

Sulfonated cyclodextrins show nontoxic, broad-spectrum virucidal activity in vitro, ex vivo, and in vivo.
Abstract
Viral infections kill millions of people and new antivirals are needed. Nontoxic drugs that irreversibly inhibit viruses (virucidal) are postulated to be ideal. Unfortunately, all virucidal molecules described to date are cytotoxic. We recently developed nontoxic, broad-spectrum virucidal gold nanoparticles. Here, we develop further the concept and describe cyclodextrins, modified with mercaptoundecane sulfonic acids, to mimic heparan sulfates and to provide the key nontoxic virucidal action. We show that the resulting macromolecules are broad-spectrum, biocompatible, and virucidal at micromolar concentrations in vitro against many viruses [including herpes simplex virus (HSV), respiratory syncytial virus (RSV), dengue virus, and Zika virus]. They are effective ex vivo against both laboratory and clinical strains of RSV and HSV-2 in respiratory and vaginal tissue culture models, respectively. Additionally, they are effective when administrated in mice before intravaginal HSV-2 inoculation. Lastly, they pass a mutation resistance test that the currently available anti-HSV drug (acyclovir) fails.
INTRODUCTION
Viruses can negatively affect society at several levels: from viral infections of food crops and livestock to the serious health impacts of viruses that infect humans, such as HIV, Ebola, and Zika virus (ZIKV). When prevention is not possible, drugs must be administered to limit viral replication and aid the immune systems fight against the infection, if they are available. Unfortunately, most existing antivirals act intracellularly, with related problems of permeability and toxicity, are virus specific (1), and/or have a reversible (virustatic) effect (2–4).
Broad-spectrum antivirals like heparin or heparin-like materials (5–8) have been developed that mimic the cell surface sugars responsible for initial viral attachment, such as heparan sulfate (HS). Unfortunately, upon dilution, the viruses are no longer bound to the drug and are still infectious; thus, the clinical efficacy remains unproven (2–4). A drug with an irreversible action, i.e., a virucidal drug, could be ideal to fight viral infection, given that it would not be subject to loss of efficacy upon dilution and have long-lasting effects. All previously identified virucidal molecules have toxic side effects that render their clinical use impossible (9). To the best of our knowledge, there is currently no approved drug that shows virucidal activity.
Recently, we have shown that highly sulfonated gold nanoparticles display broad-spectrum virucidal properties in vitro, ex vivo, and in vivo (10). In that work, a slight modification to the nanoparticle structure, relative to published work (6), altered the antiviral mode of action from virustatic to virucidal, with nanomolar median effective concentrations (EC50’s). This allowed us to maintain the broad-spectrum and nontoxic properties of virustatic nanoparticles while imparting a virucidal mechanism. Unfortunately, there are concerns with the use of gold nanoparticles as drugs due to their unknown clearance mechanism and possible long-term toxicity (11). Nonetheless, the principle of displaying multiple viral attachment ligands to create a virucidal drug remains compelling.
Cyclodextrins (CDs) are naturally occurring glucose derivatives, with a rigid cyclic structure, consisting of α(1-4)–linked glucopyranoside units. The most common CDs, referred to as α, β, and γ, have 6-, 7-, and 8-glucopyranoside units, respectively. They have found use in many commercial applications including drug delivery, air fresheners, cosmetics, and food (12, 13). Sulfonated CDs have shown antiviral properties only against HIV (14–18); however, their action was found to be reversible (virustatic) and virus specific. Here, we attach highly sulfonated chemicals to a U.S. Food and Drug Administration (FDA)–approved CD scaffold and achieve highly efficient virucidal broad-spectrum molecules, effective in vitro, ex vivo, and in an animal model.
RESULTS AND DISCUSSION
To test whether a modified CD (CD1; Fig. 1A) has antiviral activity like its nanogold counterpart (10), we used sodium undec-10-enesulfonate to synthesize a modified CD (CD1) (see figs. S1 and S2 for characterization) that exposes the sulfonate groups in a similar manner. In addition, two other modified CDs were synthesized. To alter the length of the linker, we synthesized CD2 (Fig. 1A), which bears a seven-carbon sulfonated alkyl chain (see figs. S1 and S3 for characterization). While CD1 showed strong inhibition of the growth of herpes simplex virus type 2 (HSV-2), with an EC50 of 28.51 ± 2.319 μg/ml, CD2 showed no significant effect (Fig. 1B). To alter the nature of the linker, we also synthesized a nonalkyl linker of similar overall length to sodium undec-10-enesulfonate (CD3) (see figs. S1 and S4 for characterization). We hypothesized that removing the flexibility of the linker would result in stronger binding to the virus and improved overall antiviral effect.
Fig. 1. Structures and virucidal data for modified CDs.
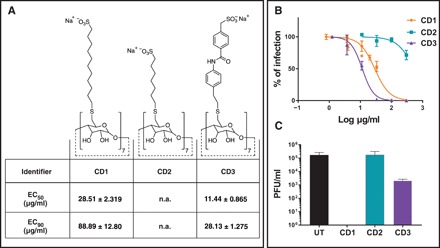
(A) Structures of modified CDs and relative effective concentrations of inhibition of HSV-2 growth. (B) Dose-response assay in Vero cells. Serial dilutions of CD1, CD2, and CD3 were incubated for 1 hour at 37°C with HSV-2 and then added on cells for 2 hours at 37°C. Subsequently, cells were washed and overlaid with medium containing methylcellulose. Plaques were counted 24 hpi. Percentages of infections were calculated by comparing the number of plaques in treated and untreated wells. (C) Virucidal assays: HSV-2 was incubated with media or CDs (CD1, CD2, and CD3) at 300 μg/ml and then serially diluted on Vero cells to a negligible concentration of compound. Results are shown as the mean and SEM of three (for CD1) and two (for all other compounds) independent experiments. UT, untreated; n.a., not assessable.
In a dose response assay, CD3 displayed enhanced antiviral effects, with an approximately threefold reduction in EC50 over the time course of a single-cycle infection compared to CD1. To determine whether the observed inhibition of HSV-2 growth was virustatic or virucidal, we pretreated viral solutions with CD1 to CD3. CD1 was highly virucidal, but this effect was markedly reduced when moving from CD1 to CD3 (Fig. 1C). These findings for the first time assert the utility of long flexible linkers to provide a virucidal mode of action.
CDs are known to extract cholesterol from membranes (19). To exclude a cholesterol-dependent antiviral effect of CD1 related to this property, we performed a cholesterol replenishment assay in cells infected with HSV-2 and treated with CD1 or methyl-β-CD at their EC90. As shown in fig. S5, the inhibitory activity of methyl-β-CD, but not of CD1, is lost in the presence of cholesterol, highlighting that CD1 is antiviral in a cholesterol-independent manner.
To establish whether our functionalized CDs exhibited a broad-spectrum of antiviral activity comparable to the gold nanoparticles (10), we investigated their inhibitory effect against several HS-dependent viruses. As shown in Table 1, CD1 displayed broad-spectrum activity against a wide range of viruses belonging to different families. The molecule is active against HSV-1 as well as laboratory-passaged strains, clinical strains (passaged only once in cells), and acyclovir-resistant HSV-2. Antiviral activity is maintained against members of the Pneumoviridae family [respiratory syncytial virus type A (RSV-A) and B (RSV-B) and human metapneumovirus (HMPV)], against members of the Paramyxoviridae family [human parainfluenzavirus 3 (HPIV3)], and against members of the Flaviviridae family [dengue virus type 2 (DENV-2), ZIKV, and hepatitis C virus (HCV)]. For the latter, both the wild-type strain and variants resistant to protease inhibitors and NS5A inhibitors like BILN-2061 and daclatasvir (HCV D168A and HCV Y93H) (20) are susceptible to CD1. In addition, the compound proved to be active also in a fusion assay between cells expressing CD4 and cells expressing gp120-gp41 of HIV (21), demonstrating an ability to prevent this viral protein–receptor interaction. Enterovirus D68 (Fermon strain) and influenza virus H3N2, two viruses that were previously shown to be dependent on sialic acid, were not inhibited, supporting the specificity of the molecule (22). In all the different cell lines tested, no toxicity of the drug was evidenced up to 300 μg/ml, which corresponds to favorable selectivity indices (SIs). In contrast, Captisol, an FDA-approved sulfonated CD solubilizing reagent, displayed no antiviral activities.
Table 1. Inhibitory activity of CD1 on different viruses.
Results are mean of two independent experiments. n.a., not assessable.
| Virus |
EC50 (μg/ml) |
EC50 (μM) | CC50 (μg/ml) | SI | |
| CD1 | HSV-1 | 46.9 | 15.3 | >300 | >6.39 |
| HSV-2 | 26.4 | 8.61 | >300 | >11.4 | |
| HSV-2 R ACVR | 19.8 | 6.46 | >300 | >15.2 | |
| HSV-2 clinical | 34.3 | 11.2 | >300 | >8.74 | |
| RSV-A | 9.11 | 2.99 | >300 | >32.9 | |
| RSV-B | 5.48 | 1.79 | >300 | >54.7 | |
| HMPV | 0.613 | 0.201 | >300 | >489 | |
| PIV3 | 1.198 | 0.394 | >300 | >250 | |
| HIV | 10.19* | 3.35 | >300 | >29.4 | |
| DENV-2 | 14.1 | 4.63 | >300 | >21.3 | |
| ZIKV | 0.61 | 0.200 | >300 | >492 | |
| HCV | 3.88 | 1.27 | >300 | >77.3 | |
| HCV Y93H | 6.26 | 2.04 | >300 | >47.9 | |
| HCV D168A | 2.87 | 0.939 | >300 | >105 | |
| EV-D68 | n.a. | n.a. | >300 | >n.a. | |
| H3N2 | n.a. | n.a. | >300 | >n.a. | |
| Captisol | All viruses |
n.a. | n.a. | >300 | >n.a. |
*The experiment was conducted on a surrogate model of infection.
After having demonstrated its broad-spectrum antiviral activity, we evaluated the mechanism of action of CD1 through a time of addition assay using three different viruses (RSV-A, DENV-2, and HSV-2) (Fig. 2A). No activity was observed when cells were preincubated with compound and then washed out before infection, ruling out an indirect cell-mediated effect. Next, we evaluated the direct effect of the drug on the virus when added before, during, or after infection. The strongest inhibitory effect was observed when the compound was preincubated with the virus for 1 hour before infection, as shown in Fig. 2A. However, CD1 was active also when added directly during infection without prior incubation or after infection, opening possibilities for a therapeutic use (see also fig. S6). We subsequently verified, through virucidal assays, that the inhibition is linked to a permanent effect, which is not lost upon dilution (Fig. 2B). A DNA exposure assay further confirmed that after incubation with CD1, the HSV-2 genome becomes accessible to degradation by deoxyribonucleases (DNases), while nontreated viruses remain protected (Fig. 2C). We also observed a virucidal action against ZIKV (Table 1 and fig. S7), whose dependence on heparan sulfate proteoglycans (HSPGs) is debated (23–25).
Fig. 2. Mechanism of action of CD1.
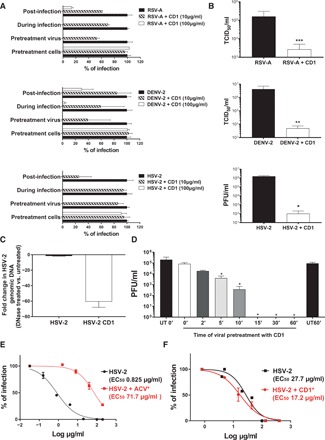
(A and B) CD1 was tested against RSV-A (top), DENV-2 (middle), and HSV-2 (bottom). (A) CD1 was either preincubated for 1 hour with the virus before addition on cells (Pretreatment virus), directly added with viruses on cells (During infection), added for 1 hour on cells and then washed out before infection (Pretreatment cells), or finally added after removal of the virus (Post-infection). (B) Virucidal assay: Viruses (in panels as above) were incubated with CD1 (30 μg) for 1 hour and then serially diluted on cells. (C) DNA exposure assay: HSV-2 was incubated in the presence or absence of CD1 (30 μg) for 1 hour at 37°C and then incubated for 30 min with Turbo DNase or only buffer and subsequently subjected to qPCR. Results are expressed in fold change of DNase treated versus untreated. (D) Time course of virucidal activity: HSV-2 and CD1 (30 μg) were incubated for different time periods and then serially diluted on Vero cells. (E and F) Drug resistance assay: HSV-2 passaged eight times in the presence of increasing concentrations of (E) acyclovir or (F) CD1, or no inhibitory compounds were subjected to a dose-response assay. The percentages of infection were calculated by comparing the number of plaques in treated and untreated wells. Results are mean and SEM of at least two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.005.
For HSV-2, we analyzed in depth the time dependency of the virucidal activity (Fig. 2D). It was possible to observe a significant reduction of viral titer after 5 min and a complete inactivation at 15 min. These results allow us to infer that because the time required to exert activity is short, CD1 is virucidal also when added after infection on the viral progeny released from infected cells and could inhibit cell-to-cell spread.
To further investigate the in vitro activity of CD1, its interaction with serum (26) was studied by comparing the EC50 in its presence or absence. As shown in fig. S8, CD1 is inhibited by fetal bovine serum (FBS), which is demonstrated by the significant shift in the EC50, both against HSV-2 and RSV-A. In the absence of serum, CD1 was shown to have nanomolar activity against HSV-2 and low micromolar activity against RSV-A. These compounds are thus optimal for topical administration, while they need further improvement to treat systemic infections.
To investigate the structural factors likely to lead to virucidal effects of this broad-spectrum antiviral, we performed atomistic molecular dynamics (MD) simulations of the three modified CDs (Fig. 3A) interacting with glycoprotein B (gB), which is located on the surface of HSV-2 (see the Supplementary Materials). The structural rationale for these comparisons was that relatively short seven-carbon sulfonated alkyl chain reduced the number of interactions between the CD and virus, leading to its observed antiviral affect. We also hypothesized that the nature of the linker plays an important role in antiviral properties, exemplified by the antiviral activity of CD3, which bears a nonalkyl linker of similar overall length to sodium undec-10-enesulfonate in CD1. Note that this linker leads to a better reversible viral inhibition but that the inhibitory effect is almost totally lost after dilution (i.e., limits to no virucidal effect). This leads us to propose that the nature of the linker, independent of the binding affinity, is key to provide a virucidal mode of action.
Fig. 3. MD simulations.
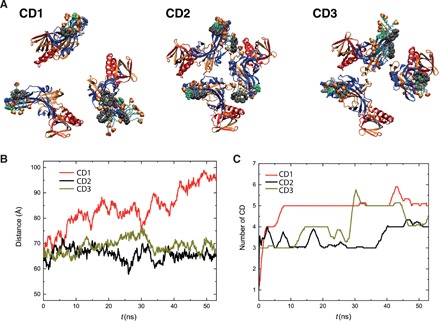
Interactions of CDs with gB proteins. (A) Representative snapshots of the modified CDs interacting with HSV-2 gB proteins after 50 ns of MD simulations. (B) Average distance between gB fusion loops, defined as the instantaneous average of the three distances labeled in fig. S9, over time. (C) Number of CDs interacting with gB proteins over time.
Figure 3 shows the results of CD1, CD2, and CD3 interacting with the gB protein, which were simulated by placing 10 CDs near the gB fusion loops (solvated in 0.15 M NaCl solution) before being released. Figure 3 (B and C) shows the number of binding CDs per gB and the distance between the gB fusion loops, respectively, after 50 ns. On average, CD1 bound to the most gB molecules over time, which induced a greater distance between gB fusion loops. Both CD2 and CD3 also interact with the fusion loops of gB, but they only cause small fluctuations in the average distance between the fusion loops. We propose that virucidal action of CDs relies on its ability to block the fusion loop, which subsequently leads to the induced gB conformational change (here, manifested by opening of the gB trimer). The stronger binding strength of CDs to gB does not guarantee larger binding numbers of CDs (fig. S9), because the interaction between CDs may cause the accumulation of CDs on the binding site.
The emergence of pathogen resistance to antimicrobial drugs is an important issue worldwide. One solution to drug resistance is the use of combined therapies, as is currently used in the treatment of HIV and HCV. Therefore, the identification of drugs with extremely high barriers to resistance is important. We compared the barrier for resistance to CD1 with that of acyclovir (an inhibitor of viral DNA polymerase, which is in clinical use) by culturing HSV-2 in the presence of increasing concentrations of the two molecules, using a procedure described previously (27). After eight passages in increasing concentrations of acyclovir (from 0.14 to 17.9 μg/ml), the EC50 value was 86.9 times higher than that of a virus passaged in the absence of the drug, when tested for its acyclovir inhibition activity, compared to a virus passaged in the absence of acyclovir (Fig. 2E). In contrast, no infectious virus was recovered in supernatants of cells treated with CD1 (160 μg/ml) by passage 5. For passages 5 to 8, the concentration of CD1 was maintained at 80 μg/ml, and still poor viral growth was observed. Therefore, as opposed to acyclovir, the EC50 value of CD1 did not change after eight passages in the presence of the drug, suggesting that under these experimental conditions, HSV-2 is unable to develop resistance to CD1.
As we have observed broad-spectrum virucidal activity and a high barrier to resistance in cultured cell lines, we tested whether the antiviral activity was maintained in three-dimensional (3D) tissue cultures derived from human biopsies and redifferentiated in vitro to mimic the 3D structure of human tissues. We evaluated the antiviral activity against RSV in an upper respiratory tissue model that reproduces the human upper airway epithelium with its characteristic mucus-secreting cells, ciliated cells, and basal cells (MucilAir, Epithelix). CD1 presented an efficiency comparable to palivizumab (an FDA-approved monoclonal antibody directed against RSV surface protein F) not only in co-treatment assays, where the drug and the virus were added simultaneously on the apical surfaces of tissues but also in therapeutic conditions where CD1 was added 24 hours post-infection (hpi), demonstrating the ability of CD1 to control the infection (Fig. 4, A and B). The complete protection in co-treatment assays was also confirmed by the absence of infected cells in treated tissues (Fig. 4D). CD1 caused neither toxicity nor release of proinflammatory cytokines even at high doses, or after repeated administrations, in this polarized respiratory tissue model (fig. S10). This tissue culture system also allows growth of viruses directly from infected specimens, thus avoiding potential artefacts that result from excessive adaptation to cell culture. We thus tested the ability of the compound to inhibit a clinical RSV-A isolate amplified only in reconstituted airway epithelia. Figure 4C shows that the inhibition is maintained also in strains currently circulating in the population. Of note, the inhibitory activity was reduced on clinical isolates compared to laboratory-adapted RSV isolates but remained comparable to that of palivizumab (Fig. 4C).
Fig. 4. Ex vivo experiments.
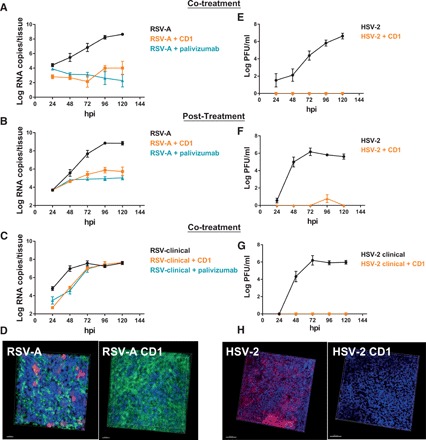
(A) Co-treatment of respiratory tissues with RSV-A and CD1 (10 μg). (B) Respiratory tissues infected with RSV-A and treated with CD1 (6 μg) 24 hpi (Post-treatment). (C) Co-treatment of respiratory tissues with a clinical strain of RSV-A and CD1 (10 μg). (D) Respiratory tissues infected with RSV-A or cotreated with RSV-A and CD1 were subjected to immunostaining at 7 days post-infection (dpi). Green, ciliated cells (tubulin); red, infected cells; blue, nuclei. Scale bars, 20 μm. (E) Co-treatment of vaginal tissues with HSV-2 and CD1 (50 μg). (F) Vaginal tissues infected with HSV-2 and treated with CD1 (30 μg) 8 hpi post-treatment. (G) Co-treatment of vaginal tissues with a clinical strain of HSV-2 and CD1 (50 μg). Results are mean and SEM of two independent experiments. (H) Vaginal tissues infected with HSV-2 or cotreated with HSV-2 and CD1 were subjected to immunostaining at 7 dpi. Red, infected cells; blue, nuclei. Scale bars, 100 μm.
An additional possible application of these modified CDs, in line with their broad-spectrum antiviral activities, is as a vaginal microbicide to prevent and treat sexually transmitted infections such as HIV, HPV (human papillomavirus), and HSV-2. Therefore, we tested the ability of CD1 to inhibit HSV-2 in ex vivo vaginal tissues. We observed a good inhibition (Fig. 4, E and F) both in co-treatment, confirmed also by immunofluorescence (Fig. 4H) and in post-treatment; in the latter, CD1 was formulated in 2.7% hydroxyethylcellulose (HEC) gel and added 8 hpi. Moreover, the compound was also able to inhibit a clinical HSV-2 strain isolated from a vaginal swab and passaged only once in cells (Fig. 4G). Because ZIKV is sexually transmissible, we also evaluated the ability to inhibit this virus in this ex vivo model. The results presented in fig. S7 show that, in the presence of CD1, the replication of the virus is abrogated, demonstrating the broad-spectrum activity also ex vivo. These data show that the activity evidenced in vitro is translated in ex vivo human models. Last, we evaluated the efficacy of CD1 formulated in HEC gel to prevent intravaginal HSV-2 infection in a murine model. BALB/c mice pretreated with the CD1 formulation exhibited only a slight weight loss and a lower increase in mean lesion scores after HSV-2 infection compared to mice that received the HEC gel (Fig. 5, A and B). The area under the curve (AUC) for mean lesion scores calculated from days 4 to 10 after infection was lower in the CD1 compared to the HEC group (39.70 ± 25.14 and 54.05 ± 17.78, respectively). No toxicity (such as weight loss, redness, and swelling in the vaginal region) was recorded following one intravaginal application of 15 μl of the HEC gel or CD1 formulation to mice. The percent survival was higher in mice that received the CD1 formulation (30%) compared to the HEC group (10%), but the difference was not statistically significant (Fig. 5C). Viral titers in vaginal mucosa of mice pretreated with the CD1 formulation were significantly lower compared to mice that received the HEC gel (Fig. 5D). These data show that the administration of CD1 formulation before intravaginal challenge with HSV-2 delayed the infection of mice. This suggests that CD1 could have an interesting potential as a vaginal microbicide.
Fig. 5. Intravaginal HSV-2 infection of mice.
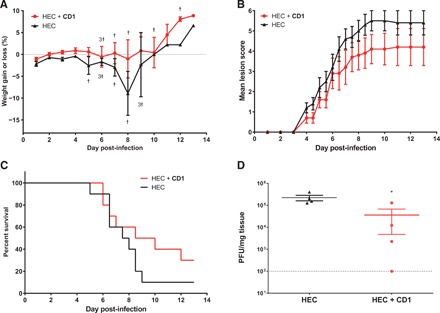
(A) Percentage of weight changes, (B) mean lesion scores, (C) survival rates, and (D) viral titers in vaginal mucosa of BALB/c mice pretreated with 15 μl of HEC gel or 0.03 mg of CD1 per mouse in HEC gel (15 μl). Five minutes later, mice were infected intravaginally with 1.2 × 105 PFU of HSV-2 strain 333 in a 5-μl volume. Vaginal mucosa was taken on day 3 after infection in a subset of mice, and viral titers were determined by plaque assay on Vero cells. Results represent the mean of 10 mice per group (A to C) or the mean and SEM of 4 mice per group (D). †, mouse death. *P < 0.05.
CONCLUSIONS
We have synthesized a biocompatible sulfonated CD that proved to be active against a large number of HS-dependent viruses. It exhibits a broad-spectrum virucidal, irreversible mechanism of action, presents a high barrier to viral resistance, and is biocompatible. We demonstrated its preventive and therapeutic activity both in cell lines and in human-derived pseudostratified and highly differentiated histocultures mimicking faithfully the upper respiratory tract and the vagina as well as in a relevant murine model of HSV-2 infection. Modified CDs are thus potent tools to fight multiple viral infections.
MATERIALS AND METHODS
All starting materials were purchased from Sigma-Aldrich and used as received unless stated otherwise. Captisol was provided by Ligand (San Diego, CA). All aqueous solutions were made in deionized water treated with a Milli-Q reagent system ensuring a resistivity of ≥15 megohm cm−1. The HEC placebo gel at pH 4.4 was obtained through the National Institutes of Health (NIH) Acquired Immunodeficiency Syndrome (AIDS) Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), NIH.
Cells
Cell lines A549, LLMCK2, BHK21, and Vero were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with heat-inactivated 10% FBS and 1% penicillin/streptomycin (Sigma-Aldrich) at 37°C in an atmosphere of 5% CO2. HeLa-P5L and HeLa-Env-Ada were gifts from O. Hartley.
Viruses
HSV-2 was provided by M. Pistello (University of Pisa, Italy) and was propagated and titrated on Vero cells with plaque assays. RSV-A [American Type Culture Collection (ATCC)], RSV-B (ATCC), and RSV-A mCherry (provided by Prof. J. F. Eleouet, Institut National de la Recherche Agronomique, France) were propagated in A549 cells in DMEM supplemented with 2.5% FBS and 1% penicillin/streptomycin and titrated by an indirect immunoperoxidase staining procedure using an RSV monoclonal antibody (Millipore, MAB5006). Influenza A/H3N2/Singapore/2004 was a gift from M. Schmolke (University of Geneva, Switzerland), and it was propagated on Madin-Darby canine kidney (MDCK) cells in DMEM supplemented with l-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK)–trypsin (0.2 μg/ml) and titrated on MDCK cells with an indirect immunoperoxidase staining procedure using a Flu A monoclonal antibody (Millipore, MAB5001). EV-D68 (strain Fermon, GenBank AY426531) was propagated on HeLa cells in DMEM supplemented with 2.5% FBS and titrated with the TCID50 (median tissue culture infectious dose) method. HMPV ATCC was propagated in Vero cells in DMEM supplemented with 1% penicillin/streptomycin and trypsin (200 ng/ml) and titrated by the indirect immunoperoxidase staining procedure using an HMPV monoclonal antibody (HMPV 24, Bio-Rad). Parainfluenza virus 3 (PIV3) (ATCC) was propagated in LLCMK2 cells in DMEM supplemented with 1% penicillin/streptomycin and trypsin (200 ng/ml) and titrated by plaque assay. An anonymized clinical isolate of RSV-A was confirmed by one-step real-time quantitative polymerase chain reaction (RT-qPCR), and subsequently, stocks were prepared by infection of MucilAir tissues with collection of supernatant from 48 to 120 hpi. JFH-1 wild type [HCV strain 2a (28)], JFH-1 D168A [HCV 2a (20)], and JFH-1 Y93H were propagated in Huh7.5.1, as described previously (20). DENV-2 was propagated from complementary DNA (cDNA) encoding cell culture–adapted strain 16681, as described previously (29). ZIKV strain PRVABC59 was provided by M. Alves (University of Bern, Switzerland). HSV-2 strain 333 was provided by L. R. Stanberry (Sealy Center for Vaccine Development, University of Texas, TX, USA) and propagated in Vero cells in minimum essential medium (MEM) supplemented with 2% FBS.
Cell viability
Cell viability was measured by the MTS assay (Promega). Confluent cell cultures seeded in 96-well plates were incubated with different concentrations of materials in triplicate under the same experimental conditions described for the antiviral assays. The 50% cytotoxic concentrations (CC50) and 95% confidence intervals (CIs) were determined using Prism software (GraphPad Software, San Diego, CA).
Inhibition assay for HSV-2, DENV-2, and ZIKV
The effect of CDs on HSV-2, DENV-2, and ZIKV infection was evaluated by a plaque reduction assay. Vero cells or BHK21 cells were seeded 24 hours in advance in 24-well plates at a density of 105 cells per well. Increasing concentrations of CDs were incubated with HSV-2, DENV-2, or ZIKV [multiplicity of infection (MOI), 0.0003 plaque-forming units (PFU)/cell] at 37°C for 1 hour, and then the mixtures were added to the cells. Following virus adsorption (2 hours at 37°C), the virus inoculum was removed and the cells were washed with medium and then overlaid with a medium containing 1.2% methylcellulose or Aquacide. After incubation with HSV-2, DENV-2, or ZIKV for 24, 72, or 120 hours, respectively, at 37°C, cells were fixed and stained with 0.1% of crystal violet in 20% ethanol and viral plaques were counted. The concentration of compound producing 50% reduction in plaque formation (EC50) was determined using the Prism software by comparing drug-treated and untreated wells.
Inhibition assay for RSV and HMPV
A549 cells (at 8000 cells per well) and Vero cells (at 12,103 cells per well) were seeded in 96-well plate. CDs were serially diluted in medium and incubated with virus (MOI, 0.01 PFU/cell) for 1 hour at 37°C, and then mixtures of compound and virus were added to cells to allow the viral adsorption for 3 hours at 37°C. The monolayers were then washed and overlaid with 1.2% methylcellulose medium; for HMPV, this medium contained trypsin (200 ng/ml). Three days after infection, cells were fixed with cold methanol and acetone for 1 min and subjected to RSV- or HMPV-specific immunostaining. Immunostained plaques were counted, and the percent inhibition of virus infectivity was determined by comparing the number of plaques in treated wells with the number in untreated control wells.
HCV inhibition assay
Huh7.5.1 cells were seeded in 12-well plates. On the following day, serial dilutions of CD1 were incubated for 1 hour with HCV at an MOI of 1 and subsequently added on cells. After 4 hours, the inoculum was removed and replaced with fresh medium. The cells were incubated for 72 hours at 37°C, and subsequently, cell lysates were made, followed by RNA extraction and qPCR analysis of viral genome.
Cell fusion assay
Cell fusion assay was conducted following previously published procedures (30). HeLa-P5L cells were seeded in 96-well plates (104 cells per well). Twenty-four hours later, medium was removed and replaced with medium containing 104 HeLa-Env-ADA cells per well and serial dilutions of CD1. After a further 24 hours, cells were washed once in phosphate-buffered saline (PBS), lysed, and assayed for β-galactosidase activity by the addition of the substrate CPRG (chlorophenol red-β-d-galactopyranoside). The percentage of fused cells was calculated subtracting the background value of only HeLa-P5L cells. Dose-inhibition curves were calculated with Prism software (GraphPad). The results are mean of two independent experiments performed in triplicate.
Evaluation of virucidal activity against HSV-2, RSV-A, and DENV-2
Viruses (105 PFU for HSV-2, 105 PFU for DENV-2, and 104 PFU for RSV-A) and CDs (300 μg/ml) were incubated for 1 hour at 37°C, and then the virucidal effect was investigated with serial dilutions of the mixtures. Viral titers were calculated at dilutions at which the CD was not effective.
Viral yield reduction assay
The assay was designed to quantify the antiviral effect of compound on the production of infectious viruses. BHK21 cells were seeded in 24-well plates at a density of 105 cells per well and infected in duplicate with DENV-2 at an MOI of 0.1 PFU/cell. Following adsorption at 37°C for 2 hours, the virus inoculum was removed and cultures were grown in the presence of serial dilutions of CD1 until control cultures displayed extensive cytopathology. Supernatants were harvested and pooled 48 hours after infection, and cell-free virus infectivity titers were determined in duplicate by TCID50 in BHK21 cell monolayers. Extents of inhibition were reported as the concentration of CDs that reduced virus yield by 50% (EC50) compared to untreated virus controls.
Time of addition assays
Materials (10 or 100 μg/ml) were added on cells 1 hour before infection, during infection, or after infection as described previously. Viral titers were then quantified by viral plaques assay. Viral titers were quantified by viral plaque assay 24 hpi for HSV-2, 72 hpi for RSV, and 120 hpi for DENV-2.
DNA exposure assay
HSV-2 (1 × 105 PFU) was incubated for 1 hour at 37°C with PBS or with PBS and 30 μg of CD1. Subsequently, the mixture was diluted in 1:20 in PBS, and 100 μl of mixtures was either exposed to 8 units of Turbo DNase (Thermo Fisher Scientific) or only incubated with Turbo DNase buffer for 30 min at 37°C. At the end of the incubation, cells were lysed and subjected to DNA extraction and qPCR amplification with TaqMan Universal PCR Master Mix (primers: 5′-CCGTCAGCACCTTCATCGA-3′ and 5′-CGCTGGACCTCCGTGTAGTC-3′; probe, 5′-FAM-CCACGAGATCAAGGACAGCGGCC-TAMRA). Fold change was calculated with the ΔCt method.
Resistance selection
HSV-2 mutants resistant to acyclovir or to CD1 were selected, as described previously (27). HSV-2 was passaged eight times in the presence of doubling concentrations of acyclovir or CD1. HSV-2 (MOI, 0.01) was used to infect Vero cells. Two hours after infection, the inoculum was removed, and the cells were washed and overlaid with medium containing increasing amounts of drugs (starting at 0.14 μg/ml of acyclovir and at 10 μg/ml of CD1). When the cultures displayed extensive cytopathic effects, the virus was collected and titrated on Vero cells. After eight passages, the viruses were subjected to a dose-response assay in parallel with HSV-2 passaged for eight times in the absence of drug and EC50 values were determined.
MucilAir antiviral assays
Tissues were obtained from Epithelix (Geneva, Switzerland) and used as described previously (31). For all experiments, epithelia were prepared with different single donor’s biopsies. All inserts were tested negative for HIV-1, mycoplasma, hepatitis B, hepatitis C, bacteria, yeast, and fungi. Before inoculation with RSV-A, MucilAir tissues were incubated in 250 μl of PBS Ca2+Mg2+ (PBS++) for 45 min at 37°C. One hundred microliters of viral suspension with or without CD in MucilAir culture medium was then applied apically. At 4 hours after incubation at 33°C, tissues were rinsed three times with MucilAir medium to remove nonadsorbed virus and cultures were continued in the air-liquid interface with 500 μl of fresh culture medium. At different times after inoculation, 200 μl of MucilAir medium was applied to the apical face of the tissue for 20 min at 33°C for sample collection. At the same time point, the basal medium was replaced with 500 μl of fresh MucilAir medium. For post-treatment experiments, CD1 was added after 24 hours of infection in 30 μl of MucilAir medium containing 200 μg/ml and fresh dilutions were added each day following the supernatant collection.
EpiVaginal antiviral assays
The EpiVaginal tissues (VEC-100/VEC-100-FT) were purchased from MatTek Corporation (Ashland, MA, USA). According to the manufacturer’s instructions, EpiVaginal cultures were seeded with the apical surface exposed to air in six-well plates containing 5 ml of MatTek assay medium (VEC-100-ASY) per well. Plates were incubated overnight at 37°C in 5% CO2. On the following day, the tissues were subjected to antiviral assays. CD1 at 500 μg/ml were mixed with HSV-2 (105 PFU) and added for 2 hours apically on EpiVaginal tissues in duplicate. At the end of incubation, any liquid remaining on top of the tissue was decanted and inserts were washed with PBS to remove any residual material. Alternatively, tissues were infected and then treated with CD1 at 1 mg/g formulated in 2.7% HEC gel at pH 4.4 [NIAID (32)] apically 8 hpi. The tissues were fed every other day with 5 ml of fresh medium basolaterally. Everyday, 200 μl of assay medium was applied to the tissue apically for 20 min, and then the collected medium was used for subsequent titration on Vero cells.
Viral load quantification
RSV RNA extracted with NucliSENS easyMAG (bioMérieux) was quantified by qPCR with the QuantiTect Kit (Qiagen, 204443) in a StepOne thermocycler (Applied Biosystems). The primer and probes were previously described (31, 33). HCV RNA was extracted from cells or from culture supernatants using Trizol of the QIAamp Viral RNA Mini Kit (Qiagen). A standard curve was generated using in vitro transcribed HCV viral RNA (vRNA). qRT-PCR was performed using the QuantiTect SYBR Green RT-PCR Kit (Qiagen) and the qRT-PCR forward (5′-CTGGCGACTGGATGCGTTTC-3′) and reverse (5′-CGCATTCCTCCATCTCATCA-3′) primers. Samples were analyzed using the 7300 RT-PCR System (Applied Biosystems).
Immunofluorescence
RSV was detected by direct mCherry expression, and β-tubulin primary rabbit antibody (Abcam) was used as a marker of ciliated cells. HSV-2 was detected by primary mouse antibody anti-gC (H1196, Santa Cruz Biotechnology). The Alexa 488 goat anti-rabbit antibody and the Alexa 594 goat anti-mouse antibody (Life Technologies) were used as secondary antibodies, and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired with a Zeiss LSM 700 Meta confocal microscope and processed by Imaris.
Murine model of HSV-2 intravaginal infection
BALB/c female mice were purchased from Charles River Canada (St-Constant, QC, Canada) at 4 weeks of age. Animals were housed four to five per cage and acclimated to standard laboratory conditions for 1 week. All animals were used in accordance to the Canadian Council on Animal Care guidelines, and the protocol was approved by the Animal Care Ethics Committee of Laval University (protocol no. 2018033). On day 7 before infection, mice received a subcutaneous injection of Depo-Provera (Pfizer Canada Inc., Kirkland, QC, Canada) in the neck region (2.5 mg in sterile physiological water). On the day of infection, vaginal secretions were removed using two Puritan Calcium alginate nasopharyngeal swabs. Mice were then pretreated intravaginally with 2.7% HEC gel (15 μl) at pH 4.0 (14 mice) or 0.03 mg of CD1 per mouse in HEC gel (15 μl; 14 mice) using a gavage needle. Five minutes later, mice were infected intravaginally with 1.2 × 105 PFUs of HSV-2 strain 333 in a 5-μl volume using a micropipette [Roy et al. (34). AAC.45:1671-1681]. Ten mice per group were weighed and examined daily for 13 days to evaluate the clinical signs of infection according to a lesion score (i.e., 0, no sign; 1, redness; 2, redness and swelling without hair loss; 3, redness and swelling with hair loss; 4, massive hair loss or ruffled fur; 5, massive hair loss and ulcerated epithelium or hunched posture; 6, hind limb paralysis or death). Animals were sacrificed when a weight loss equal to or greater than 20% were recorded. On day 3 after infection, four mice per group were euthanized. The vaginal mucosa was collected and homogenized in PBS. Infectious viral titers were determined by a standard plaque assay on Vero cells. Briefly, monolayers of Vero cells were infected with serial dilutions of vaginal mucosa homogenates in MEM supplemented with 2% FBS. Plates were centrifuged at 800g for 45 min at room temperature. Samples were removed and replaced by MEM + 2% FBS containing 0.4% SeaPlaque agarose (Lonza, Walkersville, MD) for 2 days. Cells were fixed and stained, and the number of plaques was counted. The limit of detection of the assay is 5 PFUs per well.
Data analysis
The EC50 values for inhibition curves were calculated by regression analysis using the program GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA) to fit a variable slope sigmoidal dose-response curve. The SIs were calculated dividing the CC50 for the EC50. Differences in weight changes, mean lesion scores, and viral titers in the vaginal mucosa between mice pretreated with the HEC gel or the CD1 formulation were analyzed at each day after infection by a unilateral Student’s t test using Microsoft Excel 2013 (Microsoft Corporation). The AUC values for mean lesion scores were calculated as [(score on day 4 + score on day 10)/2] + sum of all scores between days 4 and 10.
Synthesis of compounds
All solvents used were dry, and reactions were carried out under argon atmosphere. The starting materials were purchased from Sigma-Aldrich if not otherwise stated. Heptakis-(6-deoxy-6-mercapto)-β-CD and hexakis-(6-deoxy-6-mercapto)-α-CD were purchased from Cyclodextrin-Shop, Netherlands. Care was taken to use a freshly synthesized batch to minimize the presence of disulfides.
Synthesis of sodium undec-10-ene-1-sulfonate
11-Bromo-1-undecene (2.122 g, 9.1 mmol) and Na2SO3 (3.061 g, 24.3 mmol) were added into a mixture of MeOH (25 ml) and H2O (45 ml) and refluxed for 18 hours. MeOH was removed under reduced pressure vacuo, and the aqueous layer was washed with Et2O (2 × 50 ml) and dried. The white solid was extracted with hot MeOH and filtered twice, leaving the insoluble Na2SO3 salt on the filter. The filtrate was recrystallized from EtOH/H2O mixture (20/1), and the product was collected as white crystalline needles (1.983 g, 85%). 1H NMR (nuclear magnetic resonance) [400 MHz, dimethyl sulfoxide (DMSO)–d6] δ 5.78 (ddt, J = 17.0, 10.2, 6.5 Hz, 1H), 4.98 (d, J = 17.0 Hz, 1H), 4.92 (d, J = 10.2 Hz, 1H), 2.41 (dd, J = 9.4, 6.5 Hz, 2H), 1.99 (q, J = 7.1 Hz, 2H), 1.55 (p, J = 7.1 Hz, 2H), 1.25 (m, 14H). 13C NMR (101 MHz, DMSO) δ 139.27, 115.10, 51.92, 33.64, 29.35, 29.30, 28.98, 28.86, 28.86, 28.74, and 25.50. High-resolution mass spectrometry (HRMS) [electrospray ionization (ESI)/quadrupole time-of-flight (QTOF)] mass/charge ratio (m/z): [M − Na+1]− calculated for C11H21O3S− 233.1217; found 233.1214.
Synthesis of sodium hept-6-ene-1-sulfonate
7-Bromo-1-heptene (2.150 g, 12.1 mmol) and Na2SO3 (4.591 g, 36.4 mmol) were added into a mixture of MeOH (25 ml) and H2O (45 ml) and refluxed for 18 hours. MeOH was removed under reduced pressure, and the aqueous layer was washed with Et2O (2 × 50 ml) and dried. The white solid was extracted with hot MeOH and filtered (twice). The filtrate was recrystallized from EtOH/H2O mixture (20/1), and the product was collected as white crystalline needles (1.862 g, 77%). 1H NMR (400 MHz, DMSO-d6) δ 5.79 (ddt, J = 17.0, 10.3, 6.6 Hz, 1H), 4.99 (dd, J = 17.0, 2.1 Hz, 1H), 4.93 (dd, J = 10.3, 2.0 Hz, 1H), 2.47 − 2.30 (m, 1H), 2.00 (q, J = 6.8 Hz, 2H), 1.56 (p, J = 7.3 Hz, 2H), and 1.32 (m, 4H). 13C NMR (101 MHz, DMSO) δ 137.79, 112.01, 48.81, 30.58, 25.39, 25.01, and 21.62. HRMS (ESI/QTOF) m/z: [M − Na+1]− calculated for C7H13O3S− 177.0591; found 177.0594.
Synthesis of 4-(bromomethyl)-N-(4-vinylphenyl)benzamide
4-(Bromomethyl)benzoic acid (1.200 g, 5.58 mmol) was dissolved in CH2Cl2 (15 ml), dimethylformamide (DMF) (1 drop, catalyst) was added, and the mixture was cooled on ice. Diluted (COCl)2 (1.43 ml, 16.74 mmol) in CH2Cl2 (5 ml) was slowly added, and the reaction mixture was allowed to warm up to room temperature, where it was stirred for 3 hours. After the solvent was removed under reduced pressure, the yielded solid was diluted in CH2Cl2 (10 ml) and slowly added to a mixture of N,N-diisopropylethylamine (1.488 ml, 16.74 mmol) and 4-vinylaniline (732 mg, 6.14 mmol) in CH2Cl2 (15 ml) at 0°C. The reaction mixture was stirred at room temperature for 3 hours and quenched with aqueous NH4Cl (5%, 200 ml). The solution was extracted with EtOAc (200 ml), and the organic layer was washed with aqueous NH4Cl (5%, 150 ml) again. The organic layer was subsequently heated and filtered. The filtrate was reduced under pressure and recrystallized from EtOAc, and the product was collected as white crystalline solid (1.105 g, 63%). 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 7.93 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.6 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 6.70 (dd, J = 17.6, 10.9 Hz, 1H), 5.77 (d, J = 17.6 Hz, 1H), 5.20 (d, J = 10.9 Hz, 1H), and 4.78 (s,2H). 13C NMR (101 MHz, DMSO) δ 165.00, 141.50, 138.81, 136.16, 134.68, 132.59, 129.23, 128.05, 126.44, 120.21, 112.97, and 33.46. HRMS (ESI/QTOF) m/z: [M + H]+ calculated for C16H15BrNO+ 316.0332; found 316.0332.
Synthesis of sodium (4-((4-vinylphenyl)carbamoyl)phenyl)methanesulfonate
4-(Bromomethyl)-N-(4-vinylphenyl)benzamide (760 mg, 2.4 mmol) and Na2SO3 (364 mg, 2.88 mmol) were added into a mixture of MeOH (2 ml) and H2O (5 ml) and heated to 80°C for 2.5 hours. After MeOH was removed under reduced pressure, more H2O (10 ml) was added and the aqueous fraction was washed with Et2O (15 ml), before being dried under reduced pressure. The solid was recrystallized from MeOH/H2O mixture (1:1) and collected as white crystalline solid (620 mg, 76%) on filter. 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H), 7.84 (d, J = 8.3 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 7.44 (dd, J = 8.3, 4.7 Hz, 4H), 6.69 (dd, J = 17.6, 10.9 Hz, 1H), 5.76 (d, J = 17.6 Hz, 1H), 5.18 (d, J = 10.9 Hz, 1H), and 3.80 (s, 2H). 13C NMR (101 MHz, DMSO) δ 165.79, 139.38, 139.09, 136.34, 132.86, 132.65, 130.33, 127.09, 126.59, 120.45, 113.11, and 57.44. HRMS (ESI/QTOF) m/z: [M − Na+1]− calculated for C16H14NO4S− 316.0649; found 316.0652.
Synthesis of CD1
Heptakis-(6-deoxy-6-mercapto)-β-CD (50 mg, 0.040 mmol), sodium undec-10-ene-1-sulfonate (108 mg, 0.421 mmol), and 2,2-dimethoxy-2-phenylacetophenone (22 mg, 0.084 mmol) were dissolved in DMSO (5 ml). The reaction mixture was placed under ultraviolet (UV) lamp (250 W) and stirred for 18 hours. Crude product was precipitated by the addition of Et2O (45 ml) and collected by centrifugation. The off-white solid was washed by MeOH (45 ml) and EtOH (45 ml) and collected by centrifugation. The product was purified by dialysis against Milli-Q H2O for 3 days, filtered through 0.2-μm filter, and collected as a yellow solid (92 mg, 76%).
Synthesis of CD2
Heptakis-(6-deoxy-6-mercapto)-β-CD (50 mg, 0.040 mmol), sodium hept-6-ene-1-sulfonate (85 mg, 0.421 mmol), and 2,2-dimethoxy-2-phenylacetophenone (22 mg, 0.084 mmol) were dissolved in DMSO (5 ml). The reaction mixture was placed under UV lamp (250 W) and stirred for 18 hours. Crude product was precipitated by the addition of Et2O (45 ml) and collected by centrifugation. The off-white solid was washed with MeOH (45 ml) and EtOH (45 ml) and collected by centrifugation. The product was purified by dialysis against Milli-Q H2O for 3 days, filtered through 0.2-μm filter, and collected as a yellow solid (74 mg, 70%).
Synthesis of CD3
Heptakis-(6-deoxy-6-mercapto)-β-CD (50 mg, 0.040 mmol), sodium (4-((4-vinylphenyl)carbamoyl)phenyl)methanesulfonate (143 mg, 0.421 mmol), and 2,2-dimethoxy-2-phenylacetophenone (22 mg, 0.084 mmol) were dissolved in DMSO (5 ml). The reaction mixture was placed under UV lamp (250 W) and stirred for 18 hours. Crude product was precipitated by the addition of Et2O (45 ml) and collected by centrifugation. The off-white solid was washed with MeOH (45 ml) and collected by centrifugation. The product was purified by dialysis against Milli-Q H2O for 3 days, filtered through 0.2-μm filter, and collected as a yellow solid (81 mg, 56%).
Supplementary Material
Acknowledgments
We thank F. Cerini for helping with the HIV-related fusion assay. We thank N. Van Buuren for the help with HCV inhibition assays. Funding: S.T.J. was supported by Dame Kathleen Ollerenshaw Fellowship. V.C. was supported by Firmenich Foundation for EPFL-Stanford Exchange. J.P. and G.B. were financed by the Canadian Institutes of Health Research (grant no. 148361 to G.B.). L.V. was supported by the NIAID of the NIH under award number R03AI142553. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank the reviewers for their useful feedback on additional experiments especially the DNase experiment that strengthened the message in our manuscript. The work was supported by the Leenaards Foundation (grant 4390 to C.T. and F.S.) and the Swiss National Science Foundation (via a Sinergia grant CRSII5_180323 to F.S. and C.T.), and the National Center of Competence in Research (NCCR) Bio-Inspired Materials that supported M.G. Author contributions: S.T.J. developed the CD synthesis and synthesized CD1 and CD2. V.C. performed the in vitro assays and ex vivo assays. The experiments on dengue virus were performed with the help of D.A.C. under the supervision of K.K. M.J. synthesized CD2 and CD3. D.O. and N.G. characterized the CDs. J.P. and G.B. performed the in vivo experiments. M.G. formulated the CD for ex vivo and in vivo experiments. Y.H., L.V., and P.K. performed the simulation experiments. S.H. and S.C. developed the respiratory tissue model. L.K. revised the manuscript. S.T.J., V.C., F.S., and C.T. conceived and planned the experiments. S.T.J., V.C., F.S., and C.T. wrote the manuscript with the revisions of all authors. Competing interests: S.T.J. and F.S. are inventors on patent number WO 2018/015465 A1—Virucidal compounds and uses thereof. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax9318/DC1
Section S1. Synthesis, characterization, and assays
Fig. S1. Reaction scheme overview of the synthesized modified CDs.
Fig. S2. Characterization of CD1.
Fig. S3. Characterization of CD2.
Fig. S4. Characterization of CD3.
Fig. S5. Cholesterol replenishment.
Fig. S6. Viral yield reduction assay of DENV-2.
Fig. S7. Virucidal activity and ex vivo inhibition against ZIKV.
Fig. S8. Viral inhibition in the presence or absence of serum.
Fig. S9. Simulations of CDs with gB fusion loops.
Fig. S10. Toxicity of CD1 in respiratory tissues.
REFERENCES AND NOTES
- 1.De Clercq E., Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 1, 13–25 (2002). [DOI] [PubMed] [Google Scholar]
- 2.McCormack S., Ramjee G., Kamali A., Rees H., Crook A. M., Gafos M., Jentsch U., Pool R., Chisembele M., Kapiga S., Mutemwa R., Vallely A., Palanee T., Sookrajh Y., Lacey C. J., Darbyshire J., Grosskurth H., Profy A., Nunn A., Hayes R., Weber J., PRO2000 vaginal gel for prevention of hiv-1 infection (microbicides development programme 301): A phase 3, randomised, double-blind, parallel-group trial. Lancet 376, 1329–1337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pirrone V., Wigdahl B., Krebs F. C., The rise and fall of polyanionic inhibitors of the human immunodeficiency virus type 1. Antivir. Res. 90, 168–182 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Van Damme L., Govinden R., Mirembe F. M., Guédou F., Solomon S., Becker M. L., Pradeep B., Krishnan A., Alary M., Pande B., Ramjee G., Deese J., Crucitti T., Taylor D.; CS Study Group , Lack of effectiveness of cellulose sulfate gel for the prevention of vaginal hiv transmission. N. Engl. J. Med. 359, 463–472 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Rusnati M., Vicenzi E., Donalisio M., Oreste P., Landolfo S., Lembo D., Sulfated K5 Escherichia coli polysaccharide derivatives: A novel class of candidate antiviral microbicides. Pharmacol. Ther. 123, 310–322 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Baram-Pinto D., Shukla S., Gedanken A., Sarid R., Inhibition of hsv-1 attachment, entry, and cell-to-cell spread by functionalized multivalent gold nanoparticles. Small 6, 1044–1050 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Bergstrom D. E., Lin X., Wood T. D., Witvrouw M., Ikeda S., Andrei G., Snoeck R., Schols D., De Clercq E., Polysulfonates derived from metal thiolate complexes as inhibitors of hiv-1 and various other enveloped viruses in vitro. Antivir. Chem. Chemother. 13, 185–195 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Scordi-Bello I. A., Mosoian A., He C., Chen Y., Cheng Y., Jarvis G. A., Keller M. J., Hogarty K., Waller D. P., Profy A. T., Herold B. C., Klotman M. E., Candidate sulfonated and sulfated topical microbicides: Comparison of anti-human immunodeficiency virus activities and mechanisms of action. Antimicrob. Agents Chemother. 49, 3607–3615 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abe M., Kaneko K., Ueda A., Otsuka H., Shiosaki K., Nozaki C., Goto S., Effects of several virucidal agents on inactivation of influenza, newcastle disease, and avian infectious bronchitis viruses in the allantoic fluid of chicken eggs. Jpn. J. Infect. Dis. 60, 342–346 (2007). [PubMed] [Google Scholar]
- 10.Cagno V., Andreozzi P., D’Alicarnasso M., Jacob Silva P., Mueller M., Galloux M., Le Goffic R., Jones S. T., Vallino M., Hodek J., Weber J., Sen S., Janeček E.-R., Bekdemir A., Sanavio B., Martinelli C., Donalisio M., Rameix Welti M.-A., Eleouet J.-F., Han Y., Kaiser L., Vukovic L., Tapparel C., Král P., Krol S., Lembo D., Stellacci F., Broad-spectrum non-toxic antiviral nanoparticles with a virucidal inhibition mechanism. Nat. Mater. 17, 195–203 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Haute D. V., Berlin J. M., Challenges in realizing selectivity for nanoparticle biodistribution and clearance: Lessons from gold nanoparticles. Ther. Deliv. 8, 763–774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valle E. D., Cyclodextrins and their uses: A review. Process Biochem. 39, 1033–1046 (2004). [Google Scholar]
- 13.Davis M. E., Brewster M. E., Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 3, 1023–1035 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Anand R., Nayyar S., Pitha J., Merril C. R., Sulphated sugar alpha-cyclodextrin sulphate, a uniquely potent anti-hiv agent, also exhibits marked synergism with azt, and lymphoproliferative activity. Antivir. Chem. Chemother. 1, 41–46 (1990). [Google Scholar]
- 15.Moriya T., Kurita H., Matsumoto K., Otake T., Mori H., Morimoto M., Ueba N., Kunita N., Potent inhibitory effect of a series of modified cyclodextrin sulfates on the replication of hiv-1 in vitro. J. Med. Chem. 34, 2301–2304 (1991). [DOI] [PubMed] [Google Scholar]
- 16.Otake T., Schols D., Witvrouw M., Naesens L., Nakashima H., Moriya T., Kurita H., Matsumoto K., Ueba N., De Clercq E., Modified cyclodextrin sulphates(mcds11) have potent inhibitory activity against hiv and high oral bioavailabiiity. Antivir. Chem. Chemother. 5, 155–161 (1994). [Google Scholar]
- 17.Moriya T., Saito K., Kurita H., Matsumoto K., Otake T., Mori H., Morimoto M., Ueba N., Kunita N., A new candidate for an anti-hiv-1 agent: Modified cyclodextrin sulfate (mcds71). J. Med. Chem. 36, 1674–1677 (1993). [DOI] [PubMed] [Google Scholar]
- 18.Leydet A., Moullet C., Roque J. P., Witvrouw M., Pannecouque C., Andrei G., Snoeck R., Neyts J., Schols D., Clercq E. D., Polyanion inhibitors of HIV and other viruses. 7. Polyanionic compounds and polyzwitterionic compounds derived from cyclodextrins as inhibitors of HIV transmission. J. Med. Chem. 41, 4927–4932 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Nishijo J., Moriyama S., Shiota S., Interactions of cholesterol with cyclodextrins in aqueous solution. Chem. Pharm. Bull. 51, 1253–1257 (2003). [DOI] [PubMed] [Google Scholar]
- 20.van Buuren N., Tellinghuisen T. L., Richardson C. D., Kirkegaard K., Transmission genetics of drug-resistant hepatitis C virus. eLife 7, e32579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pleskoff O., Tréboute C., Brelot A., Heveker N., Seman M., Alizon M., Identification of a chemokine receptor encoded by human cytomegalovirus as a cofactor for hiv-1 entry. Science 276, 1874–1878 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Royston L., Essaidi-Laziosi M., Pérez-Rodríguez F. J., Piuz I., Geiser J., Krause K.-H., Huang S., Constant S., Kaiser L., Garcin D., Tapparel C., Viral chimeras decrypt the role of enterovirus capsid proteins in viral tropism, acid sensitivity and optimal growth temperature. PLOS Pathog. 14, e1006962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghezzi S., Cooper L., Rubio A., Pagani I., Capobianchi M. R., Ippolito G., Pelletier J., Meneghetti M. C. Z., Lima M. A., Skidmore M. A., Broccoli V., Yates E. A., Vicenzi E., Heparin prevents zika virus induced-cytopathic effects in human neural progenitor cells. Antivir. Res. 140, 13–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan C. W., Sam I.-C., Chong W. L., Lee V. S., Chan Y. F., Polysulfonate suramin inhibits zika virus infection. Antivir. Res. 143, 186–194 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Cagno V., Tseligka E. D., Jones S. T., Tapparel C., Heparan sulfate proteoglycans and viral attachment: True receptors or adaptation bias? Viruses 11, 596 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.F. W. Putnam, All About Albumin, T. Peters, Ed. (Academic Press, 1995), pp. xi–xiii. [Google Scholar]
- 27.Donalisio M., Nana H. M., Ngono Ngane R. A., Gatsing D., Tiabou Tchinda A., Rovito R., Cagno V., Cagliero C., Boyom F. F., Rubiolo P., Bicchi C., Lembo D., In vitro anti-herpes simplex virus activity of crude extract of the roots of nauclea latifolia smith (rubiaceae). BMC Complement. Altern. Med. 13, 266 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato T., Date T., Murayama A., Morikawa K., Akazawa D., Wakita T., Cell culture and infection system for hepatitis c virus. Nat. Protoc. 1, 2334–2339 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Constant D. A., Mateo R., Nagamine C. M., Kirkegaard K., Targeting intramolecular proteinase ns2b/3 cleavages for trans-dominant inhibition of dengue virus. Proc. Natl. Acad. Sci. U.S.A. 115, 10136–10141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartley O., Dorgham K., Perez-Bercoff D., Cerini F., Heimann A., Gaertner H., Offord R. E., Pancino G., Debré P., Gorochov G., Human immunodeficiency virus type 1 entry inhibitors selected on living cells from a library of phage chemokines. J. Virol. 77, 6637–6644 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essaidi-Laziosi M., Brito F., Benaoudia S., Royston L., Cagno V., Fernandes-Rocha M., Piuz I., Zdobnov E., Huang S., Constant S., Boldi M.-O., Kaiser L., Tapparel C., Propagation of respiratory viruses in human airway epithelia reveals persistent virus-specific signatures. J. Allergy Clin. Immunol. 141, 2074–2084 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tien D., Schnaare R. L., Kang F., Cohl G., McCormick T. J., Moench T. R., Doncel G., Watson K., Buckheit R. W. Jr., Lewis M. G., Schwartz J., Douville K., Romano J. W., In vitro and in vivo characterization of a potential universal placebo designed for use in vaginal microbicide clinical trials. AIDS Res. Hum. Retrovir. 21, 845–853 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Essaidi-Laziosi M., Lyon M., Mamin A., Fernandes Rocha M., Kaiser L., Tapparel C., A new real-time rt-qpcr assay for the detection, subtyping and quantification of human respiratory syncytial viruses positive- and negative-sense rnas. J. Virol. Methods 235, 9–14 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Roy S., Gourde P., Piret J., Désormeaux A., Lamontagne J., Haineault C., Omar R. F., Bergeron M. G., Thermoreversible gel formulations containing sodium lauryl sulfate or n-Lauroylsarcosine as potential topical microbicides against sexually transmitted diseases. Antimicrob. Agents Chemother. 45, 1671–1681 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansze S. M., Ortiz D., Fadaei Tirani F., Scopelliti R., Menin L., Severin K., Inflating face-capped pd6l8 coordination cages. Chem. Commun. 54, 9529–9532 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Zeev-Ben-Mordehai T., Vasishtan D., Hernández Durán A., Vollmer B., White P., Prasad Pandurangan A., Siebert C. A., Topf M., Grünewald K., Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein b. Proc. Natl. Acad. Sci. U.S.A. 113, 4176–4181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Hannah B. P., Cairns T. M., Bender F. C., Whitbeck J. C., Lou H., Eisenberg R. J., Cohen G. H., Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83, 6825–6836 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A. D. Jr., Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M., All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K., Scalable molecular dynamics with namd. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darden T., York D., Pedersen L., Particle mesh ewald: An nlog(n) method for ewald sums in large systems. J. Chem. Phys. 98, 10089 (1993). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax9318/DC1
Section S1. Synthesis, characterization, and assays
Fig. S1. Reaction scheme overview of the synthesized modified CDs.
Fig. S2. Characterization of CD1.
Fig. S3. Characterization of CD2.
Fig. S4. Characterization of CD3.
Fig. S5. Cholesterol replenishment.
Fig. S6. Viral yield reduction assay of DENV-2.
Fig. S7. Virucidal activity and ex vivo inhibition against ZIKV.
Fig. S8. Viral inhibition in the presence or absence of serum.
Fig. S9. Simulations of CDs with gB fusion loops.
Fig. S10. Toxicity of CD1 in respiratory tissues.