

Abstract
Transthyretin cardiac amyloidosis (ATTR‐CA) demonstrates progressive, potentially fatal, and infiltrative cardiomyopathy caused by extracellular deposition of transthyretin‐derived insoluble amyloid fibrils in the myocardium. Two distinct types of transthyretin (wild type or variant) become unstable, and misfolding forms aggregate, resulting in amyloid fibrils. ATTR‐CA, which has previously been underrecognized and considered to be rare, has been increasingly recognized as a cause of heart failure with preserved ejection fraction among elderly persons. With the advanced technology, the diagnostic tools have been improving for cardiac amyloidosis. Recently, the efficacy of several disease‐modifying agents focusing on the amyloidogenic process has been demonstrated. ATTR‐CA has been changing from incurable to treatable. Nevertheless, there are still no prognostic improvements due to diagnostic delay or misdiagnosis because of phenotypic heterogeneity and co‐morbidities. Thus, it is crucial for clinicians to be aware of this clinical entity for early diagnosis and proper treatment. In this mini‐review, we focus on recent advances in diagnosis and treatment of ATTR‐CA.
Keywords: Transthyretin, Amyloidosis, Cardiomyopathy, Heart failure, Diagnosis, Red‐flags, Disease‐modifying agents
1. Introduction
Transthyretin cardiac amyloidosis (ATTR‐CA) demonstrates infiltrative cardiomyopathy caused by extracellular deposition of insoluble transthyretin (TTR) amyloid fibrils in the myocardium.1 TTR is a plasma protein mainly synthesized in the liver, recognized as a transporter of thyroxine and retinol‐binding protein. Unstable changes in two different types of TTR (wild type or variant) become misfolding, aggregate, and form ultimately amyloid fibrils. Cardiac amyloidosis (CA) has been recently highlighted as a cause of heart failure with preserved ejection fraction (HFpEF) among elderly persons, and its incidence has been constantly increasing because the population is aging.2, 3 CA is progressive and life‐threatening if left untreated, and thus, early diagnosis is critical. However, a definitive diagnosis of CA is considerably difficult, because left ventricular hypertrophy (LVH) shows a similar phenotype in hypertension, hypertrophic cardiomyopathy (HCM), and aortic stenosis (AS).4, 5, 6 Moreover, the development of an appropriate strategy to reach the correct diagnosis and treatment of CA has been long overdue. In this mini‐review, we focus on the current diagnosis and treatment of ATTR‐CA.
2. Epidemiology and clinical presentation
Wild type ATTR‐CA (ATTRwt‐CA), previously known as senile systemic amyloidosis, is caused by age‐related misfolding of TTR, but its mechanism remains to be fully elucidated.1 ATTRwt‐CA will become the most frequent form of amyloidosis in the USA. ATTRwt‐CA affects male‐predominant patients over 60 years of age and typically presents HFpEF.7 Dyspnoea, fatigue, and weakness are common symptoms. However, these symptoms are often misunderstood as non‐specific symptoms due to aging. Anginal chest pain caused by microvascular amyloid infiltration in patients without obstructive epicardial coronary stenosis can also occur.8, 9 Recently, ATTRwt‐CA is increasingly recognized as a cause of various common diseases. While ATTRwt‐CA was identified in 13% of elderly patients (>60 years old) with HFpEF and 16% of patients undergoing transcatheter aortic valve replacement, respectively,2, 4 bilateral carpal tunnel syndrome (CTS) and lumbar spinal stenosis frequently present many years before the development of cardiac symptoms..10, 11
Hereditary ATTR‐CA (ATTRv‐CA) is an autosomal‐dominant disease in which gene mutations lead to changes in the protein TTR.12 More than 120 mutations have been reported in the TTR gene with considerable phenotypic and geographical heterogeneity. Their clinical symptoms vary extensively from neurological‐predominant phenotype to cardiac‐predominant phenotype. This phenotypic heterogeneity depends on many factors, such as specific TTR mutation site, geographical distribution, inheritance pattern, timing of onset, and epidemic/non‐epidemic aggregation. While the Val30Met induces progressive peripheral sensory‐motor polyneuropathy, other mutations (Thr60Ala, Ile68Leu, Leu111Met, and Val122Ile) cause exclusively infiltrative cardiomyopathy.13 In particular, a sporadic case of ATTRv‐CA is extremely rare, and its diagnosis is considerably difficult.14
The Val122Ile variant is the most frequent form of TTR amyloid in the USA and prevalent in 3.4% of African Americans.15 The clinical phenotype is late‐onset restrictive cardiomyopathy, identical to ATTRwt‐CA and often mimics hypertensive cardiomyopathy due to high morbidity of hypertension. A prospective observational Atherosclerosis Risk in Communities study, which analysed 3856 African Americans, reported a low clinical penetrance of the disease. Echocardiogram revealed that only three (7%) carriers had typical echocardiographic findings characteristic of CA. However, surprisingly, Val122Ile carriers had a significantly increased risk of heart failure (HF) during the later years of the study compared with non‐carriers (age‐stratified and sex‐stratified hazard ratio, 1.47 and 95% confidence interval, 1.03 to 2.10),16 suggesting that Val122Ile carriers are predominantly at increased risk of HF with an age‐dependent clinical penetrance. The Thr60Ala variant, the second most frequent form of TTR amyloid in the USA, affects up to 1% of the population of Northwestern Ireland.17 The Thr60Ala variant causes both neurologic and cardiac phenotypes, and symptom onset mainly occurs between 50 and 60 years of age.18 The Ile68Leu variant is endemic in central‐northern Italy and shows exclusively cardiac phenotype with male predominance and age‐dependent penetrance.13, 19 In Denmark, the Leu111Met variant, which shows exclusively cardiac phenotype, has only been found in Danish families.20 Transthyretin Amyloidosis Outcomes Survey registry verified that patients with the Leu111Met variant were significantly younger (mean age at symptom onset: 47.6 years) and less likely to be male (63.6%) than those with other known cardiac variants (Val122Ile, Thr60Ala, and Ile68Leu).21 On the other hand, the Val30Met variant, known as transthyretin familial amyloid polyneuropathy (ATTR‐FAP), is the most frequent form of TTR amyloid in Europe and other countries.12 In endemic early‐onset Val30Met patients in Portugal, Brazil, and Japan, an exclusively neurologic phenotype is common, and its disease onset frequently occurs in the third and fourth decade. On the other hand, in non‐endemic late‐onset Val30Met patients in France, Sweden, and Japan, an exclusively cardiac phenotype is common, and its disease onset occurs relatively late (>50 years old), affecting predominantly male. Median survival was notably shorter in late‐onset Val30Met patients.22 There is a bimodal distribution of onset age among patients with the Val30Met variant in Japan.13, 23 Several studies demonstrated that the non‐Val30Met variants were associated with disease severity and worse outcomes compared with those with the Val30Met variant.24, 25
3. Implications of genetic diagnosis of transthyretin gene sequencing
Early and correct identification of ATTRv amyloidosis plays a vital role for the estimation of prognosis, treatment of choice, familial screening, and genetic counselling. In a prospective multicentre study in France, genetic screening for a TTR gene revealed that 5% of 298 consecutive elderly patients (>60 years old) clinically diagnosed with HCM have ATTRv‐CA with a predominance of the Val122Ile variant.26 Moreover, the Beta‐Blocker Evaluation of Survival Trial study showed a high prevalence of the Val122Ile variant in elderly African Americans with severe HF.27 Thus, TTR gene sequencing should be systematically carried out in even elderly patients of African descent having HCM phenotype of unknown cause or unexplained HF. Regular monitoring should start for asymptomatic carriers of a variant of interest 10 years before the predicted age of onset of symptomatic disease.28
4. Diagnosis
Lack of recognition of this clinical entity, non‐specific symptoms, and co‐morbidities often leads to delayed diagnosis, resulting in disease progression.29 One descriptive study revealed that 35% had been previously misdiagnosed with other cardiovascular diseases commonly recognized in clinical practice. Among them, hypertensive cardiomyopathy (35%) was the most frequent followed by HCM (23.5%), ischaemic heart disease (11.8%), HFpEF (8.8%), and AS (8.8%), respectively.30 Thus, it is critical to make an early diagnosis for proper treatment. ATTR‐CA should be highly suspected if left ventricular (LV) wall thickening is observed in combination with one or more of red‐flags shown in Figure 1 . Newly diagnosed HFpEF patients with LVH (>12 mm) over 60 years old are good candidates for ATTR‐CA.2 Moreover, atrial fibrillation (AF) (38–67%) and symptomatic atrioventricular block requiring pacemaker (8–40%) are the most common in patients with ATTRwt‐CA (Table 1).7, 31, 32 Unexplained CTS (bilateral or intractable) should strongly suspect ATTRwt‐CA.10 Intriguingly, patients with ATTRv‐CA frequently had CTS and/or neuropathy (46% and 53%, respectively).26 In addition, a series of studies demonstrated that ATTR‐CA was prevalent in 14% to 16% of elderly patients with severe calcified AS undergoing transcatheter aortic valve replacement.4, 36 In particular, this is highly likely to be the case with low‐flow, low‐gradient severe AS. There are also warnings that should not be overlooked. Because blood pressure often falls as the disease progresses, symptomatic hypotension or resolution of hypertension in previously hypertensive patients might be other promising clues to suspect ATTR‐CA.
Figure 1.

Red‐flags that should be highly suspected of transthyretin cardiac amyloidosis. ACE‐I, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; CCB, calcium‐channel blocker; CMR, cardiac magnetic resonance; ECG, electrocardiogram; Echo, echocardiogram; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; LGE, late gadolinium enhancement; LV, left ventricle.
Table 1.
Baseline characteristics and survival of patients with different types of cardiac amyloidosis
| AL31 | ATTRwt7, 19, 31, 32 | Val122Ile33, 34 | Thr60Ala18 | Ile68Leu19 | Late Val30Met35 | |
|---|---|---|---|---|---|---|
| Age at diagnosis (year) | 63 | 73‐76 | 69‐74 | 66 | 71 | 67.3 |
| Male (%) | 69.4 | 89‐98 | 76‐85 | 70 | 78 | 86 |
| Common ethnicity | Variable | Caucasian | Afro‐Caribbean | Irish | Caucasian | Japanese |
| Family history (%) | NA | NA | NA | 37 | 63 | 48 |
| NYHA Class III–IV (%) | 60 | 62‐85 | 47‐55 | NA | 27 | 14 |
| CTS (%) | 8 | 39‐48 | 29‐46 | NA | 43 | NA |
| Peripheral neuropathy (%) | 8 | 3‐9 | 38 | 54 | 19 | 80 |
| Autonomic symptoms (%) | NA | NA | 10 | 75 | 6 | 10 |
| SBP/DBP (mmHg) | 107/72 | 116/74 | 112/69 | NA | 120/80 | NA |
| Atrial fibrillation (%) | 11 | 38‐67 | 31‐52 | NA | 30 | 3.8 |
| Pacemakers (%) | 5 | 8‐40 | 8.7‐11 | NA | 9 | 12 |
| IVSd/LVPWd (mm) | 15/15 | 17/17 | 17/17 | 17/17 | 17/16 | 16/14 |
| LVEF (%) | 42 | 47‐51 | 39‐51 | 53 | 51 | 64 |
| NT‐ proBNP (pg/mL) | 6038 | 3361 | 2734 | 2528 | 3287 | 1116 |
| (3615–13 302)a | ±845 | (2307–4467)a | (42–18 148)b | (1745–5658)a | ±1384 | |
| Diagnosis to death (year) | 0.9 | 2.7‐3.9 | 2.62 | 3.4 | 5‐year survival (37%) | 4.3 |
The numerical values of circulating NT‐proBNP levels are described in mean ± standard deviation,,7, 19, 31, 32, 35 median (range, min–max),18 or median (Q1–Q3 percentile),19, 31, 33, 34 according to the description in the original literature.
AL, light‐chain amyloidosis; ATTRwt, wild type transthyretin amyloidosis; CTS, carpal tunnel syndrome; DBP, diastolic blood pressure; IVSd, interventricular septum thickness at end‐diastole; LVEF, left ventricular ejection fraction; LVPWd, left ventricular posterior wall thickness at end‐diastole; NA, not available; NT‐proBNP, N‐terminal pro b‐type natriuretic peptide; NYHA class, New York Heart Association class; SBP, systolic blood pressure.
Median (Q1–Q3 percentile).
Median (range, min–max).
5. Electrocardiography
Patients may develop low QRS voltage on electrocardiogram or pseudoinfarction pattern even in the absence of epicardial coronary stenosis as the disease progresses. Unexplained LVH, characterized by low QRS voltage on electrocardiogram despite LVH on echocardiogram, provides valuable clues for the suspicion of CA. However, these electrocardiographic features are actually not sensitive enough to identify CA.37, 38
6. Echocardiography
Echocardiogram plays an important key role in diagnosing CA. Echocardiogram can recognize diastolic dysfunction at early stages and systolic dysfunction at later stages. The most common CA phenotype is the presence of increased LV wall thickness, small LV chamber size with systolic impairment, atrial enlargement, and signs of elevated filling pressures caused by restrictive diastolic filling. Left ventricular ejection fraction (LVEF) declines as the disease progresses, resulting in a transition from preserved LVEF to reduced LVEF at an advanced stage. Other clues include right ventricular hypertrophy, pericardial effusions, atrial septal or cardiac valve thickening, and intra‐cardiac thrombus,39 which are not pathognomonic of ATTR‐CA.
With recent advanced technology, strain echocardiography has become a new imaging modality to measure myocardial deformation. Relative apical‐sparing of longitudinal strain (LS) is considered as a hallmark for CA. A relative apical LS index, which is the ratio of average apical LS/(average basal LS + average mid‐LS), was useful for differentiating CA from other causes of LVH (93% sensitivity and 82% specificity).40 Another study evaluated the diagnostic accuracy of strain imaging. A cut‐off value of septal apical to basal longitudinal systolic strain ratio > 2.1 differentiated CA from other causes of LVH such as hypertension, Fabry disease, or Friedreich ataxia (88% sensitivity, 85% specificity, 67% positive predictive value, and 96% negative predictive value).41 In addition, a combination of the systolic strain gradient and deceleration time of early filling (<200 ms) could further improve the diagnostic accuracy for detecting CA (88% sensitivity, 100% specificity, 100% positive predictive value, 96% negative predictive value). The ratio of ejection fraction/global LS, which is another specific echo parameter, is effective for distinguishing CA from HCM.42
7. Cardiac magnetic resonance
Cardiac magnetic resonance can provide unique information on myocardial tissue properties. CMR shows various characteristic patterns of late gadolinium enhancement (LGE) in CA: global subendocardial LGE, global transmural LGE, atrial LGE, and suboptimal myocardial nulling. CMR with LGE was very useful for diagnosing CA (80% sensitivity and 94% specificity).43 Although speckle‐tracking imaging can reliably recognize and differentiate CA from the non‐amyloid cause of cardiomyopathy, echocardiogram alone cannot distinguish the CA subtypes. An original scoring system, the Query Amyloid Late Enhancement (QALE) score, has been designed to distinguish ATTR‐CA from immunoglobulin light‐chain cardiac amyloidosis (AL‐CA) with 82% sensitivity and 76% specificity.44 The QALE score is a semi‐quantitative index of measuring the extent of amyloid burden in both ventricles identified by LGE. However, the value of the QALE score remains inconsistent because another study has failed to prove its usefulness.45 Actually, LGE cannot detect diffuse myocardial fibrosis due to significant extracellular amyloid infiltration, and patients with renal dysfunction cannot utilize this technique. Recently, T1‐mapping technique can overcome these limitations, identify early disease, and quantitatively evaluate disease progression without contrast agents.46
8. Radionuclide bone scintigraphy
Nuclear imaging technique employing technetium pyrophosphate (99mTc‐PYP), once used as bone scintigraphy, has recently been reported as a reliable diagnostic method for ATTR‐CA, which is distinguished from AL‐CA, or other wall thickening disease with high specificity of 100%. Cardiac TTR deposition can be detected at an asymptomatic stage.47, 48 In addition, multicentre study validated that bone‐avid tracers provided excellent diagnostic performance for diagnosis of ATTR‐CA when used in combination without evidence of monoclonal protein (100% specificity and 100% positive predictive value). Moreover, marked myocardial uptake of 99mTc‐PYP was associated with poor prognosis in ATTR‐CA.49 Thus, cardiac radioisotope examination has established a valuable position as indispensable tools for the diagnosis, severity, and treatment planning.
9. Histology
Tissue diagnosis remains the golden standard for making the diagnosis of amyloidosis. Congo red or Direct Fast Scarlet 4BS staining binds to deposit amyloid fibrils and yields to characteristic apple‐green birefringence under polarized light microscopy. Note that apple‐green birefringence should be observed in the same amyloid deposits stained with Congo red dye. In addition, electron microscopy demonstrates randomly oriented and non‐branching fibrils with a diameter of approximately 7.5–10 nm. Subsequently, subtyping of amyloid fibril can be performed by immunohistochemistry or laser microdissection/mass spectroscopy. Laser microdissection/mass spectroscopy can allow accurate diagnosis, typing, and variants of amyloidosis.50, 51 Traditionally, a conventional endomyocardial biopsy was performed to confirm the diagnosis and typing of CA with high sensitivity. However, it was not always practical and limited by a risky and invasive procedure. Recently, a paradigm shift in diagnosing CA has occurred with technological advances in diagnostic imaging. Positive nuclear imaging with bone‐avid tracers (Grade 2 or 3 tracer uptake) in the absence of detectable monoclonal protein in serum or urine allows non‐histological diagnosis of ATTR‐CA reliably (Figure 2 ).48 However, conventional histopathology and subsequent amyloid typing are still essential in all cases that do not meet such diagnostic criteria. Poor sensitivity of abdominal fat pad aspiration leads to delayed diagnosis (84% for AL, 15% for ATTRwt, and 45% for ATTRv, respectively).52 Moreover, the prevalence of monoclonal gammopathy of undetermined significance in patients with ATTR amyloidosis has been reported to be high.31, 53 Because treatment methods of each disease are totally different, endomyocardial biopsy should be carried out for definitive diagnosis.
Figure 2.
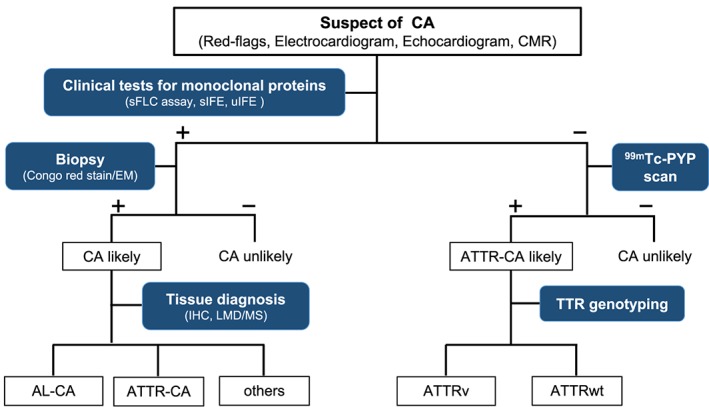
Diagnostic algorithm for patients with suspected cardiac amyloidosis. −, negative test; +, positive test; 99mTc‐PYP, technetium pyrophosphate; AL‐CA, light‐chain cardiac amyloidosis; ATTR‐CA, transthyretin cardiac amyloidosis; ATTRv, hereditary transthyretin amyloidosis; ATTRwt, wild type transthyretin amyloidosis; CA, cardiac amyloidosis; CMR, cardiovascular magnetic resonance; EM, electron microscopy; IHC, immunohistochemistry; LMD/MS, Laser microdissection and mass spectrometry; sFLC, serum‐free light chain; sIFE, serum immunofixation electrophoresis; uIFE, urine immunofixation electrophoresis.
10. Treatment of transthyretin cardiac amyloidosis
A treatment strategy for CA includes both managing cardiovascular complications and treating the underlying disease process.
11. Management of cardiovascular complications
In CA, both the interstitial amyloid deposition and the subendocardial fibrosis due to ischaemia cause morphological and functional abnormalities. Amyloid infiltration, not myocyte hypertrophy, leads to the thickening of ventricular walls concomitant with normal to small cavity size with diastolic filling impairment due to reduced contractility. Moreover, atrial dysfunction due to amyloid deposit may further reduce diastolic filling. These pathological changes result in a reduced stroke volume concomitant with considerable elevated intra‐cardiac pressures.
Cardiac amyloidosis initially exhibits a clinical phenotype similar to HFpEF.54 Most of the well‐recognized anti‐HF drugs may be harmful due to its unique pathology. Angiotensin‐converting enzyme inhibitors, or angiotensin II receptor blockers, have the risk of producing profound hypotension because of activation of the renin–angiotensin–aldosterone system due to autonomic dysfunction. Moreover, beta‐blockers may be detrimental because they cause cardiac output reduction due to lowering heart rate and negative inotropic effect, resulting in profound hypotension. Calcium‐channel blocker or digitalis should be avoided because they bind irreversibly to amyloid fibrils and might cause serious side effects.55, 56 Diuretics remain the first‐line treatment for congestion in HF. A combination of loop diuretics and mineralocorticoid receptor antagonists is effective. In addition, the combined use of vasopressin V2 receptor antagonists (aquaretics) is useful to safely reach euvolaemia because the excessive use of loop diuretics (natriuretics) easily causes intravascular dehydration, resulting in pre‐renal azotaemia or symptomatic hypotension.57 Nevertheless, uncontrollable pleural effusion requiring frequent fluid drainage may suggest amyloid infiltration, which requires pleurodesis.58
Atrial fibrillation, which is very common in CA, may worsen HF, and its treatment is challenging. Rate and rhythm control in AF is very important because patients with CA have fixed stroke volumes and the cardiac output extremely depends on the heart rate. However, rate control options including beta‐blocker and calcium‐channel blocker are limited as the aforementioned reason. Amiodarone can be used to restore and maintain sinus rhythm safely. Catheter ablation for AF is not effective due to high recurrence rate.59
In CA, increased LV filling pressure, atrial contractile dysfunction caused by atrial amyloid infiltration, and AF highly cause intra‐cardiac thrombus, leading to an increased risk of thromboembolism.39, 60 Spontaneous echo contrast or decreased atrial appendage Doppler velocities (<40 cm/s) on transesophageal echocardiography is strongly suggestive of atrial contractile dysfunction.61 Thus, anticoagulation should be warranted in cases of AF or even sinus rhythm with evidence of atrial contractile dysfunction.
12. Pacemaker implantation
Many ATTR‐CA patients are facing at high risk of conduction system disorders requiring cardiac pacing for symptomatic atrioventricular block or bradycardia, and its indication follows current standard guidelines. However, there are no guidelines for the indication or optimal timing of prophylactic pacemaker implantation for asymptomatic ATTR‐CA patients. Although cardiac pacing can be helpful in symptomatic relief, there are no significant beneficial effects on patient survival.62, 63, 64, 65 Moreover, ventricular dyssynchrony or lead‐induced tricuspid regurgitation caused by right ventricular pacing might have deleterious impacts on haemodynamics.
13. Implantable cardioverter‐defibrillator
Sudden cardiac death (SCD) is a major problem in CA patients accounting for one quarter of all‐cause death.66 Although implantable cardioverter‐defibrillator (ICD) therapy can be a promising therapeutic option for high‐risk CA patients, the efficacy of ICD is controversial because electromechanical dissociation seems to be a dominant cause of SCD. Most clinical studies targeted AL‐CA patients with poor prognosis and a high frequency of SCD. Several cases reported that ICD has successfully terminated sustained ventricular arrhythmia in selected patients with AL‐CA.67 Based on such limited data, 2015 European Society of Cardiology guidelines recommend that ICDs should be considered in patients with AL‐CA or ATTR‐CA with a history of sustained ventricular arrhythmia and an estimated life expectancy of >1 year (Class IIa and Level C).68 On the other hand, ICD therapy is not recommended for primary prevention of SCD in ATTR‐CA patients because its effectiveness is questionable.69
14. Disease‐modifying treatments
Variants and aging cause dissociation of the TTR tetramer into non‐native monomers with low conformational stability, which misfolds into an amyloidogenic form and aggregates, leading to insoluble amyloid fibril formation.70 Subsequently, the extracellular deposition of amyloid fibrils in the myocardium causes ATTR‐CA. Thus, treatment options of the underlying misfolding protein disease include organ transplantation and investigational agents focusing on the TTR amyloidogenic pathway.
15. Organ transplantation
Liver transplantation (LT), which can remove the main source of circulating pathogenic TTR protein, has been considered a promising therapeutic choice to cure ATTR‐FAP.71 Unfortunately, there might be a serious risk of the development of progressive cardiomyopathy following LT among patients.72, 73 Autopsy findings revealed the preponderance of wild type TTR in cardiac amyloid deposits after LT,74 suggesting that paradoxical progressive deposition of wild type TTR on pre‐existing variant TTR amyloid deposits acting as templates. Thus, combined heart and liver transplant (CHLT) may be the most preferable alternative for advanced HF patients with ATTRv‐CA. According to the International Society for Heart and Lung Transplantation guideline, while young patients should be considered for CHLT to prevent systemic disease progression, elderly patients with cardiac‐dominant manifestations such as ATTR‐CA (wild type or Val122Ile variant) should be considered for isolated heart transplantation.75 Patients who have a definitive diagnosis of ATTR‐CA should perform an evaluation for CHLT or isolated heart transplantation as soon as possible to undergo organ transplantation in the earlier stage.
16. Transthyretin gene silencers
16.1. siRNA (patisiran)
A novel TTR‐targeted small‐interfering ribonucleic acid encapsulated lipid nanoparticle, namely, ALN‐TTR02 (patisiran), has developed. A Phase 3 APOLLO trial (NCT01960348), a randomized 18‐month trial, evaluated the efficacy and safety of the patisiran (0.3 mg/kg i.v., once every 3 weeks) in 225 ATTR‐FAP patients compared with the placebo group, demonstrated that patisiran significantly improved the quality of life (QOL) and clinical neuropathy scores, and ameliorated the disease progression in ATTR‐FAP patients.76 The drug was generally well tolerated, and the frequency of serious adverse effects (SAEs) was similar between the two groups. In a cardiac subgroup, patisiran significantly improved LV basal LS, lowered N‐terminal pro b‐type natriuretic peptide levels, and ameliorated abnormal LV geometric patterns including LVH compared with placebo groups.77, 78
16.2. Antisense oligonucleotides (inotersen)
Inotersen is a TTR‐directed antisense oligonucleotide, which interferes with hepatic TTR synthesis. A Phase 3 NEURO‐TTR trial (NCT01737398), a randomized 66‐week trial, evaluated the efficacy and safety of the subcutaneous inotersen (300 mg s.c., once weekly), in 172 ATTR‐FAP patients in the presence of cardiomyopathy and demonstrated that inotersen improved clinical manifestations and the neurological clinical scores in ATTR‐FAP patients. The most common SAEs were glomerulonephritis (3%) and life‐threatening thrombocytopaenia (3%). Thus, frequent laboratory monitoring is necessary to avoid these potential side effects.79
17. Transthyretin tetramer stabilizers
17.1. Tafamidis
Tafamidis is a novel molecule that can bind a thyroxine‐binding site of the TTR tetramer, resulting in inhibition of its dissociation into monomers, an important step in the TTR amyloid‐forming cascade.80 In a Phase 3 multicentre randomized ATTR‐ACT trial (NCT01994889), 441 patients with ATTR‐CA (wild type or variant) were randomly assigned in a 2:1:2 ratio to receive tafamidis 80 mg, 20 mg, or placebo orally every 24 h for 30 months.81 Patients with New York Heart Association (NYHA) class IV HF or an estimated glomerular filtration rate of <25 mL/min/1.73 m2 were excluded. Tafamidis reduced all‐cause mortality and cardiovascular‐related hospitalizations compared with placebo. Also, tafamidis significantly delayed decline in functional capacity assessed by the 6‐min walk distance and in QOL assessed by the Kansas City Cardiomyopathy Questionnaire‐Overall Summary/KCCQ‐OS. In sub‐analysis, tafamidis steadily improved all‐cause mortality and cardiovascular‐related hospitalizations in all subgroup (TTR genotype, NYHA class baseline, and tafamidis dosage) except for frequency of cardiovascular‐related hospitalizations in NYHA class III. This trial suggests that the severity of cardiac dysfunction at baseline drives the outcome yet leaves several questions to be solved. The preliminary experimental study demonstrated the effects of tafamidis on plasma TTR tetramer stability in a dose‐dependent manner.82 Thus, tailoring of the tafamidis dose to the patients in NYHA class III might need to achieve maximum kinetic stabilization. Because many randomized‐control trials for patients with systolic HF have suggested the need of months to recognize anti‐remodelling effects on the LV during the therapeutic intervention,83, 84 the observation period might not be sufficient to determine the drug treatment efficacy. The Phase 3 trial has further been extended up to 60 months and will end in 2021.
17.2. Diflunisal
Diflunisal, a non‐steroidal anti‐inflammatory drug, can stabilize TTR tetramers in vitro. In a Phase 3 randomized trial, 130 ATTR‐FAP patients with symptomatic neuropathy were randomly assigned to diflunisal 250 mg or placebo orally twice daily for 2 years.85 This trial demonstrated that diflunisal significantly reduced the progression of neurologic impairment and preserved QOL, compared with placebo. However, diflunisal had no beneficial effects on the improvement in cardiac status. Although the dosage of diflunisal generally were well tolerated in this trial, its cyclooxygenase inhibitory activity can cause renal and gastrointestinal damage, such as renal failure, gastric mucosal injury, volume overload, and hypertension. The potential for life‐threatening SAEs remains a major concern. Thus, the diflunisal remains to be used off‐label in the treatment of ATTR‐FAP.
17.3. AG10
Many pathologic TTR variants, such as Val30Met or Val122Ile, are of the loss‐of‐function type causing TTR destabilization and increase the risk of ATTR amyloidosis. However, a certain TTR variant, Thr119Met, is known as the gain‐of‐function type that super‐stabilizes a TTR tetramer, resulting in the prevention of ATTR amyloidosis. AG10 is a novel stabilizing compound that has a similar motif to the thyroxine‐binding site of the Thr119Met variant, specifically binding to TTR tetramer to inhibit TTR dissociation. In vitro study proved that AG10 had a stronger TTR tetrameric stability, compared with tafamidis and diflunisal.86 A randomized, double‐blind, placebo‐controlled, Phase 2 trial (NCT03458130) confirmed the safety and efficacy of AG10 in ATTR‐CA patients (wild type or variant).87 A Phase 3 trial (NCT03860935) in ATTR‐CA has been initiated.
18. Transthyretin amyloid disruptors
18.1. Doxycycline/tauroursodeoxycholic acid
Both the tetracycline antibiotic doxycycline and the antiapoptotic agent tauroursodeoxycholic acid (TUDCA) are effective in degrading non‐fibrillar TTR deposits.88 The concurrent treatment has been verified to attenuate disease progression in ATTR amyloidosis patients in limited clinical trials.89 An open‐label Phase 1/2 trial (NCT01855360) evaluated tolerability and efficacy of a combination of doxycycline and TUDCA on the disease progression in patients with ATTR‐CA (wild type or variant). Thirty‐eight ATTR‐CA patients were treated with the combination of TUDCA (250 mg orally three times daily) and doxycycline (100 mg orally twice daily) for 18 months. This trial has recently been completed, but the results have not been released yet.
18.2. Green tea
Epigallocatechin‐gallate, a well‐known major polyphenol in green tea, can inhibit TTR amyloid fibril formation and disaggregate amyloid deposits.90 Two observational studies revealed that 12 months of green tea consumption significantly reduced the LV mass by 6–13% evaluated by CMR in ATTRwt‐CA patients, suggesting that green tea consumption extracts have an inhibitory effect on the disease progression.91, 92 However, these studies are open‐label, observational studies and limited to small sample sizes.
18.3. Serum amyloid P component
Serum amyloid P component (SAP) is a normal plasma protein found in all types of amyloid deposits and stabilizes the amyloid deposit formation. The drug (R)‐1‐[6‐[(R)‐2‐carboxy‐pyrrolidin‐1‐yl]‐6‐oxo‐hexanoyl]pyrrolidine‐2‐carboxylic acid (CPHPC) has been developed to work as a competitive inhibitor of SAP binding to amyloid fibrils and efficiently depletes circulating SAP yet leaves some residual SAP in amyloid deposition.93 Subsequently, sequential treatment with CPHPC followed by anti‐SAP monoclonal antibodies efficiently elicited immunotherapeutic removal of amyloid deposits from key organs including liver in animal studies and in patients with systemic amyloidosis in early phase clinical trials (NCT01777243).94, 95, 96, 97
Conclusions
Both diagnosing and treating ATTR‐CA remain challenging. ATTR‐CA has been increasingly recognized as a cause of HFpEF. Large cohort study indicated that most patients diagnosed with ATTR‐CA were in NYHA class II (>70%).98 Also, with the rise of nuclear imaging techniques, the early non‐invasive diagnosis has become possible with high diagnostic accuracy.48 Moreover, the results of the ATTR‐ACT trial highlight the significance of high clinical suspicion and early diagnosis of ATTR‐CA given that tafamidis provides huge benefits to patients with early‐stage ATTR‐CA.81 For example, early screening for ATTR‐CA might be useful for initial evaluation of elderly patients with newly diagnosed HFpEF with LVH or elderly patients hospitalized for HF.2, 99 With the emergence of new efficient agents including tafamidis, tailor‐made medicine will be needed according to the individual disease stage and clinical phenotype. However, there are several problems to be solved: what should we do with treatment strategies for non‐responders? Or which is the most effective treatment for ATTR‐CA, a monotherapy or dual therapies (gene silencer and stabilizer)? Further studies are needed to address these unanswered questions. In addition, other novel innovative agents focused on the amyloidogenic process are ongoing. Thus, ATTR‐CA is becoming a treatable disease. However, misdiagnosis and delayed diagnosis still interfere with such benefits. Clinicians should be aware of the clinical entity and make much effort to identify ATTR‐CA as soon as possible.
Conflict of interest
None declared.
Author contributions
H.Y. and T.Y. drafted and revised the manuscript. Both authors discussed, read, and approved the submission of this manuscript to the journal.
Funding
No source of funding was declared for this study.
Yamamoto, H. , and Yokochi, T. (2019) Transthyretin cardiac amyloidosis: an update on diagnosis and treatment. ESC Heart Failure, 6: 1128–1139. 10.1002/ehf2.12518.
References
- 1. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012; 126: 1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gonzalez‐Lopez E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐Del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, Garcia‐Pavia P. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585–2594. [DOI] [PubMed] [Google Scholar]
- 3. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, Roger VL, Gertz MA, Dispenzieri A, Zeldenrust SR, Redfield MM. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014; 2: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Castano A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, Rubin J, Chiuzan C, Nazif T, Vahl T, George I, Kodali S, Leon MB, Hahn R, Bokhari S, Maurer MS. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38: 2879–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galat A, Guellich A, Bodez D, Slama M, Dijos M, Zeitoun DM, Milleron O, Attias D, Dubois‐Rande JL, Mohty D, Audureau E, Teiger E, Rosso J, Monin JL, Damy T. Aortic stenosis and transthyretin cardiac amyloidosis: the chicken or the egg? Eur Heart J 2016; 37: 3525–3531. [DOI] [PubMed] [Google Scholar]
- 6. Damy T, Jaccard A, Guellich A, Lavergne D, Galat A, Deux JF, Hittinger L, Dupuis J, Frenkel V, Rigaud C, Plante‐Bordeneuve V, Bodez D, Mohty D. Identification of prognostic markers in transthyretin and AL cardiac amyloidosis. Amyloid 2016; 23: 194–202. [DOI] [PubMed] [Google Scholar]
- 7. Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC. Heart failure resulting from age‐related cardiac amyloid disease associated with wild‐type transthyretin: a prospective, observational cohort study. Circulation 2016; 133: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neben‐Wittich MA, Wittich CM, Mueller PS, Larson DR, Gertz MA, Edwards WD. Obstructive intramural coronary amyloidosis and myocardial ischemia are common in primary amyloidosis. Am J Med 2005; 118: 1287. [DOI] [PubMed] [Google Scholar]
- 9. Al Suwaidi J, Velianou JL, Gertz MA, Cannon RO 3rd, Higano ST, Holmes DR Jr, Lerman A. Systemic amyloidosis presenting with angina pectoris. Ann Intern Med 1999; 131: 838–841. [DOI] [PubMed] [Google Scholar]
- 10. Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, Doden T, Koyama J, Yanagisawa S, Ikeda S. Carpal tunnel syndrome: a common initial symptom of systemic wild‐type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23: 58–63. [DOI] [PubMed] [Google Scholar]
- 11. Westermark P, Westermark GT, Suhr OB, Berg S. Transthyretin‐derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci 2014; 119: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Plante‐Bordeneuve V, Rapezzi C, Said G, Salvi F. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013; 8: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rapezzi C, Quarta CC, Obici L, Perfetto F, Longhi S, Salvi F, Biagini E, Lorenzini M, Grigioni F, Leone O, Cappelli F, Palladini G, Rimessi P, Ferlini A, Arpesella G, Pinna AD, Merlini G, Perlini S. Disease profile and differential diagnosis of hereditary transthyretin‐related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 2013; 34: 520–528. [DOI] [PubMed] [Google Scholar]
- 14. Yamamoto H, Hashimoto T, Kawamura S, Hiroe M, Yamashita T, Ando Y, Yokochi T. Hereditary cardiac amyloidosis associated with Pro24Ser transthyretin mutation: a case report. J Med Case Reports 2018; 12: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African‐Americans. Amyloid 2015; 22: 171–174. [DOI] [PubMed] [Google Scholar]
- 16. Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, Mosley TH, Butler KR, Boerwinkle E, Solomon SD. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med 2015; 372: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reilly MM, Staunton H, Harding AE. Familial amyloid polyneuropathy (TTR ala 60) in north west Ireland: a clinical, genetic, and epidemiological study. J Neurol Neurosurg Psychiatry 1995; 59: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SD, Pinney JH, Stangou AJ, Rowczenio D, Pflugfelder PW, Fox Z, Lachmann HJ, Wechalekar AD, Hawkins PN, Gillmore JD. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J 2012; 33: 1120–1127. [DOI] [PubMed] [Google Scholar]
- 19. Gagliardi C, Perfetto F, Lorenzini M, Ferlini A, Salvi F, Milandri A, Quarta CC, Taborchi G, Bartolini S, Frusconi S, Martone R, Cinelli MM, Foffi S, Reggiani MLB, Fabbri G, Cataldo P, Cappelli F, Rapezzi C. Phenotypic profile of Ile68Leu transthyretin amyloidosis: an underdiagnosed cause of heart failure. Eur J Heart Fail 2018; 20: 1417–1425. [DOI] [PubMed] [Google Scholar]
- 20. Suhr OB, Svendsen IH, Andersson R, Danielsson A, Holmgren G, Ranlov PJ. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med 2003; 254: 225–235. [DOI] [PubMed] [Google Scholar]
- 21. Damy T, Kristen AV, Suhr OB, Maurer MS, Plante‐Bordeneuve V, Yu CR, Ong ML, Coelho T, Rapezzi C, THAOS Investigators. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J 2019; pii: ehz173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mariani LL, Lozeron P, Theaudin M, Mincheva Z, Signate A, Ducot B, Algalarrondo V, Denier C, Adam C, Nicolas G, Samuel D, Slama MS, Lacroix C, Misrahi M, Adams D, Familial F. Amyloid Polyneuropathies Network Study G. Genotype‐phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol 2015; 78: 901–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red‐flag symptom clusters and treatment algorithm. Orphanet J Rare Dis 2018; 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T. investigators T. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis—report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS ONE 2017; 12: e0173086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arruda‐Olson AM, Zeldenrust SR, Dispenzieri A, Gertz MA, Miller FA, Bielinski SJ, Klarich KW, Scott CG, Grogan M. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid 2013; 20: 263–268. [DOI] [PubMed] [Google Scholar]
- 26. Damy T, Costes B, Hagege AA, Donal E, Eicher JC, Slama M, Guellich A, Rappeneau S, Gueffet JP, Logeart D, Plante‐Bordeneuve V, Bouvaist H, Huttin O, Mulak G, Dubois‐Rande JL, Goossens M, Canoui‐Poitrine F, Buxbaum JN. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J 2016; 37: 1826–1834. [DOI] [PubMed] [Google Scholar]
- 27. Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit‐Chen S, Lavori P. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol 2006; 47: 1724–1725. [DOI] [PubMed] [Google Scholar]
- 28. Conceicao I, Damy T, Romero M, Galan L, Attarian S, Luigetti M, Sadeh M, Sarafov S, Tournev I, Ueda M. Early diagnosis of ATTR amyloidosis through targeted follow‐up of identified carriers of TTR gene mutations. Amyloid 2019; 26: 3–9. [DOI] [PubMed] [Google Scholar]
- 29. Bishop E, Brown EE, Fajardo J, Barouch LA, Judge DP, Halushka MK. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid 2018; 25: 174–179. [DOI] [PubMed] [Google Scholar]
- 30. Gonzalez‐Lopez E, Gagliardi C, Dominguez F, Quarta CC, de Haro‐Del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo‐Marcos M, Lorenzini M, Lara‐Pezzi E, Foffi S, Alonso‐Pulpon L, Rapezzi C, Garcia‐Pavia P. Clinical characteristics of wild‐type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 2017; 38: 1895–1904. [DOI] [PubMed] [Google Scholar]
- 31. Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2013; 2: e000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, Klarich KW, Miller WL, Maleszewski JJ, Dispenzieri A. Natural history of wild‐type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68: 1014–1020. [DOI] [PubMed] [Google Scholar]
- 33. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Plante‐Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C, Investigators T. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol 2016; 68: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dungu JN, Papadopoulou SA, Wykes K, Mahmood I, Marshall J, Valencia O, Fontana M, Whelan CJ, Gillmore JD, Hawkins PN, Anderson LJ. Afro‐Caribbean heart failure in the United Kingdom: cause, outcomes, and ATTR V122I cardiac amyloidosis. Circ Heart Fail 2016; 9: pii: e003352. [DOI] [PubMed] [Google Scholar]
- 35. Koike H, Tanaka F, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, Fujitake J, Kawanami T, Kato T, Yamamoto M, Sobue G. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late‐onset cases from non‐endemic areas. J Neurol Neurosurg Psychiatry 2012; 83: 152–158. [DOI] [PubMed] [Google Scholar]
- 36. Scully PR, Treibel TA, Fontana M, Lloyd G, Mullen M, Pugliese F, Hartman N, Hawkins PN, Menezes LJ, Moon JC. Prevalence of cardiac amyloidosis in patients referred for transcatheter aortic valve replacement. J Am Coll Cardiol 2018; 71: 463–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014; 114: 1089–1093. [DOI] [PubMed] [Google Scholar]
- 38. Sperry BW, Vranian MN, Hachamovitch R, Joshi H, McCarthy M, Ikram A, Hanna M. Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol 2016; 214: 477–481. [DOI] [PubMed] [Google Scholar]
- 39. Feng D, Syed IS, Martinez M, Oh JK, Jaffe AS, Grogan M, Edwards WD, Gertz MA, Klarich KW. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation 2009; 119: 2490–2497. [DOI] [PubMed] [Google Scholar]
- 40. Phelan D, Collier P, Thavendiranathan P, Popovic ZB, Hanna M, Plana JC, Marwick TH, Thomas JD. Relative apical sparing of longitudinal strain using two‐dimensional speckle‐tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012; 98: 1442–1448. [DOI] [PubMed] [Google Scholar]
- 41. Liu D, Hu K, Niemann M, Herrmann S, Cikes M, Stork S, Gaudron PD, Knop S, Ertl G, Bijnens B, Weidemann F. Effect of combined systolic and diastolic functional parameter assessment for differentiation of cardiac amyloidosis from other causes of concentric left ventricular hypertrophy. Circ Cardiovasc Imaging 2013; 6: 1066–1072. [DOI] [PubMed] [Google Scholar]
- 42. Pagourelias ED, Mirea O, Duchenne J, Van Cleemput J, Delforge M, Bogaert J, Kuznetsova T, Voigt JU. Echo parameters for differential diagnosis in cardiac amyloidosis: a head‐to‐head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging 2017; 10: e005588. [DOI] [PubMed] [Google Scholar]
- 43. Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, Greulich S, Klingel K, Kandolf R, Sechtem U. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol 2008; 51: 1022–1030. [DOI] [PubMed] [Google Scholar]
- 44. Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, Lachmann HJ, Wechalekar A, Gillmore JD, Whelan CJ, Hawkins PN, Anderson LJ. CMR‐based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7: 133–142. [DOI] [PubMed] [Google Scholar]
- 45. Ridouani F, Damy T, Tacher V, Derbel H, Legou F, Sifaoui I, Audureau E, Bodez D, Rahmouni A, Deux JF. Myocardial native T2 measurement to differentiate light‐chain and transthyretin cardiac amyloidosis and assess prognosis. J Cardiovasc Magn Reson 2018; 20: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karamitsos TD, Piechnik SK, Banypersad SM, Fontana M, Ntusi NB, Ferreira VM, Whelan CJ, Myerson SG, Robson MD, Hawkins PN, Neubauer S, Moon JC. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2013; 6: 488–497. [DOI] [PubMed] [Google Scholar]
- 47. Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc‐pyrophosphate scintigraphy for differentiating light‐chain cardiac amyloidosis from the transthyretin‐related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013; 6: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AW, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C, Hawkins PN. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404–2412. [DOI] [PubMed] [Google Scholar]
- 49. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, Pozniakoff T, Ruberg FL, Miller EJ, Berk JL, Dispenzieri A, Grogan M, Johnson G, Bokhari S, Maurer MS. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol 2016; 1: 880–889. [DOI] [PubMed] [Google Scholar]
- 50. Sethi S, Vrana JA, Theis JD, Leung N, Sethi A, Nasr SH, Fervenza FC, Cornell LD, Fidler ME, Dogan A. Laser microdissection and mass spectrometry‐based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int 2012; 82: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry‐based proteomic analysis in clinical biopsy specimens. Blood 2009; 114: 4957–4959. [DOI] [PubMed] [Google Scholar]
- 52. Quarta CC, Gonzalez‐Lopez E, Gilbertson JA, Botcher N, Rowczenio D, Petrie A, Rezk T, Youngstein T, Mahmood S, Sachchithanantham S, Lachmann HJ, Fontana M, Whelan CJ, Wechalekar AD, Hawkins PN, Gillmore JD. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017; 38: 1905–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, Sarosiek S. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid 2018; 25: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112: 2047–2060. [DOI] [PubMed] [Google Scholar]
- 55. Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest 1993; 104: 618–620. [DOI] [PubMed] [Google Scholar]
- 56. Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981; 63: 1285–1288. [DOI] [PubMed] [Google Scholar]
- 57. Vaduganathan M, Mentz RJ, Greene SJ, Senni M, Sato N, Nodari S, Butler J, Gheorghiade M. Combination decongestion therapy in hospitalized heart failure: loop diuretics, mineralocorticoid receptor antagonists and vasopressin antagonists. Expert Rev Cardiovasc Ther 2015; 13: 799–809. [DOI] [PubMed] [Google Scholar]
- 58. Berk JL, Keane J, Seldin DC, Sanchorawala V, Koyama J, Dember LM, Falk RH. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 2003; 124: 969–977. [DOI] [PubMed] [Google Scholar]
- 59. Tan NY, Mohsin Y, Hodge DO, Lacy MQ, Packer DL, Dispenzieri A, Grogan M, Asirvatham SJ, Madhavan M, Mc LC. Catheter ablation for atrial arrhythmias in patients with cardiac amyloidosis. J Cardiovasc Electrophysiol 2016; 27: 1167–1173. [DOI] [PubMed] [Google Scholar]
- 60. Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, Syed IS, Hughes DA, Lust JA, Jaffe AS, Gertz MA, Klarich KW. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation 2007; 116: 2420–2426. [DOI] [PubMed] [Google Scholar]
- 61. Santarone M, Corrado G, Tagliagambe LM, Manzillo GF, Tadeo G, Spata M, Longhi M. Atrial thrombosis in cardiac amyloidosis: diagnostic contribution of transesophageal echocardiography. J Am Soc Echocardiogr 1999; 12: 533–536. [DOI] [PubMed] [Google Scholar]
- 62. Barreiros AP, Post F, Hoppe‐Lotichius M, Linke RP, Vahl CF, Schafers HJ, Galle PR, Otto G. Liver transplantation and combined liver‐heart transplantation in patients with familial amyloid polyneuropathy: a single‐center experience. Liver Transpl 2010; 16: 314–323. [DOI] [PubMed] [Google Scholar]
- 63. Okamoto S, Hornsten R, Obayashi K, Wijayatunga P, Suhr OB. Continuous development of arrhythmia is observed in Swedish transplant patients with familial amyloidotic polyneuropathy (amyloidogenic transthyretin Val30Met variant). Liver Transpl 2011; 17: 122–128. [DOI] [PubMed] [Google Scholar]
- 64. Algalarrondo V, Dinanian S, Juin C, Chemla D, Bennani SL, Sebag C, Plante V, Le Guludec D, Samuel D, Adams D, Slama MS. Prophylactic pacemaker implantation in familial amyloid polyneuropathy. Heart Rhythm 2012; 9: 1069–1075. [DOI] [PubMed] [Google Scholar]
- 65. Milner J, Teixeira RN, Marinho AV, Silva N, Calretas S, Ferrao J, Furtado E, Telo MJ, Ventura M, Cristovao J, Elvas L, Pego GM, Antonio N. Pacemaker implantation in familial amyloid polyneuropathy: when and for whom? J Interv Card Electrophysiol 2019; 55: 207–211. [DOI] [PubMed] [Google Scholar]
- 66. Falk RH, Rubinow A, Cohen AS. Cardiac arrhythmias in systemic amyloidosis: correlation with echocardiographic abnormalities. J Am Coll Cardiol 1984; 3: 107–113. [DOI] [PubMed] [Google Scholar]
- 67. Kristen AV, Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, Sack FU, Voss F, Becker R, Katus HA, Bauer A. Prophylactic implantation of cardioverter‐defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm 2008; 5: 235–240. [DOI] [PubMed] [Google Scholar]
- 68.19.Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ, Group ESCSD . ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015; 36: 2793–2867. [DOI] [PubMed] [Google Scholar]
- 69. Lin G, Dispenzieri A, Kyle R, Grogan M, Brady PA. Implantable cardioverter defibrillators in patients with cardiac amyloidosis. J Cardiovasc Electrophysiol 2013; 24: 793–798. [DOI] [PubMed] [Google Scholar]
- 70. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349: 583–596. [DOI] [PubMed] [Google Scholar]
- 71. Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, Andersen O, Karlberg I, Norden G, Nakazato M, Hawkins P, Richardson S, Pepys M. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP‐met30). Clin Genet 1991; 40: 242–246. [DOI] [PubMed] [Google Scholar]
- 72. Dubrey SW, Davidoff R, Skinner M, Bergethon P, Lewis D, Falk RH. Progression of ventricular wall thickening after liver transplantation for familial amyloidosis. Transplantation 1997; 64: 74–80. [DOI] [PubMed] [Google Scholar]
- 73. Olofsson BO, Backman C, Karp K, Suhr OB. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation 2002; 73: 745–751. [DOI] [PubMed] [Google Scholar]
- 74. Yazaki M, Mitsuhashi S, Tokuda T, Kametani F, Takei YI, Koyama J, Kawamorita A, Kanno H, Ikeda SI. Progressive wild‐type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant 2007; 7: 235–242. [DOI] [PubMed] [Google Scholar]
- 75. Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, Danziger‐Isakov L, Kirklin JK, Kirk R, Kushwaha SS, Lund LH, Potena L, Ross HJ, Taylor DO, Verschuuren EA, Zuckermann A. International Society for Heart Lung Transplantation Infectious Diseases C, International Society for Heart Lung Transplantation Pediatric Transplantation C, International Society for Heart Lung Transplantation Heart F, Transplantation C. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10‐year update. J Heart Lung Transplant 2016; 35: 1–23. [DOI] [PubMed] [Google Scholar]
- 76. Adams D, Gonzalez‐Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante‐Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11–21. [DOI] [PubMed] [Google Scholar]
- 77. Minamisawa M, Claggett B, Adams D, Kristen AV, Merlini G, Slama MS, Dispenzieri A, Shah AM, Falk RH, Karsten V, Sweetser MT, Chen J, Riese R, Vest J, Solomon SD. Association of patisiran, an RNA interference therapeutic, with regional left ventricular myocardial strain in hereditary transthyretin amyloidosis: the APOLLO study. JAMA Cardiol 2019; 4: 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Solomon SD, Adams D, Kristen A, Grogan M, Gonzalez‐Duarte A, Maurer MS, Merlini G, Damy T, Slama MS, Brannagan TH 3rd, Dispenzieri A, Berk JL, Shah AM, Garg P, Vaishnaw A, Karsten V, Chen J, Gollob J, Vest J, Suhr O. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin‐mediated amyloidosis. Circulation 2019; 139: 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Benson MD, Waddington‐Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Plante‐Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceicao I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A 2012; 109: 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C, Investigators A‐AS. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 82. Cho Y, Baranczak A, Helmke S, Teruya S, Horn EM, Maurer MS, Kelly JW. Personalized medicine approach for optimizing the dose of tafamidis to potentially ameliorate wild‐type transthyretin amyloidosis (cardiomyopathy). Amyloid 2015; 22: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Groenning BA, Nilsson JC, Sondergaard L, Fritz‐Hansen T, Larsson HB, Hildebrandt PR. Antiremodeling effects on the left ventricle during beta‐blockade with metoprolol in the treatment of chronic heart failure. J Am Coll Cardiol 2000; 36: 2072–2080. [DOI] [PubMed] [Google Scholar]
- 84. Solomon SD, Foster E, Bourgoun M, Shah A, Viloria E, Brown MW, Hall WJ, Pfeffer MA, Moss AJ, Investigators M‐C. Effect of cardiac resynchronization therapy on reverse remodeling and relation to outcome: multicenter automatic defibrillator implantation trial: cardiac resynchronization therapy. Circulation 2010; 122: 985–992. [DOI] [PubMed] [Google Scholar]
- 85. Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, Nordh E, Corato M, Lozza A, Cortese A, Robinson‐Papp J, Colton T, Rybin DV, Bisbee AB, Ando Y, Ikeda S, Seldin DC, Merlini G, Skinner M, Kelly JW, Dyck PJ, Diflunisal Trial C. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013; 310: 2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, Powers ET, Reixach N, Chan WK, Wilson IA, Kelly JW, Graef IA, Alhamadsheh MM. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy‐associated V122I transthyretin. Proc Natl Acad Sci U S A 2013; 110: 9992–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, Selby VN, Jacoby D, Hanna M, Nativi‐Nicolau J, Patel J, Rao S, Sinha U, Turtle CW, Fox JC. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol 2019; 74: 285–295. [DOI] [PubMed] [Google Scholar]
- 88. Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J 2006; 20: 234–239. [DOI] [PubMed] [Google Scholar]
- 89. Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, Perlini S, Saraiva MJ, Merlini G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012; 19: 34–36. [DOI] [PubMed] [Google Scholar]
- 90. Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin‐3‐gallate as a potential therapeutic drug for TTR‐related amyloidosis: “in vivo” evidence from FAP mice models. PLoS ONE 2012; 7: e29933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. aus dem Siepen F, Bauer R, Aurich M, Buss SJ, Steen H, Altland K, Katus HA, Kristen AV. Green tea extract as a treatment for patients with wild‐type transthyretin amyloidosis: an observational study. Drug Des Devel Ther 2015; 9: 6319–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kristen AV, Lehrke S, Buss S, Mereles D, Steen H, Ehlermann P, Hardt S, Giannitsis E, Schreiner R, Haberkorn U, Schnabel PA, Linke RP, Rocken C, Wanker EE, Dengler TJ, Altland K, Katus HA. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol 2012; 101: 805–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, Lovat LB, Bartfai T, Alanine A, Hertel C, Hoffmann T, Jakob‐Roetne R, Norcross RD, Kemp JA, Yamamura K, Suzuki M, Taylor GW, Murray S, Thompson D, Purvis A, Kolstoe S, Wood SP, Hawkins PN. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 2002; 417: 254–259. [DOI] [PubMed] [Google Scholar]
- 94. Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, Mangione PP, Gallimore JR, Millar DJ, Minogue S, Dhillon AP, Taylor GW, Bradwell AR, Petrie A, Gillmore JD, Bellotti V, Botto M, Hawkins PN, Pepys MB. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 2010; 468: 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gillmore JD, Tennent GA, Hutchinson WL, Gallimore JR, Lachmann HJ, Goodman HJ, Offer M, Millar DJ, Petrie A, Hawkins PN, Pepys MB. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br J Haematol 2010; 148: 760–767. [DOI] [PubMed] [Google Scholar]
- 96. Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, Fontana M, Moon JC, Pinzani M, Gillmore JD, Hawkins PN, Pepys MB. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med 2015; 373: 1106–1114. [DOI] [PubMed] [Google Scholar]
- 97. Richards DB, Cookson LM, Barton SV, Liefaard L, Lane T, Hutt DF, Ritter JM, Fontana M, Moon JC, Gillmore JD, Wechalekar A, Hawkins PN, Pepys MB. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci Transl Med 2018; 10: pii: eaan3128. [DOI] [PubMed] [Google Scholar]
- 98. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez‐Naharro A, Quarta CC, Rezk T, Whelan CJ, Gonzalez‐Lopez E, Lane T, Gilbertson JA, Rowczenio D, Petrie A, Hawkins PN. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018; 39: 2799–2806. [DOI] [PubMed] [Google Scholar]
- 99. Gilstrap LG, Dominici F, Wang Y, El‐Sady MS, Singh A, Di Carli MF, Falk RH, Dorbala S. Epidemiology of cardiac amyloidosis‐associated heart failure hospitalizations among fee‐for‐service medicare beneficiaries in the United States. Circ Heart Fail 2019; 12: e005407. [DOI] [PMC free article] [PubMed] [Google Scholar]