

Abstract
Aspergillus flavus is best known for producing the family of potent carcinogenic secondary metabolites known as aflatoxins. However, this opportunistic plant and animal pathogen also produces numerous other secondary metabolites, many of which have also been shown to be toxic. While about forty of these secondary metabolites have been identified from A. flavus cultures, analysis of the genome has predicted the existence of at least 56 secondary metabolite gene clusters. Many of these gene clusters are not expressed during growth of the fungus on standard laboratory media. This presents researchers with a major challenge of devising novel strategies to manipulate the fungus and its genome so as to activate secondary metabolite gene expression and allow identification of associated cluster metabolites. In this review, we discuss the genetic, biochemical and bioinformatic methods that are being used to identify previously uncharacterized secondary metabolite gene clusters and their associated metabolites. It is important to identify as many of these compounds as possible to determine their bioactivity with respect to fungal development, survival, virulence and especially with respect to any potential synergistic toxic effects with aflatoxin.
Key words: : Aspergillus flavus, gene cluster, mycotoxins, secondary metabolite
1. General Introduction
Chemical analysis of fungal developmental structures such as hyphae, conidia, fruiting bodies and sclerotia has uncovered a plethora of secondary metabolites, many of which are secreted into the environment1). Secondary metabolites (SMs) are low molecular weight natural products that often display bioactive properties and are produced from precursors derived from primary metabolism. Secondary metabolites are, by definition, not essential to the producing organism and for the most part are characterized based on their impact on human and animal health. Fungal SMs are quite diverse in their chemistry and their bioactivities, being sources of beneficial therapeutic compounds such as penicillins and lovastatin as well as harmful toxins such as aflatoxins and fumonisins. The evolutionary advantages of the production, sequestration and secretion of bioactive SMs by fungi has been the subject of decades of scientific speculation. Though not essential for growth like primary metabolites, SMs do impart advantages to the producing organism. Fungi are in a constant battle with both predator and competitor organisms that they must be able to coexist, compete and interact with in their particular environmental niche. These niches, be they the human body or an agricultural field are dynamic environments subject to change where fungi must be able to adapt in order to compete for available nutrients allowing them to reproduce and disseminate thus ensuring survival. Fungi have evolved the capability to produce a wide array of SMs in response to their environments many of which play important biological roles such as virulence factors, chemical defense agents, insect attractants, protection from abiotic stressors, developmental regulators, and as chemical signals for communication with other organisms.
Aspergillus flavus is a saprophyte in soils worldwide where it survives by obtaining nutrients from dead or decaying organic matter. In soils, A. flavus can survive as propagules in the form of as asexual conidia or overwintering structures termed sclerotia that can germinate when environmental conditions become conducive to growth to form hyphae and conidiophores. With the discovery of a sexual stage, A. flavus sclerotia were shown to function as fruiting bodies, termed stromata, that harbor the sexual ascospores2). A. flavus is a frequent contaminant of agricultural crops such as maize, peanut, cottonseed, and a number of tree nuts, both pre- and post-harvest, where it produces the toxic and carcinogenic group of SMs known as aflatoxins (AFs)3). Contamination of susceptible crops with AFs poses serious health risks in developing countries and significant economic losses in the U.S. and other developed countries4,5). AF contamination of crops usually results from opportunistic invasion by the fungus due, in most cases, to mechanical or insect damage of the plant cell wall6).
The A. flavus genome is estimated to be approximately 37 Mb and capable of encoding about 13,000 predicted functional genes7). Though best known for its production of AFs, in silico analysis of the A. flavus 3357 genome sequence utilizing the web-based Secondary Metabolite Unique Regions Finder (SMURF) algorithm (http://www.jcvi.org/smurf) has shown that it has the potential to produce numerous other SMs. Using SMURF, Georgianna et al.8) predicted the genome of A. flavus to contain 55 SM gene clusters. The total number of SM clusters in A. flavus can be increased to 56 when including the kojic acid (KA) gene cluster identified originally in A. oryzae by Marui et al.9). However this may be an underestimation, as other SM gene cluster prediction programs have identified even greater numbers of gene clusters in A. flavus10). Although about 40 SMs have been identified from A. flavus cultures11–13) (Table 1), metabolites have only been experimentally assigned to twelve of the predicted 56 A. flavus SM clusters (see section 5). Elucidation of the products of the remaining SM gene clusters is confounded by the fact that many are transcriptionally silent, inactive due to gene mutations or expressed at extremely low levels thus making identification of their respective products problematic under laboratory growth conditions. These types of gene clusters are termed cryptic or orphan and identification of their metabolite(s) cannot be achieved using classical knockout of a cluster gene followed by comparative metabolic analysis of the mutant and the wild-type by liquid chromatography-mass spectrometry (LC-MS). Analysis of ribonucleic acid sequencing (RNA-Seq) data from a number of A. flavus strains growing on artificial media indicates that approximately half of the gene clusters are silent or expressed at very low levels18). A number of methods have been used to overcome silent gene clusters thereby enabling identification of some previously unknown cluster metabolites19–21). Determination of the chemical structure and biological activities of metabolites produced by these uncharacterized gene clusters will undoubtedly provide invaluable insights into the ecology of A. flavus as well as other fungal plant pathogens harboring similar gene clusters. It is important to identify as many of these SMs as possible to determine their bioactivity with respect to fungal development, survival, virulence and especially with respect to any potential synergistic toxic effects with AF.
Table 1. A. flavus secondary metabolites.
| Compound | Class | Reference |
|---|---|---|
| 3-nitropropionic acid | propionate | 12 |
| 4-hydroxymethyl-5-hydroxy-2H-pyran-2-one | pyranone | 13 |
| aflatoxins | difurocoumarolactone | 12 |
| aflatrem | indole diterpene | 12 |
| aflavarin | bicoumarin | 13 |
| aflavazole | indole diterpene | 12 |
| aflavinines | indole diterpene | 12 |
| asparasone | anthraquinone | 13 |
| asperflavin | anthrone | 12 |
| aspergillic acids | pyrazinone-hydroxamic acid | 12 |
| asperipin-2a | cyclic peptide | 14 |
| aspirochlorines | epithiodiketopiperazine | 13 |
| cladosporin | isocoumarin | 12 |
| cyclo(D-N-methyl-Leu-L-Trp) | diketopiperazine | 13 |
| cyclopiazonic acid | indole-tetramic acid | 12 |
| ditryptophenaline | diketopiperazine | 12 |
| ferriaspergillin | pyrazinone-hydroxamic acid | 12 |
| flavoglaucin | quinol | 13 |
| flufuran | furan | 13 |
| gliotoxin | epithiodiketopiperazine | 13 |
| indole | indole | 13 |
| isoflavipucine | pyridone | 12 |
| itaconic acid | succinate | 13 |
| kojic acid | pyranone | 13 |
| kotanin | bicoumarin | 13 |
| leporins | pyridone | 15 |
| lna and lnb piperazines | piperazine | 16 |
| lovastatin | nonaketide | 13 |
| mellein | isocoumarin | 13 |
| miyakamides | didehydrotryptamine-dipeptide | 13 |
| oxylipins | fatty acid | 13 |
| parasiticol | difurocoumarolactone | 13 |
| parasiticolide A | terpene | 13 |
| paspaline | indole diterpene | 12 |
| paspalinine | indole diterpene | 12 |
| penicillin | beta-lactam | 13 |
| sterigmatocystins | difuroxanthone | 13 |
| ustiloxin B | cyclic peptide | 17 |
In this review, we present information on the identification and characterization of SM gene clusters and their concomitant metabolites from A. flavus. A number of gene clusters in A. flavus are highly homologous to gene clusters from other filamentous fungi for which the associated metabolite has been identified. However, this review will only discuss those SM gene clusters for which the attendant metabolite(s) have been experimentally confirmed in A. flavus. Information will be presented on the genomic, biochemical, bioinformatic and molecular methods used to elucidate the genes and their encoded enzymes/proteins that are responsible for biosynthesis of SMs in A. flavus. Due to the many comprehensive reports available on AFs3,22,23) and cyclopiazonic acid (CPA)24,25), these two SMs will not be covered in this review. Instead, we will focus on recently characterized A. flavus SM gene clusters and their metabolites. In addition to discussion of the genetics and chemistry of A. flavus SMs and their gene clusters, the potential roles these metabolites play in the biology of the fungus will also be presented.
2. Functional Genomics and Secondary Metabolism
Research approaches in fungi utilizing functional genomics may entail a range of molecular, biochemical or bioinformatic tools, however, most often the term describes research aimed at determining gene or protein function and interaction utilizing a genome-wide analysis of data. Such experiments include a bottom-up approach where specific genes are targeted for disruption or ectopic expression and the resulting effect on transcriptional regulation or biological processes is determined26–29). Other common approaches include the use of chemical treatment, environmental conditioning, host-pathogen interaction or other stimuli relevant to the investigation to identify novel genes, proteins, interactions, expression profiles, and entire processes responding to the stimuli30–33). Alternatively, functional genomics can utilize genome-wide information to elucidate gene function with techniques such as such as gain-of-function cDNA screens34), small interfering RNA (siRNA)-based knockdown genome libraries35), or clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 approaches designed to target a majority of the transcriptome36). In addition, proteomics and metabolomics have incorporated functional genomics as a component of large scale screening approaches, such as mass-spec analysis of 2D protein gels combined with genome data37,38), high-throughput yeast two hybrid interaction assays39), carbon flux algorithms correlated with gene expression data40) and transcriptome-metabolome co-expression analysis41).
Through the use of knockout technologies, microarray analysis, RNA-sequencing, and in silico analysis, SM cluster prediction is continually evolving to become more targeted and efficient in its predictive power. Section 2 of this review will focus on the functional genomic tools using genome-wide expression analysis to elucidate secondary metabolic gene cluster activation and regulation in A. flavus.
2-1. Microarray Technology
Microarray technology was among early approaches characterizing A. flavus gene function42), natural antisense transcripts43), and secondary metabolism, with an initial focus on AF regulation44–48). Subsequent to this, the examination of other SM gene clusters using customized microarrays were reported8,29,49,50). An early effort at this was by Georgianna et al.8) whereby a collection of microarray data from 28 different treatments was used to comprehensively associate cluster expression patterns depending on treatment and environmental conditions. Here four clades, or expression patterns, of the 55 SMURF-identified clusters was described, with about half of the clusters determined to be low or non-expressing at all conditions analyzed. Another clade was characterized by high expression at most conditions and included clusters 23 (leporins), 55 (CPA), 26 (unknown) and 36 (unknown). Cluster 26 was also identified by Ehrlich and Mack18) and Wang et al.51) as differentially expressed on solid media and during pathogen-host interaction in peanut, respectively. The study by Georgianna et al.8) was limited in that it could not account for the effect of accessory enzymes, but provided one of the first comprehensive profiling of secondary metabolic genes in A. flavus.
In 2013, Reverberi et al.49) examined 24 SMURF-identified gene clusters that were described to be up or down regulated in two separate comparisons. In the first, A. flavus grown in liquid media with and without autoclaved maize kernels indicated up regulation of what is now known to be the gene clusters producing a pyrazinone (cluster 11), aflatrem (cluster 15), leporins (cluster 23), and down regulation of the AF (cluster 54) cluster. A second comparison whereby A. flavus was grown on autoclaved kernels and live kernels in the field identified effects of 16 different clusters, including the pyrazinone, leporin, CPA and AF clusters. Cary et al. observed the altered co-expression of clustered genes using microarray analysis of a veA knockout strain to identify the sclerotia-related clusters that synthesize aflavarin29) and asparasone52). Both aflavarin (cluster 39) and asparasone (cluster 27) were among the clusters predicted to be sclerotia-related in the Georgianna et al.8) microarray analysis and by Ehrlich and Mack18) based on the temporal correlation of sclerotia development and cluster expression.
2-2. RNA Sequencing
RNA-sequencing technologies are quickly replacing most other means of high-throughput transcriptomics, given their decreasing cost, high accuracy, and the increased availability of bioinformatic services53–55). Ehrlich and Mack18) was among the first to utilize RNA-sequencing to characterize secondary metabolic gene clusters in A. flavus, primarily for annotation purposes in identifying absolute expression activity and defining cluster boundaries. The authors identified eight clusters demonstrating high expression levels under their conditions and eleven clusters correlating with sclerotial development. In addition to asparasone and aflavarin mentioned previously, the cluster for leporin production was also among those predicted15).
To date there has not been numerous use of RNA-seq combined with gene knockout technology in A. flavus, however RNA-seq was conducted on the small sclerotia producing A. flavus strain AF70 to examine the effects of knocking out the zinc-finger transcription factor nsdC. Inactivation of nsdC resulted in decreased aflatrem production, as well as decreased expression of genes encoding the backbone enzymes for penicillin, asparasone, ustiloxin B, and AF biosynthesis26). Clusters 5, 20, 41, 44, and others also showed altered transcriptional profiles in the nsdC knockout strain. Metabolites produced by these clusters remain to be identified.
In addition to gene knockout strategies, chemical treatment and the manipulation of environmental conditions combined with RNA-seq technology has successfully characterized various gene clusters in A. flavus. Treatment of A. flavus with resveratrol, a naturally occurring phytoalexin and antifungal compound, affected 17 genes in 13 clusters, 2 of which were nonribosomal peptide synthetases (NRPSs) or NRPS-like enzymes (cluster 3 and 45, respectively)56). Although fungi are thought to not undergo DNA methylation57) the DNA methyltransferase inhibitor 5-azacytidine has been speculated to interfere with toxisome formation50). Upon treatment of A. flavus with 5-azacytidine, RNA-seq revealed an increase in expression of several genes of cluster 27 (asparasone), cluster 35 (piperazine), cluster 55 (CPA) and moderately decreased expression of AF cluster genes58). In another RNA-seq study, the plant volatile, decanal, impacted expression of genes present in 26 of the 55 clusters59). Finally, because drought-stress leads to increases in reactive oxygen species, a RNA-seq study examined the effects of oxidative stress (exposure to H2O2) on 3 strains of A. flavus synthesizing varying levels of AF. The focus of this study on secondary metabolic gene products besides AF was limited, however the authors did indicate genes in the aflatrem and KA gene clusters were affected60).
Recently the use of functional genomics, and in particular RNA-sequencing, to measure the genome-wide response to changing environmental conditions has become more widespread30,61), and whereas early experiments focused on the AF gene cluster, secondary metabolic cluster prediction and transcriptome-wide profiling has allowed for more complete analysis. The influence of temperature or water activity combined with RNA-seq has been used to examine small non-coding RNAs62), development63) and SM production64,65). Yu et al.65) showed 10 of the 55 gene clusters had higher expression at 30°C as compared to 37°C (cluster 1, 10, 11, 19, 20, 21, 24, 45, 54, and 55) and cluster 3 decreasing at lower temperatures. Medina et al.64) found genome-wide, a decreased transcriptome response to changing environmental conditions correlating with low water activity or high temperature, with water being the primary predictor. As an example, the CPA cluster (cluster 55), was lower in every condition examined due to low water availability, regardless of temperature conditions. Preliminary or small-scale studies have indicated that other environmental conditions, for example, those related to climate change also impact secondary metabolism61,66,67). Studies are ongoing to examine this impact on a genome-wide scale.
2-3. Bioinformatic Tools Used for Cluster Prediction in A. flavus
The most commonly used bioinformatic tools for RNA-seq analysis such as KEGG (Kyoto Encyclopedia of Genes)68), gene co-expression network analysis69) and gene ontology (GO) enrichment analysis70) have the potential to facilitate the analysis of large data sets and have been used in functional genomics analysis of A. flavus26,52,56,58). While these methods will categorize genes, determine categories significantly enriched, and identify similar gene expression patterns, the identification of cluster function is generally beyond these tools. To facilitate cluster prediction in A. flavus multiple computational approaches have been developed71)(Table 2). During development and testing, A. flavus clusters were used as test sequences for the motif-dependent program SMURF, and the motif-independent programs: Motif-Independent De Novo Detection of Secondary Metabolite Gene Clusters (MIDDAS-M), and Motif-Independent Prediction without Core Genes (MIPS-CG). Among the more popular programs, SMURF, developed by Khaldi et al.72) at the J. Craig Venter Institute, uses hidden Markov Model (HMM) searches to predict the presence of TIGRFAM and Pfam conserved protein domains associated with various SM backbone enzyme genes. These include NRPSs, polyketide synthases (PKSs), hybrid NRPS-PKS enzymes and prenyltransferase enzymes (DMATS, dimethylallyltryptophan synthases). SMURF predictions are limited in the type of cluster predicted, but are generally regarded as consistent with other forms of prediction73,74). While SMURF has demonstrated significant effectiveness as evidenced by the publications it has contributed to, the last update was April 2015 and technical support to resolve discrepancies appears unavailable75,76).
Table 2. Comparison of categories of A. flavus predicted secondary metabolite gene clusters using different web-based computational programs.
| Category | SMURFa | antiSMASHb | MIDDAS-Mc |
|---|---|---|---|
| PKS (Type I and III) | 25 | 20 | 15 |
| NRPS | 18 | 11 | 12 |
| PKS-NRPS hybrid | 2 | 3 | 3 |
| DMAT/Terpenesd | 8 | 13 | 7 |
| NRPS-Like | 14 | − | 20 |
| PKS-Like | 3 | 2 | − |
| Indolee | − | 5 | 4 |
| RiPS | − | − | 1f |
aPre-computed data from www.jcvi.org/smurf. Differences from Georgianna et al. (2010)8) data could be contributed to changes in the SMURF algorithm or sequences used.
bObtained by running antiSMASH v. 4.0.0 (www.fungismash.secondarymetabolites.org) using A. flavus NRRL 3357 genome sequence (EQ963472).
cSupplementary data obtained from Umemura et al. (2013)10).
dantiSMASH categorizes DMAT's as terpenes, while SMURF defines them independently.
eIndole clusters in antiSMASH determined by hidden Markov modeling using the StaD-like chromopyrrolic acid synthase domain (Medema et al. 2011)78).
fMIDDAS-M identified the ustiloxin B cluster. A. flavus genome analysis has identified an additional 7 potential Ri
Alternatively, the popular algorithm antibiotics and Secondary Metabolite Analysis Shell (antiSMASH), is subject to frequent upgrades and user input, with versions made available as recently as April 201777–79). The software serves as a pipeline that integrates multiple published analysis types to predict a wide range of cluster types and enzyme domains, including PKS/NRPS’s, terpenes, indoles, siderophores, and in recent versions, Ribosomally-synthesized and Post-translationally modified Peptides (RiPPs). At the core of antiSMASH, like SMURF, amino acid sequence translations are identified as containing signature domains using hidden Markov Models, and regions up to 20kb away are searched for accessory enzymes. Other programs within antiSMASH utilize trained and phylogentic-based algorithms to predict substrate structure based on motifs within acyltransferase or adenylation domains, from which a final structure is derived based on other domains present. In early versions the “greedy” approach of boundary prediction led to excessively large clusters being predicted, however recent versions have attempted to optimize this element by incorporating sequence-based algorithims (CASSIS and ClusterFinder) that use known regulatory motifs and protein domains to assist in border prediction. Additionally, antiSMASH incorporates data from the Minimum Information about a Biosynthetic Gene cluster (MiBIG) database which attempts to incorporate experimentally-determined SM data.
Finally, MIDDAS-M does not utilize characterized motifs as a basis for prediction10). Rather, it utilizes both genomic and transcriptomic data to calculate expression ratios of neighboring genes, and large induction ratios to indicate cluster activity. This is beneficial when attempting to identify novel clusters that do not contain motifs searched for by other programs. The authors claim that even SM biosynthetic genes not co-localized, such as those synthesizing KA in A. oryzae can be detected80). The ustiloxin B biosynthetic gene cluster is unique in that the peptide is ribosomally synthesized. The ustiloxin B cluster was detected by MIDDAS-M, which was later experimentally-validated17). A comprehensive comparison of MIDDAS, antiSMASH and SMURF using A. flavus was conducted by Umemura et al80). We have not found other software programs utilized extensively for cluster predictions in A. flavus. In our own experience, this is sometimes due to difficulty of use, inaccurate cluster predictions, a failure to maintain basic but essential updates beyond publication, or technical defects (bugs) with no technical support.
2-4. Cluster Prediction in the Age of Genomics
With the recent increase in large SM and genomic data sets and databases such as ClusterMine360, IMG-ABC, and MIBiG, strategies using comparative and computational genomics and transcriptomics for cluster prediction are becoming more effective73,74,81). These approaches must go beyond using genes in other species of Aspergillus to predict A. flavus SM gene clusters, primarily because the degree of homology needed and role of accessory proteins and “hypotheticals” is unknown. Certainly more than backbone gene identity is important. For example, PKS enzymes often share high degrees of similarity with unrelated PKSs. The PKS-encoding gene afvB (AFLA_108550) in A. flavus shares identity (E value =0, 43.8% aa identity) to the A. nidulans PKS encoded by mdpG (AN0150), however the A. nidulans product monodictyphenone is structurally dissimilar to aflavarin82), and none of the aflavarin cluster accessory enzymes are thought to be located near mdpG. Thus Basic Local Alignment Search Tool (BLAST) or bioinformatic approaches must involve a multigene approach.
In A. flavus, one example of such an approach is the discovery that ustiloxin B is ribosomally synthesized17) and that two genes in the cluster, ustYa/Yb, are found in 19 loci in the A. flavus genome, 9 of which are associated with a ustiloxin-like precursor sequence (ust-RiPS)14). In the case of one of these loci, knockout of the ustYa/Yb genes led to the identification of the novel compound, asperipin-2a14). The authors here developed a pipeline utilizing PSI-BLAST, which iteratively refines searches based on conserved amino acids, signal peptide detection software, and BLASTClust to further sort precursor proteins. Another method utilized in A. flavus by our group involves using known eukaryotic clusters in the MIBiG database83) as queries against sequenced strains of A. flavus. The software program multigeneblast84) will search a genome or set of sequences using multiple sequences as the query, thus allowing for gene rearrangement and differing degrees of identity. Hits can then be associated with the known metabolite from the database. Using this approach we can putatively identify several uncharacterized clusters in A. flavus. For example, all 22 of the genes in the experimentally verified A. oryzae aspirochlorine cluster are present as a cluster in A. flavus (cluster 21). Similar results are obtained for tenellin, chaetoglobosin, and trans-resorcyclide with varying levels of identity. As databases such as this increase in user contribution, predictive powers of such tools is likewise expected to increase.
3. Regulation of Secondary Metabolism in A. flavus
3-1. Global Regulation
The genetic mechanisms that regulate secondary metabolism in fungi have been the subject of intense investigation21,85). In this section, we focus on the regulation of SM production in A. flavus along with recently published literature that has not been presented in previous review articles. The role of LaeA, a putative histone methyltransferase, as a global regulator of development and SM production in A. flavus as well as other Aspergillus species, has been the subject of several reports86,87). Deletion of laeA in A. flavus resulted in complete loss of aflatoxin B1 (AFB1), aflatoxin B2 (AFB2), and CPA production, whereas, over-expression of laeA did not have a significant effect on production of these SMs86). The presence of other SMs including paspaline, paspalinine, aflatrem, and aflavinines (see section 5–5) were difficult to detect in wild-type A. flavus, however, in the A. flavus laeA over-expressor strain, levels were increased such that they could be readily detected86). Kojic acid (KA) production was lost in A. flavus ΔlaeA mutants and levels were not increased due to over-expression of laeA. Kojic acid production was also abrogated in an A. oryzae ΔlaeA mutant due to down-regulation of KA biosynthesis genes88). In addition, laeA has also been shown to regulate the A. flavus lna and lnb gene clusters (see section 5–4) involved in the production of piperazine and morpholine SMs16).
The light-responsive protein, VeA is a component of a heterotrimeric, nuclear-localized protein complex, designated the velvet complex, which also includes LaeA and the velvet-like protein VelB. The velvet complex is responsible for regulation of development and SM production in numerous filamentous fungi and was shown to positively regulate the production of AF, CPA, and aflatrem in A. flavus89,90). Production of CPA was reduced by 48–66% in the ΔveA mutant (vs. control) depending upon growth in light or dark conditions. An A. flavus ΔvelB mutant was unable to produce sclerotia and AFs28). A transcriptomic study in A. flavus showed that 28 SM gene clusters contain at least one gene whose expression is differentially regulated by the presence or absence of veA29). Characterization of the aflavarin gene cluster (see section 5–3) showed that veA directly regulates the production of aflavarin with all of the cluster genes significantly down-regulated in a ΔveA mutant, demonstrating direct regulation of this cluster by VeA. Functional characterization of the asparasone A gene cluster (see section 5–1) in A. flavus showed a significant down-regulation of the polyketide synthase gene (pks27) and the Zn(2)-Cys(6)-type transcription factor gene znf27 in a ΔveA mutant resulting in loss of asparasone production52).
Another global regulatory gene, nsdC, mainly known for its role in development in A. nidulans91), was also shown to be required for AF production in A. flavus27). In addition to loss of AF biosynthesis, A. flavus ΔnsdC mutants also demonstrated reduced aflatrem content compared to a control strain. RNA-seq analysis of the ΔnsdC mutant revealed differential expression of genes associated with development and production of SMs such as AFs, penicillin, and aflatrem26). The studies presented here suggest a coordinated action between global regulators and cluster specific transcription factor genes in regulating SM production in A. flavus.
The Far transcription factor was shown to play an important role in fungal virulence and AF production through its involvement in lipid metabolism. The A. nidulans farA and farB genes were shown to regulate expression of genes associated with fatty acid catabolism92). Further characterization of far genes in A. flavus showed their essential roles in mitochondrial beta oxidation of fatty acids and regulation of genes involved in unsaturated fatty acid biosynthesis93). Deletion mutants of A. flavus farA and farB showed aberrant growth phenotypes when grown on medium-chain fatty acids. Aflatoxin production was significantly decreased in a ΔfarB mutant (vs. control) both in vitro and during maize seed infection accompanied by reduced sporulation, suggesting a role of farB in lipid metabolism contributing to fungal pathogenesis and AF production. Another Zn(2)-Cys(6) transcription factor gene, aswA, has been demonstrated to affect sclerotial production and associated sclerotial SMs in A. flavus94). The ΔaswA mutant produced higher numbers of deformed, non-pigmented sclerotia and no asparasone A, aflatrem, dihydroxyaflavinine, or aflavarin could be detected from sclerotia as compared to the control strain. Aflatoxin and CPA content were not affected by the aswA mutation. In a recently published study, Cary et al.95) demonstrated that a homeobox transcription factor gene, hbx1, is required for production of conidiophores, sclerotia, and the SMs AF, CPA, and aflatrem in A. flavus. Upon forced overexpression, a globally-acting C2H2 zinc-finger transcription factor, MtfA, was shown to be required for normal development and AF production in A. flavus96).
Besides DNA-binding transcription factors, other regulatory factors with diverse functions have been found to regulate secondary metabolism in A. flavus. Examples include RmtA, a putative arginine methyltransferase, shown to be required for production of normal levels of AFB1 and other uncharacterized SMs97). Similar regulation of SM production in A. flavus was also observed by RtfA, a putative RNA-Pol II transcription elongation factor, in coordination with VeA and LaeA98). The ΔrtfA mutant exhibited a significant decrease in sclerotia production along with reduction in AF and other unknown SMs.
3-2. Cluster-specific Regulation
In addition to global regulation of SM gene clusters, cluster-specific transcription factors are also involved in regulation of SM production in A. flavus. One of the most studied cluster specific transcriptional regulators is the aflR gene present in the AF gene cluster. The aflR gene is a DNA-binding, Gal4-type Zn(2)-Cys(6) transcriptional activator that regulates expression of most AF cluster genes99–101). A. flavus aflR deletion mutants do not make AFs due to lack of expression of cluster genes. AflR binds to the palindromic sequence 5ʹ-TCGN5CGA-3ʹ present in the promoter region of most AF biosynthetic genes102,103). Aflatoxin biosynthesis is also regulated by aflS (aflJ) located adjacent to and divergently transcribed from aflR104). The exact role of aflS in AF biosynthesis is not clear despite intense investigation. Deletion of aflS in A. flavus did not appear to affect expression of AF cluster genes however aflS deletion mutants did not produce AF104).
Inactivation of the A. flavus leporin cluster (see section 5–2) Zn(2)-Cys(6) transcription factor gene, lepE, resulted in significant down-regulation of all the cluster genes15). Over-expression of lepE produced orange-red pigmented hyphae due to the synthesis of the 2-pyridone, leporin B. The asparasone A gene cluster contains a Zn(2)-Cys(6) transcription factor gene, znf27, that when inactivated resulted in transformants that produced lighter, reddish-brown sclerotia compared to the dark brown sclerotia of the control strain. The reduced sclerotial pigmentation was due to lower levels of asparasone A production in the znf27 mutant. Gene expression analysis showed a significant reduction in expression of asparasone cluster genes in the ∆znf27 mutant compared to the control indicating that znf27 functions as a pathway-specific transcription factor. A pathway-specific C6-type transcriptional regulator gene present in the ustiloxin B gene cluster (see section 5–6), encoded by ustR, was identified and its overexpression resulted in an increase of ustiloxin B production by about 5-fold while deletion abrogated ustiloxin production14,17). Disruption of KA gene cluster (see section 5–8) Zn(2)-Cys(6) transcriptional activator gene, kojR, resulted in a significant down-regulation of expression of KA biosynthetic genes kojA and kojT accompanied by a complete loss of KA production9). Over-expression of kojR resulted in >3-fold higher expression of KA biosynthetic genes along with >3-fold increase in KA production compared to the control strain demonstrating direct regulation of KA biosynthesis by KojR.
3-3. Methods to Activate Silent Secondary Metabolite Gene Clusters
In fungi and other micro-organisms, considerable interest lies in understanding the production of SMs by cryptic or silent gene clusters that only produce their associated metabolites when exposed to specific environmental conditions not normally duplicated during growth on standard laboratory media. Several methods have been used to activate silent SM gene clusters in fungi and these have been summarized in a number of excellent reviews21,105–107). These include over-expression of global or pathway-specific transcriptional regulators, expression of SM cluster genes in heterologous hosts, co-culture of a fungal strain with another micro-organism, proteomics, exposure to epigenetic modifiers and antibiotics and alteration of media composition. In the genus Aspergillus, most experiments to activate silent SM gene clusters have been conducted in the model species, A. nidulans, so there is a need to extend these approaches to A. flavus.
4. Chemistry of Secondary Metabolites
4-1. Chemistry of Cluster Backbone Genes
Secondary metabolites produced by fungi exhibit a broad range of chemical diversity. Several classes of biosynthetic backbone genes give rise to a plethora of complex chemical structures that impact human health and agriculture. These backbone genes encode for enzymes, most of which are multifunctional megasynthases, that biosynthesize the metabolites’ core structure. Tailoring enzymes then operate on the core structure to afford a diverse suite of SMs with varying functions, many of which have been shown to enhance the fitness of the fungi. Table 3 provides a list of features associated with experimentally characterized secondary metabolite gene clusters in A. flavus.
Table 3. Features of A. flavus secondary metabolite gene clusters.
| Metabolitea | Cluster numberb |
Backbone gene classification |
Cluster-specific transcriptional regulator | Number of genes in clusterc | Putative biological
function |
Representative structure |
Reference |
|---|---|---|---|---|---|---|---|
| Aflatoxin | 54 | PKS | Yes | 30 | antiinsectan/oxidative stress resistance? | 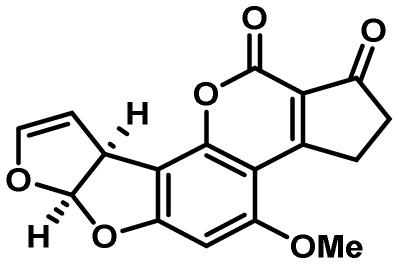 |
3,22,23 |
| Aflatrem | 15 and 32 | DMATs/GGPPs | No | 5 and 3 | antiinsectan/ antifeedant |
 |
108,109 |
| Aflavarin | 39 | PKS | No | 5 | antiinsectan/sclerotial development | 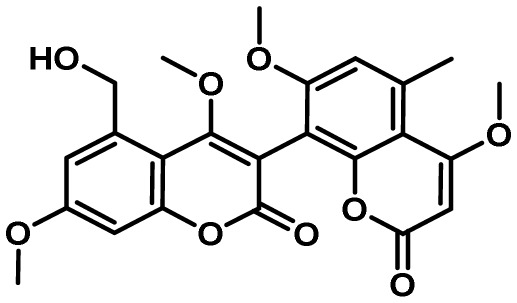 |
29 |
| Asparasone A | 27 | PKS | Yes | 4 | antiinsectan/sclerotial pigment/abiotic stress resistance | 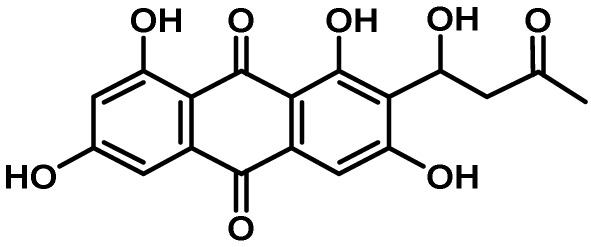 |
52 |
| Cyclopiazonic acid | 55 | DMATs/hybrid PKS-NRPS | No | 5 | mammalian antifeedant? |
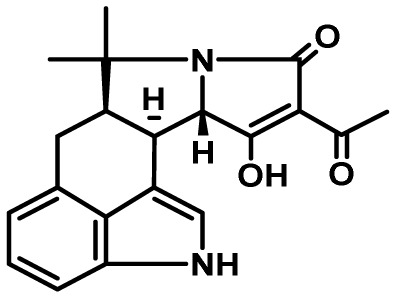 |
24,25 |
| Ditryptophenaline | 4 | NRPS | No | 3 | ? | 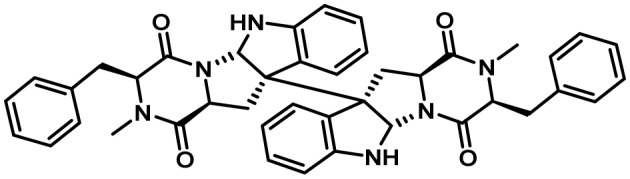 |
110 |
| Kojic acid | NA | oxidoreductase | Yes | 3 | insect
antifeedant/ antioxidant? |
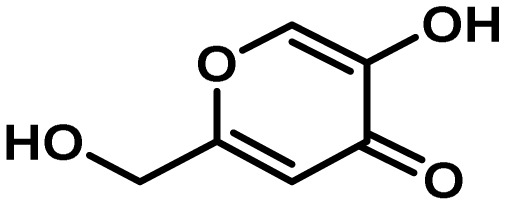 |
111 |
| Leporin | 23 | hybrid PKS-NRPS | Yes | 9 | insect antifeedant/ sclerotial development |
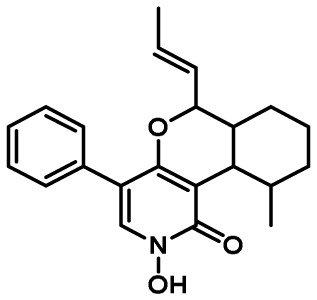 |
15 |
| Piperazine | 35 and 48 | NRPS-like | No | 6 and 6 | sclerotial development |
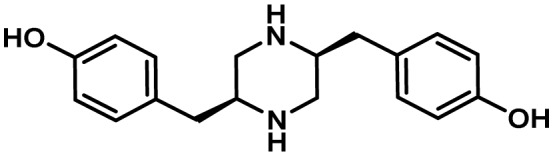 |
16 |
| Ustiloxin B | 31 | RiPPS | Yes | 15 | ? | 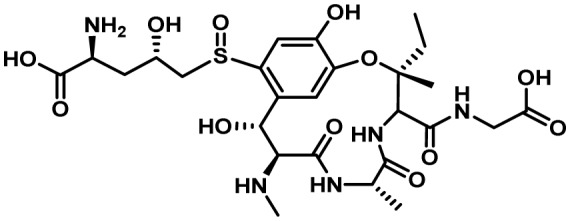 |
10,17 |
4-1-1. Polyketide synthases (PKSs)
Fungal polyketide metabolites are biosynthesized by iterative, type I PKSs and have highly diverse chemical architecture112–114). A type I PKS contains multiple modular catalytic domains encoded by one gene. Three domains are necessary for polyketide biosynthesis: the acyltransferase (AT) domain binds acetyl-CoA or malonyl-CoA starter units, the ketosynthase (KS) domain catalyzes a Claisen condensation that extends the polyketide chain with additional malonyl-CoA or methylmalonyl-CoA units, while the acyl carrier protein (ACP) tethers the elongating polyketide chain to the PKS through a phosphopantetheine linkage. Iterative PKSs use the same AT and KS catalytic domains to add multiple malonate units to the growing polyketide. Additional reductive tailoring of the growing polyketide chain can occur if the PKS harbors functional ketoreductase (KR), dehydratase (DH), or enoyl reductase (ER) domains. Typically, a thioesterase (TE) domain severs the thioester-linked PKS-polyketide bond, liberating the nascent polyketide as a linear molecule or a macrolactone. After release from the PKS, the polyketide is further transformed by oxidases and oxygenases resulting in SMs with abundant structural complexity115). PKSs in A. flavus produce AFs, aflavarin, and asparasone A (see sections 5–1 and 5–3).
4-1-2. Nonribosomal peptide synthetases (NRPSs)
Fungal nonribosomal peptides are biosynthesized by NRPSs from proteinogenic and non-proteinogenic amino acids113,116). NRPSs contain multiple catalytic domains arranged in modules. Each module contains an adenylation (A) domain that activates a specific amino acid using ATP, a peptidyl carrier (P) domain, which utilizes a phosphopantetheine linker analogous to ACP domains in PKSs, and a condensation (C) domain that catalyzes peptide bond formation. Multiple modules are arranged in an assembly-line fashion whereby each module adds one amino acid to the growing peptide chain. Many NRPSs contain a TE termination domain that catalyzes peptide release. In some fungal NRPSs, the peptide chain is liberated from the NRPS by a condensation-like termination domain (CT), which affords a cyclic peptide117,118). “NRPS-like” proteins contain A and T domains but lack C domains and terminate with a thioester reductase (R) domain that liberates the tethered amino acid or peptide as an aldehyde. NRPSs in A. flavus produce ditryptophenaline and NRPS-like enzymes produce several piperazine natural products (see sections 5–4 and 5–7).
4-1-3. Hybrid backbone genes
Hybrid PKS-NRPSs biosynthesize SMs the have both polyketide and amino acid components. A. flavus utilizes a hybrid PKS-NRPS enzyme (KS-AT-ACP-C-A-T-R) that forms an intermediate in CPA acid biosynthesis from a mevalonate and two acetate moieties using PKS domains and a tryptophan residue using NRPS domains119). A. flavus also produces leporins using a hybrid PKS-NRPS (see section 5–2).
4-1-4. Dimethylallyl tryptophan synthases (DMATs)
Fungal dimethylallyl tryptophan synthases alkylate tryptophan or other aromatic moieties with the isoprenoid substrate dimethylally pyrophospate120). A DMATS in A. flavus prenylates an intermediate in the biosynthesis of CPA119,121). The biosynthetic pathway of aflatrem in A. flavus also contains a DMATS-catalyzed prenylation (see section 5–5).
4-1-5. Meroterpenes
Meroterpenes are SMs that are partially derived from terpenoid biosynthesis pathways. Indole diterpenes are meroterpenes that contain a cyclic diterpene skeleton derived from geranylgeranyl diphosphate (GGPP) and an indole moiety derived from indole-3-glycerol phosphate122). Biosynthesis of these molecules is complex and involves a GGPP synthase as well as variety of oxidases. A. flavus produces an assortment of indole diterpenes including aflatrem and aflavinines (see section 5–5).
4-2. Methods of Analysis
Robust chromatographic techniques have been used extensively to detect mycotoxins and other SMs produced by fungi. Thin-layer chromatography (TLC) is the most accessible and, due to its simplicity and low cost, the most widely-used technique, especially for preliminary metabolite investigation123). Normal-phase TLC is commonly used for mycotoxin analysis and utilizes non-polar solvent systems to separate metabolites over silica gel adsorbent. TLC plates pretreated with a fluorescent indicator allow UV absorbing compounds to be easily detected. Non-UV active and non-fluorescent compounds can be detected by treating the plate with various developing reagents124). High-performance liquid chromatography (HPLC) affords better separation, usually on a C18 reverse-phase column, and when paired to a diode-array detector (DAD) provides diagnostic UV spectra for each peak125,126). Fungi can be identified using chemotaxinomic data acquired from organic fungal extracts run on an HPLC-DAD instrument. Coupling HPLC-DAD to a mass spectrometer (MS) provides more information for identification127,128). High resolution mass spectrometry (HRMS) allows molecular formula estimation and simplifies dereplication129). Databases of characterized natural products searchable by monoisotopic mass, such as Antibase, can assist in dereplication130). HPLC-DAD-MS dereplication of SM production produced by genome-sequenced strains of A. oryzae (RIB40) and A. flavus (NRRL 3357) revealed that, although bioinformatics analysis shows 99.5% gene homology between the strains, the overall chemical profiles differed significantly131). Orbitrap HRMS and multiple stage mass spectrometry (MSn) was used in conjunction with SIEVE software to identify differentially expressed metabolites in control and mutant A. flavus strains (see section 5–1)132). Similar analytical methodology was developed using ultra-high-performance liquid chromatography (UHPLC)-Orbitrap HRMS that allows untargeted determination of fungal metabolites and led to the characterization of A. flavus cluster 23 metabolites (see section 5–2)133).
One-dimensional (1D)-nuclear magnetic resonance (NMR) data, usually 1H and 13C spectra, are often used to confirm metabolite identification. Structure elucidation, particularly atom connectivity, of new natural products is reliant on various 2D-NMR techniques134). Purification of metabolites is usually required for structure elucidation, however, recently, several metabolites have been identified and structurally characterized using NMR without having been isolated in pure form135). 2D-NMR analysis of unpurified extracts can also be used to investigate an organisms’ metabolome136). Differential analysis by 2D-NMR spectroscopy (DANS)137,138) was employed to examine the gliotoxin biosynthesis pathway (gli) in A. fumigatus, which revealed many new gli-dependent metabolites139).
5. Experimentally Identified A. flavus Secondary Metabolite Gene Clusters
5-1. Asparasone Cluster
Asparasone A, a PKS-derived 1,3,6,8-tetrahydroxy-2-(1′-hydroxy-3′-oxobutyl)-anthraquinone was shown to be a precursor molecule required for the formation of the dark pigment associated with the outer rind of sclerotia52). Asparasone A was first described in A. parasiticus extracts as a pigment that was structurally related to versicolorin intermediates involved in the biosynthesis of AFs and demonstrated to possess antiinsectan properties108). The asparasone A gene cluster consists of four genes, a polyketide synthase (AFLA_082150, pks27), a Zn(2)-Cys(6)-type transcription factor (AFLA_082140, znf27) and a putative high-affinity glucose transporter (AFLA_082160, mfs1) and MFS transporter (AFLA_082170, mfs2)52). A. flavus Δpks27 mutants produced greyish-yellow sclerotia rather than having the dark brown pigment presented by control strains (Fig. 1). Complementation of the ∆pks27 mutant with a wild-type copy of pks27 restored production of dark-pigmented sclerotia. Production of sclerotia was significantly reduced in the ∆pks27 mutant compared to that of the wild-type. Ultra-high performance liquid chromatography and mass spectrometry revealed that asparasone A (m/z (M-H)=357.0612) was still being produced by the ∆znf27 mutant though at significantly reduced levels which probably explained the reduction in observed sclerotial pigmentation. While function of the putative mfs1 and mfs2 transporters was not confirmed, they were considered to be a part of the asparasone cluster due to their coordinate regulation with pks27 by znf27.
Fig. 1.

Microscopic examination of sclerotia. Sclerotia production in A. flavus SRRC1582 wild-type (WT) and a ∆pks27 mutant was observed using an Olympus SZH10 stereomicroscope (x96 magnification) and Nikon DS-Qi1 camera. Strains were grown on GMMS agar for 10 days in the dark at 30°C. Note loss of dark pigmentation in sclerotia of the ∆pks27 mutant due to abrogation of production of asparasone A.
The characteristic dark pigmentation associated with most fungal sclerotia was previously thought to be due to a process of melanization. However, the study of Cary et al.52) showed the dark brown pigments present in A. flavus sclerotia are the product of PKS27-derived anthraquinones rather than typical tetrahydronapthalene-based melanins (Fig. 2). The presence of the asparasone A-based anthraquinone pigment in A. flavus sclerotia was shown to provide resistance to both biotic and abiotic stressors. In both choice and no choice feeding assays with the sap beetle Carpophilus freemani, a preference for consumption of sclerotia produced by the ∆pks27 mutant compared to control sclerotia was observed indicating that the dark pigment functions as an insect antifeedant. Exposure of sclerotia to UV irradiation or high temperatures resulted in decreased viability of sclerotia from the ∆pks27 mutant compared to the control strain.
Fig. 2.

Proposed biosynthesis of asparasone A in A. flavus. Pks27 assembles the linear nonaketide from an acyl started unit and eight malonyl units. Cyclization and release from the PKS affords the anthrone. The anthrone is oxidized to the anthraquinone and one ketone moiety reduced by unknown enzymes that are predicted to be similar to those in the aflatoxin biosynthesis gene cluster. Adapted from Cary et al52).
5-2. Leporin Cluster
Of the 55 A. flavus SM gene clusters identified by SMURF analysis, two were found to harbor hybrid PKS-NRPS backbone genes. One of these clusters was shown to be responsible for the production of the indole tetramic acid, cyclopiazonic acid140) while the second cluster produced a family of 2-pyridones known as leporins15). TePaske et al.141) reported on the production of the antiinsectan compound, leporin A, in the fungus Aspergillus leporis, however there had been no previous reports of leporin production in A. flavus. In addition to the PKS-NRPS (AFLA_066840, lepA), the leporin cluster was predicted to encode an additional 14 genes that could be involved in metabolite production15). An A. flavus ∆lepA mutant showed no significant differences in metabolite production compared to that of an isogenic control strain, indicating that the genes responsible for leporin biosynthesis were not being expressed or expressed at extremely low levels. However, overexpression of a putative Zn(2)-Cys(6) cluster-specific transcriptional regulatory gene, lepE (AFLA_066900) resulted in transformants producing a distinctive orange-red pigment (Fig. 3). Liquid chromatography and high resolution mass spectrometry (LC-HRMS) analysis of the OE::lepE extracts revealed a metabolite of m/z (M+H)=352.1902 that represented a demethylated leporin A precursor designated leporin B15). Additionally, a metabolite of m/z (M+H)=336.1959 was also produced that represented the N-demethoxy derivative of leporin A, designated leporin C. While leporin B had been previously reported to be produced by an unidentified fungus142), this was the first report of production of leporin C. Interestingly, metabolites were identified in the OE::lepE strain that were consistent with homodimers of both leporin B and C.
Fig. 3.

Production of leporins by A. flavus. Images of broth cultures of an A. flavus CA14 OE::lepE strain and CA14 wild-type (WT) following 2 days growth on Czapek-Dox broth with agitation in the dark at 30°C. Note orange-red pigmentation of the OE::lepE culture broth indicative of the formation of leporin B-iron complexes.
The biosynthesis of leporin B and C was elucidated using LC-HRMS analysis of extracts from knockout mutants of the leporin cluster genes in the A. flavus OE::lepE background15) (Fig. 4). The products of both lepA (PKS-NRPS) and lepG (enoyl reductase) are responsible for fusion of phenylalanine with a hexaketide and subsequent release of the stable tetramic acid precursor, pre-leporin C. The putative cytochrome P450, LepH, an enzyme demonstrating significant homology to the ring expansion cytochrome P450s found in the 2-pyridones, tenellin and bassianin, was shown to be required for the ring expansion step. LepF, a putative short chain dehydrogenase/reductase, was hypothesized to reduce the acyl carbonyl to an alcohol143). Interestingly, although lepI was predicted to encode a putative methyltransferase, elegant in vitro studies by Tang and coworkers143) showed that LepI catalyzed an S-adenosyl-L-methionine (SAM)-dependent inverse electron demand hetero-Diels–Alder (HDA) reaction forming leporin C. In addition to the HDA reaction, LepI was shown to possess retro-Claisen activity, which allows the enzyme to convert a byproduct of the Diels–Alder reactions to leporin C. Leporin C is then further oxidized by the N-hydroxylase, LepD, to form leporin B. The function of LepA, G, H, F, I and D were confirmed by heterologous reconstitution in A. nidulans; the function of LepF and LepI were investigated in vitro using recombinant protein expressed from S. cerevisiae and E. coli, respectively143).
Fig. 4.

Proposed biosynthesis of leporin B. The hybrid PKS-NRPS LepA and the LepG enoyl reductase form a tetramic acid intermediate (not shown), which is converted to a 4-hydroxypyridinone intermediate by the ring-expanding P450 LepH. LepF reduces the ketone in the intermediate to an alcohol. The S-adenosyl-L-methionine (SAM)-dependent enzyme LepI catalyzes a stereoselective dehydration of the alcohol followed by an inverse electron demand hetero-Diels–Alder reaction forming leporin C. The LepD P450 forms the hydroxamic acid moiety through N-hydroxylation resulting in leporin B. Adapted from Cary et al.15) and Ohashi et al.143).
While 2-pyridones are normally colorless, it is believed that leporin B, in the presence of an atom of Fe(III) is oxidized to form a highly stable leporin B trimer with an atom of iron (0) (m/z = 1107, iron(0)-trioxyleporin B) which results in the observed orange-red colony pigmentation. It was not determined if oxidation of leporin B to form the trimer is self-catalyzed or catalyzed by another enzyme residing within or outside of the leporin gene cluster. Such a trimeric iron chelate had not been previously described for a 2-pyridone and unlike typical iron-chelating siderophores, the leporin B-iron complex was highly soluble in organic solvents thus representing the first siderophore-like molecule with this property to be produced by a PKS-NRPS.
Though leporin A from A. leporis was shown to have antiinsectan properties there were no previous reports on the insecticidal properties of leporins B or C. In no-choice insect feeding studies performed with a preparation of the leporin B-iron chelate (LIC) compound extracted from the OE::lepE strain, a significant reduction in growth of first instar larvae of Fall armyworms (Spodoptera frugiperda) and Corn earworms (Helicoverpa zea) was observed15). In choice assays, both caterpillar species were observed feeding more frequently on the control disks than 0.1% LIC disks, and consumed significantly less of the LIC diet disks than the control diet disks. These results indicated that the LIC was acting as an antifeedant with little evidence of antiinsectan properties. Alterations in the developmental programs were also noted between the OE::lepE strain compared to the control15). A 10-fold reduction in conidia production was observed in the OE::lepE strain and sclerotial production was either reduced or delayed depending on the growth medium used compared to the control. Studies have shown that conidiation and sclerotial differentiation in filamentous fungi including A. flavus are induced by oxidative stress144,145). Cary et al.15) speculated that the large quantities of leporin B produced by the OE::lepE strain may have led to an increase in sequestration of iron, in the form of the leporin B-iron chelate, thus reducing oxidative stress caused by excess free iron. The reduction in oxidative stress levels, in turn, resulted in a decrease in the production of both conidia and sclerotia.
5-3. Aflavarin Cluster
Comparative transcriptomics of an A. flavus ΔveA mutant and isogenic control strain led to the identification of the aflavarin gene cluster in which all but one of the genes were significantly down-regulated in the mutant in at least one of the conditions assayed29). Like many previously characterized A. flavus SMs, aflavarin was isolated from sclerotia and shown to exhibit antiinsectan activity however the biosynthetic genes responsible for its production were unknown146). SMURF analysis predicted the aflavarin cluster to be composed of a backbone PKS gene (AFLA_108550, afvB) encoding an N terminal beta-ketoacyl synthase (KS) domain with an adjacent acyl transferase (AT) domain and a C-terminal malonyl CoA-acyl carrier protein transacylase (ACP) domain. Other veA-dependent genes present in this cluster included an O-methyl transferase (AFLA_108560, afvC), methyl transferase (AFLA_108570, afvD), cytochrome P450 (AFLA_108580, afvE), and a NADH oxidase (AFLA_108540, afvA). A blastp search of the NCBI and the Broad Aspergillus Comparative Genome databases indicated that the genes constituting the aflavarin gene cluster in A. flavus were highly conserved in A. oryzae, A. niger and partially conserved in A. clavatus and A. terreus.
Comparative analysis of the metabolomes of an A. flavus control and ∆afvB PKS mutant using high resolution mass spectrometry allowed identification of a series of bicoumarins that were present in the control and absent in the mutant29). The major differentially produced metabolite had a calculated mass of 455 which was identical to the mass of aflavarin and this was subsequently confirmed by LC/MS comparison to an authentic sample of aflavarin. Knockout of the cluster biosynthetic genes allowed determination of their roles in aflavarin synthesis (Fig. 5). Deletion of the O-methyltransferase (afvC) gene, involved in the O-methylation of the PKS-derived pentaketide, didesmethyl-siderin, totally abolished the biosynthesis of the monomeric siderin precursors that are subsequently dimerized to form the final bicoumarin products. Disruption of the cytochrome P450 monooxygenase (afvE) led to accumulation of the monomeric coumarins and loss of bicoumarin production, proving that the cytochrome P450 monooxygenase is responsible for phenolic coupling reactions. Inactivation of the afvD methyltransferase was found to be responsible for the final O-methylation steps following formation of the bicoumarins. The putative NADH oxidase (afvA) was proposed to be involved in hydroxylation of siderin and 7-O-demethyl siderin to their respective hydroxylated analogues.
Fig. 5.

Proposed biosynthesis of aflavarin. The PKS AfvB assembles 4,7-didesmethyl-siderin from an acyl started unit and four malonyl units. AfvC can perform subsequent methylations affording siderin. Hydroxylation by AfvA gives hydroxy-siderin, which can be oxidatively coupled with siderin by the AfvE P450 to form aflavarin. An alternate pathway to aflavarin is possible by an AfvE-catalyzed coupling of hydroxydesmethyl-siderin and siderin followed by a methylation of desmethylaflavarin by AfvD. Adapted from Cary et al.29).
In addition to its previously demonstrated antiinsectan properties, it was also shown that production of aflavarin is necessary for normal levels of sclerotial production. While no cluster-specific transcriptional activator gene was present in aflavarin cluster, there were a number of putative binding sites for stress-responsive DNA binding proteins as well the AF cluster-specific transcriptional activator, AflR. This suggested that there may be regulatory cross-talk between these 2 clusters. (Fig. 6)
Fig. 6.

Proposed biosynthesis of piperazines. The NRPS-like enzymes LnaA and LnaB catalyze the reduction of tyrosine and form an imine dimer. LnaA and LnaB can then reduce the imine dimer to a piperazine. LnaB and LnbB can form a morpholine analog from the imine dimer intermediate, while LnaC and LnbC are proposed to oxidize the imine dimer to a pyrazine analog (not shown). Adapted from Forseth et al.16).
5-4. Piperazine Clusters
Two clusters were identified in A. flavus named lna (chromosome 6) and lnb (chromosome 8) that were shown to be primarily involved in the biosynthesis of two piperazines as well as a number of other derivatives16). The genetic makeup of the two clusters is quite similar being composed of genes encoding a putative NRPS protein (lnaA/lnbA), and a number of genes likely to encode tailoring enzymes such as an NMR-like protein (lnaB/lnbB), cytochrome P450s (lnaC/lnbC), alcohol dehydrogenase/NADH:flavin oxidoreductase (lnaE/lnbE), and a transporter (lnaF/lnbF). The lna cluster also contains a second putative P450 enzyme gene (lnaD) while the lnb cluster contains a gene encoding a hypothetical protein not present in the lna cluster. The LnaA and LnbA NRPSs share a fairly high level of amino acid identity (58%) and canonical adenylation, acyl carrier protein and thioester reductase domains but lack a condensation domain. No cluster-specific transcriptional regulator genes were found in either the lna or lnb clusters. However, both clusters were shown to be under the control of LaeA, a global regulator of fungal development, secondary metabolism and virulence86,147). Searches of fungal genome databases indicated that LnaA-like proteins are present a number of Aspergillus species (though not specified) and in the genomes of the basidiomycetes, Serpula lacrymans and Heterobasidion annosum16).
Comparative metabolomics of A. flavus wild-type, deletion, and lnaA overexpression (OE) strains allowed identification of two major piperazine metabolites and eight other lna cluster-derived metabolites that had not been previously reported in aspergilli or other fungi16). Both LnaA and LnbA proteins were shown to activate tyrosine but because they lacked condensation domains, there was no peptide bond formation as confirmed by their inability to be activate an L-Tyr-L-Tyr dipeptide. Instead it was postulated that the C-terminal reductase domains of LnaA and LnbA act to reduce L-Tyr to its aldehyde. This suggested that the lna cluster NRPSs function as amino acid reductases characteristic of Lys2-type reductases involved in fungal L-lysine biosynthesis148). The resulting L-Tyr aldehyde was proposed to dimerize and be subsequently further reduced to piperazines and morpholine compounds by the activities of the NMR-like reductase LnaB/LnbB. While the function of the LnaC, LnaD or LnbC oxidases was not determined it was speculated that one or all may be involved in the conversion of the Tyr dimer to a pyrazine. Of particular interest was the finding that LnaB can participate in the reduction of LnaA-derived intermediates indicating that the lna and lnb clusters encode partially redundant biosynthetic pathways capable of sharing biosynthetic intermediates.
Suppression of lnaA and lnbA expression in A. flavus resulted in significant reduction in sclerotial formation compared to the wild-type suggesting a role for the SM products of these clusters in fungal development16).
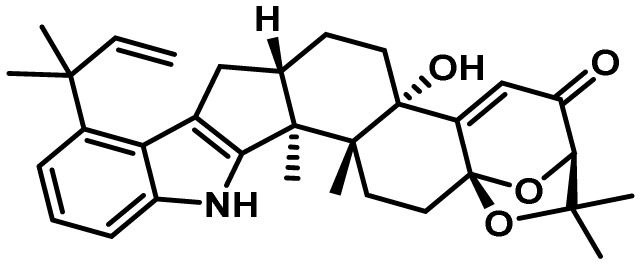
5-5. Aflatrem Clusters
Similar to piperazine biosynthesis, the genes required for the synthesis of the potent tremorgenic toxin, aflatrem, are also located at loci present on two different chromosomes (5 and 8) in A. flavus140). Aflatrem and its isomer, β-aflatrem, are examples of indole-diterpenes (IDTs), a class of structurally-diverse SMs that are found mainly in Aspergillus and Penicillium spp. as well as Epichloë. Identification of the genes required for aflatrem biosynthesis (atm) in A. flavus and A. oryzae was made possible through comparative genomics with genes from Penicillium paxilli required for the production of the indole-ditepene, paxilline109,149). As observed in A. flavus, the genes required for paxilline biosynthesis (pax) are clustered in 2 loci present on separate chromosomes. A total of three pax gene homologs (atmG, atmM and atmC) comprise the A. flavus ATM1 locus while atmD, atmQ, atmB, atmA and atmP comprise the ATM2 gene cluster locus. The presence of these pax gene homologs in A. flavus provided support for the biosynthesis of aflatrem proceeding via a paxilline intermediate.
Indole-diterpenes are all derived from a common core structure consisting of a tetracyclic diterpene derived from the activities of a geranylgeranyl diphosphate synthase (GGPPS, PaxG/AtmG) and geranylgeranyl transferase (GGT, PaxC/AtmC) that combines GGPP with an indole moiety derived from indole-3-glycerol-phosphate to form 3-geranylgeranyl indole (GGI)140). GGI is then converted to an epoxide by the action of an epoxidase (PaxM/AtmM) and subsequently transformed by an IDT cyclase (PaxB/AtmB) to the first stable intermediate, paspaline150,151). In A. flavus, paspaline is then converted to paspalinine by the action of the cytochrome P450 monooxygenases AtmP and AtmQ followed by subsequent conversion to aflatrem by the prenyltransferase AtmD. Interestingly, the chemical diversity that is characteristic of IDTs is due in large part to the IDT cyclases (IDTCs). Using a yeast host, Tang et al.151) were able to propose mechanisms by which A. flavus utilizes unique IDTCs to produce both aflatrems and the IDT, aflavinine (Fig. 7). They proposed that AtmG/C/M produces a common epoxide intermediate which is then converted to paspaline by the aflatrem cluster IDTC, AtmB, thus leading to subsequent formation of aflatrem. Based on the ability of A. flavus to also produce aflavinine, Tang et al.151) performed a phylogenetic analysis of all atmB homologs in A. flavus to identify candidate IDTCs that may be responsible for formation of aflavinine. When they introduced the standalone (not associated with a typical SM gene cluster) atmB homolog, afB, into a yeast host expressing atmG/C/M, the major product was shown to be aflavinine. These findings indicated that the standalone IDTC, AfB, catalyzes an alternate set of cyclization reactions leading to the production of aflavinine. How the fungus regulates expression of IDTCs such as atmB and afB to selectively control the ratios of aflatrems to aflavinines produced is unclear but it may have to do with the desired biological activity inherent in the particular IDT. Interestingly, the yeast studies indicate that the product of the atmA gene is not required for aflatrem or aflavinine biosynthesis.
Fig. 7.

Proposed biosynthesis of indole diterpenes in A. flavus. Two indole diterpene cyclases (AtmB and AfB) work in conjunction with the epoxidase AtmM to produce diverse chemical structures from a common intermediate. Genes encoding for biosynthesis of the epoxide intermediate (AtmG, AtmC, and AtmM) are clustered together (ATM1). Genes encoding for biosynthesis of paspaline, paspalinine, and aflatrem (AtmB, AtmP, AtmQ and AtmD) are also clustered together (ATM2), while the gene encoding AfB, responsible for aflavinine biosynthesis, is not clustered with either ATM1 or ATM2. Adapted from Nicholson et al.140) and Tang et al.151).
Expression of atm genes at both ATM loci were shown to be coordinately controlled despite their presence on separate chromosomes. However, no cluster-specific transcriptional regulatory genes were found in or near either of the ATM loci and this was also the case for the pax loci in P. paxilli140). This indicated that control of expression of atm genes probably occurs at the global level. In fact, it was shown that the global regulators of A. flavus development and secondary metabolism, VeA and LaeA, are both required for production of wild-type levels of aflatrem86,90).
Aflatrems and aflavinines are both associated with sclerotia in A. flavus152). Aflatrem was shown to be a potent mammalian tremorgen153) while aflavinines demonstrated antiinsectan properties152). The biological advantage that aflatrem imparts on the fungus is not clear though β-aflatrem has been shown to have some antiinsectan activity146). The mechanism of action of aflatrems and aflavinines remains to be determined but their antifeedant/antiinsectan properties suggest a role to increase survival of sclerotial propagules in the field from insect predation, thus allowing eventual germination to produce more fungal spores for dissemination.
5-6. Ustiloxin B Cluster
The proliferation of next generation sequencing of fungal genomes coupled with web-based bioinformatics tools (e.g. SMURF and antiSMASH) has been a major driver in the identification of secondary metabolic gene clusters in filamentous fungi. However, these prediction algorithms are based in large part on the presence of typical SM cluster backbone genes such as PKSs and NRPSs. A novel algorithm created by Umemura et al.10), designated MIDDAS-M was able to predict fungal SM gene clusters lacking a backbone gene based on integration of annotated genome and transcriptomic data. Using this approach, they were able to identify a gene cluster in A. flavus predicted to consist of 18 genes, none of which were identified as a typical cluster backbone gene. Comparative metabolomics of gene knockout mutants of this cluster compared to a control strain identified a metabolite of m/z (M+H)=646.240 that corresponded to the antimitotic cyclic peptide, ustiloxin B.
Ustiloxins were originally identified in the rice pathogen, Ustilaginoidea virens, but were not known to be produced by A. flavus154,155). These compounds are tetrapeptides with ustiloxin B consisting of the amino acids Tyr-Ala-Ile-Gly (YAIG). Enzymes encoded by the gene cluster cyclize the tetrapeptide at the side chains of Tyr and Ile and catalyze side chain modifications and linking of a non-proteinogenic amino acid, norvaline, to the Tyr moiety17) (Fig. 8). Ustiloxin B was initially thought to be the product of a NRPS-like enzyme, but no NRPS-like genes were present in or near the gene cluster. Upon further examination, the cluster gene ustA, was predicted to encode a protein containing an N-terminal signal sequence and 16-fold repeated peptide region containing YAIG repeats which represent the exact tetrapeptide sequence of ustiloxin B. Biosynthesis was proposed to proceed via processing of the UstA precursor protein by a Kex2 protease not present in the cluster that cleaves at nearby Lys-Arg residues on the C-terminal side of the YAIG tetrapeptide17). Subsequent work in A. oryzae confirmed that inactivation of the kexB endopeptidase gene resulted in almost complete loss of ustiloxin B156). The partially processed pre-protein is then believed to be processed in a stepwise manner by the products of ustP and ustH, both encoding peptidases, resulting in generation of the mature YAIG tetrapeptide. Prior to removal of the N-terminal leader peptide by UstP/UstH, the pre-protein is believed to be modified and cyclized by the action of the monooxygenases UstF1/UstF2 and UstQ (tyrosinase), respectively, to yield the intermediate ustiloxin F. The intermediate is then further modified by methylation (UstM), linkage of the norvaline to the Tyr residue (UstF and UstS) and oxygenation of the sulfur group (UstC). The functions of a number of genes considered as part of the ustiloxin B gene cluster were not determined in the study of Umemura et al.17). The absence of an NRPS-like gene in the cluster along with a predicted protein (UstA) harboring 16 short YAIG repeat peptide regions and an N-terminal signal peptide suggested that ustiloxin B is biosynthesized through a ribosomal peptide synthetic pathway (RiPS) not previously described in A. flavus or other Ascomycetes17).
Fig. 8.

Proposed biosynthesis of ustiloxin B. UstA is processed by the endoproteinase Kex2 at the C-terminus in the Golgi apparatus resulting in 16 copies of the precursor peptide. The C-terminus of each precursor peptide is then cleaved by peptidases UstP and UstH to afford a nonapeptide containing a leader and core peptide (underlined). While still attached to the leader peptide, the core YAIG peptide is cyclized. The leader peptide is then cleaved by UstP and UstH affording ustiloxin F. Methylation by UstM is follow by attachment and modification of norvaline by UstF, S, D, and C yielding ustiloxin F. Adapted from Umemura et al.17).
A subsequent study by Nagano et al.14) showed that deletion mutants of ustQ or ustYa/ustYb failed to produce the pathway intermediate ustiloxin F, suggesting that all three gene products are involved in cyclization of the intermediate. Interestingly, Nagano et al.14) screened Aspergillus genome sequences for the presence of the ustYa/ustYb and ustA gene homologs and were able to predict the presence of 94 ribosomal peptide precursor candidate genes that could be classified into 40 types having a variety of core peptide sequences. From this computational analysis they were able to experimentally identify a novel bicyclic peptide designated asperipin-2a in A. flavus.
Despite their ability to inhibit polymerization of purified porcine microtubule proteins as well as mitosis in human cell lines but not bacteria or fungi, the role of ustiloxin B in the biology of A. flavus remains to be determined154).
5-7. Ditryptophenaline Cluster
The ditryptophenaline (DTP) cluster is responsible for the production of the dimeric diketopiperazine alkaloid, (−)-ditryptophenaline, that is derived from two molecules of tryptophan and phenylalanine157). Comparative metabolomics of putative A. flavus DTP cluster gene (dtp) mutants with a control strain enabled determination of the function of the cluster genes. The cluster was found to be composed of three genes encoding a NRPS (dtpA) that condenses L-tryptophan and L-phenylalanine together to form the diketopiperazines core; an N-methyltransferase (dtpB) that methylates the core structure; and a cytochrome P450 (dtpC) that is likely responsible for cyclization and dimerization of the methylated diketopiperazine to form DTP (Fig. 9). Though DTP has been shown to have weak activity as an inhibitor of the neurotransmitter, substance P, in animal models, its function in the biology of A. flavus is not known110).
Fig. 9.

Proposed biosynthesis of dityrptophenaline. The NRPS DtpA forms the diketopiperazine cyclo(L-Phe-L-Trp) from phenylalanine and tryptophan. DtpB methylates cyclo(L-Phe-L-Trp) and the P450 DtpC catalyzes dimerization affording ditryptophenaline. Adapted from Saruwatari et al.157).
5-8. Kojic Acid (KA) Cluster
Though not originally confirmed in A. flavus, the KA gene cluster was experimentally identified in its close relative, A. oryzae111). Use of comparative transcriptomics of A. oryzae cultures grown under KA-inducing and non-conducing conditions, allowed the identification of a limited number of genes that may be involved in KA biosynthesis. Deletion of these candidate genes in A. oryzae followed by growth on media that is diagnostic for production of KA allowed identification of two deletion mutants that could no longer produce KA. The genes knocked out in these two mutants mapped within 3 kb of one another on the A. oryzae genome suggesting that a small gene cluster was responsible for KA biosynthesis. One of the genes (AO090113000136; A. flavus AFLA_096040) designated kojA, encodes a predicted FAD-dependent oxidoreductase while the other, kojT (AO090113000138; A. flavus AFLA_096060) is a major facilitator superfamily transporter. KojA is believed to function in the oxidation of glucose to KA though other as of yet unidentified genes may be required for complete conversion of glucose to KA111) (Fig. 10). Located between kojA and kojT is a gene designated kojR (AO090113000137; A. flavus AFLA_096050) that encodes a Zn(2)-Cys(6) transcription factor.
Fig. 10.

Proposed biosynthesis of kojic acid. The putative FAD-dependent oxidoreductase KojA is predicted to form kojic acid from glucose, possibly in conjunction with other enzymes not encoded in the gene cluster. Adapted from Terabayashi et al.111).
The earlier studies of Terabayashi et al.111) had shown when sodium nitrate was added to KA production medium, KA biosynthesis was inhibited in A. oryzae. The negative impact of sodium nitrate on KA production was supported by deletion of nrtA, encoding a transporter essential for sodium nitrate uptake in A. oryzae158). Deletion strains of nrtA were still be able to produce KA even in the presence of sodium nitrate and the expression of kojA and kojT were significantly higher in the nrtA mutant than the control in the presence of sodium nitrate.
Though the contribution of KA to the biology of the producing fungus is not clear at this time it appears that it may be involved as an insect antifeedant or perhaps as an antioxidant that reduces the ability of insect oxidative enzymes to detoxify AFB1 or host plant-based antiinsectan compounds such as nicotine159). Compared to AFB1, kojic acid was shown to be relatively non-toxic to mice but in insect feeding studies in combination with AFB1, it appeared to act synergistically causing increased mortality compared to feeding of either metabolite alone159). The ability of KA to chelate iron and other cations may indicate that it is involved in homeostasis of these metals.
6. Conclusions
The advent of fungal genomics, bioinformatics and advanced analytical chemical techniques has resulted in a rapid expansion of the numbers of identified A. flavus SM gene clusters and their affiliated metabolites. While it took the better part of two decades to identify the aflatoxin gene cluster and the enzymes required for production of aflatoxins, genome sequencing and annotation coupled with modern molecular biological, bioinformatic and analytical techniques have allowed an additional nine A. flavus metabolites and their associated gene clusters and enzymes/regulatory genes to be described, all within a period of less than a decade. Though metabolites have been linked to most of the more readily tractable SM gene clusters in A. flavus, there remains a number of silent gene clusters for which characterization of their SM product’s form and function is lacking.
There is a growing body of evidence that points to significant roles of SMs in A. flavus biology and therefore it is critical that approaches are developed to activate silent SM gene clusters so that their impact on the physiology of the fungus can also be ascertained. While a number of techniques have been used successfully to activate silent gene clusters in the model fungus, A. nidulans, as well as other Aspergillus spp., experiments of this nature are lacking for the most part in A. flavus. More information must be gleaned on regulatory mechanisms of SM production in A. flavus such as global and cluster-specific regulators, signaling pathways, epigenetic regulation, and how these respond to the fungus’ environment. Evidence for regulatory cross-talk between A. flavus gene clusters and their ability to share biosynthetic intermediates has opened up new avenues of investigation into the regulation of SM gene cluster biosynthetic machinery. It is imperative that laboratory observations on the biological function of SMs also be translated to the field with respect to the interaction of A. flavus with the environment, especially during the host plant-fungus interaction. Additionally, little is known about the role of SMs on the interaction of A. flavus with other microbes that constitute the microbiome of the host plant and surrounding soil environment. These types of studies will provide invaluable insights into the roles of A. flavus SMs with respect to fungal virulence, survival, toxigenic potential and modulation of microbial and insect communities, all with the goal of controlling contamination of susceptible crops with A. flavus mycotoxins, particularly aflatoxins.
Abbreviations: ACP: acyl carrier protein, AFs: aflatoxins, AFB1: aflatoxin B1, AFB2: aflatoxin B2, antiSMASH: antibiotics and secondary metabolite analysis shell, AT: acyltransferase, BLAST: Basic Local Alignment Search Tool, CASSIS: Cluster Assignment by Islands of Sites, CRISPR: clustered regularly interspaced short palindromic repeats, CPA: cyclopiazonic acid, DAD: diode-array detector, DANS: differential analysis by 2D-NMR spectroscopy, DH: dehydratase, DMATS: dimethylallyltryptophan synthases, DTP: ditryptophenaline, ER: enoyl reductase, GGI: 3-geranylgeranyl indole, GGPP: geranylgeranyl diphosphate, GGT: geranylgeranyl transferase, GO: gene ontology, HAD: hetero-Diels–Alder, HMM: hidden Markov Model, HPLC: high-performance liquid chromatography, HRMS: high resolution mass spectrometry, IDT: indole-diterpene, IDTC: indole-diterpene cyclase, KEGG: Kyoto Encyclopedia of Genes, KA: kojic acid, KR: ketoreductase, KS: ketosynthase, LC-HRMS: liquid chromatography and high resolution mass spectrometry, LC-MS: liquid chromatography-mass spectrometry, LIC: leporin B-iron chelate, MiBIG: Minimum Information about a Biosynthetic Gene cluster, MIDDAS-M: motif-independent de novo detection of secondary metabolite gene clusters, MIPS-CG: motif-independent prediction without core genes, MS: mass spectrometer, NMR: nuclear magnetic resonance, NRPS: nonribosomal peptide synthetase, OE: overexpression, PKS: polyketide synthase, PSI-BLAST: Position Specific Iterated-BLAST, RiPPs: ribosomally-synthesized and post-translationally modified peptides, RNA-Seq: ribonucleic acid sequencing, SAM: S-adenosyl-L-methionine, siRNA: small interfering RNA, SMs: secondary metabolites, SMURF: secondary metabolite unique regions finder, TE: thioesterase, TLC: thin-layer chromatography, UHPLC: ultra-high-performance liquid chromatography, YAIG: Tyr-Ala-Ile-Gly
Footnotes
Conflict of interest: The authors have no conflict of interest.
References
- 1.Gloer JB. Applications of fungal ecology in the search for new bioactive natural products. In: Kubicek CP, Druzhinina IS, eds. The Mycota, Volume IV. 2nd ed. New York: Springer-Verlag; 2007:257–283. [Google Scholar]
- 2.Horn BW, Moore GG, Carbone I. Sexual reproduction in Aspergillus flavus. Mycologia. 2009; 101: 423–429. 10.3852/09-011 [DOI] [PubMed] [Google Scholar]
- 3.Kumar P, Mahato DK, Kamle M, Mohanta TK, Kang SG. Aflatoxins: A Global Concern for Food Safety, Human Health and Their Management. Frontiers in Microbiology. 2017; 07: 2170. 10.3389/fmicb.2016.02170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu F, Liu Y, Bhatnagar D. Cost-effectiveness of aflatoxin control methods: economic incentives. Toxin Reviews. 2008; 27: 203–225. 10.1080/15569540802393690 [DOI] [Google Scholar]
- 5.Robens J, Cardwell K. The Costs of Mycotoxin Management to the USA: Management of Aflatoxins in the United States. Journal of Toxicology: Toxin Reviews. 2003; 22: 139–152. 10.1081/TXR-120024089 [DOI] [Google Scholar]
- 6.Mc Millian WW, Wilson DM, Widstrom NW. Aflatoxin contamination of preharvest corn in Georgia: A six-year study of insect damage and visible Aspergillus flavus. Journal of Environment Quality. 1985; 14: 200–202. 10.2134/jeq1985.00472425001400020010x [DOI] [Google Scholar]
- 7.Nierman WC, Yu J, Fedorova-Abrams ND, et al. Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed. Genome Announcements. 2015; 3: e00168-15. 10.1128/genomeA.00168-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgianna DR, Fedorova ND, Burroughs JL, et al. Beyond aflatoxin: four distinct expression patterns and functional roles associated with Aspergillus flavus secondary metabolism gene clusters. Molecular Plant Pathology. 2010; 11: 213–226. 10.1111/j.1364-3703.2009.00594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marui J, Yamane N, Ohashi-Kunihiro S, et al. Kojic acid biosynthesis in Aspergillus oryzae is regulated by a Zn(II)2Cys6 transcriptional activator and induced by kojic acid at the transcriptional level. Journal of Bioscience and Bioengineering. 2011; 112: 40–43. 10.1016/j.jbiosc.2011.03.010 [DOI] [PubMed] [Google Scholar]
- 10.Umemura M, Koike H, Nagano N, et al. MIDDAS-M: motif-independent de novo detection of secondary metabolite gene clusters through the integration of genome sequencing and transcriptome data. PLoS ONE. 2013; 8: e84028. 10.1371/journal.pone.0084028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole RJ, Schweikert MA, Jarvis BB. Handbook of Secondary Fungal Metabolites, 3-Volume Set. Elsevier Science; 2003. [Google Scholar]
- 12.Turner WB, Aldridge DC. Fungal metabolites. Academic Press; 1983. [Google Scholar]
- 13.Vadlapudi V, Borah N, Yellusani KR, et al. Aspergillus Secondary Metabolite Database, a resource to understand the Secondary metabolome of Aspergillus genus. Scientific Reports. 2017; 7: 7325. 10.1038/s41598-017-07436-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagano N, Umemura M, Izumikawa M, et al. Class of cyclic ribosomal peptide synthetic genes in filamentous fungi. Fungal Genetics and Biology. 2016; 86: 58–70. 10.1016/j.fgb.2015.12.010 [DOI] [PubMed] [Google Scholar]
- 15.Cary JW, Uka V, Han Z, et al. An Aspergillus flavus secondary metabolic gene cluster containing a hybrid PKS–NRPS is necessary for synthesis of the 2-pyridones, leporins. Fungal Genetics and Biology. 2015; 81: 88–97. 10.1016/j.fgb.2015.05.010 [DOI] [PubMed] [Google Scholar]
- 16.Forseth RR, Amaike S, Schwenk D, et al. Homologous NRPS-like gene clusters mediate redundant small-molecule biosynthesis in Aspergillus flavus. Angew Chem Int Ed. 2012; 51: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umemura M, Nagano N, Koike H, et al. Characterization of the biosynthetic gene cluster for the ribosomally synthesized cyclic peptide ustiloxin B in Aspergillus flavus. Fungal Genetics and Biology. 2014; 68: 23–30. 10.1016/j.fgb.2014.04.011 [DOI] [PubMed] [Google Scholar]
- 18.Ehrlich K, Mack B. Comparison of expression of secondary metabolite biosynthesis cluster genes in Aspergillus flavus, A. parasiticus, and A. oryzae. Toxins. 2014; 6: 1916–1928. 10.3390/toxins6061916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strauss J, Reyes-Dominguez Y. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genetics and Biology. 2011; 48: 62–69. 10.1016/j.fgb.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brakhage AA, Schroeckh V. Fungal secondary metabolites – Strategies to activate silent gene clusters. Fungal Genetics and Biology. 2011; 48: 15–22. 10.1016/j.fgb.2010.04.004 [DOI] [PubMed] [Google Scholar]
- 21.Brakhage AA. Regulation of fungal secondary metabolism. Nature Reviews Microbiology. 2013; 11: 21–32. 10.1038/nrmicro2916 [DOI] [PubMed] [Google Scholar]
- 22.Yu J. Current understanding on aflatoxin biosynthesis and future perspective in reducing aflatoxin contamination. Toxins. 2012; 4: 1024–1057. 10.3390/toxins4111024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amaike S, Keller NP. Aspergillus flavus. Annual Review of Phytopathology. 2011; 49: 107–133. 10.1146/annurev-phyto-072910-095221 [DOI] [PubMed] [Google Scholar]
- 24.Chang PK, Horn BW, Dorner JW. Clustered genes involved in cyclopiazonic acid production are next to the aflatoxin biosynthesis gene cluster in Aspergillus flavus. Fungal Genetics and Biology. 2009; 46: 176–182. 10.1016/j.fgb.2008.11.002 [DOI] [PubMed] [Google Scholar]
- 25.Tokuoka M, Seshime Y, Fujii I, Kitamoto K, Takahashi T, Koyama Y. Identification of a novel polyketide synthase–nonribosomal peptide synthetase (PKS–NRPS) gene required for the biosynthesis of cyclopiazonic acid in Aspergillus oryzae. Fungal Genetics and Biology. 2008; 45: 1608–1615. 10.1016/j.fgb.2008.09.006 [DOI] [PubMed] [Google Scholar]
- 26.Gilbert MK, Mack BM, Wei Q, Bland JM, Bhatnagar D, Cary JW. RNA sequencing of an nsdC mutant reveals global regulation of secondary metabolic gene clusters in Aspergillus flavus. Microbiological Research. 2016; 182: 150–161. 10.1016/j.micres.2015.08.007 [DOI] [PubMed] [Google Scholar]
- 27.Cary JW, Harris-Coward PY, Ehrlich KC, et al. NsdC and NsdD affect Aspergillus flavus morphogenesis and aflatoxin production. Eukaryotic Cell. 2012; 11: 1104–1111. 10.1128/EC.00069-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang PK, Scharfenstein LL, Li P, Ehrlich KC. Aspergillus flavus VelB acts distinctly from VeA in conidiation and may coordinate with FluG to modulate sclerotial production. Fungal Genetics and Biology. 2013; 58-59: 71–79. 10.1016/j.fgb.2013.08.009 [DOI] [PubMed] [Google Scholar]
- 29.Cary JW, Han Z, Yin Y, et al. Transcriptome Analysis of Aspergillus flavus Reveals veA -Dependent Regulation of Secondary Metabolite Gene Clusters, Including the Novel Aflavarin Cluster. Eukaryotic Cell. 2015; 14: 983–997. 10.1128/EC.00092-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert MK, Mack BM, Payne GA, Bhatnagar D. Use of functional genomics to assess the climate change impact on Aspergillus flavus and aflatoxin production. World Mycotoxin Journal. 2016; 9: 665–672. 10.3920/WMJ2016.2049 [DOI] [Google Scholar]
- 31.Alves PC, Hartmann DO, Núñez O, et al. Transcriptomic and metabolomic profiling of ionic liquid stimuli unveils enhanced secondary metabolism in Aspergillus nidulans. BMC Genomics. 2016; 17: 284. 10.1186/s12864-016-2577-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen ZY, Warburton ML, Hawkins L, et al. Production of the 14 kDa trypsin inhibitor protein is important for maize resistance against Aspergillus flavus infection/aflatoxin accumulation. World Mycotoxin Journal. 2016; 9: 215–228. 10.3920/WMJ2015.1890 [DOI] [Google Scholar]
- 33.Brown RL, Chen Z, Menkir A, Cleveland TE. Proteomics to identify resistance factors in corn-a review. Mycotoxin Research. 2006; 22: 22–26. 10.1007/BF02954553 [DOI] [PubMed] [Google Scholar]
- 34.Heynen-Genel S, Pache L, Chanda SK, Rosen J. Functional genomic and high-content screening for target discovery and deconvolution. Expert Opinion on Drug Discovery. 2012; 7: 955–968. 10.1517/17460441.2012.711311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao Z, Petschnigg J, Ketteler R, Stagljar I. Application guide for omics approaches to cell signaling. Nature Chemical Biology. 2015; 11: 387–397. 10.1038/nchembio.1809 [DOI] [PubMed] [Google Scholar]
- 36.Agrotis A, Ketteler R. A new age in functional genomics using CRISPR/Cas9 in arrayed library screening. Frontiers in Genetics. 2015; 6: 300. 10.3389/fgene.2015.00300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliver S, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends in Biotechnology. 1998; 16: 373–378. 10.1016/S0167-7799(98)01214-1 [DOI] [PubMed] [Google Scholar]
- 38.Pechanova O, Pechan T, Rodriguez JM, Paul Williams W, Brown AE. A two-dimensional proteome map of the aflatoxigenic fungus Aspergillus flavus. PROTEOMICS. 2013; 13: 1513–1518. 10.1002/pmic.201100659 [DOI] [PubMed] [Google Scholar]
- 39.Fromont-Racine M, Rain JC, Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nature Genetics. 1997; 16: 277–282. 10.1038/ng0797-277 [DOI] [PubMed] [Google Scholar]
- 40.Panagiotou G, Andersen MR, Grotkjaer T, Regueira TB, Nielsen J, Olsson L. Studies of the production of fungal polyketides in Aspergillus nidulans by using systems biology tools. Applied and Environmental Microbiology. 2009; 75: 2212–2220. 10.1128/AEM.01461-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saito K, Matsuda F. Metabolomics for functional genomics, systems biology, and biotechnology. Annual Review of Plant Biology. 2010; 61: 463–489. 10.1146/annurev.arplant.043008.092035 [DOI] [PubMed] [Google Scholar]
- 42.Cary JW, OBrian GR, Nielsen DM, et al. Elucidation of veA-dependent genes associated with aflatoxin and sclerotial production in Aspergillus flavus by functional genomics. Applied Microbiology and Biotechnology. 2007; 76: 1107–1118. 10.1007/s00253-007-1081-y [DOI] [PubMed] [Google Scholar]
- 43.Smith CA, Robertson D, Yates B, et al. The effect of temperature on Natural Antisense Transcript (NAT) expression in Aspergillus flavus. Current Genetics. 2008; 54: 241–269. 10.1007/s00294-008-0215-9 [DOI] [PubMed] [Google Scholar]
- 44.Wilkinson JR, Yu J, Bland JM, Nierman WC, Bhatnagar D, Cleveland TE. Amino acid supplementation reveals differential regulation of aflatoxin biosynthesis in Aspergillus flavus NRRL 3357 and Aspergillus parasiticus SRRC 143. Applied Microbiology and Biotechnology. 2007; 74: 1308–1319. 10.1007/s00253-006-0768-9 [DOI] [PubMed] [Google Scholar]
- 45.Yu J, Cleveland TE, Wilkinson JR, et al. Aspergillus flavus expressed sequence tags and microarray as tools in understanding aflatoxin biosynthesis. Mycotoxin Research. 2006; 22: 16–21. 10.1007/BF02954552 [DOI] [PubMed] [Google Scholar]
- 46.OBrian GR, Fakhoury AM, Payne GA. Identification of genes differentially expressed during aflatoxin biosynthesis in Aspergillus flavus and Aspergillus parasiticus. Fungal Genetics and Biology. 2003; 39: 118–127. 10.1016/S1087-1845(03)00014-8 [DOI] [PubMed] [Google Scholar]
- 47.Guo BZ, Yu J, Holbrook CC, Lee RD, Lynch RE. Application of Differential Display RT‐PCR and EST/Microarray Technologies to the Analysis of Gene Expression in Response to Drought Stress and Elimination of Aflatoxin Contamination in Corn and Peanut. Journal of Toxicology: Toxin Reviews. 2003; 22: 287–312. 10.1081/TXR-120024095 [DOI] [Google Scholar]
- 48.Yu J, Ronning CM, Wilkinson JR, et al. Gene profiling for studying the mechanism of aflatoxin biosynthesis in Aspergillus flavus and A. parasiticus. Food Additives and Contaminants. 2007; 24: 1035–1042. 10.1080/02652030701513800 [DOI] [PubMed] [Google Scholar]
- 49.Reverberi M, Punelli M, Scala V, et al. Genotypic and phenotypic versatility of Aspergillus flavus during maize exploitation. PLoS ONE. 2013; 8: e68735. 10.1371/journal.pone.0068735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilkinson JR, Kale SP, Bhatnagar D, Yu J, Ehrlich KC. Expression profiling of non-aflatoxigenic Aspergillus parasiticus mutants obtained by 5-azacytosine treatment or serial mycelial transfer. Toxins. 2011; 3: 932–948. 10.3390/toxins3080932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, Lei Y, Yan L, et al. Functional genomic analysis of Aspergillus flavus interacting with resistant and susceptible peanut. Toxins. 2016; 8: 46. 10.3390/toxins8020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cary JW, Harris-Coward PY, Ehrlich KC, et al. Functional characterization of a veA-dependent polyketide synthase gene in Aspergillus flavus necessary for the synthesis of asparasone, a sclerotium-specific pigment. Fungal Genetics and Biology. 2014; 64 (Supplement C): 25–35. 10.1016/j.fgb.2014.01.001 [DOI] [PubMed] [Google Scholar]
- 53.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. 2016;17:333. [DOI] [PMC free article] [PubMed]
- 54.Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE. 2014; 9: e78644. 10.1371/journal.pone.0078644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.’t Hoen PAC, Ariyurek Y, Thygesen HH, et al. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Research. 2008; 36: e141–e141. 10.1093/nar/gkn705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang H, Lei Y, Yan L, et al. Deep sequencing analysis of transcriptomes in Aspergillus flavus in response to resveratrol. BMC Microbiology. 2015; 15: 182. 10.1186/s12866-015-0513-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu SY, Lin JQ, Wu HL, et al. Bisulfite sequencing reveals that Aspergillus flavus holds a hollow in DNA methylation. PLoS ONE. 2012; 7: e30349. 10.1371/journal.pone.0030349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin JQ, Zhao XX, Zhi QQ, Zhao M, He ZM. Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genetics and Biology. 2013; 56 (Supplement C): 78–86. 10.1016/j.fgb.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 59.Chang PK, Scharfenstein LL, Mack B, Yu J, Ehrlich KC. Transcriptomic profiles of Aspergillus flavus CA42, a strain that produces small sclerotia, by decanal treatment and after recovery. Fungal Genetics and Biology. 2014; 68 (Supplement C): 39–47. 10.1016/j.fgb.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 60.Fountain JC, Bajaj P, Nayak SN, et al. Responses of Aspergillus flavus to Oxidative Stress Are Related to Fungal Development Regulator, Antioxidant Enzyme, and Secondary Metabolite Biosynthetic Gene Expression. Frontiers in Microbiology. 2016; 7: 2048. 10.3389/fmicb.2016.02048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Medina A, Rodriguez A, Magan N. Effect of climate change on Aspergillus flavus and aflatoxin B1 production. Frontiers in Microbiology. 2014; 5: 348. 10.3389/fmicb.2014.00348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bai Y, Lan F, Yang W, et al. sRNA profiling in Aspergillus flavus reveals differentially expressed miRNA-like RNAs response to water activity and temperature. Fungal Genetics and Biology. 2015; 81 (Supplement C): 113–119. 10.1016/j.fgb.2015.03.004 [DOI] [PubMed] [Google Scholar]
- 63.Zhang F, Guo Z, Zhong H, et al. RNA-Seq-based transcriptome analysis of aflatoxigenic Aspergillus flavus in response to water activity. Toxins. 2014; 6: 3187–3207. 10.3390/toxins6113187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Medina A, Gilbert MK, Mack BM, et al. Interactions between water activity and temperature on the Aspergillus flavus transcriptome and aflatoxin B 1 production. International Journal of Food Microbiology. 2017; 256: 36–44. 10.1016/j.ijfoodmicro.2017.05.020 [DOI] [PubMed] [Google Scholar]
- 65.Yu J, Fedorova ND, Montalbano BG, et al. Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-Seq. FEMS Microbiology Letters. 2011; 322: 145–149. 10.1111/j.1574-6968.2011.02345.x [DOI] [PubMed] [Google Scholar]
- 66.Medina A, Schmidt-Heydt M, Rodríguez A, Parra R, Geisen R, Magan N. Impacts of environmental stress on growth, secondary metabolite biosynthetic gene clusters and metabolite production of xerotolerant/xerophilic fungi. Current Genetics. 2015; 61: 325–334. 10.1007/s00294-014-0455-9 [DOI] [PubMed] [Google Scholar]
- 67.Medina Á, Rodríguez A, Sultan Y, Magan N. Climate change factors and Aspergillus flavus : effects on gene expression, growth and aflatoxin production. World Mycotoxin Journal. 2015; 8: 171–179. 10.3920/WMJ2014.1726 [DOI] [Google Scholar]
- 68.Aoki-Kinoshita KF, Kanehisa M. Gene annotation and pathway mapping in KEGG. Methods in Molecular Biology. 2007; 396: 71–91. 10.1007/978-1-59745-515-2_6 [DOI] [PubMed] [Google Scholar]
- 69.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9: 559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology. 2010; 11: R14. 10.1186/gb-2010-11-2-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Inglis DO, Binkley J, Skrzypek MS, et al. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiology. 2013; 13: 91. 10.1186/1471-2180-13-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khaldi N, Seifuddin FT, Turner G, et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genetics and Biology. 2010; 47: 736–741. 10.1016/j.fgb.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andersen MR, Nielsen JB, Klitgaard A, et al. Accurate prediction of secondary metabolite gene clusters in filamentous fungi. Proceedings of the National Academy of Sciences. 2013; 110: E99–E107. 10.1073/pnas.1205532110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weber T, Kim HU. The secondary metabolite bioinformatics portal: Computational tools to facilitate synthetic biology of secondary metabolite production. Synthetic and Systems Biotechnology. 2016; 1: 69–79. 10.1016/j.synbio.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.JCVI Smurf − Secondary Metabolite Unique Regions Finder 2010; http://www.jcvi.org/smurf/index.php. Accessed 10/25/2017.
- 76.JCVI Storage of all code of the SMURF tool (smurf.jcvi.org). 2015; https://github.com/rsanka/smurf. Accessed 10/25/2017.
- 77.Blin K, Wolf T, Chevrette MG, et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Research. 2017; 45 (W1): W36–W41. 10.1093/nar/gkx319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Medema MH, Blin K, Cimermancic P, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(Web Server issue):W339-W346. [DOI] [PMC free article] [PubMed]
- 79.Weber T, Blin K, Duddela S, et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Research. 2015;43(Web Server issue):W237-W243. [DOI] [PMC free article] [PubMed]
- 80.Umemura M, Koike H, Machida M. Motif-independent de novo detection of secondary metabolite gene clusters—toward identification from filamentous fungi. Frontiers in Microbiology. 2015; 6: 371. 10.3389/fmicb.2015.00371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vesth TC, Brandl J, Andersen MR. FunGeneClusterS: Predicting fungal gene clusters from genome and transcriptome data. Synthetic and Systems Biotechnology. 2016; 1: 122–129. 10.1016/j.synbio.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chiang YM, Szewczyk E, Davidson AD, et al. Characterization of the Aspergillus nidulans monodictyphenone gene cluster. Applied and Environmental Microbiology. 2010; 76: 2067–2074. 10.1128/AEM.02187-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Medema MH, Kottmann R, Yilmaz P, et al. Minimum Information about a Biosynthetic Gene cluster. Nature Chemical Biology. 2015; 11: 625–631. 10.1038/nchembio.1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Medema MH, Takano E, Breitling R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Molecular Biology and Evolution. 2013; 30: 1218–1223. 10.1093/molbev/mst025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Macheleidt J, Mattern DJ, Fischer J, et al. Regulation and role of fungal secondary metabolites. Annual Review of Genetics. 2016; 50: 371–392. 10.1146/annurev-genet-120215-035203 [DOI] [PubMed] [Google Scholar]
- 86.Kale SP, Milde L, Trapp MK, Frisvad JC, Keller NP, Bok JW. Requirement of LaeA for secondary metabolism and sclerotial production in Aspergillus flavus. Fungal Genetics and Biology. 2008; 45: 1422–1429. 10.1016/j.fgb.2008.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bok JW, Keller NP. Insight into fungal secondary metabolism from ten years of LaeA research. In: Hoffmeister D, ed. The Mycota: Biochemistry and Molecular Biology III. 3rd Edition ed. Cham: Springer International Publishing; 2016:21–29. [Google Scholar]
- 88.Oda K, Kobayashi A, Ohashi S, Sano M. Aspergillus oryzae laeA regulates kojic acid synthesis genes. Bioscience, Biotechnology, and Biochemistry. 2011; 75: 1832–1834. 10.1271/bbb.110235 [DOI] [PubMed] [Google Scholar]
- 89.Calvo AM. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genetics and Biology. 2008; 45: 1053–1061. 10.1016/j.fgb.2008.03.014 [DOI] [PubMed] [Google Scholar]
- 90.Duran RM, Cary JW, Calvo AM. Production of cyclopiazonic acid, aflatrem, and aflatoxin by Aspergillus flavus is regulated by veA, a gene necessary for sclerotial formation. Applied Microbiology and Biotechnology. 2006; 73: 1158–1168. 10.1007/s00253-006-0581-5 [DOI] [PubMed] [Google Scholar]
- 91.Kim HR, Chae KS, Han KH, Han DM. The nsdC gene encoding a putative C2H2-type transcription factor is a key activator of sexual development in Aspergillus nidulans. Genetics. 2009; 182: 771–783. 10.1534/genetics.109.101667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hynes MJ, Murray SL, Duncan A, Khew GS, Davis MA. Regulatory genes controlling fatty acid catabolism and peroxisomal functions in the filamentous fungus Aspergillus nidulans. Eukaryotic Cell. 2006; 5: 794–805. 10.1128/EC.5.5.794-805.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luo X, Affeldt KJ, Keller NP. Characterization of the Far transcription factor family in Aspergillus flavus. G3: Genes|Genomes|Genetics. 2016;6(10):3269–3281. [DOI] [PMC free article] [PubMed]
- 94.Chang PK, Scharfenstein LL, Li RW, Arroyo-Manzanares N, De Saeger S, Diana Di Mavungu J. Aspergillus flavus aswA, a gene homolog of Aspergillus nidulans oefC, regulates sclerotial development and biosynthesis of sclerotium-associated secondary metabolites. Fungal Genetics and Biology. 2017; 104 (Supplement C): 29–37. 10.1016/j.fgb.2017.04.006 [DOI] [PubMed] [Google Scholar]
- 95.Cary J, Harris-Coward P, Scharfenstein L, et al. The Aspergillus flavus homeobox gene, hbx1, is required for development and aflatoxin production. Toxins. 2017; 9: 315. 10.3390/toxins9100315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhuang Z, Lohmar J, Satterlee T, Cary J, Calvo A. The master transcription factor mtfA governs aflatoxin production, morphological development and pathogenicity in the fungus Aspergillus flavus. Toxins. 2016; 8: 29. 10.3390/toxins8010029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Satterlee T, Cary JW, Calvo AM. RmtA, a putative arginine methyltransferase, regulates secondary metabolism and development in Aspergillus flavus. PLOS ONE. 2016; 11: e0155575. 10.1371/journal.pone.0155575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lohmar JM, Harris-Coward PY, Cary JW, Dhingra S, Calvo AM. rtfA, a putative RNA-Pol II transcription elongation factor gene, is necessary for normal morphological and chemical development in Aspergillus flavus. Applied Microbiology and Biotechnology. 2016; 100: 5029–5041. 10.1007/s00253-016-7418-7 [DOI] [PubMed] [Google Scholar]
- 99.Woloshuk CP, Foutz KR, Brewer JF, Bhatnagar D, Cleveland TE, Payne GA. Molecular characterization of aflR, a regulatory locus for aflatoxin biosynthesis. Appl Environ Microbiol. 1994; 60: 2408–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu JH, Butchko RAE, Fernandes M, Keller NP, Leonard TJ, Adams TH. Conservation of structure and function of the aflatoxin regulatory geneaflR fromAspergillus nidulans andA. flavus. Current Genetics. 1996; 29: 549–555. 10.1007/BF02426959 [DOI] [PubMed] [Google Scholar]
- 101.Flaherty JE, Weaver MA, Payne GA, Woloshuk CPA. A β-glucuronidase reporter gene construct for monitoring aflatoxin biosynthesis in Aspergillus flavus. Appl Environ Microbiol. 1995; 61: 2482–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fernandes M, Keller NP, Adams TH. Sequence-specific binding by Aspergillus nidulans AflR, a C6 zinc cluster protein regulating mycotoxin biosynthesis. Molecular Microbiology. 1998; 28: 1355–1365. 10.1046/j.1365-2958.1998.00907.x [DOI] [PubMed] [Google Scholar]
- 103.Ehrlich KC, Montalbano BG, Cary JW. Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene. 1999; 230: 249–257. 10.1016/S0378-1119(99)00075-X [DOI] [PubMed] [Google Scholar]
- 104.Meyers DM, Obrian G, Du WL, Bhatnagar D, Payne GA. Characterization of aflJ, a gene required for conversion of pathway intermediates to aflatoxin. Appl Environ Microbiol. 1998; 64: 3713–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lim FY, Sanchez JF, Wang CCC, Keller NP. Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Methods in Enzymology. 2012; 517: 303–324. 10.1016/B978-0-12-404634-4.00015-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Netzker T, Fischer J, Weber J, et al. Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Frontiers in Microbiology. 2015; 6: 299. 10.3389/fmicb.2015.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deepika VB, Murali TS, Satyamoorthy K. Modulation of genetic clusters for synthesis of bioactive molecules in fungal endophytes: A review. Microbiological Research. 2016; 182 (Supplement C): 125–140. 10.1016/j.micres.2015.10.009 [DOI] [PubMed] [Google Scholar]
- 108.Sobolev VS, Cole RJ, Dorner JW, Horn BW, Harrigan GG, Gloer JB. Isolation and structure elucidation of a new metabolite produced by Aspergillus parasiticus. Journal of Natural Products. 1997; 60: 847–850. 10.1021/np970131m [DOI] [Google Scholar]
- 109.Zhang S, Monahan BJ, Tkacz JS, Scott B. Indole-diterpene gene cluster from Aspergillus flavus. Applied and Environmental Microbiology. 2004; 70: 6875–6883. 10.1128/AEM.70.11.6875-6883.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barrow CJ, Sedlock DM. 1′-(2-Phenyl-ethylene)-ditryptophenaline, a new dimeric diketopiperazine from Aspergillus flavus. Journal of Natural Products. 1994; 57: 1239–1244. 10.1021/np50111a008 [DOI] [PubMed] [Google Scholar]
- 111.Terabayashi Y, Sano M, Yamane N, et al. Identification and characterization of genes responsible for biosynthesis of kojic acid, an industrially important compound from Aspergillus oryzae. Fungal Genetics and Biology. 2010; 47: 953–961. 10.1016/j.fgb.2010.08.014 [DOI] [PubMed] [Google Scholar]
- 112.Hang L, Liu N, Tang Y. Coordinated and Iterative Enzyme Catalysis in Fungal Polyketide Biosynthesis. ACS Catalysis. 2016; 6: 5935–5945. 10.1021/acscatal.6b01559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Keller NP, Turner G, Bennett JW. Fungal secondary metabolism — from biochemistry to genomics. Nature Reviews Microbiology. 2005; 3: 937–947. 10.1038/nrmicro1286 [DOI] [PubMed] [Google Scholar]
- 114.Cox RJ, Simpson TJ. Fungal type I polyketide synthases. Methods in Enzymology. 2009; 459: 49–78. 10.1016/S0076-6879(09)04603-5 [DOI] [PubMed] [Google Scholar]
- 115.Walsh CT, Tang Y. Carbon-based Radicals in C-C bond Formations in Natural Products A. Oxidases B. Oxygenases. In: Natural Product Biosynthesis: Chemical Logic and Enzymatic Machinery. London: Royal Society of Chemistry; 2017:457–522. [Google Scholar]
- 116.Eisfeld K. Non-Ribosomal Peptide Synthetases of Fungi. In: Anke T, Weber D, eds. Physiology and Genetics: Selected Basic and Applied Aspects. Berlin, Heidelberg: Springer Berlin Heidelberg; 2009:305–330. [Google Scholar]
- 117.Haynes SW, Ames BD, Gao X, Tang Y, Walsh CT. Unraveling terminal C-domain-mediated condensation in fungal biosynthesis of imidazoindolone metabolites. Biochemistry. 2011; 50: 5668–5679. 10.1021/bi2004922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gao X, Haynes SW, Ames BD, et al. Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nature Chemical Biology. 2012; 8: 823–830. 10.1038/nchembio.1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chang PK, Ehrlich K, Fujii I. Cyclopiazonic acid biosynthesis of Aspergillus flavus and Aspergillus oryzae. Toxins. 2009; 1: 74–99. 10.3390/toxins1020074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Metzger U, Schall C, Zocher G, et al. The structure of dimethylallyl tryptophan synthase reveals a common architecture of aromatic prenyltransferases in fungi and bacteria. Proceedings of the National Academy of Sciences. 2009; 106: 14309–14314. 10.1073/pnas.0904897106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xu W, Gavia DJ, Tang Y. Biosynthesis of fungal indole alkaloids. Nat. Prod. Rep. 2014; 31: 1474–1487. 10.1039/C4NP00073K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saikia S, Nicholson MJ, Young C, Parker EJ, Scott B. The genetic basis for indole-diterpene chemical diversity in filamentous fungi. Mycological Research. 2008; 112: 184–199. 10.1016/j.mycres.2007.06.015 [DOI] [PubMed] [Google Scholar]
- 123.Betina V. Thin-layer chromatography of mycotoxins. Journal of Chromatography A. 1985; 334: 211–276. 10.1016/S0021-9673(00)80272-1 [DOI] [PubMed] [Google Scholar]
- 124.Jork H, Funk W, Fischer WR, Wimmer H. Physical and Chemical Detection Methods: Fundamentals, Reagents I, Volume 1a, Thin-Layer Chromatography: Reagents and Detection Methods. Wiley-VCH; 1989. [Google Scholar]
- 125.Frisvad JC, Thrane U. Standardized high-performance liquid chromatography of 182 mycotoxins and other fungal metabolites based on alkylphenone retention indices and UV—VIS spectra (diodearray detection). Journal of Chromatography A. 1987; 404: 195–214. 10.1016/S0021-9673(01)86850-3 [DOI] [PubMed] [Google Scholar]
- 126.Smedsgaard J. Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. Journal of Chromatography A. 1997; 760: 264–270. 10.1016/S0021-9673(96)00803-5 [DOI] [PubMed] [Google Scholar]
- 127.Nielsen K, Smedsgaard J. Fungal metabolite screening: database of 474 mycotoxins and fungal metabolites for dereplication by standardised liquid chromatography–UV–mass spectrometry methodology. Journal of Chromatography A. 2003; 1002: 111–136. 10.1016/S0021-9673(03)00490-4 [DOI] [PubMed] [Google Scholar]
- 128.Sulyok M, Krska R, Schuhmacher R. A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Analytical and Bioanalytical Chemistry. 2007; 389: 1505–1523. 10.1007/s00216-007-1542-2 [DOI] [PubMed] [Google Scholar]
- 129.Klitgaard A, Iversen A, Andersen MR, Larsen TO, Frisvad JC, Nielsen KF. Aggressive dereplication using UHPLC–DAD–QTOF: screening extracts for up to 3000 fungal secondary metabolites. Analytical and Bioanalytical Chemistry. 2014; 406: 1933–1943. 10.1007/s00216-013-7582-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nielsen KF, Månsson M, Rank C, Frisvad JC, Larsen TO. Dereplication of microbial natural products by LC-DAD-TOFMS. Journal of Natural Products. 2011; 74: 2338–2348. 10.1021/np200254t [DOI] [PubMed] [Google Scholar]
- 131.Rank C, Klejnstrup ML, Petersen LM, et al. Comparative Chemistry of Aspergillus oryzae (RIB40) and A. flavus (NRRL 3357). Metabolites. 2012; 2: 39–56. 10.3390/metabo2010039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Malysheva SV, Arroyo-Manzanares N, Cary JW, et al. Identification of novel metabolites from Aspergillus flavus by high resolution and multiple stage mass spectrometry. Food Additives & Contaminants: Part A. 2014; 31: 111–120. 10.1080/19440049.2013.859743 [DOI] [PubMed] [Google Scholar]
- 133.Arroyo-Manzanares N, Diana Di Mavungu J, Uka V, et al. Use of UHPLC high-resolution Orbitrap mass spectrometry to investigate the genes involved in the production of secondary metabolites in Aspergillus flavus. Food Additives & Contaminants: Part A. 2015; 32: 1656–1673. 10.1080/19440049.2015.1071499 [DOI] [PubMed] [Google Scholar]
- 134.Reynolds WF, Mazzola EP. Nuclear Magnetic Resonance in the Structural Elucidation of Natural Products. In: Kinghorn AD, Falk H, Kobayashi J, eds. Progress in the Chemistry of Organic Natural Products 100. Cham: Springer International Publishing; 2015:223–309. [DOI] [PubMed] [Google Scholar]
- 135.Forseth RR, Schroeder FC. NMR-spectroscopic analysis of mixtures: from structure to function. Current Opinion in Chemical Biology. 2011; 15: 38–47. 10.1016/j.cbpa.2010.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Robinette SL, Brüschweiler R, Schroeder FC, Edison AS. NMR in metabolomics and natural products research: two sides of the same coin. Accounts of Chemical Research. 2012; 45: 288–297. 10.1021/ar2001606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Forseth RR, Schroeder FC. Correlating Secondary Metabolite Production with Genetic Changes Using Differential Analysis of 2D NMR Spectra. In: Keller NP, Turner G, eds. Fungal Secondary Metabolism: Methods and Protocols. Totowa, NJ: Humana Press; 2012:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schroeder FC, Gibson DM, Churchill ACL, et al. Differential analysis of 2D NMR spectra: new natural products from a pilot-scale fungal extract library. Angewandte Chemie International Edition. 2007; 46: 901–904. 10.1002/anie.200603821 [DOI] [PubMed] [Google Scholar]
- 139.Forseth RR, Fox EM, Chung D, Howlett BJ, Keller NP, Schroeder FC. Identification of cryptic products of the gliotoxin gene cluster using NMR-based comparative metabolomics and a model for gliotoxin biosynthesis. Journal of the American Chemical Society. 2011; 133: 9678–9681. 10.1021/ja2029987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nicholson MJ, Koulman A, Monahan BJ, Pritchard BL, Payne GA, Scott B. Identification of two aflatrem biosynthesis gene loci in Aspergillus flavus and metabolic engineering of Penicillium paxilli to elucidate their function. Applied and Environmental Microbiology. 2009; 75: 7469–7481. 10.1128/AEM.02146-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.TePaske MR, Gloer JB, Wicklow DT, Dowd PF, Leporin A. Leporin A: an antiinsectan N-alkoxypyridone from the sclerotia of Aspergillus leporis. Tetrahedron Letters. 1991; 32: 5687–5690. 10.1016/S0040-4039(00)93530-5 [DOI] [Google Scholar]
- 142.Zhang C, Jin L, Mondie B, et al. Leporin B: a novel hexokinase II gene inducing agent from an unidentified fungus. Bioorganic & Medicinal Chemistry Letters. 2003; 13: 1433–1435. 10.1016/S0960-894X(03)00153-7 [DOI] [PubMed] [Google Scholar]
- 143.Ohashi M, Liu F, Hai Y, et al. SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature. 2017; 549: 502–506. 10.1038/nature23882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aguirre J, Ríos-Momberg M, Hewitt D, Hansberg W. Reactive oxygen species and development in microbial eukaryotes. Trends in Microbiology. 2005; 13: 111–118. 10.1016/j.tim.2005.01.007 [DOI] [PubMed] [Google Scholar]
- 145.Grintzalis K, Vernardis SI, Klapa MI, Georgiou CD. Role of oxidative stress in Sclerotial differentiation and aflatoxin B1 biosynthesis in Aspergillus flavus. Applied and Environmental Microbiology. 2014; 80: 5561–5571. 10.1128/AEM.01282-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.TePaske MR, Gloer JB, Wicklow DT, Dowd PF. Aflavarin and beta-aflatrem: new anti-insectan metabolites from the sclerotia of Aspergillus flavus. Journal of Natural Products. 1992; 55: 1080–1086. 10.1021/np50086a008 [DOI] [Google Scholar]
- 147.Bok JW, Keller NP. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryotic Cell. 2004; 3: 527–535. 10.1128/EC.3.2.527-535.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ehmann DE, Gehring AM, Walsh CT. Lysine biosynthesis in Saccharomyces cerevisiae: mechanism of α-aminoadipate reductase (Lys2) involves posttranslational phosphopantetheinylation by Lys5. Biochemistry. 1999; 38: 6171–6177. 10.1021/bi9829940 [DOI] [PubMed] [Google Scholar]
- 149.Saikia S, Parker EJ, Koulman A, Scott B. Four gene products are required for the fungal synthesis of the indole-diterpene, paspaline. FEBS Letters. 2006; 580: 1625–1630. 10.1016/j.febslet.2006.02.008 [DOI] [PubMed] [Google Scholar]
- 150.Tagami K, Minami A, Fujii R, et al. Rapid reconstitution of biosynthetic machinery for fungal metabolites in Aspergillus oryzae: total biosynthesis of aflatrem. ChemBioChem. 2014; 15: 2076–2080. 10.1002/cbic.201402195 [DOI] [PubMed] [Google Scholar]
- 151.Tang MC, Lin HC, Li D, et al. Discovery of unclustered fungal indole diterpene biosynthetic pathways through combinatorial pathway reassembly in engineered yeast. Journal of the American Chemical Society. 2015; 137: 13724–13727. 10.1021/jacs.5b06108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gloer JB, TePaske MR, Sima JS, Wicklow DT, Dowd PF. Antiinsectan aflavinine derivatives from the sclerotia of Aspergillus flavus. The Journal of Organic Chemistry. 1988; 53: 5457–5460. 10.1021/jo00258a011 [DOI] [Google Scholar]
- 153.Gallagher RT, Wilson BJ. Aflatrem, the tremorgenic mycotoxin from aspergillus flavus. Mycopathologia. 1979; 66: 183–185. 10.1007/BF00683969 [DOI] [PubMed] [Google Scholar]
- 154.Koiso Y, Li Y, Iwasaki S, et al. Ustiloxins, antimitotic cydic peptides from false smut balls on rice panicles caused by Ustilaginoidea virens. The Journal of Antibiotics. 1994; 47: 765–773. 10.7164/antibiotics.47.765 [DOI] [PubMed] [Google Scholar]
- 155.Koiso Y, Morisaki N, Yamashita Y, et al. Isolation and structure of an antimitotic cyclic peptide, ustiloxin F: chemical interrelation with a homologous peptide, ustiloxin B. The Journal of Antibiotics. 1998; 51: 418–422. 10.7164/antibiotics.51.418 [DOI] [PubMed] [Google Scholar]
- 156.Yoshimi A, Umemura M, Nagano N, Koike H, Machida M, Abe K. Expression of ustR and the Golgi protease KexB are required for ustiloxin B biosynthesis in Aspergillus oryzae. AMB Express. 2016; 6: 9. 10.1186/s13568-016-0181-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Saruwatari T, Yagishita F, Mino T, Noguchi H, Hotta K, Watanabe K. Cytochrome P450 as dimerization catalyst in diketopiperazine alkaloid biosynthesis. ChemBioChem. 2014; 15: 656–659. 10.1002/cbic.201300751 [DOI] [PubMed] [Google Scholar]
- 158.Sano M. Aspergillus oryzae nrtA affects kojic acid production. Bioscience, Biotechnology, and Biochemistry. 2016; 80: 1776–1780. 10.1080/09168451.2016.1176517 [DOI] [PubMed] [Google Scholar]
- 159.Dowd PF. Synergism of aflatoxin B1 toxicity with the co-occurring fungal metabolite kojic acid to two caterpillars. Entomologia Experimentalis et Applicata. 1988; 47: 69–71. 10.1111/j.1570-7458.1988.tb02283.x [DOI] [Google Scholar]