

Summary
The host must develop tolerance to commensal microbes and protective responses to infectious pathogens, yet the mechanisms enabling a privileged relationship with commensals remain largely unknown. Skin colonization by commensal Staphylococcus epidermidis facilitates immune tolerance preferentially in neonates via induction of antigen-specific regulatory T cells (Tregs). Here we demonstrate that this tolerance is not indiscriminately extended to all bacteria encountered in this early window. Rather, neonatal colonization by Staphylococcus aureus minimally enriches for antigen-specific Tregs and does not prevent skin inflammation upon later life exposure. S. aureus α-toxin contributes to this response by stimulating myeloid cell production of IL-1β, which limits S. aureus-specific Tregs. Loss of α-toxin or the IL-1 receptor increases Treg enrichment, whereas topical application of IL-1β or α-toxin diminishes tolerogenic responses S. epidermidis. Thus, preferential activation of a key alarmin pathway facilitates early discrimination of microbial foe from friend, thereby preventing tolerance to a common skin pathogen.
Keywords: Neonatal, Skin Immunity, Regulatory T cells, Commensal, Pathogen
Graphical Abstract
eTOC Summary
Mechanisms enabling the host to establish to a privileged relationship with commensal bacteria while preserving protective responses against pathogens remain largely unknown. Leech et al. demonstrate an early life capacity to distinguish between commensal and pathogenic skin bacteria which preferentially facilitates accumulation of commensal-specific Tregs and immune tolerance.
Introduction
Our bodies harbor many more bacterial than human cells. To survive in this microbe-rich environment, our immune system must reconcile two fundamental yet seemingly discordant needs: one, establish immune tolerance to commensal microbes and two, mount a protective immune response against infectious pathogens. Achieving these dueling objectives is especially critical at body barrier sites. In these tissues, eliminating invasive pathogens is paramount but dysregulated immune responses to commensal microbes can fuel a growing burden of chronic inflammatory disease.
The skin is the body’s largest organ and serves both as a physical protective barrier against environmental insults and as a dynamic interface for host-microbe interaction (Clark, 2010; Clark et al., 2006; Grice and Segre, 2011). The communities of bacteria, fungi, viruses, and archaea inhabiting human skin are notably diverse and spatially complex (Byrd et al., 2018). Commensal bacteria, in particular, have been shown to promote skin health both by restricting pathogen colonization and fine-tuning the function of skin-resident immune cells (Belkaid and Harrison, 2017; Clark, 2010; Naik et al., 2015; Scharschmidt et al., 2015). In contrast, bacterial skin pathogens contribute to significant worldwide morbidity via skin and soft tissue infections. Mechanisms that allow us to distinguish bacterial “friend” from “foe” in the context of skin colonization and establish a privileged relationship with the former remain largely undefined. Elucidating these is of fundamental importance to understanding skin immune homeostasis and the microbial contributions to inflammatory skin diseases, such as atopic dermatitis, acne vulgaris and others.
Regulatory T cells (Tregs) are essential to establish and maintain immune homeostasis in peripheral tissues. This subset of CD4+ T cells, largely defined by expression of the canonical transcription factor Forkhead box P3 (Foxp3), also plays a vital role in mediating tolerance to commensal microbes. In the intestinal lamina propria, this tolerance is supported by a combination of thymically-derived Tregs and those that are peripherally-induced in response to commensal bacteria and their products (Cebula et al., 2013). Tregs in skin are found in equally high abundance (Clark et al., 2006; Sanchez Rodriguez et al., 2014; Scharschmidt et al., 2015). Here they promote tolerance to self and commensal antigens as well as facilitate tissue-specific functions such as wound repair and hair growth (Ali and Rosenblum, 2017). Of note, Tregs accumulating in tissues early in life demonstrate heightened activation and suppressive capacity (Thome et al., 2016; Yang et al., 2015).
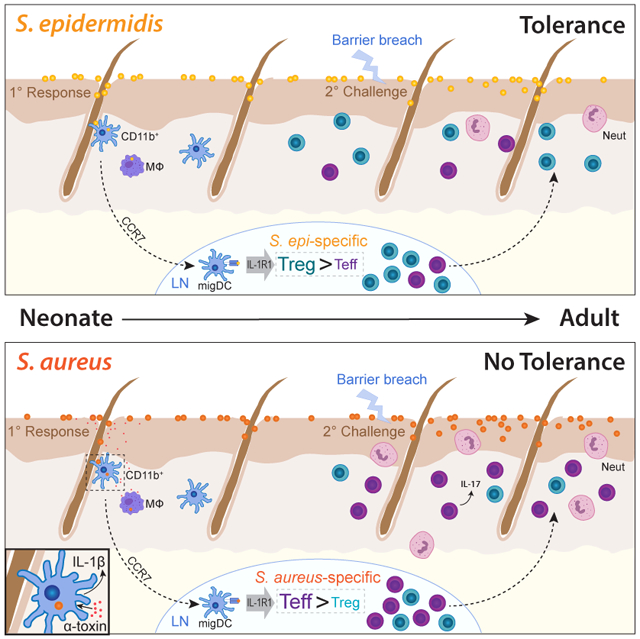
We have previously demonstrated that neonatal life represents a key developmental window for establishment of immune tolerance to skin commensals. In these studies, colonization of neonatal but not adult mice with Staphylococcus epidermidis (S. epi) was associated with protection against skin inflammation and expansion of a population of S. epi-specific Tregs upon subsequent challenge (Scharschmidt et al., 2015). This preferential capacity for tolerance in early life was facilitated by an abundance of highly activated Tregs in neonatal skin, the accumulation of which was dependent on both hair follicle development and microbial colonization (Scharschmidt et al., 2017). Not addressed in these studies, however, was whether the immune environment in neonatal skin facilitates tolerance to any microbe or, conversely, if the host retains mechanisms to prevent inopportune tolerance to pathogens encountered during this period.
Here, we use a system to track bacteria-specific CD4+ T cells in the context of a polyclonal T cell repertoire and complex microbiota. By design, our system allows direct comparison of commensal versus pathogen-specific responses independent of variables recognized to influence T cell activation and differentiation, such as antigen load, T cell receptor interaction strength and T cell precursor frequency. Controlling for these factors, we show that the host is capable of differentiating between antigens expressed by a commensal versus pathogenic bacterium during early life skin colonization and mounts a preferentially tolerogenic response to the former. Moreover, we demonstrate that toxin-mediated activation of IL-1R signaling upon pathogen encounter plays a key role in limiting enrichment of pathogen-specific Tregs. Thus, differential T cell priming upon initial microbial antigen exposure in skin sets the course for tolerance or immunity.
Results
Early life colonization by Staphylococcus epidermidis preferentially promotes antigen-specific immune tolerance versus colonization by Staphylococcus aureus
We previously developed a murine model of cutaneous commensalism in which bacteria-specific CD4+ T cells can be assayed longitudinally in the context of both a complex microbiome and a polyclonal immune repertoire (Scharschmidt et al., 2015). In this system, colonization of C57BL/6 mice with S. epi engineered to express the 2w peptide (Moon et al., 2007) (S. epi-2w) results in activation and expansion of endogenous CD4+ T cells with T cell receptors specific for the 2w antigen. These 2w+ cells can be detected using a fluorescent 2w-loaded major histocompatibility complex class II (MHCII) tetramer and studied by flow cytometry in the skin and secondary lymphoid organs of colonized mice. Using this model, we demonstrated that skin colonization by S. epi-2w in neonatal life is protective against tissue inflammation upon later life exposure and facilitates enrichment of antigen-specific Tregs (Scharschmidt et al., 2015).
Here we developed analogous tools to enable us to study the antigen-specific response to Staphylococcus aureus (S. aureus), a common skin pathobiont and the leading cause of skin and soft tissue infections (Brugger et al., 2016). Linking of the 2w peptide to a fluorescent mcherry protein in our plasmid-based system allows for quantification of bacterial antigen expression (Figure S1A). Transformation of this plasmid into S. aureus SF8300, a wound isolate of the USA300 subtype (Diep et al., 2008), (S. aureus-2w) and S. epi Tü3298 (S. epi-2w) resulted in equivalent 2w expression as measured by mcherry intensity on flow cytometry (Figure S1B). Colonization of neonatal pups with either S. epi-2w or S. aureus-2w also resulted in comparable bacterial skin persistence (Figure S1C).
To investigate whether bacteria-specific tolerance is established preferentially to a commensal versus a pathogenic bacteria present on neonatal skin, we colonized neonatal mice with either S. epi-2w or S. aureus-2w for 1 week beginning on postnatal day 7 and challenged them 3-4 weeks later with the same bacteria in combination with gentle tape-stripping to minimally abrade the epidermis (pre-colonized groups). Separated littermates or age-matched pups that were not colonized neonatally but challenged in adulthood with S. epi-2w or S. aureus-2w (no pre-colonization) served as controls (Figure S1D). As previously demonstrated (Scharschmidt et al., 2015), the skin of mice colonized with S. epi-2w in the neonatal period demonstrated reduced histologic evidence of skin inflammation and contained fewer neutrophils following S. epi-2w challenge compared with the skin of control animals (Figure 1A, 1C and S1E). In contrast, histologic skin inflammation and numbers of skin neutrophils were greater in both groups challenged with S. aureus-2w, and neither parameter was attenuated by preceding neonatal S. aureus-2w colonization (Figure 1B, 1C and S1F). This suggests that whereas early exposure to a commensal, S. epi, limits skin inflammation upon subsequent exposure, early colonization by a skin pathogen, S. aureus, does not.
Figure 1: Early life colonization by S. epi but not S. aureus facilitates adaptive immune tolerance upon subsequent challenge.

Neonatal mice were colonized with S. epi-2w or S. aureus-2w (Precol) on postnatal day 7, 10 and 13 or left uncolonized (No Precol) before challenge 3-4 weeks later with either bacteria in the context superficial skin abrasion. (A) Representative histology and disease scoring of S. epi or (B) S. aureus treated mice. (C) Representative flow cytometry plots and graphs of % skin neutrophils (gated on a live CD45+CD3neg cells). (D) Representative flow plots and graphs of %Treg in 2w-specific cells in SDLNs and (E) skin of all groups. Plots in D are gated on the live DUMPnegCD45+CD3+CD4+CD44+2w+ cells in the tetramer-enriched fraction and in E on the same population in total (unenriched) skin. Each point represents an individual mouse. Data are representative of three independent experiments with at least 4 mice per group. See also Figure S1.
We next examined the impact of neonatal S. epi-2w or S. aureus-2w colonization on bacteria-specific CD4+ T cells. Challenge with either strain resulted in equivalent numbers of 2w+CD44+CD4+ in the skin-draining lymph nodes (SDLN), suggesting comparable exposure to 2w antigen when expressed by either bacteria (Figure S1G). Possibly consistent with deletion of 2w+CD4+ upon early life antigen exposure, both groups of pre-colonized mice had lower absolute numbers of bacteria-specific cells in the SDLN post-challenge as compared to mice not exposed as pups (Figure S1H). The percentage of Tregs within a population of antigen-specific CD4+ T cells is directly related to host propensity for tolerance to that antigen (Sakaguchi et al., 2008). Thus, we next examined the proportion of 2w-specific Tregs in mice colonized with either bacteria. As anticipated, 2w-specific T cells in the SDLN and skin of S. epi-2w pre-colonized mice were significantly enriched for Tregs. In comparison, S. aureus-2w pre-colonization resulted in a significantly lower percentage of antigen-specific Tregs in the SDLN (Figure 1D) with a similar trend present among antigen-specific Tregs in the skin (Figure 1E). This preferential Treg enrichment in S. epi-2w versus S. aureus-2w pre-colonized mice was restricted to bacteria-specific cells, as polyclonal Treg percentages in the skin and SDLN were largely equivalent across groups (Figure S1I and S1J). Collectively, these results demonstrate that skin colonization by commensal S. epi-2w in the neonatal period preferentially promotes adaptive immune tolerance as compared to the pathogen S. aureus, and that this is closely linked with percentages of commensal versus pathogen-specific Tregs during inflammatory re-exposure.
Host discrimination of S. epidermidis versus S. aureus during neonatal colonization results in differential enrichment of antigen-specific Tregs.
The observation that neonatal colonization followed by adult challenge with S. aureus-2w yielded reduced antigen-specific Tregs as compared to S. epi-2w, could result from limited enrichment of S. aureus-specific Tregs upon initial exposure and/or their increased suppression during inflammatory re-challenge. As early life interactions have been shown to have a critical role in shaping adaptive immune responses to microbial antigens (Belkaid and Harrison, 2017; Scharschmidt et al., 2017), we hypothesized that initial neonatal exposure to either bacteria would be sufficient to replicate divergent Treg responses. To test this, we modified our model to isolate bacterial antigen exposure to this early window and recalled the antigen-specific response in mice neonatally colonized with S. epi-2w or S. aureus-2w via intradermal injection of 2w peptide in Incomplete Freund’s adjuvant (IFA:2w). Mice that were not colonized with either bacteria but similarly challenged with IFA:2w served as controls. Consistent with our hypothesis, 2w+CD44+CD4+ T cells in the SDLN of mice colonized with S. aureus-2w contained lower percentages of Tregs compared with those colonized by S. epi-2w (Figure 2A). Notably, mice colonized neonatally with S. aureus-2w demonstrated a higher percentage of 2w-specific Tregs in the SDLN following 2w:IFA as compared with uncolonized controls. This suggests a baseline propensity for early life antigen exposure to facilitate antigen-specific Tregs.
Figure 2: Discrimination between commensal and pathogen occurs during initial neonatal skin colonization and facilitates enrichment of S. epi-specific versus S. aureus-specific Tregs.

(A) Neonatal mice colonized with S. epi-2w or S. aureus-2w on postnatal day 7, 10 and 13 or left uncolonized (Naïve) and then re-challenged 2 weeks later with intradermal 2w peptide in Incomplete Fruend’s Adjuvant (IFA). Representative flow cytometry plots and percentages of 2w-specific Tregs in the SDLNs 8 days later. (B) Neonatal mice were colonized as above and the primary 2w response in the SDLN assessed at weaning age. Representative flow cytometry plots, percentages and absolute numbers of 2w-specific Tregs are shown. All plots are gated on live DUMPnegTCRb+CD4+CD44+2w+ cells in the tetramer-enriched fraction. Data are representative of three independent experiments with at least 3 mice per group. See also Figure S2.
To exclude the possibility that the reduced percentage of S. aureus-specific versus S. epi-specific Tregs was somehow attributable to differential re-expansion in response to IFA, we next examined the primary response to these bacteria in the SDLN of weaning age mice. Challenges inherent to measuring a small primary antigen-specific response during this dynamic developmental period led to variable absolute numbers of 2w+CD44+CD4+ T cells among individual neonates. Across experiments, however, 2w-specific responses to S. epi-2w or S. aureus-2w in the SDLN were revealed to be comparably-sized (Figure S2A and S2B). As anticipated, the percentages of bacteria-specific Tregs were significantly lower in S. aureus-2w versus S. epi-2w colonized mice (Figure 2B). To test the robustness of this finding, we engineered additional clinical isolates of S. epi and S. aureus to express the 2w peptide. Assessment of the primary antigen-specific response following neonatal colonization with this broader panel of strains again demonstrated preferential Treg enrichment in response to S. epi versus S. aureus across all isolates tested (Figure S2C). These differences were restricted, as before, to bacteria-specific CD4+ Tregs as polyclonal Tregs in the SDLN at weaning age (Figure S2D and S2E) and in skin at postnatal day 13 (Figure S2F), the peak of early life Treg recruitment, were comparable in S. epi and S. aureus colonized mice. Variability in the total number of 2w+ Tregs and 2w+Foxp3neg effector CD4+ T cells (Teffs) across mice in both groups limited our ability to determine if preferential expansion of Tregs, reduced expansion of Teffs or both contributed to the observed differences in Treg percentage among 2w+CD4+CD44+ T cells (Figure S2G and S2H). These experiments support a model by which the host’s ability to distinguish S. epi from S. aureus during neonatal skin colonization facilitates preferential enrichment of commensal-specific versus pathogen-specific Tregs.
Priming of bacteria-specific T cells in the SDLN is dependent on CCR7-mediated transport of microbial antigen from skin.
Dendritic cells (DCs) are professional antigen presenting cells responsible for priming CD4+ T cell expansion and their differentiation towards distinct subsets upon integration of various external stimuli (Eisenbarth, 2019). The reduction in antigen-specific Tregs in response to S. aureus-2w as compared to S. epi-2w prompted us to examine the role of DCs in these responses. We postulated that this might be explained by functionally different DC subsets preferentially acquiring commensal versus pathogen-derived antigens. To elucidate which DCs acquire bacterial antigen in the context of neonatal S. epi or S. aureus skin colonization, we engineered both bacteria to express zsgreen (zsg-S. epi and zsg-S. aureus) (Figure S3A). The brightness of this fluorophore and its resistance to low pH or lysosomal degradation (Roberts et al., 2016) enabled us to quantitate bacterial antigen uptake by different skin and SDLN DCs using flow cytometry. In neonatal skin, DCs were identified as live, CD45+CD64negMHCII+CD11c+ cells and further classified as CD103+CD11bneg (CD103+ DCs), CD103negCD11b+CD207neg (CD11b+ DCs) or CD103negCD11b+CD207+ Langerhans cells (LHCs) (Figure 3A). Following overnight colonization with either zsg-S. epi or zsg-S. aureus, zsgreen was readily detectable in skin DCs (Figure S3B and S3C). Contrary to our hypothesis, the percent and number of zsgreen+ CD103+ DCs, CD11b+ DCs and LHCs were equivalent in the skin of mice colonized with zsg-S. epi or zsg-S. aureus (Figure 3B-3D). With either strain, zsgreen was predominantly seen in CD11b+ DCs, potentially implicating a key role for this population in uptake and presentation of skin bacterial antigens.
Figure 3: S. epi and S. aureus antigens are concentrated in skin cDC2s following neonatal colonization.

Neonatal mice were colonized with zsg-S. epi or zsg-S. aureus on postnatal day 9 and skin was harvested 18 hours later. (A) Gating strategy to delineate skin dendritic cell subsets. Progressive gating identifies, CD103+, CD11b+, and Langerhans cells (LHC). (B) Representative flow cytometry plots of zsgreen+ CD11b+, CD103+ and LHCs. (C) Percentage and (D) absolute numbers of zsgreen+ DC populations in skin by subtype. Data are representative of two independent experiments with at least 4 mice per group. See also Figure S3.
We next examined transport of zsgreen+ signal to the SDLN. There, migratory DCs (migDCs) that have trafficked from the skin can be distinguished from lymphoid-resident DCs (rDCs) by their higher expression of MHCII (Figure 4A) (Anandasabapathy et al., 2014). Following overnight colonization with zsg-S. epi or zsg-S. aureus, zsgreen was preferentially contained in CD11c+MHCIIhi migDCs (Figure 4B and 4C). We therefore sought to determine whether active transport of bacterial antigen by skin-resident DCs to the SDLN, as compared with passage of antigen via lymphatic drainage, is required for priming of bacteria-specific CD4+ T cells. To test this, we employed neonatal mice deficient for CCR7, a receptor required for trafficking of leukocytes, including DCs, from the skin to SDLN (Ohl et al., 2004). As anticipated, despite equivalent zsgreen signal in the skin of Ccr7−/− mice (data not shown), their SDLN contained significantly fewer zsgreen+ CD11c+MHChi migDCs as compared with their Ccr7+/− littermates following overnight colonization with zsg-S. epi or zsg-S. aureus (Figures 4D and 4E). Irrespective of CCR7 status or bacterial strain, a small zsgreen signal was detectable in CD11c+MHCmid rDCs (Figure S4A), possibly consistent with passive drainage of soluble antigen to the SDLN via lymphatics in the context of ongoing skin barrier maturation (Kastenmüller et al., 2012). To measure to what extent priming of bacteria-specific T cells was dependent on bacterial antigen transport to the SDLN by migDCs, we colonized neonatal Ccr7−/− and Ccr7+/− littermates with S. epi-2w or S. aureus-2w before measuring the primary 2w+ response in the SDLN at weaning. Ccr7+/− mice demonstrated comparable expansion of 2w+CD44+CD4+ cells (Figure 4F) to wild-type mice (S2A and S2B) following colonization with either S. epi-2w or S. aureus-2w. In contrast, priming of bacteria-specific CD4+ cells was largely absent in Ccr7−/− mice (Figure 4F, 4G and S4B). Thus, CCR7-dependent transport of bacterial antigen to the SDLN by skin-resident DCs is required for effective priming of either commensal-specific or pathogen-specific T cells and the identity of these antigen-laden DCs does not differ in these two contexts.
Figure 4: Neonatal priming of bacteria-specific CD4+ T cells requires CCR7-dependent antigen transport to lymph node.

WT, Ccr7+/− or Ccr7−/− pups were colonized with zsgreen-expressing bacteria, zsg-S. epi or zsg-S. aureus, on postnatal day 9 and skin-draining lymph nodes (LN) were harvested 18 hours later. (A) Gating strategy to delineate LN dendritic cells (DC). Progressive gating identifies, CD11c+MHChi migratory DC (migDC) and CD11c+MHCmid resident DC (rDC). (B) Representative flow cytometry plots (gated as per A) and (C) absolute numbers of zsgreen-containing migDC and rDC following colonization of WT mice with either zsg-S. epi or zsg-S. aureus. (D) Flow plots of total migDC and (E) absolute numbers of zsgreen-containing migDC in LN of Ccr7+/− or Ccr7−/− mice colonized with zsg-S. epi or zsg-S. aureus. (F) Ccr7−/− or Ccr7−/− pups were colonized with S. epi-2w or S. aureus-2w and LN were stained for tetramer-positive cells at weaning age. Representative flow cytometry plots gated on live DUMPnegCD45+CD3+CD4+ in tetramer-enriched fraction. (G) Absolute numbers of 2W+CD4+CD44+ cells in LN of Ccr7+/− and CcrT−/− mice. Data are representative of two independent experiments with at least 4 mice per group. See also Figure S4.
Myeloid-derived IL-1β limits enrichment of S. aureus-specific Tregs.
The equivalent profile of zsgreen+ DCs following zsg-S. epi or zsg-S. aureus colonization suggested that the distinct antigen-specific Treg percentages observed in response to these bacteria may not be due to T cell priming by separate DC subsets but rather differences in the quality of priming upon commensal versus pathogen encounter. To elucidate relevant host pathways that might be differentially activated in these two contexts we performed bulk RNA sequencing on Tregs and Teffs sorted from the skin of weaning age mice colonized from birth with either S. epi or S. aureus. Principal component analysis (PCA) on these four cell populations (S. epi Tregs, S. epi Teffs, S. aureus Tregs, S. aureus Teffs) demonstrated clustering by condition and cell type (Figure 5A). Differential expression analysis was performed on S. epi versus S. aureus Tregs and S. epi versus S. aureus Teffs. For each comparison, differentially expressed genes with a corrected p-value of less than 0.05 were then taken into Ingenuity upstream regulator analysis. This tool identifies genes in a dataset known to be regulated by a particular molecule or pathway and assigns a p-value and activation score to these potential upstream regulators. Multiple upstream regulators were identified for Teffs (Figure 5B) or Tregs (Figure 5C) in S. aureus versus S. epi colonized mice, the majority of which were predicted to be activated in the context of S. aureus. To narrow this list we focused on potential regulators shared by Tregs and Teffs, of which IL-1β was of particular interest given its documented roles in immune defense against S. aureus (Miller et al., 2007), type 17 helper (Th17) T cell differentiation (Chung et al., 2009) and Foxp3 destabilization and impeded Treg induction via modulation of STAT3 versus STAT5 signaling (Basu et al., 2015). Indeed, further interrogation of our sequencing data demonstrated preferential expression of IL-1β target molecules in Teffs (Figure 5D) and Tregs (Figure 5E) from S. aureus versus S. epi colonized mice, and gene set enrichment analysis confirmed strong enrichment of the IL-1R1 signature in these populations (Figure 5F and 5G). Elevation of IL-1β, but not IL-1α or IL-1ra, transcript levels in whole skin of S. aureus versus S. epi-colonized mice (Figure S5A-C) and a strong Th17 signature in S. aureus skin Teffs (Figure S5D and S5E) further validated IL-1β as a top candidate warranting functional exploration.
Figure 5: IL-1β is a key upstream regulator of the transcriptional signature of CD4+ T cells in the skin of neonates colonized with S. aureus as compared to S. epi.

WT pups were colonized with S. epi (SE) or S. aureus (SA) every two days from birth and skin Tregs (CD25hiICOShi) and CD4+ effector (CD25negICOSneg) T cells (Teff) were sorted from the skin at weaning age and processed for RNA sequencing. (A) Principal component analysis plot of the four sorted cell populations. (B) Upstream regulatory analysis was performed in Ingenuity on genes that were differentially expressed between SA vs. SE Teff and (C) SA vs. SE Tregs. Top ten regulators identified for each comparison for which there was a predicted direction of activation are shown in descending order of statistical significance. (D) Volcano plots showing padj value and fold change for genes differentially expressed by skin Teff and (E) Tregs from S. aureus vs. S. epi colonized mice. Blue dots represent genes with padj < 0.05 and > 2-fold difference in gene expression between groups. Genes in red are those annotated in Ingenuity as being IL-1β regulated. (F) Gene set enrichment analysis for the subset of these genes that are specifically regulated by IL-1β at the transcriptional level in S. aureus vs. S. epi Teff and (G) Tregs. See also Figure S5.
To determine if signaling downstream of IL-1β was involved in shaping percentages of bacteria-specific Tregs in early life, we colonized neonatal wild-type (WT) or Il1r1−/− mice with S. aureus-2w and assayed the primary response at weaning. Despite comparable expansion of 2w+CD4+CD44+ T cells (Figure S6A) and equivalent percentages of polyclonal Tregs (Figure S6B) in the SDLN of both groups, the percentage (Figure 6A) and number (Figure S6C) of 2w+ S. aureus-specific Tregs were significantly increased in Il1r1−/− as compared to WT mice. Although S. epi has been demonstrated to elicit IL-1R signaling following adult skin colonization (Naik et al., 2012), analogous experiments with S. epi-2w did not reveal substantial differences in antigen-specific Tregs between Il1r1−/− and WT mice (Figure 6B). Together these results suggest greater activation of IL-1R1 in the context of S. aureus versus S. epi, leading to greater constraint on S. aureus-specific Tregs.
Figure 6: Myeloid-derived Il-1β during neonatal colonization constrains S. aureus-specific Tregs.

Neonatal WT and Il1r1−/− mice were colonized on postnatal day 7, 10 and 13 and the primary 2w response in the SDLN assessed at weaning age. Representative flow cytometry plots and percentages of 2w-specific Tregs are shown for (A) S. aureus or (B) S. epi colonized mice. Data in A and B are pooled from two of three replicate experiments. All plots are gated on live DUMPnegTCRb+CD4+CD44+2w+ cells in the tetramer-enriched fraction. Neonatal WT mice were colonized with S. epi as above with or without (C) topical IL-1β or (D) topical IL-1α then challenged 2 weeks later with intradermal 2w peptide in IFA. Percentages of 2w-specific Tregs in the SDLN 1 week thereafter are shown. 9-day-old mice were colonized overnight with zsg-S. epi or zsg-S. aureus and skin harvested the next day. Intracellular levels of pro-IL-1β in skin (E) macrophages and (F) CD11b+ DCs as measured by flow cytometry. (G) Levels of IL-1β by ELISA in media of BMMs exposed for 2 hours to culture supernatants of S. epi or S. aureus. See also Figure S6.
To directly test the role of IL-1β versus IL-1α in our model, we topically applied either cytokine or vehicle alone to S. epi-2w colonized neonates and examined their sufficiency to limit bacteria-specific Tregs in this window. Whereas IL-1β substantially reduced the percentage of S. epi-specific Tregs, IL-1α failed show an effect (Figures 6C and 6D). We therefore sought to better define sources of IL-1β in the skin of S. aureus-colonized neonates. To do so we colonized pups overnight with zsg-S. epi, zsg-S. aureus or no bacteria and measured pro-IL-1β expression in skin by flow cytometry. Across all conditions, myeloid cells expressed substantially more pro-IL-1β, than did CD45neg cells or CD3+ T cells (Figure S6D and S6E). Colonization by zsg-S. aureus, as compared to zsg-S. epi, led to increased pro-IL-1β in skin macrophages and CD11b+ DCs but not CD103+ DCs, specifically within zsgreen+ cells that had phagocytosed bacteria (Figure 6E & 6F and Figure S6F-S6H). To measure the impact of S. epi versus S. aureus on levels of caspase-cleaved IL-1β, we exposed bone marrow-derived myeloid cells (BMMs) to sterilely-filtered supernatants from either bacteria in vitro and measured IL-1β by ELISA after two hours. As expected, significantly more IL-1β was released in response to S. aureus as compared to S. epi supernatant (Figure 6G). This was also replicated more broadly across a larger panel of commensal versus pathobiont skin bacteria (Figure S6I). Together these results support a role for myeloid cell-derived IL-1β in limiting S. aureus-specific Tregs upon neonatal skin colonization.
Deficiency of S. aureus α-toxin leads to recovery in pathogen-specific Tregs.
Different bacteria-specific Treg responses to S. epi versus S. aureus may derive from Treg-promoting S. epi molecules, Treg-limiting S. aureus molecules or some combination thereof. Based on the identification of IL-1R1 signaling as a pathway limiting Tregs in the context of S. aureus colonization, we decided to pursue identification of S. aureus molecules that might lower the percentage of pathogen-specific Tregs via increased IL-1β production. Expression of many S. aureus virulence factors are regulated by its two-component quorum-sensing system, the accessory gene regulator, which is encoded by the agr locus. To determine the contribution of agr-regulated virulence factors in limiting S. aureus-specific Tregs, we colonized neonatal mice with an isogenic strain of SF8300 S. aureus-2w in which the agr locus was deleted (ΔagrA S. aureus-2w) (Rasigade et al., 2013). In the absence of agr quorum sensing, the 2w+ response to S. aureus was rendered more “commensal-like”, with increased percentages of 2w-specific Tregs in the SDLN of mice colonized with ΔagrA as compared to WT S. aureus-2w (Figure 7A).
Figure 7: S. aureus alpha toxin limits accumulation of antigen-specific Tregs.

(A) Neonatal mice colonized with S. aureus-2w or S. aureus-2w-ΔagrA, S. aureus-2w-Δhla or S. aureus-2w-Δpsm on postnatal day 7, 10 and 13 and then re-challenged 2 weeks later with intradermal 2w peptide in Incomplete Freund’s Adjuvant (IFA). Representative flow cytometry plots and percentages of 2w-specific Tregs in the SDLNs 8 days later. (B) Neonatal mice were colonized as above with either S. aureus-2w, S. aureus-2w-Δhla or S. aureus-2w-Δpsm and then re-challenged 2 weeks later with intradermal 2w peptide in IFA. (C) Neonatal mice were colonized with S. epi as above with or without recombinant hla and challenged 2 weeks later with intradermal 2w peptide in IFA. Representative flow cytometry and percentages of 2w-specific Tregs 8 days later. All plots are gated on live 2w+DUMPnegTCRb+CD4+CD44+ cells in the tetramer-enriched fraction. Data are representative of two independent experiments with at least 3 mice per group. See also Figure S7.
Agr-deficiency broadly affects S. aureus fitness including its capacity for skin colonization (Williams et al., 2019). As such, we sought to identify whether deletion of a single agr-regulated molecule could replicate results from the ΔagrA S. aureus-2w. Alpha-toxin, encoded by the hla gene, is a membrane-perforating toxin regulated by agr, which has been critically implicated in the pathogenesis of S. aureus bacteremia and pneumonia, as well as in eliciting host T cell responses and production of IL-1β in response to this pathogen (Berube and Bubeck Wardenburg, 2013). To determine whether there was a role for α-toxin in shaping the antigen-specific T cell response to S. aureus, we engineered 2w expression in an isogenic mutant of USA300 SF8300 lacking the gene for α-toxin (Δhla S. aureus-2w). Exposure of BMMs to supernatant from Δhla S. aureus-2w elicited significantly less IL-1β production as compared to WT S. aureus-2w supernatant (Fig S7A), a result not entirely linked to the extent of cell death (Figure S7B). Mice colonized neonatally with Δhla S. aureus-2w demonstrated significantly increased percentages of S. aureus-specific Tregs following re-challenge with 2w:IFA (Figure 7B). To understand the specificity of this response, we generated and tested in parallel a 2w-expressing SF8300 mutant in which all genes encoding the family of S. aureus phenol soluble modulins (psmα1-4, psmβ1-2, hld) were deleted (Δpsm S. aureus-2w). Despite data implicating S. aureus PSMs in modulation of DC function (Schreiner et al., 2013) and coordination of the T cell response to epicutaneous S. aureus exposure (Liu et al., 2017), percentages of 2w+ Tregs following Δpsm S. aureus-2w colonization were comparable to those seen with WT S. aureus-2w (Figure 7B). This was not due to survival defects as colonization of neonatal pups with either S. aureus-2w, Δhla S. aureus-2w or Δpsm S. aureus-2w resulted in comparable bacterial skin persistence (Figure S7C). To test sufficiency of S. aureus α-toxin in limiting bacteria-specific Tregs, we topically applied α-toxin to neonatal mice colonized with S. epi-2w. This significantly lowered the percentage of S. epi-specific Tregs, albeit not to the level observed with WT S. aureus (Figure 7C). Together these results support a central and specific role for S. aureus α-toxin in limiting the percentage of antigen-specific Tregs elicited in response to early colonization by this important skin pathogen.
Discussion
Whereas our prior studies revealed early life as a permissive window for establishment of tolerance and generation of antigen-specific Tregs to commensal skin bacteria, here we show that this Treg-dominant CD4+ response is not indiscriminately applied to any bacteria present on neonatal skin. Rather, the developing immune system distinguishes the pathobiont, S. aureus, from the commensal, S. epi, such that neonatal skin colonization by the former neither elicits a Treg-dominant CD4+ response nor attenuates tissue inflammation upon later life re-exposure. Skin CD11b+ DCs contain the majority of bacterial antigen following colonization with either S. aureus or S. epi, and active CCR7-dependent transport of antigen to the lymph node is required for priming of bacteria-specific CD4+ cells. Rather than involvement of different DC subsets, the lower percentage of antigen-specific Tregs in response to S. aureus could be explained by heightened Il-1β-dependent activation of IL-1R1 in response to the virulence factor alpha-toxin. These results reveal an early capacity to discriminate microbial friend from foe and support a model wherein IL-1R1 signaling serves as a host safeguard that can prevent tolerance to a skin pathogen during a key window of neonatal development.
To date, various mechanisms have been implicated in the process of commensal versus pathogen discrimination. These include strategic compartmentalization of pattern recognition receptors, inflammasome sensing of virulence factors, variation among pathogen-associated molecular patterns and microbial interference in NFκB signaling (Srinivasan, 2010). Our work extends this literature by establishing a distinct IL-1R1-dependent mechanism, which acts in the earliest stages of development to tailor the quality of the bacteria-specific T cell repertoire in a way that provides an enduring bias for tolerance towards commensal antigens. These findings are particularly notable in that they represent a key restraint on the neonatal immune system’s proven propensity to promote tolerogenic responses (Mold and McCune, 2012). Arginase-2 expression in fetal DCs supports preferential Treg induction (McGovern et al., 2017), and Tregs generated early in life have a unique capacity to protect tissues from autoimmune attack (Yang et al., 2015). In neonatal skin, a high density of activated Tregs facilitates the tolerogenic tone of the tissue (Scharschmidt et al., 2015). Notably, failure to enrich for S. aureus-specific Tregs occurred despite preservation of this early polyclonal Treg wave. This indicates that while a high density of Tregs in neonatal skin helps promote tolerance to peripherally encountered antigens, it alone is not sufficient. Rather, microbial cues have the capacity to reinforce or counterbalance the host’s propensity for tolerance in this window.
Prior studies have demonstrated that antigen-specific tolerance is more dependent on the percentage rather than absolute numbers of antigen-specific Tregs (Su et al., 2016). Indeed, in our experiments the percentage of bacteria-specific Tregs strongly correlated with preferential tolerance to S. epi as compared to S. aureus, as evidenced by protection against tissue inflammation during secondary bacterial encounter. Low numbers of tetramer-positive cells and their variable expansion among neonatal mice limited our ability to definitively determine whether reduced numbers of Tregs, increased numbers of Teffs or both were responsible for driving down percentages of S. aureus Tregs. Additionally, because naïve 2w-specific CD4+ T cells in C57BL/6 mice contain both Tregs and Teffs (Pagán et al., 2013), our model cannot disentangle the contribution of peripheral conversion of Teff into Tregs versus relative proliferation of these two populations. Future development of T cell receptor transgenic systems to study the CD4+ response to these two bacteria in adoptive transfer models should help elucidate these outstanding questions both by augmenting numbers of antigen-specific cells and enabling selective transfer of Tregs or Teffs.
Our work also provides insight into where initial priming of T cells specific for skin bacteria occurs, namely in the SDLN, presumably with subsequent migration into skin for establishment of resident memory populations. Intriguing reports suggest that passive antigen transfer from the skin to the LN may play a role in priming the T cell response (Liao and Weid, 2015; Randolph et al., 2017). In this context, it was tempting to consider that S. aureus expression of proteases (Nakatsuji et al., 2016) and other virulence factors might enable deeper penetration into neonatal skin and increased passive transfer of antigen to the SDLN. However, failure of S. epi or S. aureus-specific CD4+ T cell expansion in Ccr7−/− mice, in which leukocyte trafficking is impeded, suggests that active cellular transport of antigen to the LN is required during bacterial skin colonization. Engineering zsgreen-expressing S. epi and S. aureus strains to track antigen uptake, enabled the interesting observation that a large proportion of skin DCs sampling these bacteria belong to the CD11b+ subset. CD103+ DCs and Langerhans cells also acquired bacterial antigen to a lesser degree. Functional specialization of these three subsets has been proposed with CD103+ conventional DC1s (cDC1s) stimulating CD8+ T cells and Th1 responses to intracellular pathogens (Martínez-López et al., 2015; Mashayekhi et al., 2011) and CD11b+ cDC2s inducing Th2 and Th17 responses to extracellular pathogens (Gao et al., 2013; Persson et al., 2013). Treg-dominant responses have been credited to different subsets of skin DCs, including CD11b+ cDC2s that express high levels of RALDH or PDL2 (Guilliams et al., 2010), as well as langerin+ dermal DCs and Langerhans cells (Seneschal et al., 2012). Further work is required to delineate whether CD11b+ DCs are primarily responsible for priming of CD4+ T cells, and specifically Tregs, to bacteria colonizing neonatal skin or if significant redundancy exists among DC subsets in this window as previously described for priming of commensal-specific CD8+ T cells in adult skin (Naik et al., 2015).
Various mechanisms have been implicated in limiting Treg responses following pathogen encounter. In the context of LCMV infection, type I interferons were shown to inhibit co-stimulation-dependent Treg activation and proliferation (Srivastava et al., 2014). In Mycobacterium tuberculosis-infected mice, antigen-specific Tregs dwindled in late-stage infection due, in part, to their IL-12-mediated expression of T-bet (Shafiani et al., 2013). Finally, a highly Th1-polarized environment during Toxoplasma gondii infection was shown to precipitate a drop in Treg numbers via a combination of mechanisms that included IL-2 shutdown (Oldenhove et al., 2009). In our own studies, we identified a central role for IL-1β and signaling through its receptor, IL-1R1, in facilitating host discrimination of S. aureus from S. epi and in limiting antigen-specific Tregs to the former. Multiple cellular sources of IL-1β, including neutrophils, keratinocytes, macrophages and dendritic cells, have been identified in the context of S. aureus skin infection (Miller et al., 2007; Wang et al., 2017). In our neonatal model of S. aureus skin colonization, we found myeloid cells to be the major source of IL-1β.
The functional relevance of IL-1R1 signaling was intriguing to pursue in our model based on prior literature implicating it in the developmental balance between Tregs and Th17 cells. IL-1β has been shown to promote Th17 cell differentiation at the expense of Tregs via various T cell intrinsic mechanisms that include augmented phosphorylated STAT3 activity downstream of IL-6, suppression of TGF-β-mediated Foxp3 induction and alternative splicing of Foxp3 transcript (Basu et al., 2015; Chewning and Weaver, 2014; Chung et al., 2009; Ikeda et al., 2014; Mailer et al., 2015). IL-1R1 signaling on DCs themselves can also favor priming of distinct T cell subsets by promoting DC activation and cytokine production (Deng et al., 2019; Eriksson et al., 2003; Pang:2013dc; 2003; Pang et al., 2013). Our RNA sequencing of skin Teffs from S. aureus-colonized mice revealed a strong Th17 signature (Chang et al., 2018). This suggests that described mechanisms by which IL-1β favors Th17 over Treg differentiation, might also be active here. However, the relative contribution of these mechanisms and the extent to which our result in Il1r1−/− mice reflects T cell-intrinsic versus extrinsic IL-1R1 signaling remains to be determined.
In S. aureus, the agr system controls production of enzymes necessary for growth on host substrates (Kolar et al., 2013; Olson et al., 2014) (Kolar:2013gy; Olson et al., 2014) as well as exoproteins that impact bacterial motility, biofilm restructuring and host cell lysis (Peschel and Otto, 2013). Emerging data also suggests that a functional agr system facilitates S. aureus skin colonization (Paharik et al., 2017). Alpha toxin, also known as alpha-hemolysin or hla, is a pore-forming cytotoxin that is expressed under agr-regulation by the vast majority of clinical S. aureus isolates (Sharma-Kuinkel et al., 2015). Alpha toxin plays a major role in S. aureus virulence via a myriad of well-described mechanisms that particularly target hematopoietic and epithelial cells. These include cell death via membrane lysis or apoptosis, platelet aggregation, inflammasome activation and barrier disruption via the above mechanisms and direct binding to the host receptor ADAM10 (Berube and Bubeck Wardenburg, 2013). Here we show that alpha toxin has relevance not only during skin infection but also colonization. Namely its expression in this context appears to significantly limit early life enrichment of S. aureus-specific Tregs that might unduly facilitate adaptive immune tolerance to this pathogen. Additional work is needed to decipher the most relevant alpha toxin-dependent mechanisms and host cell targets contributing to this phenotype. However, prior studies documenting role for alpha toxin in NLRP3 inflammasome-dependent IL-1β production (Craven et al., 2009) and induction of Th17 cells may provide early clues (Frank et al., 2012).
The implications of our findings are interesting to consider in the context of a lifetime demarcated by intermittent and repeat encounters with environmental pathogens. In these studies, neonatal colonization with S. aureus resulted in a modest but statistically significant increase in the percentage of antigen-specific Tregs elicited upon adult cutaneous re-exposure. Whether the immune footprint left by this early colonization would prove to be protective or crippling in the face of a more invasive later life infection is worthy of future study. On the other hand, it will be important to decipher what impact, if any, early pathogen encounter has on the establishment of tolerance to commensal bacteria present at that site. Can the host simultaneously distinguish friend from foe or does pathogen activation of innate alarmins, such as IL-1β, undermine establishment of tolerance to other “benign” antigens? If preferential tolerance to commensal microbes is maintained in the presence of pathogen colonization what are the mechanisms the host has evolved to compartmentalize these responses? Continued investigation along these lines will better inform any future efforts to tune host-microbe interactions for therapeutic benefit in the context of either inflammatory or infectious diseases.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tiffany Scharschmidt (Tiffany.Scharschmidt@ucsf.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Experimental animals
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) then bred and maintained in the UCSF specific pathogen-free facility on the Parnassus campus. Il1r1−/− and Ccr7−/− mice were purchased from Jackson. All mice used in experiments were socially housed under a 12 hour light/dark cycle. Animals were 6 days to 10 weeks old at the time of experiments. Littermates were used as controls whenever possible. When experimental design precluded exclusive use of littermates, mice from age-matched litters were used and cross-fostered to minimize cage effects. Both male and female mice were included in experiments, with equal distribution of sexes across groups. All animals were used in experiments for the first time. Animal work was performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the guidelines of the Laboratory Animal Resource Center and Institutional Animal Care and Use Committee of the University of California, San Francisco.
Bacterial strains and culture conditions
Staphylococcus epidermidis (S. epi) strain Tü3298 (Allgaier et al., 1986; Augustin and Götz, 1990) and Staphylococcus aureus (S. aureus) USA300 strain SF8300 (Diep et al., 2006) were used for the majority of experiments in this study. In Figures S2C and S6I additional strains were used as listed in the key resources table. To derive an erythromycin-susceptible SF8300 strain, the erythromycin-resistant SF8300 parental strain was passaged overnight in tryptic soy broth and colonies were replica-plated onto tryptic soy agar and tryptic soy agar supplemented with 5 μg/mL erythromycin to select for erythromycin-susceptible colonies that lost the ermB-encoding plasmid. Isogenic mutants of SF8300 (ΔagrA, Δhla, Δpsmα/psmβ/hld) were generated by allelic replacement as previously described (Bae and Schneewind, 2006; Diep et al., 2016). The pan-PSM mutant was created from the SF8300Δpsmα1-4 mutant strain (Hodille et al., 2016) by sequential deletion of psmβ1-psmβ2 and then introduction of a nonsense mutation into the start codon of hld gene to abolish its translation (see Supplemental Table 1 for primers used in allelic replacement mutagenesis using pKOR1) (Diep et al., 2016). All strains were grown in tryptic soy broth at 37°C, with erythromycin or chloramphenicol for plasmid selection where appropriate.
Method Details
Generation of plasmids for Staphylococcal expression of 2w-mcherry and 2w-zsgreen
In published work, S. epi was engineered to express the 2w antigen linked to the fluorophore mcherry under control of the agr promoter via plasmid pJL71-2w-gpmcherry (Scharschmidt et al., 2015). Here, we generated a plasmid, pJL74-2w-gpmcherry, in which expression of the 2w-mcherry fusion peptide is driven by the staphylococcal sarA P1 promoter, which we found more conducive to facilitating consistently high and equivalent antigen expression across S. epi and S. aureus (Figure S2A). Specifically, the 2w-gpmcherry ORF was excised from pJL71-2w-gpmcherry at the EcoRI and AscI restriction sites and cloned into the pJL74 backbone (Liese et al., 2013) at the corresponding sites downstream of the sarA promoter sequence, generating pJL74-2w-gpmcherry. Because pJL74–2w-gpmcherry relies on erythromycin selection for transformation and maintenance and certain strains used here are erythromycin resistant, it was further modified to enable selection with chloramphenicol. To generate this pJL74-2w-gpmcherry-cat plasmid, pJL74-2w-gpmcherry was digested with SacII and StyI to linearize and remove the ermC cassette. In parallel, the cat (chloramphenicol acetyltransferase) element was amplified from pIMAY (Monk et al., 2012) with primers TCS149 CATTGATTGCTTTTATTGGGCCCACCTAGGCCCATTATGCTTTGGCAG and TCS150 AACTATGCGGCCGCTCGAGCGCCGCGGTTATAAAAGCCAGTCATTAGG, which also contained overlap sequence to the linearized pJL74. These elements were then joined using the NEBuilder® HiFi DNA Assembly kit (New England Biosciences). Finally, to allow for better tracking of fluorescent antigen uptake, we generated pJL74-2w-zsgreen. The genetic sequence for zsgreen was optimized for expression in Staphylococcus and synthesized with an upstream 2w peptide and linker sequence as well as flanking EcoRI and AscI restriction sites (ATUM) prior to cloning into pJL74 via restriction digest as described above.
Transformation of S. epi, S. aureus and isogenic mutants
Plasmids described above were electroporated into Escherichia coli DC10B, which facilitates subsequent direct transformation into S. epidermidis via direct electroporation (Monk et al., 2012). Electroporation protocol was adapted from (Cui et al., 2015; Löfblom et al., 2007; Sato'o et al., 2018). Briefly, competent bacteria were prepared as follows: bacteria were grown overnight, diluted in B2low medium (2.5% yeast extract, 1% Tryptone, 1% NaCl, 0.5% glucose and 0.1% H2HPO4, wt/vol, pH 7.5), cultured until exponential phase, washed in NaCL 1.5M then distilled water and resuspended in 10% glycerol and stored at −80C. Bacteria were thawed, incubated 5 min at 25C, centrifuged and resuspended with equal volume of 0.5M sucrose in 10% glycerol. 3μg of plasmid was added to competent bacteria and electroporated (2kV, 25μF). 1 mL of B2 medium (2.5% yeast extract, 1% Tryptone, 2.5% NaCl, 0.5% glucose and 0.1% H2HPO4, wt/vol, pH 7.5) was added, bacteria were heat-shocked at 55C for 1min, incubated at 37C with shaking for 5h and plated on tryptic soy agar with appropriate antibiotics for selection. For transformation of S. aureus SF8300 or isogenic mutants thereof, plasmids were electroporated into the restriction deficient S. aureus strain RN4220 and then transferred via Φ11 phage into SF8300. An erythromycin-susceptible SF8300 strain, as described above, was used with the erythromycin-selectable plasmids, pJL74-2w-gpmcherry and pJL74-2w-zsgreen. To enable 2w expression in isogenic mutants of SF8300 (ΔagrA, Δhla, Δpsmα/psmβ/hld) in which ermC is preserved, plasmid pJL74-2w-gpmcherry-cat was used with chloramphenicol selection. In experiments using these mutant strains, wild-type SF8300 containing pJL74-2w-gpmcherry-cat was used as the control.
Bacterial skin colonization and skin abrasion models
S. epi-2w or S. aureus-2w was cultured for 48 hours to achieve comparable, high mCherry expression by flow cytometry. Cells were washed and re-suspended in PBS, and 108-109 colony-forming units (CFUs) were applied via pipette and a sterile PBS-soaked cotton-tipped swab to the back skin of mice starting at 1 week of age. This procedure was repeated every 3 days for a total of three applications to constitute one round of neonatal colonization. To assess the primary immune response to S. epi-2w or S. aureus-2w, mice were colonized as above and then SDLNs (axillary, brachial, and inguinal) were harvested at weaning age (approx. day 21-25), and processed for tetramer staining. In experiments examining bacterial zsgreen uptake by myeloid populations, as well as their expression of pro-IL-1β, neonatal mice were colonized overnight with zsg-S. epi or zsg-S. aureus and harvested 16 hours later.
In certain experiments as noted in the text, mice were colonized with bacteria and then re-challenged two weeks later via intradermal injection into the back skin of 50 μg of 2w1S peptide (EAWGALANWAVDSA; Genscript) in 100uL of Incomplete Freund’s Adjuvant (IFA). SDLNs were harvested 7 days after re-challenge. To replicate physiologic exposure to skin bacteria in the context of skin abrasion, clippers and depilatory cream (Nair™ Hair Remover Body Cream) were first used to remove back hair. The upper layers of epidermis were then disrupted via repeated application and removal of adhesive tape (Shurtape HP-500), and 108–109 CFUs of S. epi-2w or S. aureus-2w were applied as above. This procedure was repeated every 3 days for a total of three times to constitute one round of challenge and to ensure equivalent bacterial burden across all mice. Back skin and SDLNs were harvested 10 days after initiation of the challenge.
Tissue processing and flow cytometry
Isolation of lymph nodes cells was performed either by mashing over sterile wire mesh in 2ml of PBS (for tetramer staining) or via mechanical and enzymatic digest (for dendritic cell staining). For the latter, SDLN were pierced and torn with sharp forceps in 24-well plates and incubated for 15 min at 37°C in 1 ml of digestion media (2mg/ml collagenase I, 2mg/ml collagenase IV, 0.1mg/ml DNase in RPMI with 1% HEPES, 1% penicillin-streptomycin and 10% fetal calf serum). After the first 15 min of incubation, cells were pipetted up and down repeatedly, then returned for a second 15 min incubation. For cell isolation from skin, the entire trunk skin was harvested, lightly defatted, then minced with scissors and re-suspended in a 50ml conical with 1-3ml of digestion media (2mg/ml collagenase XI, 0.5mg/ml hyaluronidase and 0.1mg/ml DNase in RPMI with 1% HEPES, 1% penicillin-streptomycin and 10% fetal calf serum). Following 45 min of shaking incubation at 37°C, an additional 15ml of media was added and the suspension shaken vigorously by hand for 30 sec. It was then filtered through sterile cell strainers (100 μm cell followed by 40 μm), pelleted and re-suspended in PBS for cell counting. Cells were stained in PBS for 30 min at 4°C with surface antibodies and a live dead marker (Ghost Dye™ Violet 510, Tonbo Biosciences). For intracellular staining, cells were fixed and permeabilized using the Foxp3 staining kit (eBioscience). Fluorophore-conjugated antibodies were purchased from eBioscience, BD Biosciences or BioLegend as detailed in the Key Resources Table. Samples were run on a Fortessa (BD Biosciences) in the UCSF Flow Cytometry Core. Accucheck counting beads (Invitrogen) were used calculate absolute numbers of cells. Flow cytometry data was analyzed using FlowJo software (FlowJo, LLC).
Tissue processing for histopathology and scoring of histologic skin inflammation
For histopathology, skin tissue was fixed in 10% formalin, followed by 70% ethanol and embedded in paraffin, sectioned, and stained with H&E by UCSF Mouse Pathology Core. The inflammatory response following tape-stripping was scored as follows: three parameters (overlying crust, dermal neutrophils, and neutrophilic fat infiltration) were scored separately and then added together. For scoring of crust formation, 0, no crust; 1, little crust present; 2, severe crust formation; for scoring of dermal neutrophils, 0, none present; 1, scarce infiltrate; 2, moderate infiltrate; 3, abundant infiltrate; for scoring of subcutaneous fat infiltration, 0, healthy fat; 1, scarce neutrophilic infiltrate; 2, moderate infiltrate; 3, dense infiltrate partially obliterating fat architecture. Scores were independently corroborated by two reviewers who were unblinded to the experimental set-up. Slides were digitally imaged at 20x with the Aperio AT2 scanner (Leica Biosystems, Vista, CA) using a 20x/0.75NA Plan Apo objective with a 2x optical mag changer.
Tetramer staining and enrichment
For identification of 2w+ cells, cell suspensions were stained for 1 hour in the dark at room temperature (15–25°C) with 2W1S:I-Ab–streptavidin-phycoerythrin (PE) at a concentration 10nM. Skin was then directly stained for other surface and intracellular markers as above. For SDLN samples, enrichment for the tetramer-bound fraction was performed via an adapted protocol of the EasySep PE selection kit (Stemcell Technologies) developed by Marc Jenkins’ lab. In brief, 6.25μl of EasySep PE selection cocktail was added to each sample in a total volume of 500μl and the cells were incubated in the dark at room temperature for 15 min. Subsequently, 25 μl of EasySep magnetic particles were added and the cells were incubated at room temperature for an additional 10 min. Finally, cell suspensions were brought to a total volume of 2.5 ml and placed into the EasySep magnet for 5 min. The supernatant (unbound fraction) was poured off and collected in another tube and this process of washing and enriching for magnetically-bound cells was repeated three times until the positively-selected cells (bound fraction) and pooled unbound fraction for each sample were taken for cell counting and staining.
RNA sequencing of neonatal skin, CD4+ Teff and Treg cells from S. epi or S. aureus colonized mice
Newborn C57BL6 mice were colonized with either S. epi Tu3298 or S. aureus SF8300 starting on day 3 of life and every other day thereafter until post-natal day 19. Mice were sacrificed at 21 days of age. An 8mm punch biopsy was obtained from each mouse and placed in RNAlater, and the remainder of the trunk skin was processed for flow staining as above. Single cell suspensions were pelleted and re-suspended in sort buffer (RPMI, 2 mM EDTA, 25mM HEPES, 2% FBS with U/ml RiboLock RNase inhibitor (Thermo Scientific) and stained for 30 min at 4°C with fluorophore-conjugated antibodies specific for CD3, CD4, CD8, CD25, CD45, ICOS, TCRβ, and Tonbo Live-dead Ghost Dye. Teffs (Live CD45+CD3intCD4+CD8negTCRβ+CD25negICOSneg) and Tregs (Live CD45+CD3intCD4+CD8negTCRβ+CD25hiICOShi) were then isolated via cell sorting on a MoFlo XDP (Beckman Coulter) in the UCSF Flow Cytometry Core. Cell pellets were flash frozen and RNA subsequently isolated by Expression Analysis Q2 Solutions using QIAGEN RNeasy Spin columns and quantified via Nanodrop ND-8000 spectrophotometer. For whole skin, RNA was extracted using the QIAGEN RNeasy Fibrous Tissue Mini Kit. RNA quality was checked by Agilent Bioanalyzer Pico Chip. The SMARTer Ultra Low input kit was used to generate cDNA libraries from the sorted cells while Illumina TruSeq Stranded mRNA library preparation was used for whole skin RNA. Libraries were then sequenced to a 25M read depth with Illumina RNASeq. Reads were aligned to UCSC GRCm38/mm10 reference genome with TopHat software (v. 2.0.12) (Trapnell et al., 2009). SAM files were generated with SAMtools from alignment results (Li et al., 2009). Read counts were obtained with htseq-count (0.6.1p1) with the union option (Anders et al., 2015). Differential gene expression was determined using the R/Bioconductor package DESeq2 (Anders and Huber, 2010). Data were further analyzed through the use of IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) (Krämer et al., 2014).
For gene set enrichment analysis, a pre-ranked gene list was generated for each comparison (S. aureus Tregs versus S. epi Tregs; S. aureus Teff versus S. epi Teff) with DESeq2, and GseaPreranked analysis was performed using the GSEA v3.0 tool (Mootha et al., 2003; Subramanian et al., 2005) with default settings using either the subset of genes annotated in IPA Ingenuity software as regulated transcriptionally by IL-1β {ADAMTS4, ADAMTS5, APP, CASP8, COL3A1, CSF2, CXCL2, IFNG, IL17A, IL1A, IL1B, IL22, IL6, MAPK8, MMP13, MMP14, MMP3, MMP8, MMP9, NOS2, S1008, SELE, SMAD6, TNF} or a core Th17 signature identified via in vitro differentiation of murine CD4+ T cells with IL-1β, IL-6 and IL-23 (Ghoreschi et al., 2010).
Bacterial enumeration
For CFU enumeration in Fig S1C, an 8 mm punch biopsy of skin was homogenized in 1 mL of sterile PBS via automated tissue dissociation as above. For enumerations in Fig S1D a cotton swab pre-wetted in PBS was used to sample the back skin, it was then incubated in PBS with shaking for 30 minutes to dissociate the bacteria. In both instances, the cellular suspensions were then plated on antibiotic-containing TSA and Staphylococcal colonies enumerated to quantify total CFUs.
Bone marrow-derived myeloid cell assays
Bone marrow-derived myeloid cells were obtained from culture of bone marrow cells from adult C57BL/6 mice in BMM media (RPMI with 10% FBS, 50 U/mL Penicillin/Streptomycin 10mM HEPES, 1mM Sodium Pyruvate, 50 μM β-mercaptoethanol and 20 ng/mL mGM-CSF (PeproTech)). Cells were grown in 100 mm x 20 mm tissue-culture dishes. At D3, D6 and D8 the media was renewed. After 9 days, non-adherent cells were collected, washed, counted and re-plated at a density of 250,000 per well in 96-well plates. They were then incubated at 37 degrees with sterilely-filtered bacterial supernatants, which were diluted in BMM media to 30% of the total volume. Cells incubated with the same percentage of corresponding bacterial media (TSB for Staphylcoccus spp, BHI for Corynebacterium spp) in BMM media served as controls. After 2 hours of incubation, media supernatant was collected from each well. Lactate dehydrogenase (LDH) was measured with the Pierce LDH cytotoxicity assay kit (Thermo Scientific) according to manufacturer’s instructions to assess cell death. Values are reported as ΔLDH, meaning the LDH activity of the condition subtracted by the LDH activity of corresponding media control. The concentration of IL-1β was measured in cell culture supernatants using enzyme-linked immunosorbent assay (ELISA) (R&D Systems) according to the manufacturer’s instructions.
Quantification and Statistical Analysis
The number of mice per group is annotated in corresponding figure legends. Data followed a Gaussian distribution and variation was similar between groups for each condition analyzed. Significance was assessed using the unpaired Student’s t test or one-way ANOVA with a Tukey post-test in GraphPad Prism software (GraphPad). In all figures, the mean value is visually depicted. P values correlate with symbols as follows: ns = not significant, p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Mice were allocated randomly into experimental groups after matching for age and gender.
Data and Software Availability
RNA sequencing data from Teff from skin of S. epi and S. aureus colonized mice was previously published and is available in NCBI GEO (GSE114398). RNA sequencing data for skin Tregs and whole skin is available in NCBI GEO (GSE130987). Sequences for plasmids pJL74-2w-gpmcherry, pJL74-2w-zsgreen and pJL74-2w-gpmcherry-cat have been deposited in GenBank with IDs MK928252, MK928253 and MK928254.
Supplementary Material
Key Resources Table
| Reagent | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Per-Cp-Cy5.5 anti-mouse TCR-beta (clone H57-597) | Biolegend | Cat# 109228; RRID: AB_1575173 |
| Per-Cp-Cy5.5 anti-mouse Ly6G (clone 1A8) | Biolegend | Cat# 127616; RRID: AB_1877271 |
| Per-Cp-Cy5.5 anti-mouse CD24 (clone M1/69) | Biolegend | Cat# 101823; RRID: AB_1595596 |
| FITC anti-mouse/Rat ICOS (clone C398.4A) | Thermo Fisher Scientific | Cat# 11-9949-82; RRID: AB_465458 |
| FITC anti-mouse CD3 (clone 17A2) | BD Biosciences | Cat# 555274; RRID: AB_395698 |
| FITC anti-mouse GL3 (clone eBioGL3) | Thermo Fisher Scientific | Cat# 11-5711-82; RRID: AB_465238 |
| Pe-Cy7 anti-mouse CD8 (clone 53-6.7) | BD Biosciences | Cat# 552877; RRID: AB_394506 |
| Pe-Cy7 anti-mouse CD11c (clone HL3) | BD Biosciences | Cat# 558079; RRID: AB_647251 |
| Pe-Cy7 anti – mouse pro-IL-1 beta (NJTEN3) | Thermo Fisher Scientific | Cat# 25-7114-82; RRID: AB_2573526 |
| PE anti-mouse CD4 (clone GK1.5) | Biolegend | Cat# 100407; RRID: AB_312692 |
| PE anti-mouse CD207 (clone eBioL31) | Thermo Fisher Scientific | Cat# 12-2075-82; RRID: AB_763452 |
| APC-efluor 780 anti-mouse B220 (clone RA3-6B2) | Thermo Fisher Scientific | Cat# 47-0452-80; RRID: AB_1518811 |
| APC-efluor 780 anti-mouse CD3 (clone 145-2C11) | Thermo Fisher Scientific | Cat# 47-0031-82; RRID: AB_11149861 |
| APC-efluor 780 anti-mouse CD11b (clone M1/70) | Thermo Fisher Scientific | Cat# 47-0112-82; RRID: AB_1603193 |
| APC-efluor 780 anti-mouse CD11c (clone N4818) | Thermo Fisher Scientific | Cat# 47-0114-82; RRID: AB_1548652 |
| APC-efluor 780 anti-mouse F4/80 (clone BM8) | Invitrogen | Cat# 47-4801-82; RRID: AB_2735036 |
| AlexaFluor700 anti-mouse CD45 (clone 30-F11) | Invitrogen | Cat# 56-0451-82; RRID:AB_891454 |
| APC anti-mouse CD44 (clone IM7) | Thermo Fisher Scientific | Cat# 17-0441-82; RRID: AB_469390 |
| APC anti-mouse MHCII (clone M5/114.15.2) | Thermo Fisher Scientific | Cat# 17-5321-82; RRID: AB_469455 |
| APC anti-mouse CD4 (clone GK1.5) | Thermo Fisher Scientific | Cat# 47-0041-82; RRID:AB_11218896 |
| APC anti-mouse CD103 (clone 2E7) | Thermo Fisher Scientific | Cat# 17-1031-82; RRID: AB_1106992 |
| e450 anti-mouse MHCII (clone M5/114.15.2) | Thermo Fisher Scientific | Cat#48-5321-82; RRID: AB_1272204 |
| e450 anti-mouse Foxp3 (clone FJK-16S) | Thermo Fisher Scientific | Cat#48-5773-82; RRID: AB_1518812 |
| BV480 anti-mouse CD44 (clone IM7) | BD Biosciences | Cat# 566116; RRID:AB_655116 |
| BV605 anti-mouse Ly6C (clone HK1.4) | Biolegend | Cat#128035; RRID: RRID:AB_2562352 |
| BV605 anti-mouse CD8 (clone 53-6.7) | Biolegend | Cat# 100744; RRID: AB_2562609 |
| BV605 anti-mouse TCR-beta (clone H57-597) | BD Biosciences | Cat# 562840; RRID: AB_2687544 |
| BV650 anti-mouse CD11c (clone N418) | Biolegend | Cat# 117339; RRID:AB_2562414 |
| BV650 anti-mouse CD4 (clone RM4-5) | BD Biosciences | Cat#563747; RRID: AB_2716859 |
| BV711 anti-mouse CD11b (clone M1/70) | Biolegend | Cat#101241; RRID:AB_11218791 |
| BV711 anti-mouse CD3 (clone 145-2C11) | BD Biosciences | Cat#563123; RRID: AB_2687954 |
| BV786 anti-mouse CD64 (clone X54-5/7.1) | BD Biosciences | Cat#741024; RRID:AB_2740644 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase from Clostridiumhistolyticum, Type XI | Sigma-Aldrich | Cat#C9407 |
| Collagenase from Clostridiumhistolyticum, Type I | Sigma-Aldrich | Cat#SCR103 |
| Collagenase from Clostridiumhistolyticum, Type IV | Sigma-Aldrich | Cat#lS00418 |
| DNase | Sigma-Aldrich | Cat#DN25 |
| Hyaluronidase from bovine testes | Sigma-Aldrich | Cat#H3506 |
| α-hemolysin from Staphylococcus aureus | Sigma-Aldrich | Cat#H9395 |
| Tryptic Soy Medium | BD-Bacto | Cat#211825 |
| Brain Heart Infusion Medium | BD-Bacto | Cat#221570 |
| Recombinant Murine IL-1β | Peprotech | Cat#211-11B |
| Recombinant Murine IL-1α | Peprotech | Cat#211-11A |
| PE-conjugated 2w-loaded I-A(b) Tetramer | Provided by James Moon | N/A |
| Fetal bovine serum | Fisher scientific | Cat#SH3054103 |
| Fetal calf serum | Fisher scientific | Cat#SH3007303 |
| mGM-CSF | PeproTech | Cat#315-03 |
| Critical Commercial Assays | ||
| Pierce LDH cytotoxicity assay kit | Thermo Scientific | Cat#88953 |
| IL1β ELISA kit | R&D systems | Cat#DY401-05 |
| RNeasy Fibrous Tissue Mini Kit | Qiagen | Cat#74704 |
| gentleMACS M Tubes | Miltenyi Biotec | Cat#130-093-236 |
| RNAlater Stabilization Solution | Life Technologies | Cat#AM7020 |
| EasySep™ Mouse PE Positive Selection Kit | StemCell Technologies | Cat# 18554 |
| Mouse FoxP3 Buffer Set | eBiosciences | Cat# 00-5523-00 |
| Ghost Dye™ Violet 510 Live/Dead Stain | Tonbo Biosciences | Cat# 13-0870-T100 |
| Ghost Dye Violet 780 Live/Dead Stain | Tonbo | Cat# 13-0865-T100 |
| Experimental Models: Organisms/Strains | ||
| SPF B6 mice | Jackson Laboratory | C57BL/6J Cat#000664 |
| Ccr7−/− | Jackson Laboratory | B6.129P2(C)-Ccr7tm1Rfor/J Cat#006621 |
| Il1r1−/− | Jackson Laboratory | B6.129S7-Il1r1tm1Imx/J Cat#003245 |
| Experimental Models: Organisms/Strains | ||
| Staphylococcus epidermidis Tü3298 | Provided by Michael Otto (Augustin and Gotz, 1990) | N/A |
| Staphylococcus aureus SF8300 parent strain | Provided by Binh Diep (Diep et al., 2008b) | N/A |
| Staphylococcus aureus SF8300 lacking ermB-encoding plasmid | Provided by Binh Diep | N/A |
| Staphylococcus aureus SF8300 Δhla | Provided by Binh Diep (Diep et al., 2016) | N/A |
| Staphylococcus aureus SF8300 Δpsm | Generated for these studies | N/A |
| Staphylococcus aureus USA300 NRS384 | NARSA | N/A |
| Staphylococcus aureus AD04.E17 | Provided by Heidi Kong & Julia Segre | N/A |
| Staphylococcus epidermidis NIHLM015- 2D12 | Provided by Heidi Kong & Julia Segre | N/A |
| Staphylococcus epidermidis NIHLM037- 5A02 | Provided by Heidi Kong & Julia Segre | N/A |
| Staphylococcus aureus Mu50φ | Provided by Paul Sullam | N/A |
| Staphylococcus epidermidis BCM060 | BEI | N/A |
| Staphylococcus epidermidis W23144 | BEI | Cat#HM-142 |
| Staphylococcus epidermidis SK135 | BEI | Cat#HM-118 |
| Staphylococcus hominis SK119 | BEI | Cat#HM-119 |
| Corynebacterium accolens ATCC 49725 | ATCC | N/A |
| Corynebacterium propinquum DSM44285 | Provider by Katherine Lemon | N/A |
Highlights.
Neonatal colonization enables preferential tolerance to a skin commensal vs. pathogen
Antigen-laden DC migration to lymph nodes leads to bacteria-specific T cell expansion
Discrimination of S. epidermidis vs. S. aureus increases commensal-specific Tregs
S. aureus α-toxin increases IL-1β, which limits S. aureus-specific Tregs.
Acknowledgements
We want to thank James Moon for contribution of tetramer reagents and expertise, Heidi Kong, Katherine Lemon, Michael Otto, Julia Segre, and Paul Sullam for sharing of bacterial strains, Michael Rosenblum, Niro Anandasabapathy, Kelly Remedios, Wilson Liao, Jared Liu, Richard Ahn, and Ella Chang for helpful discussions, Yongmei Hu for mouse husbandry, Jarish Cohen for assistance with slide imaging, Sepideh Nozzari for mouse genotyping and other technical lab support, and Michael Rosenblum, Rachel McLoughlin and Bruce Scharschmidt for critical review of the manuscript. Flow cytometry data were generated in the UCSF Parnassus Flow Cytometry Core, which is supported by the Diabetes Research Center (DRC) grant, NIH P30 DK063720. Paraffin-embedding of tissue, sectioning and H&E staining was performed by the UCSF Mouse Pathology Core which is supported by NIH 5P30CA082103-15. This work was primarily funded by T.C.S. grants: K08AR068409, DP2AI144968, Leo Foundation Grant LF18G+S, and Burroughs Wellcome Fund CAMS-1015631.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali N, Rosenblum MD, 2017. Regulatory T cells in Skin. Immunology. doi: 10.1111/imm.12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allgaier H, Jung G, Werner RG, Schneider U, Zähner H, 1986. Epidermin: sequencing of a heterodetic tetracyclic 21-peptide amide antibiotic. Eur. J. Biochem 160, 9–22. [DOI] [PubMed] [Google Scholar]
- Anandasabapathy N, Feder R, Mollah S, Tse S-W, Longhi MP, Mehandru S, Matos I, Cheong C, Ruane D, Brane L, Teixeira A, Dobrin J, Mizenina O, Park CG, Meredith M, Clausen BE, Nussenzweig MC, Steinman RM, 2014. Classical Flt3L-dependent dendritic cells control immunity to protein vaccine. J. Exp. Med 211, 1875–1891. doi: 10.1084/jem.20131397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W, 2010. Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W, 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin J, Götz F, 1990. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol. Lett 54, 203–207. [DOI] [PubMed] [Google Scholar]
- Bae T, Schneewind O, 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58–63. doi: 10.1016/j.plasmid.2005.05.005 [DOI] [PubMed] [Google Scholar]
- Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, Pear WS, Hatton RD, Weaver CT, 2015. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol 16, 286–295. doi: 10.1038/ni.3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Harrison OJ, 2017. Homeostatic Immunity and the Microbiota. Immunity 46, 562–576. doi: 10.1016/j.immuni.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube BJ, Bubeck Wardenburg J, 2013. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel) 5, 1140–1166. doi: 10.3390/toxins5061140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger SD, Bomar L, Lemon KP, 2016. Commensal-Pathogen Interactions along the Human Nasal Passages. PLoS Pathog. 12, e1005633. doi: 10.1371/journal.ppat.1005633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, Belkaid Y, Segre JA, 2018. The human skin microbiome. Nat. Rev. Microbiol 16, 143–155. doi: 10.1038/nrmicro.2017.157 [DOI] [PubMed] [Google Scholar]
- Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, Bry L, Kraj P, Kisielow P, Ignatowicz L, 2013. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497, 258–262. doi: 10.1038/nature12079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-W, Yan D, Singh R, Liu J, Lu X, Ucmak D, Lee K, Afifi L, Fadrosh D, Leech J, Vasquez KS, Lowe MM, Rosenblum MD, Scharschmidt TC, Lynch SV, Liao W, 2018. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 6, 154. doi: 10.1186/s40168-018-0533-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chewning JH, Weaver CT, 2014. Development and survival of Th17 cells within the intestines: the influence of microbiome- and diet-derived signals. J. Immunol 193, 4769–4777. doi: 10.4049/jimmunol.1401835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C, 2009. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587. doi: 10.1016/j.immuni.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, 2010. Skin-resident T cells: the ups and downs of on site immunity. J. Invest. Dermatol 130, 362–370. doi: 10.1038/jid.2009.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K-I, Dowgiert RK, Kupper TS, 2006. The vast majority of CLA+ T cells are resident in normal skin. The Journal of Immunology 176, 4431–4439. [DOI] [PubMed] [Google Scholar]
- Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA, 2009. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4, e7446. doi: 10.1371/journal.pone.0007446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui B, Smooker PM, Rouch DA, Deighton MA, 2015. Enhancing DNA electro-transformation efficiency on a clinical Staphylococcus capitis isolate. J. Microbiol. Methods 109, 25–30. doi: 10.1016/j.mimet.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Deng J, Yu X-Q, Wang P-H, 2019. Inflammasome activation and Th17 responses. Mol. Immunol 107, 142–164. doi: 10.1016/j.molimm.2018.12.024 [DOI] [PubMed] [Google Scholar]
- Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F, 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/S0140-6736(06)68231-7 [DOI] [PubMed] [Google Scholar]
- Diep BA, Le VTM, Badiou C, Le HN, Pinheiro MG, Duong AH, Wang X, Dip EC, Aguiar-Alves F, Basuino L, Marbach H, Mai TT, Sarda MN, Kajikawa O, Matute-Bello G, Tkaczyk C, Rasigade J-P, Sellman BR, Chambers HF, Lina G, 2016. IVIG-mediated protection against necrotizing pneumonia caused by MRSA. Sci Transl Med 8, 357ra124–357ra124. doi: 10.1126/scitranslmed.aag1153 [DOI] [PubMed] [Google Scholar]
- Diep BA, Stone GG, Basuino L, Graber CJ, Miller A, Etages, des S-A, Jones A, Palazzolo-Ballance AM, Perdreau-Remington F, Sensabaugh GF, DeLeo FR, Chambers HF, 2008. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis 197, 1523–1530. doi: 10.1086/587907 [DOI] [PubMed] [Google Scholar]
- Eisenbarth SC, 2019. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol 19, 89–103. doi: 10.1038/s41577-018-0088-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson U, Kurrer MO, Sonderegger I, Iezzi G, Tafuri A, Hunziker L, Suzuki S, Bachmaier K, Bingisser RM, Penninger JM, Kopf M, 2003. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J. Exp. Med 197, 323–331. doi: 10.1084/jem.20021788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Zhou T, Moreno-Vinasco L, Hollett B, Garcia JGN, Bubeck Wardenburg J, 2012. Host response signature to Staphylococcus aureus alpha-hemolysin implicates pulmonary Th17 response. Infect. Immun 80, 3161–3169. doi: 10.1128/IAI.00191-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nish SA, Jiang R, Hou L, Licona-Limón P, Weinstein JS, Zhao H, Medzhitov R, 2013. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 39, 722–732. doi: 10.1016/j.immuni.2013.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang X-P, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun H-W, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ, 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971. doi: 10.1038/nature09447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA, 2011. The skin microbiome. Nat. Rev. Microbiol 9, 244–253. doi: 10.1038/nrmicro2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Crozat K, Henri S, Tamoutounour S, Grenot P, Devilard E, de Bovis B, Alexopoulou L, Dalod M, Malissen B, 2010. Skin-draining lymph nodes contain dermis-derived CD103(−) dendritic cells that constitutively produce retinoic acid and induce Foxp3(+) regulatory T cells. Blood 115, 1958–1968. doi: 10.1182/blood-2009-09-245274 [DOI] [PubMed] [Google Scholar]
- Hodille E, Cuerq C, Badiou C, Bienvenu F, Steghens J-P, Cartier R, Bes M, Tristan A, Plesa A, Le VTM, Diep BA, Lina G, Dumitrescu O, 2016. Delta Hemolysin and Phenol-Soluble Modulins, but Not Alpha Hemolysin or Panton-Valentine Leukocidin, Induce Mast Cell Activation. Front Cell Infect Microbiol 6, 180. doi: 10.3389/fcimb.2016.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y, 2014. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-β-induced Foxp3 expression. J. Immunol 192, 1449–1458. doi: 10.4049/jimmunol.1300387 [DOI] [PubMed] [Google Scholar]
- Kastenmüller W, Torabi-Parizi P, Subramanian N, Lämmermann T, Germain RN, 2012. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150, 1235–1248. doi: 10.1016/j.cell.2012.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar SL, Ibarra JA, Rivera FE, Mootz JM, Davenport JE, Stevens SM, Horswill AR, Shaw LN, 2013. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen 2, 18–34. doi: 10.1002/mbo3.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, Tugendreich S, 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. doi: 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Weid, von der PY, 2015. Lymphatic system: an active pathway for immune protection. Semin. Cell Dev. Biol 38, 83–89. doi: 10.1016/j.semcdb.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liese J, Rooijakkers SHM, van Strijp JAG, Novick RP, Dustin ML, 2013. Intravital two-photon microscopy of host-pathogen interactions in a mouse model of Staphylococcus aureus skin abscess formation. Cell. Microbiol 15, 891–909. doi: 10.1111/cmi.12085 [DOI] [PubMed] [Google Scholar]
- Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, Kao T, Lee SK, Cai SS, Miller RJ, Marchitto MC, Zhang E, Riggins DP, Plaut RD, Stibitz S, Geha RS, Miller LS, 2017. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe 22, 653–666.e5. doi: 10.1016/j.chom.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löfblom J, Kronqvist N, Uhlén M, Ståhl S, Wernérus H, 2007. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J. Appl. Microbiol 102, 736–747. doi: 10.1111/j.1365-2672.2006.03127.x [DOI] [PubMed] [Google Scholar]
- Mailer RKW, Joly A-L, Liu S, Elias S, Tegner J, Andersson J, 2015. IL-1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep 5, 14674. doi: 10.1038/srep14674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-López M, Iborra S, Conde-Garrosa R, Sancho D, 2015. Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur. J. Immunol 45, 119–129. doi: 10.1002/eji.201444651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, Ploegh HL, Murphy TL, Sibley LD, Murphy KM, 2011. CD8α(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 35, 249–259. doi: 10.1016/j.immuni.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, Msallam R, Low I, Shadan NB, Sumatoh HR, Soon E, Lum J, Mok E, Hubert S, See P, Kunxiang EH, Lee YH, Janela B, Choolani M, Mattar CNZ, Fan Y, Lim TKH, Chan DKH, Tan K-K, Tam JKC, Schuster C, Elbe-Bürger A, Wang X-N, Bigley V, Collin M, Haniffa M, Schlitzer A, Poidinger M, Albani S, Larbi A, Newell EW, Chan JKY, Ginhoux F, 2017. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 546, 662–666. doi: 10.1038/nature22795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, O'Connell RM, Iwakura Y, Cheung AL, Cheng G, Modlin RL, 2007. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. The Journal of Immunology 179, 6933–6942. [DOI] [PubMed] [Google Scholar]
- Mold JE, McCune JM, 2012. Immunological tolerance during fetal development: from mouse to man. Adv. Immunol 115, 73–111. doi: 10.1016/B978-0-12-394299-9.00003-5 [DOI] [PubMed] [Google Scholar]
- Monk IR, Shah IM, Xu M, Tan M-W, Foster TJ, 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio 3, e00277–11. doi: 10.1128/mBio.00277-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK, 2007. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. doi: 10.1016/j.immuni.2007.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. doi: 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han S-J, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, Quinones M, Brenchley JM, Kong HH, Tussiwand R, Murphy KM, Merad M, Segre JA, Belkaid Y, 2015. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520, 104–108. doi: 10.1038/nature14052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmüller W, Deming C, Quinones M, Koo L, Conlan S, Spencer S, Hall JA, Dzutsev A, Kong H, Campbell DJ, Trinchieri G, Segre JA, Belkaid Y, 2012. Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119. doi: 10.1126/science.1225152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, Hata TR, Gallo RL, 2016. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Invest. Dermatol 136, 2192–2200. doi: 10.1016/j.jid.2016.05.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Förster R, 2004. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21, 279–288. doi: 10.1016/j.immuni.2004.06.014 [DOI] [PubMed] [Google Scholar]
- Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Santos, Dos L, O'Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y, 2009. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786. doi: 10.1016/j.immuni.2009.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Büttner H, Dunman PM, Rohde H, Cech NB, Fey PD, Horswill AR, 2014. Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J. Bacteriol 196, 3482–3493. doi: 10.1128/JB.01882-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagán AJ, Peters NC, Debrabant A, Ribeiro-Gomes F, Pepper M, Karp CL, Jenkins MK, Sacks DL, 2013. Tracking antigen-specific CD4+ T cells throughout the course of chronic Leishmania major infection in resistant mice. Eur. J. Immunol 43, 427–438. doi: 10.1002/eji.201242715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paharik AE, Parlet CP, Chung N, Todd DA, Rodriguez EI, Van Dyke MJ, Cech NB, Horswill AR, 2017. Coagulase-Negative Staphylococcal Strain Prevents Staphylococcus aureus Colonization and Skin Infection by Blocking Quorum Sensing. Cell Host Microbe 22, 746–756.e5. doi: 10.1016/j.chom.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang IK, Ichinohe T, Iwasaki A, 2013. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8+ T cell responses to influenza A virus. Nat Immunol 14, 246–253. doi: 10.1038/ni.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, Gudjonsson S, Håkansson U, Reizis B, Kotarsky K, Agace WW, 2013. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38, 958–969. doi: 10.1016/j.immuni.2013.03.009 [DOI] [PubMed] [Google Scholar]
- Peschel A, Otto M, 2013. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol 11, 667–673. doi: 10.1038/nrmicro3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP, 2017. The Lymphatic System: Integral Roles in Immunity. Annu. Rev. Immunol 35, 31–52. doi: 10.1146/annurev-immunol-041015-055354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasigade J-P, Trouillet-Assant S, Ferry T, Diep BA, Sapin A, Lhoste Y, Ranfaing J, Badiou C, Benito Y, Bes M, Couzon F, Tigaud S, Lina G, Etienne J, Vandenesch F, Laurent F, 2013. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS ONE 8, e63176. doi: 10.1371/journal.pone.0063176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF, 2016. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. doi: 10.1016/j.ccell.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M, 2008. Regulatory T cells and immune tolerance. Cell 133, 775–787. doi: 10.1016/j.cell.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, Yang SH-Y, Anthony BA, Sverdrup FM, Krow-Lucal E, MacKenzie TC, Johnson DS, Meyer EH, Lohr A, Hsu A, Koo J, Liao W, Gupta R, Debbaneh MG, Butler D, Huynh M, Levin EC, Leon A, Hoffman WY, McGrath MH, Alvarado MD, Ludwig CH, Truong H-A, Maurano MM, Gratz IK, Abbas AK, Rosenblum MD, 2014. Memory regulatory T cells reside in human skin. J. Clin. Invest 124, 1027–1036. doi: 10.1172/JCI72932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato'o Y, Aiba Y, Kiga K, Watanabe S, Sasahara T, Hayakawa Y, Cui L, 2018. Optimized universal protocol for electroporation of both coagulase-positive and -negative Staphylococci. J. Microbiol. Methods 146, 25–32. doi: 10.1016/j.mimet.2018.01.006 [DOI] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong H-A, Lowe MM, Sanchez Rodriguez R, Ali N, Laszik ZG, Sonnenburg JL, Millar SE, Rosenblum MD, 2017. Commensal Microbes and Hair Follicle Morphogenesis Coordinately Drive Treg Migration into Neonatal Skin. Cell Host Microbe. doi: 10.1016/j.chom.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Truong H-A, Gearty SV, Pauli ML, Nosbaum A, Gratz IK, Otto M, Moon JJ, Liese J, Abbas AK, Fischbach MA, Rosenblum MD, 2015. A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity 43, 1011–1021. doi: 10.1016/j.immuni.2015.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner J, Kretschmer D, Klenk J, Otto M, Bühring H-J, Stevanovic S, Wang JM, Beer-Hammer S, Peschel A, Autenrieth SE, 2013. Staphylococcus aureus phenol-soluble modulin peptides modulate dendritic cell functions and increase in vitro priming of regulatory T cells. J. Immunol 190, 3417–3426. doi: 10.4049/jimmunol.1202563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS, 2012. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 36, 873–884. doi: 10.1016/j.immuni.2012.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiani S, Dinh C, Ertelt JM, Moguche AO, Siddiqui I, Smigiel KS, Sharma P, Campbell DJ, Way SS, Urdahl KB, 2013. Pathogen-specific Treg cells expand early during mycobacterium tuberculosis infection but are later eliminated in response to Interleukin-12. Immunity 38, 1261–1270. doi: 10.1016/j.immuni.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma-Kuinkel BK, Wu Y, Tabor DE, Mok H, Sellman BR, Jenkins A, Yu L, Jafri HS, Rude TH, Ruffin F, Schell WA, Park LP, Yan Q, Thaden JT, Messina JA, Fowler VG, Esser MT, 2015. Characterization of alpha-toxin hla gene variants, alpha-toxin expression levels, and levels of antibody to alpha-toxin in hemodialysis and postsurgical patients with Staphylococcus aureus bacteremia. J. Clin. Microbiol. 53, 227–236. doi: 10.1128/JCM.02023-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan N, 2010. Telling apart friend from foe: discriminating between commensals and pathogens at mucosal sites. Innate Immun 16, 391–404. doi: 10.1177/1753425909357577 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Koch MA, Pepper M, Campbell DJ, 2014. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J. Exp. Med 211, 961–974. doi: 10.1084/jem.20131556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LF, del Alcazar D, Stelekati E, Wherry EJ, Davis MM, 2016. Antigen exposure shapes the ratio between antigen-specific Tregs and conventional T cells in human peripheral blood. Proc Natl Acad Sci USA 113, E6192–E6198. doi: 10.1073/pnas.1611723113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJC, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, Granot T, Griesemer A, Lerner H, Kato T, Farber DL, 2016. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med 22, 72–77. doi: 10.1038/nm.4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL, 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, McHugh BJ, Qureshi A, Campopiano DJ, Clarke DJ, Fitzgerald JR, Dorin JR, Weller R, Davidson DJ, 2017. IL-1β-Induced Protection of Keratinocytes against Staphylococcus aureus-Secreted Proteases Is Mediated by Human β-Defensin 2. J. Invest. Dermatol 137, 95–105. doi: 10.1016/j.jid.2016.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, Sanford JA, O'Neill AM, Liggins MC, Nakatsuji T, Cech NB, Cheung AL, Zengler K, Horswill AR, Gallo RL, 2019. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci Transl Med 11, eaat8329. doi: 10.1126/scitranslmed.aat8329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D, 2015. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594. doi: 10.1126/science.aaa7017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data from Teff from skin of S. epi and S. aureus colonized mice was previously published and is available in NCBI GEO (GSE114398). RNA sequencing data for skin Tregs and whole skin is available in NCBI GEO (GSE130987). Sequences for plasmids pJL74-2w-gpmcherry, pJL74-2w-zsgreen and pJL74-2w-gpmcherry-cat have been deposited in GenBank with IDs MK928252, MK928253 and MK928254.