

Abstract
Optimal management of systemic lupus erythematosus (SLE)‐associated pulmonary arterial hypertension (PAH) remains unclear. Our observation describes the case of a 31‐year‐old SLE patient presenting with cardiogenic shock revealing severe PAH, in which a therapeutic scheme combining immunosuppressants (pulse cyclophosphamide and corticosteroids) and PAH‐specific drugs (bosentan, tadalafil, and epoprostenol) led to a complete normalization of pulmonary haemodynamics and allowed a progressive weaning of PAH vasodilators. This case report supports the efficacy of immunosuppressants and use of PAH‐specific therapy as a bridge therapy in severe SLE‐PAH. Further studies on larger population are required to confirm these findings.
Keywords: Pulmonary arterial hypertension, Systemic lupus erythematosus, Immunosuppressants
1. Introduction
Pulmonary arterial hypertension (PAH) is a rare condition characterized by a proliferation and remodelling of the small pulmonary arteries, leading to a progressive increase in pulmonary vascular resistance and right heart failure.1 Several causes of PAH have been described, the most frequent of which are connective tissue diseases and particularly systemic sclerosis (SSc).2 Although more scarce than in SSc, PAH can also occur during systemic lupus erythematosus (SLE).3 In that setting, the best therapeutic strategy remains elusive, especially regarding the respective places of immunosuppressants (IS) and PAH‐specific therapy. We wanted to share our experience of a patient managed with an upfront combined treatment with a remarkable outcome.
2. Case report
A 31‐year‐old woman was referred to our tertiary care centre in September 2014 for acute right heart failure. In 2002, she had been diagnosed with SLE as manifested by skin features (malar rash), joint involvement (distal polyarthritis), kidney disease (class II nephritis), serositis (pleural and pericardial effusions), cytopenias (pure red cell aplasia and leucopenia), and immunological features [low complement levels, antinuclear antibodies with anti‐double strand (ds) DNA, anti‐U1 ribonucleoprotein, and anti‐Sm specificities]. In 2012, a diagnosis of antiphospholipid syndrome was made when a kidney biopsy performed because of persistent proteinuria revealed glomerular microthromboses associated with a positive lupus anticoagulant test, with no previous history of venous thromboembolism. Since then, she had remained in clinical and biological remission under hydroxychloroquine, prednisone, azathioprine, and warfarin.
At referral, she presented with resting dyspnoea (staged in class IV of the New York Heart Association functional classification) and signs of right heart failure. While she displayed no clinical symptom of a lupus flare, laboratory tests revealed low complement levels and high titers of anti‐dsDNA antibodies, suggesting that the disease was active again. Serum brain natriuretic peptide levels were also elevated at 1051 ng/L. Chest computed tomography angiography showed no feature of lung parenchymal involvement, veno‐occlusive disease, acute pulmonary embolism, or chronic thromboembolic disease. Pulmonary function tests found an isolated decrease of the diffusing capacity of the lung for carbon monoxide (DLCO) at 58% of its predicted value, with normal respiratory volumes. Transthoracic echocardiography exhibited signs suggestive of pulmonary hypertension (PH) (peak tricuspid regurgitant jet 4.33 m/s), right ventricle dilation (right‐to‐left ventricle diameter ratio 1.45 with interventricular septum systolic flattening), and pericardial effusion, with no sign of diastolic or systolic left heart dysfunction. A right heart catheterization was thus performed and confirmed a severe pre‐capillary PH (systolic/diastolic/mean pulmonary artery pressure 77/35/51 mmHg, pulmonary vascular resistance 14.9 Wood units, pulmonary arterial wedge pressure 1 mmHg, and right atrial pressure 7 mmHg) with an altered cardiac function (cardiac output 3.4 L/min and index 2.1 L/min/m2) and no hepatic venous pressure gradient. Within a few days, the patient progressed to cardiogenic shock that required dobutamine therapy.
After a multidisciplinary evaluation, she was diagnosed with severe PAH occurring in a context of SLE flare. PH was classified as group 1 PAH, as it was a severe pre‐capillary PH with no evidence of chronic lung disease (group 3) or chronic thromboembolic disease (group 4). We did not find other causes of PAH (such as drugs, familial history of PAH, congenital heart disease, portopulmonary hypertension, or of pulmonary veno‐occlusive disease).1, 2 She was rapidly started on an intensive IS treatment (monthly intravenous pulses of cyclophosphamide 0.6 g/m2, intravenous pulses of methylprednisolone 15 mg/kg/day for 3 days followed by oral prednisone 1 mg/kg/day) and PAH‐specific therapy (intravenous epoprostenol, oral bosentan, and tadalafil).
This treatment led to a dramatic clinical, functional, and haemodynamic improvement. Within only a few days, the patient was weaned from dobutamine. During the following months, this favourable trend continued (Figure 1 and Table 1), allowing switch to mycophenolate mofetil maintenance therapy in February 2015, epoprostenol withdrawal in August 2015, and bosentan cessation in December 2015. The last right heart catheterization performed on tadalafil monotherapy in December 2015 showed normal haemodynamic parameters (systolic/diastolic/mean pulmonary artery pressure 28/7/12 mmHg, pulmonary vascular resistance 1.18 Wood units, and cardiac index 4.2 L/min/m2).
Figure 1.
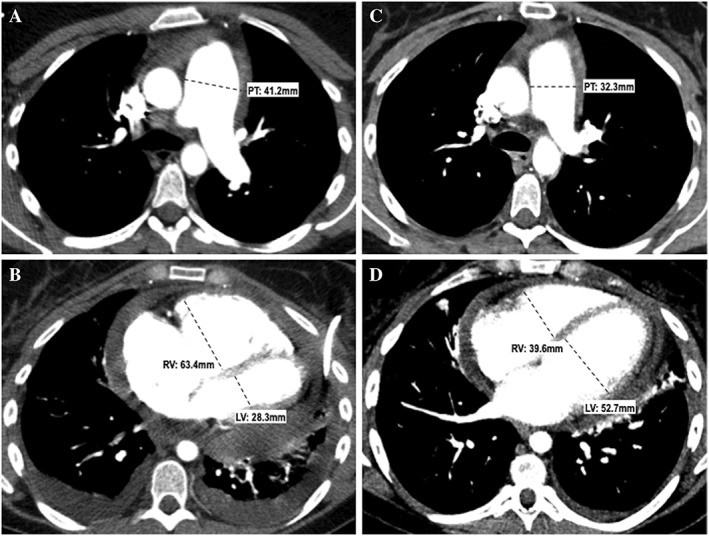
Chest computed tomography scans of our patient at diagnosis (A, B) and 6 months after treatment (C, D). Top row (A, C): Transverse computed tomography sections obtained at the level of the pulmonary trunk (A) and cardiac cavities (C) showing dilatation of the pulmonary trunk (41.2 mm) and right ventricular enlargement (63.4 mm) with a right ventricle/left ventricle ratio >1. Note the additional presence of pericardial and pleural effusion. Bottom row (B, D): Same anatomical levels as those shown on the top row, obtained 6 months later. Note the dramatic reduction in size of the pulmonary trunk (32.3 mm) (B) and right ventricle (39.6 mm) with normalization of the RV/LV ratio (D). Improvement of pericardial and left pleural effusion; resolution of right pleural effusion. PT, pulmonary trunk; RV, right ventricle; LV, left ventricle.
Table 1.
Clinical course of our patient after treatment onset
| 22 Sep 2014 | 06 Oct 2014 | 26 Feb 2015 | 08 Aug 2015 | 10 Dec 2015 | 08 Jun 2016 | ||
|---|---|---|---|---|---|---|---|
| Clinical signs | |||||||
| NYHA class | IV | II | I/II | I | I | I | |
| RHF signs | Yes | No | No | No | No | No | |
| Laboratory results | |||||||
| BNP (ng/L) | 1051 | 17 | 33 | 25 | |||
| Anti‐dsDNA (UI/L) | 48 | 17 | 12 | 15 | |||
| Complement levels | Low | Normal | Normal | Normal | |||
| Transthoracic echocardiography | |||||||
| Estimated sPAP (mmHg) | 75 + 10 | 25 + 5 | 15 + 5 | ||||
| RV dilation | Yes (major) | Yes (minor) | No | No | |||
| RV/LV ratio | 1.45 | 0.93 | |||||
| TAPSE (mm) | 12 | 13 | 19 | 17 | |||
| RV S‐wave (cm/s) | 9 | 10.5 | 12 | ||||
| IVC dilation | Yes | No | No | No | |||
| Pericardial effusion | Yes | No | No | No | |||
| Right heart catheterization | |||||||
| mPAP (mmHg) | 51 | 42 | 21 | 15 | 12 | ||
| sPAP (mmHg) | 77 | 32 | 19 | 28 | |||
| dPAP (mmHg) | 35 | 14 | 11 | 7 | |||
| PAWP (mmHg) | 1 | 12 | 9 | 7 | 4 | ||
| RAP (mmHg) | 7 | 10 | 5 | 4 | 1 | ||
| PVR (WU) | 14.9 | 4.69 | 1.23 | 0.95 | 1.18 | ||
| SvO2 (%) | 44 | 81 | 82 | ||||
| CO (mL/min) | 3.35 | 6.68 | 9.77 | 8.4 | 6.77 | ||
| CI (mL/min/m2) | 2.1 | 4.19 | 6.08 | 5.16 | 4.15 | ||
| Exercise tests | |||||||
| 6MWT | Distance (m) | 522 | 576 | 558 | 598 | ||
| Desaturation | No | Yes | No | No | |||
| Borg score | 2 | 3 | 2 | ||||
| VO2peak | (mL/kg/min) | 23 | 21 | ||||
| (% predicted) | 80 | 73 | |||||
| Treatments | |||||||
| Corticosteroids | |||||||
| Prednisone | 2 mg/day | 60 mg/day | 15 mg/day | 15 mg/day | 8 mg/day | 4 mg/day | |
| Methylprednisolone |

|
||||||
| Immunosuppressants | |||||||
| Azathioprine | 150 mg/day | ||||||
| Cyclophosphamide |

|

|
|||||
| Mycophenolate mofetil | 3 g/day | 3 g/day | 3 g/day | ||||
| Vasoactive molecules | |||||||
| Dobutamine | 7.5 μg/kg/min | ||||||
| Epoprostenol | 9 ng/kg/min | 16 ng/kg/min | 1 ng/kg/min | ||||
| Bosentan | 125 mg/day | 250 mg/day | 250 mg/day | 250 mg/day | |||
| Tadalafil | 40 mg/day | 40 mg/day | 40 mg/day | 40 mg/day | 40 mg/day | ||
6MWT, 6 min walk test; anti‐dsDNA, anti‐double strand DNA antibodies; BNP, brain natriuretic peptide; CI, cardiac index; CO, cardiac output; IVC, inferior vena cava; LV, left ventricle; NYHA, New York Heart Association; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrium pressure; RHF, right heart failure; RV, right ventricle; s/d/mPAP, systolic/diastolic/mean pulmonary arterial pressure; SvO2, venous saturation in oxygen; TAPSE, tricuspid annular plane systolic excursion.
3. Discussion
PAH (pulmonary hypertension group 1) is a rare complication of SLE, whose optimal management is poorly defined.3 In particular, the relative place of IS and PAH‐specific therapies in the therapeutic strategy of SLE‐PAH is controversial. Several works suggested that, conversely to SSc‐associated PAH, immunosuppression could be indicated as a first‐line treatment in this setting.4, 5, 6
A previous study by our group5 suggested that autoantibody positivity (especially anti‐dsDNA and anti‐Sm specificities) and high disease activity (assessed by the SLEDAI‐2K score) could be predictors of response to IS treatment, both alone and combined with PAH‐specific therapy. It is debated whether anti‐U1RNP antibodies can be associated with survival in SLE‐PAH; they were associated with better prognosis in the French SLE‐PAH cohort.3 It is interesting to note that these findings are supported by our present observation.
Optimal modalities of immunosuppression in the context of SLE‐PAH are unknown. Usual regimens generally include high‐dose corticosteroids therapy associated with intravenous pulses of cyclophosphamide,5, 7 as is the case in our observation. Interestingly, our patient was also under hydroxychloroquine at the time of PAH diagnosis. Antimalarial drug therapy was associated with better survival in the French cohort.3
Previous studies5, 7 did not show a clear benefit of adding PAH‐specific molecules to first‐line IS therapy but suggested that an upfront combined treatment should be considered in patients with serious haemodynamic impairment. Due to the extreme severity of her condition, we chose to add PAH‐specific molecules to IS treatment in our patient, using a maximal and combined vasodilator therapy (phosphodiesterase‐5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues). The favourable outcome supports this therapeutic strategy. Interestingly, it suggests also that PAH‐specific treatments could eventually be withdrawn once IS have become effective.
In conclusion, we believe that this observation adequately illustrates the interest of initially combining IS with a bridge PAH‐specific therapy in patients with severe SLE‐PAH.
Conflict of interest
S.S., N.L., M.R.J., and V.S. report no conflict of interest in relation to this work. L.S. consults and received grants from Actelion, MSD, and GlaxoSmithKline. M.H. received personal fees from Actelion Pharmaceuticals Ltd, grants and personal fees from Bayer, grants and personal fees from GSK, personal fees from Pfizer, and personal fees from Merck.
Funding
None.
Sanges, S. , Savale, L. , Lamblin, N. , Rémy‐Jardin, M. , Humbert, M. , and Sobanski, V. (2019) Efficacy of immunosuppressants with bridge vasodilator therapy in severe lupus erythematosus‐associated pulmonary arterial hypertension. ESC Heart Failure, 6: 1322–1325. 10.1002/ehf2.12507.
References
- 1. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019; 53: 1801887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hachulla E, Jais X, Cinquetti G, Clerson P, Rottat L, Launay D, Cottin V, Habib G, Prevot G, Chabanne C, Foïs E, Amoura Z, Mouthon L, Le Guern V, Montani D, Simonneau G, Humbert M, Sobanski V, Sitbon O. French Collaborators Recruiting Members(*). Pulmonary arterial hypertension associated with systemic lupus erythematosus: results from the French Pulmonary Hypertension Registry. Chest 2018; 153: 143–151. [DOI] [PubMed] [Google Scholar]
- 4. Sanchez O, Sitbon O, Jaïs X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases‐associated pulmonary arterial hypertension. Chest 2006; 130: 182–189. [DOI] [PubMed] [Google Scholar]
- 5. Jais X, Launay D, Yaici A, Le Pavec J, Tchérakian C, Sitbon O, Simonneau G, Humbert M. Immunosuppressive therapy in lupus‐ and mixed connective tissue disease‐associated pulmonary arterial hypertension: a retrospective analysis of twenty‐three cases. Arthritis & Rheumatism 2008; 58: 521–531. [DOI] [PubMed] [Google Scholar]
- 6. Sobanski V, Launay D, Hachulla E, Humbert M. Current approaches to the treatment of systemic‐sclerosis‐associated pulmonary arterial hypertension (SSc‐PAH). Curr Rheumatol Rep 2016; 18: 10. [DOI] [PubMed] [Google Scholar]
- 7. Yasuoka H, Shirai Y, Tamura Y, Takeuchi T, Kuwana M. Predictors of favorable responses to immunosuppressive treatment in pulmonary arterial hypertension associated with connective tissue disease. Circulation Journal 2018; 82: 546–554. [DOI] [PubMed] [Google Scholar]