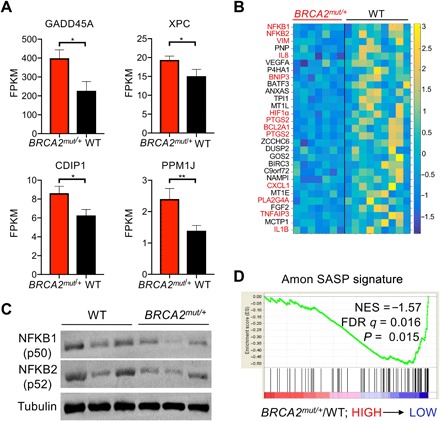
(A) Bar charts show the mean expression levels of canonical TP53 target genes in freshly sorted BRCA2 carrier LP cells (n = 7 patients) compared to controls (WT; n = 9 patients), assessed by RNA-seq. Error bars denote SEM. P values are determined by Mann-Whitney test. *P < 0.05 and **P < 0.01. XPC, Xeroderma pigmentosum group C-complementing protein; FPKM, Fragments Per Kilobase of transcript per Million mapped reads. (B) Heat map depicts down-regulation of NF-κB/SASP pathway genes in BRCA2 carrier LP cells compared to controls (WT), assessed by RNA-seq as in (A). Columns correspond to individual patients. Direct NF-κB target genes are highlighted in red. (C) Western blot analysis shows that NFKB1 (p50) and NFKB2 (p52) subunits are expressed at lower levels in BRCA2mut/+ breast tissues compared to control (WT) tissues (n = 3 patients per genotype). β-Tubulin serves as a loading control. (D) Negative enrichment of a SASP signature in GSEA of RNA-seq data from freshly sorted LP cells of BRCA2 carriers (n = 7 patients) and controls (WT; n = 9 patients). NES, normalized enrichment score; FDR, false discovery rate.
Age-associated deregulation of breast epithelial cell proportions in BRCA2 carriers suggests expansion of a damaged LP cell population over time
Deregulated DNA damage and senescence/SASP responses in BRCA2 LP cells might be expected to alter the proportion of these cells over time (36). We thus sought to address whether there were differences in the proportions of progenitor or other epithelial subpopulations in BRCA2mut/+ tissues compared to controls. We collected a larger cohort of tissues from BRCA2 carriers (n = 26) and controls (n = 28), then performed flow cytometric analysis on these specimens, and plotted the proportions of each epithelial subpopulation as a function of age for each cohort (Fig. 5A). In noncarrier controls, no significant age-associated changes in the prevalence of these subpopulations were noted. In contrast, BRCA2 carriers showed an age-associated expansion in the proportion of LP cells and a decline in the basal cell fraction (Fig. 5B and fig. S5, A and B). These differences were not accounted for by demographic factors such as parity or menopausal status, as these factors were not associated with significant differences in epithelial cell proportions (fig. S5, C and D). Thus, DNA damage and suppression of a senescence-associated program in BRCA2mut/+ LP cells are accompanied by an age-associated expansion of this progenitor cell compartment (36).
Fig. 5. Noncancerous breast tissues of BRCA2 mutation carriers demonstrate age-associated deregulation of epithelial cell proportions compared to controls.
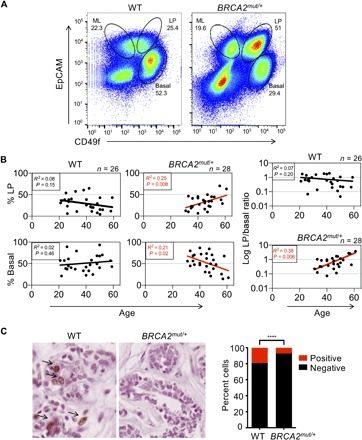
(A) Representative flow cytometry analysis showing distinct epithelial subpopulations (basal, LP, and ML) isolated from breast tissues of control (WT) and BRCA2 mutation carriers following sorting via CD49f and EpCAM staining. Numbers indicate percentages of each epithelial cell subpopulation. (B) Linear regression analysis of LP and basal cell proportions by age for controls (WT) (n = 26 patients) and BRCA2 carriers (n = 28 patients). The LP/basal ratio by patient provides additional validation as it accounts for technical factors that may have subtle effects on absolute cell numbers. (C) TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining of representative control (WT) and BRCA2 carrier tissues. Summary data obtained by counting four fields for five patients per genotype are shown. ****P < 0.0001 by Fisher’s exact test.
Last, we hypothesized that altered epithelial cell proportions and a deregulated NF-κB/SASP program in BRCA2 carrier tissues may be associated with differences in cell proliferation and/or survival in vivo (36). We did not observe strong differences in proliferation assessed by Ki67 staining between these BRCA2 carrier breast tissues and controls, prompting us to ask whether differences in cell survival might contribute to the age-associated expansion of the LP population in this context. We therefore carried out TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining, an established marker of apoptosis, in BRCA2 carrier tissues and controls. The proportion of TUNEL-positive cells is well documented in normal human breast epithelial tissues, and we observed a similar prevalence of these cells in the control tissues we tested (Fig. 5C) (37). In contrast, however, BRCA2 mutation carrier tissues consistently showed a paucity of TUNEL-positive luminal epithelial cells across all patients tested, in keeping with established links between checkpoint and NF-κB suppression and a defective apoptotic response (Fig. 5C) (38, 39). Collectively, our findings suggest that noncancerous BRCA2mut/+ breast tissues exhibit BRCA2 haploinsufficiency and an age-associated accumulation of DNA-damaged luminal epithelial progenitor cells bearing altered checkpoint and survival responses (Fig. 6).
Fig. 6. Summary of findings in primary BRCA2mut/+ breast tissues.
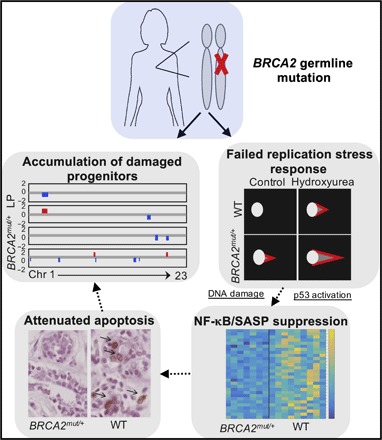
Epithelial progenitor cells of heterozygous germline BRCA2 carriers exhibit DNA damage, failed replication stress, and damage responses, together with attenuated apoptosis. LOH analyses suggest that these findings may reflect a haploinsufficient phenotype for BRCA2 in vivo.
DISCUSSION
This study advances our understanding of early changes in BRCA2mut/+ breast tissues, defining unanticipated phenotypes in this setting with implications for both cancer risk assessment and prevention. Most of the tissues we studied were deemed to be histologically normal by highly experienced breast pathologists, suggesting that the alterations we report precede clinically defined cellular abnormalities (tables S1 and S3). We present evidence that a failed replication stress response and DNA damage in BRCA2mut/+ tissues result from haploinsufficiency for BRCA2 rather than homozygous loss of function. While the presence of haploinsufficiency for either BRCA1 or BRCA2 in vivo has been controversial, our findings are in accord with data suggesting that LOH for the wild-type BRCA2 is not universal in BRCA2-associated cancers (40). Our observations are also in keeping with a recent report that the BRCA2 protein is selectively susceptible to degradation by environmental aldehydes (41), an effect that could contribute to a haploinsufficient phenotype in cells with only one functional BRCA2 allele. Nonetheless, we analyzed a relatively small number of cells and tissues, and it is difficult to definitively rule out LOH in a subset of cells. Thus, our study suggests, rather than confirms, haploinsufficiency for BRCA2 as a potential initiating event for these cancers.
A prominent feature of the phenotype we have uncovered is frequent subchromosomal aneuploidy, most prevalent within the LP cell population. LP cells are a potential target cell for BRCA2-associated carcinogenesis in the breast, and indeed we observe instances of apparently clonally related genomic alterations among these cells. While our study does not prove that they are direct cancer precursors, these alterations could conceivably represent the earliest somatic genetic abnormalities that underlie these malignancies. Notably, all the CNVs we identified were subchromosomal and therefore are to be distinguished from whole-chromosome gains and losses that are typically later events and associated with TP53 inactivation (42).
Although the early genomic changes we observed are likely to include many passenger events, they, nevertheless, may provide a quantifiable hallmark of the preneoplastic BRCA2 carrier state. Tracking the prevalence of DNA-damaged cells in the clinical setting could possibly improve risk prediction for these women, who are faced with the difficult choice of whether to undergo mastectomy long before cancer develops. Last, the BRCA2 haploinsufficient phenotype we report may portend particular vulnerabilities of certain BRCA2mut/+ cancer precursor cells. Accordingly, this work provides a foundation for future studies seeking to identify improved pharmacologic approaches to cancer prevention in this setting.
MATERIALS AND METHODS
Human breast tissues
Fresh human breast tissues were obtained from the Massachusetts General Hospital with approval by the local Institutional Review Board and signed informed patient consent (protocols 93-085 and 2008-P-1789). Samples were either normal breast tissues from reduction mammoplasties (confirmed by pathology) or noncancerous breast tissues from prophylactic mastectomies of known BRCA1 or BRCA2 mutation carriers. All BRCA1/2 carrier status was determined through clinical germline genetic testing performed by commercial providers before tissue collection.
Mammary cell preparations
Tissue samples were minced and digested with collagenase/hyaluronidase (STEMCELL Technologies) in complete EpiCult-B medium supplemented with hydrocortisone (0.48 μg/ml; STEMCELL Technologies) overnight at 37°C. The resulting suspensions were either cryopreserved or further sequentially digested with 0.25% trypsin, dispase (5 mg/ml), and deoxyribonuclease I (1 mg/ml). Single-cell suspensions were collected by filtration through a 40-μm cell strainer.
Cell staining and sorting
Cells were blocked with rat immunoglobulin (Jackson ImmunoLabs) and antibody to Fc receptor–binding inhibitor (eBioscience) before incubation with the following primary antibodies: phycoerythrin (PE)–conjugated anti-human CD31 (BD Pharmingen), PE-conjugated anti-human CD45 (BD Pharmingen), PE-conjugated anti-human CD235a (BD Pharmingen), BV650-conjugated anti-human epithelial cell adhesion molecule (EpCAM) CD326 (BioLegend), and biotin-conjugated anti-human ITGA6 (eBioscience). Where required, cells were incubated with allophycocyanin-Cy7–conjugated streptavidin (BD Pharmingen). Cells were either stained with 4′,6-diamidino-2-phenylindole (DAPI) for viability or fixed with 1% paraformaldehyde and stained with the Zombie Aqua Fixable Viability Kit (BioLegend). Viable cells were sorted on a FACSAria flow cytometer (Becton Dickinson). Data were analyzed using FlowJo software (Tree Star).
Single-cell PCR for allele-specific LOH analysis
Microaspirated single cells were transferred into PCR tubes containing lysis buffer [water + proteinase K (400 ng/μl) + 17 μM SDS], and DNA was amplified by nested PCR using primers flanking BRCA2 mutations. Sanger sequencing was performed by the Center for Computational and Integrative Biology DNA Core Facility at the Massachusetts General Hospital.
Primer sequences for patient 128 (Val3079PhefsX4) are as follows: first PCR, TGGCGTCCATCATCAGATTT (forward) and TCAGAGGTTCAAAGAGGCTTAC (reverse); second PCR, CAGATTTACCAGCCACGGGA (forward) and GCCAACTGGTAGCTCCAACTAA (reverse). Primer sequences for patient 140 (6027del4) are as follows: first PCR, GGGCCACCTGCATTTAGGAT (forward) and TGAGCTGGTCTGAATGTTCGT (reverse); second PCR, GCAGGTTGTTACGAGGCATT (forward) and CCTGGACAGATTTTCCACTTGC (reverse).
Comet assays
Single-cell suspensions from patient samples were plated in ultralow adherence plates in Dulbecco’s modified Eagle’s medium/F12 medium containing insulin (5 μg/ml), epidermal growth factor (10 ng/ml), basic fibroblast growth factor (5 ng/ml), heparin (4 μg/ml), hydrocortisone (500 ng/ml), B27, GlutaMAX, and penicillin-streptomycin. Cells were either treated with HU (10 mM; Sigma) for 4 hours or left untreated and washed with phosphate-buffered saline (PBS), and alkaline comet assays were performed using a Trevigen Comet Assay kit according to the manufacturer’s instructions. Olive tail movement was quantified with ImageJ, and 50 individual cells were quantified per condition.
Immunostaining
Immunofluorescence for paraffin sections and TUNEL staining were performed by Dana-Farber/Harvard Cancer Center Specialized Histopathology Core. For immunofluorescence in cells, fixation was performed with methanol for 10 min, followed by permeabilization in 0.1% Triton X-100 for 2 min. Blocking was performed with 10% horse serum for 30 min, and cells were further incubated with primary p-CHEK1 antibody (Novus Biologicals) for 2 hours, washed with wash buffer (PBS + 10% horse serum + 0.1% Triton X-100), incubated with appropriate secondary antibody for 1 hour, and stained with DAPI. All immunofluorescence images were captured by a confocal microscope (Leica TCS SP8) and were analyzed by ImageJ.
Western blotting
Snap-frozen tissues were homogenized using Precellys 24 homogenizer (Bertin Technologies). For total protein extraction, cells were lysed in radioimmunoprecipitation assay buffer [10 mM tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% sodium deoxycholate, 0.1% (w/v) SDS, 1% (v/v) NP-40, proteinase inhibitor cocktail, and phosphatase inhibitor cocktail] for 30 min on ice. Western blotting was performed using NFKB1 p50 (Santa Cruz Biotechnology) and NFKB2 p52 (Millipore) antibodies by standard protocol.
Single-cell copy number analysis
Fresh tissues were dissociated as described above, and single cells were isolated by microaspiration. Genomic DNA was amplified and sequenced as described in (16). Fastqs were aligned using bwa-mem, with resulting bams sorted and duplicates marked using Picard. Coverage was then computed over 500-kb bins across the entire genome. The count for each bin was then divided by the sum across all bins for the relative sample (to correct for library size) and then by the median for that genomic bin across all samples from the same batch. The coverage profiles were then transformed into .wig files and fed into the R package HMMcopy for segmentation and CNV calling. HMMCopy was run with e = 0.9999999 and nu = 5, with all other parameters set to default. A noise statistic termed “VS” was computed in the same manner as (16), with cells with values greater than or equal to 0.5 being excluded from the analysis. CNVs that mapped to the Y chromosome were less than 10 Mb in size or had an absolute log2 ratio of less than 0.4 that were excluded from the analysis.
RNA-seq analysis
Total RNA from sorted cell populations was extracted using an RNeasy FFPE kit according to the manufacturer’s instructions. Libraries for ribosomally reduced RNA were prepared by the Harvard Biopolymers Facility using directional RNA-seq Wafergen protocol. Libraries were sequenced on Illumina HiSeq 2000 at Next-Generation Sequencing Core at Massachusetts General Hospital. Transcripts per million (TPM) values were computed using Salmon and batch-corrected using ComBat. The two samples with the lowest total counts were excluded from the analysis. GSEA was run on the ComBat-corrected TPM values using phenotype permutation and default parameters. The heat maps in Fig. 4 (B and D) were made using the ComBat-corrected TPM values, subset to the comparison of interest, and transformed into z scores by gene.
Other statistical methods
P values were determined using Student’s unpaired t test, unless indicated otherwise.
Supplementary Material
Acknowledgments
We thank the HSCI-CRM Flow Cytometry Core Facility for assistance with cell sorting and the MGH DF/HCC Specialized Histopathology Service Core for immunostaining experiments. We thank the Biopolymers Facility at Harvard Medical School for library processing of RNA samples and the MGH Next Generation Sequencing Core for performing RNA-seq. We are grateful to the MIT BioMicroCenter for performing genome sequencing reactions. Funding: This work was supported by DOD/CDMRP grant BC140903 (to L.W.E.), the Tracey Davis Breast Cancer Research Fund (to L.W.E.), the Weissman Family MGH Research Scholar grant (to L.W.E.), the Susan G. Komen Foundation grant PDF16380794 (to M.K.-Y.), a Terri Brodeur Breast Cancer Foundation grant 2016D001483 (to M.K.-Y.), and the Howard Hughes Medical Institute (to A.A.). Author contributions: M.K.-Y., A.A., and L.W.E. conceived and designed the study. M.C.S. contributed patient samples. M.K.-Y., R.E.S., and S.V.S. designed and performed the experiments and interpreted the data. H.R., R.M., and V.V. performed the experiments. M.K.-Y., S.V.S., E.Z., A.D., and M.Y. performed the data analysis. M.K.-Y., A.L., K.N.R., S.R., and M.L. performed the bioinformatic analysis and interpreted the data. L.W.E., A.A., and M.L. conceived the experiments, interpreted the data, and provided the funding. L.W.E. and M.K.-Y. wrote the manuscript. All authors approved the final submitted manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaay2611/DC1
Supplementary Materials and Methods
Fig. S1. Analysis of single-cell WGS data from normal human skin and brain cells.
Fig. S2. Identification and characterization of CNVs in freshly collected BRCA2mut/+ breast epithelial cells.
Fig. S3. Characterization of replication stress response deficiency and haploinsufficiency in BRCA2mut/+ breast epithelial cells.
Fig. S4. Suppression of NF-κB/SASP response associated with loss of BRCA2.
Fig. S5. Proportions of mammary epithelial cell subsets in BRCA2 carrier and control tissues.
Table S1. Characteristics of patients undergoing WGS of breast tissues.
Table S2. Summary of single-cell site-specific PCR/sequencing data.
Table S3. Characteristics of patients undergoing RNA-seq of breast tissues.
Reference (43)
REFERENCES AND NOTES
- 1.Davies H., Glodzik D., Morganella S., Yates L. R., Staaf J., Zou X., Ramakrishna M., Martin S., Boyault S., Sieuwerts A. M., Simpson P. T., King T. A., Raine K., Eyfjord J. E., Kong G., Borg Å., Birney E., Stunnenberg H. G., van de Vijver M. J., Børresen-Dale A.-L., Martens J. W. M., Span P. N., Lakhani S. R., Vincent-Salomon A., Sotiriou C., Tutt A., Thompson A. M., van Laere S., Richardson A. L., Viari A., Campbell P. J., Stratton M. R., Nik-Zainal S., HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couch F. J., Nathanson K. L., Offit K., Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science 343, 1466–1470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ludwig T., Chapman D. L., Papaioannou V. E., Efstratiadis A., Targeted mutations of breast cancer susceptibility gene homologs in mice: Lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 11, 1226–1241 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Sharan S. K., Morimatsu M., Albrecht U., Lim D.-S., Regel E., Dinh C., Sands A., Eichele G., Hasty P., Bradley A., Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386, 804–810 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Suzuki A., de la Pompa J. L., Hakem R., Elia A., Yoshida R., Mo R., Nishina H., Chuang T., Wakeham A., Itie A., Koo W., Billia P., Ho A., Fukumoto M., Hui C. C., Mak T. W., Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev. 11, 1242–1252 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Tutt A., Connor F., Bertwistle D., Kerr P., Peacock J., Ross G., Ashworth A., Cell cycle and genetic background dependence of the effect of loss of BRCA2 on ionizing radiation sensitivity. Oncogene 22, 2926–2931 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Feng W., Jasin M., BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 8, 525 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connor F., Bertwistle D., Mee P. J., Ross G. M., Swift S., Grigorieva E., Tybulewicz V. L. J., Ashworth A., Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat. Genet. 17, 423–430 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Gowen L. C., Johnson B. L., Latour A. M., Sulik K. K., Koller B. H., Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat. Genet. 12, 191–194 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Hakem R., de la Pompa J. L., Sirard C., Mo R., Woo M., Hakem A., Wakeham A., Potter J., Reitmair A., Billia F., Firpo E., Hui C. C., Roberts J., Rossant J., Mak T. W., The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 85, 1009–1023 (1996). [DOI] [PubMed] [Google Scholar]
- 11. Pathology of familial breast cancer: Differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases. Breast Cancer Linkage Consortium. Lancet 349, 1505–1510 (1997). [PubMed] [Google Scholar]
- 12.Molyneux G., Geyer F. C., Magnay F. A., McCarthy A., Kendrick H., Natrajan R., MacKay A., Grigoriadis A., Tutt A., Ashworth A., Reis-Filho J. S., Smalley M. J., BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7, 403–417 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Lim E., Vaillant F., Wu D., Forrest N. C., Pal B., Hart A. H., Asselin-Labat M.-L., Gyorki D. E., Ward T., Partanen A., Feleppa F., Huschtscha L. I., Thorne H. J.; kConFab, Fox S. B., Yan M., French J. D., Brown M. A., Smyth G. K., Visvader J. E., Lindeman G. J., Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 15, 907–913 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Visvader J. E., Stingl J., Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 28, 1143–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knouse K. A., Wu J., Amon A., Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 26, 376–384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knouse K. A., Wu J., Whittaker C. A., Amon A., Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. U.S.A. 111, 13409–13414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Network , Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L. B., Martin S., Wedge D. C., van Loo P., Ju Y. S., Smid M., Brinkman A. B., Morganella S., Aure M. R., Lingjærde O. C., Langerød A., Ringnér M., Ahn S. M., Boyault S., Brock J. E., Broeks A., Butler A., Desmedt C., Dirix L., Dronov S., Fatima A., Foekens J. A., Gerstung M., Hooijer G. K. J., Jang S. J., Jones D. R., Kim H. Y., King T. A., Krishnamurthy S., Lee H. J., Lee J. Y., Li Y., McLaren S., Menzies A., Mustonen V., O’Meara S., Pauporté I., Pivot X., Purdie C. A., Raine K., Ramakrishnan K., Rodríguez-González F. G., Romieu G., Sieuwerts A. M., Simpson P. T., Shepherd R., Stebbings L., Stefansson O. A., Teague J., Tommasi S., Treilleux I., van den Eynden G. G., Vermeulen P., Vincent-Salomon A., Yates L., Caldas C., van’t Veer L., Tutt A., Knappskog S., Tan B. K. T., Jonkers J., Borg Å., Ueno N. T., Sotiriou C., Viari A., Futreal P. A., Campbell P. J., Span P. N., van Laere S., Lakhani S. R., Eyfjord J. E., Thompson A. M., Birney E., Stunnenberg H. G., van de Vijver M. J., Martens J. W. M., Børresen-Dale A. L., Richardson A. L., Kong G., Thomas G., Stratton M. R., Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song X., Mosby N., Yang J., Xu A., Abdel-Malek Z., Kadekaro A. L., α-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res. 22, 809–818 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Nolan E., Vaillant F., Branstetter D., Pal B., Giner G., Whitehead L., Lok S. W., Mann G. B.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab), Rohrbach K., Huang L. Y., Soriano R., Smyth G. K., Dougall W. C., Visvader J. E., Lindeman G. J., RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 22, 933–939 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Feng W., Jasin M., Homologous recombination and replication fork protection: BRCA2 and More! Cold Spring Harb. Symp. Quant. Biol. 82, 329–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walworth N., Davey S., Beach D., Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 363, 368–371 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Bartek J., Lukas J., Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429 (2003). [DOI] [PubMed] [Google Scholar]
- 24.McGrail D. J., Lin C. C. J., Dai H., Mo W., Li Y., Stephan C., Davies P., Lu Z., Mills G. B., Lee J. S., Lin S. Y., Defective replication stress response is inherently linked to the cancer stem cell phenotype. Cell Rep. 23, 2095–2106 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Mai T., Zan H., Zhang J., Hawkins J. S., Xu Z., Casali P., Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. J. Biol. Chem. 285, 37797–37810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robert N. M., Tremblay J. J., Viger R. S., Friend of GATA (FOG)-1 and FOG-2 differentially repress the GATA-dependent activity of multiple gonadal promoters. Endocrinology 143, 3963–3973 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Ghandhi S. A., Yaghoubian B., Amundson S. A., Global gene expression analyses of bystander and alpha particle irradiated normal human lung fibroblasts: Synchronous and differential responses. BMC Med. Genomics 1, 63 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jonkers J., Meuwissen R., van der Gulden H., Peterse H., van der Valk M., Berns A., Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 29, 418–425 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Smith J., Tho L. M., Xu N., Gillespie D. A., The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112 (2010). [DOI] [PubMed] [Google Scholar]
- 30.van Jaarsveld M. T. M., Deng D., Wiemer E. A. C., Zi Z., Tissue-specific Chk1 activation determines apoptosis by regulating the balance of p53 and p21. iScience 12, 27–40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDade S. S., Patel D., Moran M., Campbell J., Fenwick K., Kozarewa I., Orr N. J., Lord C. J., Ashworth A. A., McCance D. J., Genome-wide characterization reveals complex interplay between TP53 and TP63 in response to genotoxic stress. Nucleic Acids Res. 42, 6270–6285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Younger S. T., Kenzelmann-Broz D., Jung H., Attardi L. D., Rinn J. L., Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 43, 4447–4462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coppé J.-P., Patil C. K., Rodier F., Sun Y., Muñoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., Campisi J., Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLOS Biol. 6, 2853–2868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiley C. D., Schaum N., Alimirah F., Lopez-Dominguez J. A., Orjalo A. V., Scott G., Desprez P.-Y., Benz C., Davalos A. R., Campisi J., Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 8, 2410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sieben C. J., Sturmlechner I., van de Sluis B., van Deursen J. M., Two-step senescence-focused cancer therapies. Trends Cell Biol. 28, 723–737 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Childs B. G., Baker D. J., Kirkland J. L., Campisi J., van Deursen J. M., Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 15, 1139–1153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feuerhake F., Sigg W., Hofter E. A., Unterberger P., Welsch U., Cell proliferation, apoptosis, and expression of Bcl-2 and Bax in non-lactating human breast epithelium in relation to the menstrual cycle and reproductive history. Breast Cancer Res. Treat. 77, 37–48 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Kastan M. B., Bartek J., Cell-cycle checkpoints and cancer. Nature 432, 316–323 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Veuger S. J., Durkacz B. W., Persistence of unrepaired DNA double strand breaks caused by inhibition of ATM does not lead to radio-sensitisation in the absence of NF-κB activation. DNA Repair 10, 235–244 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Maxwell K. N., Wubbenhorst B., Wenz B. M., de Sloover D., Pluta J., Emery L., Barrett A., Kraya A. A., Anastopoulos I. N., Yu S., Jiang Y., Chen H., Zhang N. R., Hackman N., D’Andrea K., Daber R., Morrissette J. J. D., Mitra N., Feldman M., Domchek S. M., Nathanson K. L., BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 8, 319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan S. L. W., Chadha S., Liu Y., Gabasova E., Perera D., Ahmed K., Constantinou S., Renaudin X., Lee M., Aebersold R., Venkitaraman A. R., A class of environmental and endogenous toxins induces BRCA2 haploinsufficiency and genome instability. Cell 169, 1105–1118.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sansregret L., Swanton C., The role of aneuploidy in cancer evolution. Cold Spring Harb. Perspect. Med. 7, a028373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramsey M. R., Wilson C., Ory B., Rothenberg S. M., Faquin W., Mills A. A., Ellisen L. W., FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Invest. 123, 3525–3538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaay2611/DC1
Supplementary Materials and Methods
Fig. S1. Analysis of single-cell WGS data from normal human skin and brain cells.
Fig. S2. Identification and characterization of CNVs in freshly collected BRCA2mut/+ breast epithelial cells.
Fig. S3. Characterization of replication stress response deficiency and haploinsufficiency in BRCA2mut/+ breast epithelial cells.
Fig. S4. Suppression of NF-κB/SASP response associated with loss of BRCA2.
Fig. S5. Proportions of mammary epithelial cell subsets in BRCA2 carrier and control tissues.
Table S1. Characteristics of patients undergoing WGS of breast tissues.
Table S2. Summary of single-cell site-specific PCR/sequencing data.
Table S3. Characteristics of patients undergoing RNA-seq of breast tissues.
Reference (43)