

Abstract
Developmental origins of adult disease (DoHAD) refers to critical gestational ages during human fetal development and beyond when the endocrine-metabolic status of the mother can permanently program the physiology and/or morphology of the fetus, modifying its susceptibility to disease after birth. The aim of this review is to address how DoHAD plays an important role in the phenotypic expression of polycystic ovary syndrome (PCOS), the most common endocrinopathy of women characterized by hyperandrogenism, oligo-anovulation and polycystic ovarian morphology. Clinical studies of PCOS women are integrated with findings from relevant animal models to show how intergenerational transmission of these central components of PCOS are programmed through an altered maternal endocrine-metabolic environment that adversely affects the female fetus and long-term offspring health. Prenatal testosterone-treatment in monkeys and sheep have been particularly crucial in our understanding of developmental programming of PCOS because organ system differentiation in these species, as in humans, occurs during fetal life. These animal models, along with altricial rodents, produce permanent PCOS–like phenotypes variably characterized by LH hypersecretion from reduced steroid negative feedback, hyperandrogenism, ovulatory dysfunction, increased adiposity, impaired glucose-insulin homeostasis and other metabolic abnormalities. The review concludes that DoHAD underlies the phenotypic expression of PCOS through an altered maternal endocrine-metabolic environment that can induce epigenetic modifications of fetal genetic susceptibility to PCOS after birth. It calls for improved maternal endocrine-metabolic health of PCOS women to lower their risks of pregnancy-related complications and to potentially reduce intergenerational susceptibility to PCOS and its metabolic derangements in offspring.
Keywords: developmental programming, polycystic ovary syndrome, hyperandrogenism
Introduction
Alterations in maternal physiology can permanently program adult disease in offspring (Dumesic et al., 2014). Known as the developmental origins of adult disease (DoHAD), this concept originates from the Barker hypothesis, which posits how adverse influences on fetal development cause permanent changes in offspring physiology and metabolism after birth, increasing the risk for disease in adulthood (Barker, 2002, Neel, 1962). Accordingly, fetal stress from maternal undernutrition favors survival of offspring with the greatest capacity for energy storage to survive prolonged nutrient deprivation after birth, with low birth weight followed by infant catch-up growth predisposing individuals to chronic adult disease in today’s nutrient plentiful environment (Desai et al., 2013). Teleologically, fetal undernutrition favors genes important for energy conservation (i.e., thrifty genotype), which would be beneficial in times of food scarcity, but would lead to obesity and diabetes when food becomes abundant later in life (Neel, 1962). Original findings (Barker and Osmond, 1986) that regions of England with the highest infant mortality rates in the early 20th century subsequently had the highest mortality rates from coronary heart disease decades later were supported by later studies showing an association between low birth weight and adult development of cardiovascular disease (CVD), hypertension, insulin resistance and type 2 diabetes mellitus (Barker, 2002).
Maternal overnutrition, on the other hand, also can predispose offspring to adult disease. A “U-shaped curve” in infant birthweight relative to obesity, hypertension and insulin resistance shows that birthweight deviation from an optimal in utero environment, regardless of direction, increases the risk of developing metabolic syndrome in later life (Desai et al., 2013). Collectively, therefore, impaired fetal nutrient availability from placental insufficiency accompanies low infant birth weight, while conversely successful fetal adaptation to maternal nutrient overabundance (i.e. obesity) favors large for gestational age infants (Dumesic et al., 2014).
These different endocrine-metabolic environments in pregnancy have important developmental implications for women with polycystic ovary syndrome (PCOS), a heterogeneous syndrome characterized by luteinizing hormone (LH) hypersecretion, hyperandrogenism, menstrual irregularity and polyfollicular ovarian morphology (Chang and Dumesic, 2018). The aim of this review is to examine how an altered maternal-fetal environment can program adult disease through epigenetic modifications of fetal genetic susceptibility to PCOS after birth, as evidenced by clinical studies of PCOS women integrated with animal models using discrete experimentally-induced prenatal testosterone (T) excess to program a permanent PCOS-like phenotype (Dumesic et al., 2014, Dumesic et al., 2007).
PCOS
Women with PCOS by NIH criteria (i.e., hyperandrogenism with oligo-anovulation, excluding other endocrinopathies) have insulin resistance from abnormal insulin signaling and metabolic dysfunction in insulin-responsive tissues, with increased total abdominal and visceral fat independent of obesity (Chang and Dumesic, 2018, Dumesic et al., 2016). One-third to one-half of these PCOS women in the United States have metabolic syndrome, which is 2–3 times greater in prevalence than that of age-matched women without PCOS and worsened by obesity (Moran et al., 2010). The prevalence of metabolic syndrome is also lower in PCOS women from countries where obesity is less frequent (Moran et al., 2010, Carmina et al., 2006). Morever, 40% of PCOS women develop glucose intolerance or type 2 diabetes by the fourth decade of life (Legro et al., 1999), increasing their risk for pregnancy complications, including diabetes, pregnancy-induced hypertension, pre-eclampsia, preterm birth, impaired endovascular trophoblast invasion and abnormal placentation, with potential fetal programming effects on the offspring (Dumesic et al., 2014).
Genetic contributions to PCOS
Twin studies estimate the heritability of PCOS as 71%, and 38% for monozygotic vs. dizygotic twins, respectively (Chang and Dumesic, 2018), and are supported by a recent longitudinal study of daughters born to women with PCOS and followed from infancy to post-menarche, suggesting a 62% transmission of PCOS phenotype (Crisosto et al., 2019). Similarly, reproductive-aged sisters of women with PCOS have an increased prevalence of a hyperandrogenic PCOS phenotype (Chang and Dumesic, 2018), as do parents of PCOS women with a higher risk of metabolic dysfunction (Sir-Petermann et al., 2002a). Nevertheless, genetic susceptibility to PCOS by gene-wide association studies accounts for < 10% of its heritability to date, with at least 17 replicated PCOS risk genes from different human populations involving reproduction (FSHB, LHCGR, FSHR, DENND1A, RAB5/SUOX, HMGA2, C9orf3, YAP1, TOX3, RAD50, FBN3 and AMH) and metabolism (THADA, GATA/NEIL2, ERBB4, SUMOP11, INSR and KRR1) (Abbott et al., 2019). Consequently, PCOS appears to have complex polygenic, pathogenic origins that are susceptible to endocrine-metabolic influences during pregnancy to account for its remaining heritability and variable phenotypic expression (Abbott et al., 2019). Shared genetic traits across all PCOS phenotypes; whether clinically diagnosed by NIH, Rotterdam criteria, or self-reported; implicate common underlying pathophysiological mechanisms involving neuroendocrine, metabolic and reproductive contributions (Day et al., 2018)
The maternal environment in PCOS
Understanding how the maternal environment of PCOS women affects their offspring must consider obesity as a covariable since two-thirds of women in the United States are overweight or obese, as are one-half of Australian and one-third of Danish women (ASRM, 2015). Maternal obesity is strongly associated with several pregnancy and perinatal complications, including gestational diabetes and hypertension, preeclampsia, preterm delivery, stillbirth, early neonatal death, and small- as well as large-for-gestational age infants (ASRM, 2015). Obese women also are more likely than average-weight women to have infants with heart defects, ventral wall defects, neural tube defects and multiple anomalies (ASRM, 2015). From a DoHAD perspective, maternal obesity increases the risk of premature mortality from a cardiovascular event in their adult offspring, adjusting for maternal age, social class, gestational age, infant sex and birth weight, implying intergenerational programming of metabolic disease (Reynolds et al., 2013).
An important question is whether PCOS women are at increased risk for metabolic dysfunction in pregnancy compared to other pregnant women. Preconception hyperandrogenemia and glucose intolerance in PCOS women predict several suboptimal maternal and neonatal outcomes, including preeclampsia (Christ et al., 2019). During pregnancy, PCOS women continue to have greater serum androgen levels, higher fasting and 2-hour post-prandial insulin values (Kent et al., 2018, Sir-Petermann et al., 2002b) and elevated anti-mullerian hormone (AMH) levels (Kent et al., 2018, Tata et al., 2018) compared to normal women. Subsequently, the prevalence of gestational diabetes, glucose intolerance and type 2 diabetes in PCOS women is 3- to 5-fold higher than that of other women, independent of age or body mass index (BMI), and is worsened by obesity (Teede et al., 2018). Contributing to these events, normal- and over-weight PCOS women gain more weight by mid-gestation than pregnant women without PCOS (Kent et al., 2018), with about 40% of PCOS women developing gestational diabetes and other pregnancy complications (Abbott et al., 2019). During pregnancy, moreover, PCOS versus normal women exhibit exaggerated dyslipidemia and elevated circulating inflammatory markers that predict greater risks of gestational diabetes, hypertensive disorders and adverse obstetrical/neonatal outcomes, respectively (Palomba et al., 2014a, Palomba et al., 2014b).
From a mechanistic perspective, maternal hyperandrogenemia from PCOS is unlikely to directly program PCOS in offspring if placental aromatization is normal (Hickey et al., 2009), although reduced aromatase activity in term placenta from PCOS women has been reported (Maliqueo et al., 2013). Nevertheless, mid-gestation maternal T levels positively associate with high AMH levels in adolescent female offspring, suggesting that elevations in mid-gestation maternal T perturb subsequent ovarian function in daughters (Hart et al., 2010). More probable, however, maternal metabolic dysfunction in PCOS mothers compromises placental function of a female fetus with a genetic susceptibility to PCOS, promoting fetal hyperinsulinemia as a cause for hyperandrogenism and altered folliculogenesis in utero (Abbott et al., 2019, Dumesic et al., 2007, Dumesic et al., 2014, Palomba et al., 2012, Palomba et al., 2015).
The fetal environment
Increased susceptibility to type 2 diabetes and obesity in offspring of type 1 diabetic mothers is metabolically determined in utero. Adolescent offspring of mothers with type 1 diabetes have increased body weight, dyslipidemia and insulin resistance predicted by their previous exposure to hyperinsulinemia in utero (Weiss et al., 2000). As adults, offspring of mothers with gestational or type 1 diabetes who were previously exposed to fetal hyperglycemia exhibit epigenetic changes in subcutaneous (SC) adipose, including decreased leptin promoter methylation, increased leptin gene and protein expression, decreased mitochondrial function and impaired fat storage (Hansen et al., 2017).
Therefore, maternal metabolic abnormalities likely affect the human fetal ovary during the second trimester of development when the primordial follicular pool gets established. Mid-trimester human fetal ovaries have several steroidogenic enzymes, genes encoding steroid-signalling pathways, and receptors to steroids, insulin and insulin-like growth factors (Chang and Dumesic, 2018, Cole et al., 2006). They also can metabolize pregnenolone sulfate to dehydroepiandrosterone (DHEA) and androstenedione, and secrete DHEA, progesterone and estrone, with lesser amounts of androstenedione, estradiol and T in vitro (Chang and Dumesic, 2018). Lacking functional LH-like receptors (Wilson and Jawad, 1979), however, mid-trimester human fetal ovaries are likely less responsive than testes to the transient mid-gestational rise in fetal gonadotropins, which causes a transient sexual dimorphism in amniotic androgen levels that disappears by birth (Chang and Dumesic, 2018, Palomba et al., 2012, Beck-Peccoz et al., 1991).
Instead, human fetal ovaries probably produce androgens in response to hyperinsulinemia in vivo, contributing to a transient mid-gestational rise of serum androgen levels into the normal male range in 40% of female fetuses that diminish below male levels in term umbilical cord blood (Beck-Peccoz et al., 1991). As evidence for mid-gestational fetal hyperandrogenism accompanying an altered endocrine-metabolic maternal environment, amniotic fluid T levels are elevated in female fetuses of PCOS (Palomba et al., 2012) and diabetic (Barbieri et al., 1986) mothers alike, while hyperplasia of theca and pancreatic beta cells accompany ovarian theca-lutein cysts in hirsute female stillbirth offspring of diabetic women (Driscoll et al., 1960, Hultquist and Olding, 1981). Umbilical cord testosterone levels at birth also are elevated in some (Barry et al., 2010), but not all (Anderson et al., 2010), female infants of PCOS mothers. Consequently, elongated anogenital distance, a reliable postnatal biomarker of mid-gestational fetal hyperandrogenism (Barrett et al., 2018), can be found in both female infants of PCOS mothers and in women with PCOS (Wu et al., 2017, Sanchez-Ferrer et al., 2017).
Animal models for PCOS
Reproduction
To understand causal mechanisms underlying these human maternal-fetal relationships, animal models have employed prenatal exposure to androgen excess in rodents, monkeys and sheep to induce metabolic and reproductive abnormalities resembling PCOS (Dumesic et al., 2007, Abbott et al., 2019, Padmanabhan and Veiga-Lopez, 2013). Prenatally T-treated monkeys and sheep have been essential animal models used to program a permanent PCOS–like phenotype of LH hypersecretion, ovarian/adrenal hyperandrogenism (monkeys) or functional hyperandrogenism (androgen receptor upregulation, sheep), ovulatory dysfunction and impaired glucose-insulin homeostasis (Dumesic et al., 2007) (Table 1). This is because tissue differentiation in these species is completed during fetal life (i.e., precocial species), as in humans (Figure 1A–C), unlike mice in which only partial differentiation is completed before birth (i.e., altricial species) (Padmanabhan et al., 2007).
Table 1.
PCOS Traits in Humans Compared to the Prenatally Androgenized Monkey and Sheep Model
| PCOS TRAIT | HUMAN | PRENATALLY ANDROGENIZED RHESUS MONKEYS | PRENATALLY ANDROGENIZED FEMALE SHEEP | |
|---|---|---|---|---|
| EARLY TREATED | LATE TREATED | |||
| Reproductive | ||||
| Ovarian hyperandrogenism | Yes | Yes | Yes | Ovarian androgen receptor upregulation |
| Anovulation | Yes | Yes | Yes | Yes |
| Enlarged polyfollicular ovaries | Yes | Yes | Yes | Yes |
| LH hypersecretion | Yes | Yes | No | Yes |
| Reduced steroid negative feedback on LH | Yes | Yes | Yes | Yes |
| Impaired fertility | Yes | Yes | Yes | Yes |
| Metabolic | ||||
| Insulin resistance | Yes | Yes | No | Yes |
| Beta cell impairment | With increasing BMI | Yes | No | ? |
| Hyperglycemia | With increasing BMI | Yes | Yes | No |
| Increased type 2 diabetes | Yes | Yes | No | Unknown |
| Increased abdominal fat | Yes | Yes | With increasing BMI | ? |
| Hypertension | Yes | Unknown | Unknown | Yes |
| Dyslipidemia | Yes | Yes | Unknown | Yes |
Figure 1.
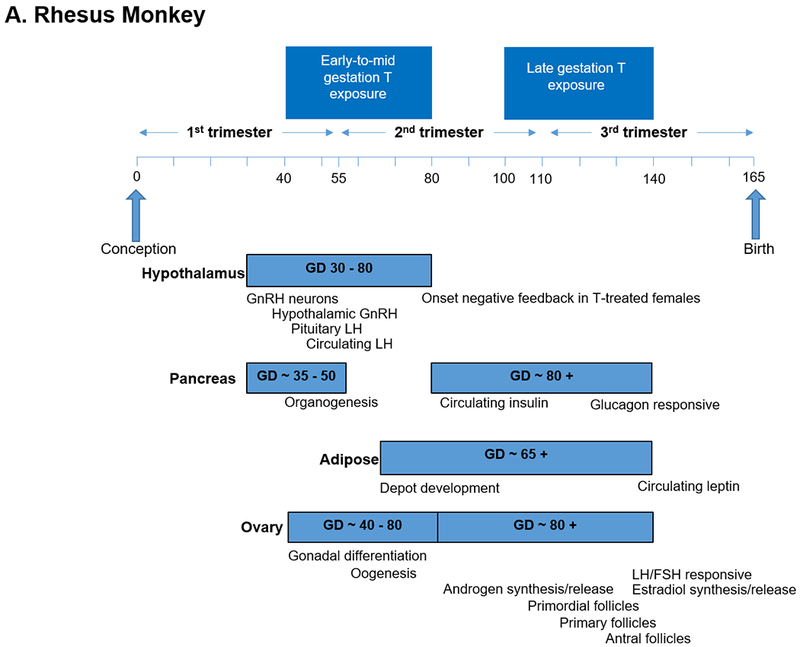
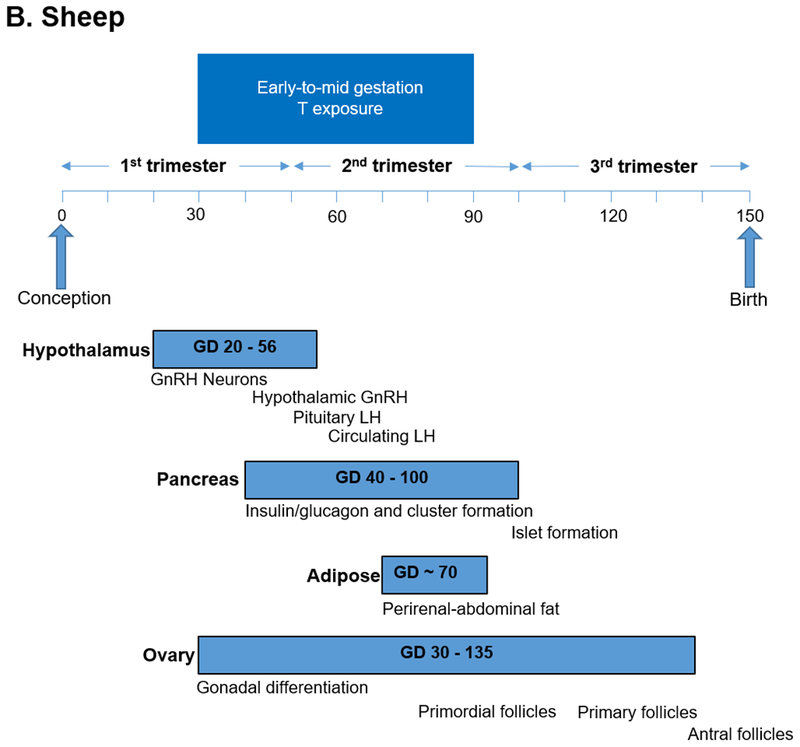
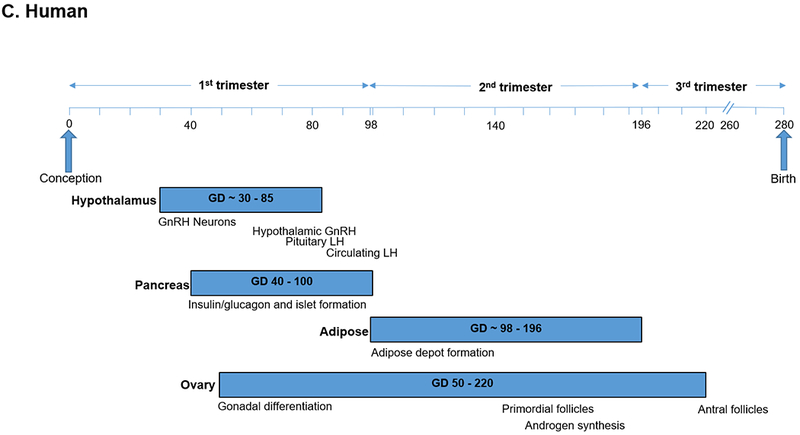
Timing of developmental programming of critical target tissues in a) rhesus monkeys, b) sheep and c) humans as precocial species (Padmanabhan and Veiga-Lopez, 2013, Abbott et al., 2005).
Neuroendocrine dysfunction
Neuroendocrine PCOS-like traits in adult prenatally T-treated female rhesus monkeys (Abbott et al., 2008) and sheep (Padmanabhan and Veiga-Lopez, 2013) are characterized by LH hypersecretion from reduced hypothalamic sensitivity to steroid negative feedback and enhanced GnRH pulsatility (Sarma et al., 2005, Robinson et al., 1999, Robinson et al., 2002, Padmanabhan et al., 2006, Abbott et al., 2018). Adult rats exposed to prenatal T also exhibit LH hypersecretion and hyperandrogenism (Tehrani et al., 2014). Furthermore, prenatal androgen antagonism with the antiandrogen, flutamide, restores normal LH surge characteristics in prenatally T-treated sheep (Padmanabhan et al., 2015) and estrous cyclicity in mice (Sullivan and Moenter, 2004).
Reduced hypothalamic sensitivity to progesterone negative feedback on LH secretion also is a hallmark of PCOS women (Chhabra et al., 2005) and is similarly restored with flutamide (Chhabra et al., 2005, Eagleson et al., 2000). Comparable neuroendocrine abnormalities are found in adolescent hyperandrogenic girls, likely representing a precursor to adult PCOS (Lundgren et al., 2018). Moreover, PCOS women show a sexually dimorphic pattern of exaggerated early LH responsiveness to GnRH analog that more closely resembles that of men or women with congenital adrenal virilizing disorders (e.g., classical CAH and adrenal virilizing carcinoma) than normal women (Rosenfield et al., 1990, Barnes et al., 1994). These common characteristics between prenatally T-treated animal models of PCOS and PCOS women implicate androgen excess during human fetal development with permanently reduced steroid negative feedback on LH and enhanced GnRH pulsatility that persists into adult life.
Ovarian dysfunction
Developmental programming of ovarian function resembling that of PCOS has been more controversial. Prenatal T-treatment in both monkeys and sheep induces female subfertility, which includes impaired quality of primate oocytes (Dumesic et al., 2002, Steckler et al., 2007b). Enlarged, polyfollicular ovaries also occur in both animal models (Abbott et al., 1997, Padmanabhan et al., 2006, Padmanabhan et al., 2012) as well as in prenatally-T treated adult rats (Tehrani et al., 2014), and are accompanied by hyperandrogenism in prenatally T-treated rhesus monkeys (Abbott et al., 2002) and rats (Tehrani et al., 2014), and by ovarian androgen receptor upregulation in prenatally T-treated sheep (Ortega et al., 2009). Polyfollicular ovaries, however, do not occur in prenatally dihydrotestosterone-treated sheep (Steckler et al., 2007a), suggesting endocrine-metabolic actions on the fetal ovaries other than androgen. Moreover, an exaggerated age-related decline in serum AMH levels from normal values in T-exposed monkeys is atypical of PCOS (Dumesic et al., 2009).
In contrast to these animal models, AMH overproduction is a characteristic of PCOS women. Elevated serum AMH levels in PCOS women represent the opposing effects of stimulatory reproductive (hyperandrogenism and increased antral follicle number) versus inhibitory metabolic (body fat) factors (Guedikian et al., 2018), although some PCOS women may have a steeper age-related decline in circulating AMH levels than normal women (Ahmad et al., 2018).
Placental aromatization
Whether maternal T can serve as a source of fetal hyperandrogenism or a programming mechanism for a PCOS-like phenotype in female offspring remains unclear. Gestational AMH excess in mice, however, can induce LH-mediated maternal T excess with reduced placental aromatization of maternal androgens, potentially programming a PCOS-like phenotype in female offspring (Tata et al., 2018).
Reduced aromatase activity in term placenta from PCOS women also has been reported (Maliqueo et al., 2013), potentially increasing exposure of female fetuses to androgen excess. PCOS-related enhancement of fetal hyperandrogenism through placental dysfunction agrees with metabolically compromised PCOS placenta exhibiting features related to hypoxia, a condition known to downregulate placental aromatase (Kumar et al., 2018, Koster et al., 2015). Maternal androgens of PCOS mothers, however, may not substantially contribute to programing PCOS in their offspring due to sufficient placental aromatization (Hickey et al., 2009). More likely, an altered maternal endocrine-metabolic environment accompanying both PCOS and placental compromise (Koster et al., 2015, Palomba et al., 2015) probably promotes fetal ovarian hyperandrogenism via hyperinsulinemia acting as a secretegogue in utero, with complex androgen-insulin interactions reprograming sensitive target tissues in susceptible female offspring (Dumesic et al., 2007, Dumesic et al., 2014, Abbott et al., 2019).
Metabolism
Prenatally T-treated monkeys and sheep as precocial species provide mechanistic links between endocrine-metabolic dysfunction in pregnancy and its long-term reproductive and metabolic consequences in offspring. In prenatally T-treated monkeys and sheep, maternal glucose intolerance causes transient hyperinsulinemia in their female fetuses. Specifically, prenatal T-treatment in rhesus monkeys impairs maternal glucose tolerance and stimulates fetal insulin release, which then potentiates insulin action within the fetus (Abbott et al., 2010). Prenatal T-treatment in sheep reduces progesterone levels and induces hyperinsulinemia (Abi Salloum et al., 2015). But when prenatally-T treated sheep are co-treated simultaneously with either flutamide or rosiglitazone, maternal progesterone levels are improved, while juvenile insulin resistance and early adult hyperleptinemia are prevented (Cardoso et al., 2016).
In addition, mid-gestation fetal sheep ovaries express LH receptors with expression levels responsive to the prevailing androgen environment (Hogg et al., 2011). These endocrine-metabolic interactions during pregnancy are also influenced by placental function, which further modifies the in utero environment to program offspring in different ways.
Metabolic dysfunction
Mid-gestational prenatal T-treatment in rhesus monkeys and sheep programs adipose dysfunction and insulin resistance in adult offspring (Dumesic et al., 2007, Padmanabhan et al., 2010). Prenatally T-treated female rhesus monkeys develop a PCOS-like phenotype characterized by progressive metabolic dysfunction with age, insulin resistance and preferential abdominal adiposity accompanied by hyperlipidemia (Eisner et al., 2000, Eisner et al., 2003, Bruns et al., 2007, Zhou et al., 2007). Early-to-mid gestation T-treated female rhesus monkeys exhibit increased visceral fat (Eisner et al., 2003), adipose insulin resistance and impaired insulin secretion (Zhou et al., 2007, Abbott et al., 2019), while late gestation T-treated females have increased total body (non-visceral) fat (Bruns et al., 2007), impaired insulin sensitivity and intact pancreatic insulin secretion, contributing to 27.3% and 11.1% rates of type 2 diabetes in the former and latter females, respectively. A TGF-β signaling pathway involving altered DNA methylation patterns of visceral fat in early-to-mid gestation T-treated female rhesus monkeys implies an epigenetic basis for reprogramming of adipose (Xu et al., 2011).
Adult prenatally T-treated sheep also develop impaired insulin sensitivity (Recabarren et al., 2005, Padmanabhan et al., 2010, Cardoso et al., 2016), along with hypertension and hypercholesterolemia after puberty (King et al., 2007) as important metabolic abnormalities, although their long-term risks of developing increased adiposity and diabetes with age remain unclear since studies were performed through only 2 years of life (i.e., prime reproductive life). Interestingly, adult rats exposed to prenatal T also exhibit insulin resistance in the absence of overt glucose intolerance (Noroozzadeh et al., 2015).
Adipogenic dysfunction
Subcutaneous (SC) adipose normally increases lipid storage capacity through adipocyte enlargement and new adipocyte formation to buffer fatty acid influx when caloric intake exceeds energy utilization (Romacho et al., 2014). Prenatally-T treated adult rhesus monkeys have an impaired ability to increase SC lipid storage relative to BMI (Bruns et al., 2007), suggesting abnormal adipogenesis, whereby multipotent adipose stem cells (ASCs) first undergo commitment to preadipocytes and then differentiate into newly-formed adipocytes (Chazenbalk et al., 2013). Using adipogenic gene markers, SC abdominal adipose of aging early T-treated females shows impaired preadipocyte differentiation into adipocytes (i.e., decreased C/EBPα) accompanying hyperandrogenemia and enhanced earlier ASC commitment to preadipocytes (i.e., increased Zfp423) possibly compensating for subsequent impaired preadipocyte differentiation (Keller et al., 2014). In agreement, prenatal ZFP423 knockout in mice fed a high fat diet exaggerates diet-induced obesity, ectopic fat deposition and insulin resistance (Shao et al., 2017). In support of developmental programming as a contributor to such events, studies of prenatally-T treated juvenile female sheep show increased ASC commitment to preadipocytes and decreased preadipocyte differentiation of visceral adipocytes, with the later prevented by dual prenatal flutamide/rosiglitazone co-treatment (Puttabyatappa et al., 2018).
In normal human SC abdominal adipose, androgen inhibits early-stage human SC abdominal adipogenesis, induces insulin resistance and impairs catecholamine-stimulated lipolysis through reduced protein expression of β2-adrenergic receptor and hormone-sensitive lipase (HSL) independent of BMI or age (Chazenbalk et al., 2013, Arner, 2005, Dicker et al., 2004, Corbould, 2007) (Figure 2). Similarly, in nonobese insulin-resistant PCOS women, SC abdominal adipose exhibits lipolytic catecholamine resistance from diminished protein levels of β2-adrenergic receptor, HSL and protein kinase A regulatory-IIβ component (PKA-RegIIβ) (Faulds et al., 2003, Ek et al., 1997). Such androgen-related events in vivo would likely constrain SC fat storage and cause lipotoxicity from ectopic lipid accumulation in non-adipose tissues (Chazenbalk et al., 2013), agreeing with findings in normal-weight PCOS women of altered SC abdominal ASC gene expression of adipogenic/angiogenic functions involving T through TGF-β signaling (Dumesic et al., 2019).
Figure 2. Hypothetical model of androgen actions on subcutaneous abdominal adipogenesis in normal-weight PCOS women.
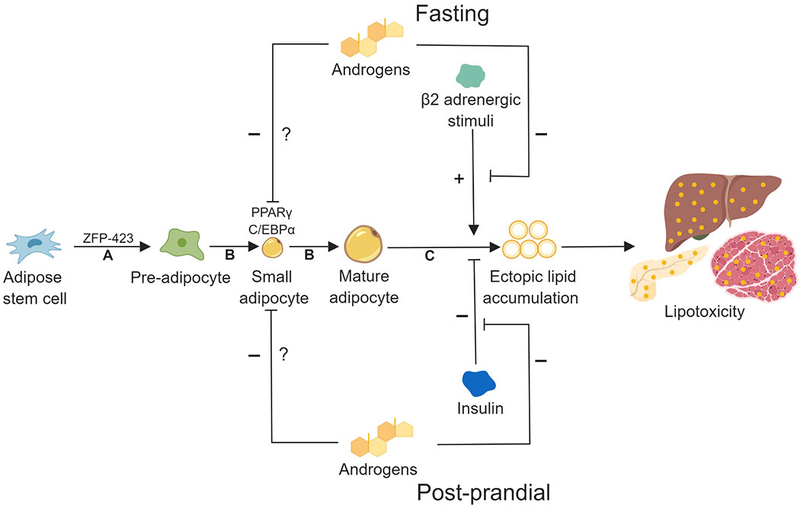
In this model, androgen 1) inhibits preadipocyte differentiation to mature adipocytes, 2) blocks catecholamine-induced lipolysis and 3) impairs insulin-inhibition of lipolysis. Exaggerated adipose stem cell (ASC) development to adipocytes also occurs via androgen-independent mechanisms. Theoretically, exaggerated ASC lipid accumulation within a hyperandrogenic environment could predispose PCOS women to lipotoxicity. A: Commitment; B: Differentiation/lipid accumulation; C: Lipolysis
In the absence of systemic T, however, cultured SC abdominal ASCs from normal-weight PCOS women show exaggerated ASC commitment to preadipocytes followed by enhanced adipocyte lipid content during mature adipocyte formation that negatively and positively correlate with circulating fasting glucose and androgen levels, respectively (Fisch et al., 2018). Such exaggerated ASC commitment to preadipocytes in cultured SC abdominal ASCs from these PCOS women, possibly accompanying intra-adipocyte hyperandrogenism (O’Reilly et al., 2017), likely represents a regulatory mechanism to maintain glucose-insulin homeostasis during accelerated fat accretion (Fisch et al., 2018), as evident in nonobese PCOS women by an altered gene expression favoring SC abdominal lipid metabolism (Chazenbalk et al., 2012).
In contrast, human visceral fat normally shows increased lipolytic activity and resistance to T inhibition of catecholamine-induced lipolysis compared to SC abdominal fat, despite both fat depots expressing androgen receptors (Dicker et al., 2004). In addition, visceral adipose of nonobese PCOS women shows exaggerated catecholamine-induced lipolysis accompanying normal antilipolytic insulin action (Ek et al., 2002, Arner, 2005). This suggests that upregulation of visceral fat lipolysis from a functional increase in the PKA-HSL complex could promote insulin resistance secondary to elevated portal free fatty acids (Ek et al., 2002, Arner, 2005). In normal-weight PCOS women, therefore, such a phenomenon could promote lipolysis for fatty acid oxidation through insulin resistance to curtail fat accretion over time, as previously shown in nondiabetic Pima Indians with a proclivity towards excess weight gain (Swinburn et al., 1991).
Altered SC abdominal adipocyte size
The size distribution of adipocytes within an adipose depot represents a balance between adipocyte enlargement and new adipocyte formation to buffer fatty acid influx (Romacho et al., 2014). An increased proportion of small SC abdominal adipocytes occurs in prenatally-T treated adult rhesus monkeys and sheep (Keller et al., 2014, Veiga-Lopez et al., 2013). This altered adipocyte morphology in early adult prenatally-T treated sheep occurs when visceral adiposity and insulin sensitivity are still normal and precedes metabolic dysfunction in later life, perhaps as a compensatory adaptation to the perturbed intrauterine environment (Cardoso et al., 2016). Interestingly, reduced adipocyte size in prenatally-T treated sheep is not reversed with flutamide cotreatment, implying endocrine-metabolic mechanisms apart from androgen, including possible estrogen action in this species (Puttabyatappa et al., 2018, Cardoso et al., 2016).
An increased proportion of small SC abdominal adipocytes also occurs in normal-weight PCOS women (Dumesic et al., 2016) and precedes the development of enlarged mature adipocytes in overweight PCOS women (Manneras-Holm et al., 2011), explaining why some, but not all, such individuals maintain normal glucose-insulin homeostasis (Chang and Dumesic, 2018). In this regard, enhanced small adipocyte formation accompanying ZFP423 upregulation and epigenetic changes in the ZFP423 promoter region protect against insulin resistance in humans (Longo et al., 2018). That regional fat depots develop in the human fetus between 14 and 28 weeks of gestation supports the hypothesis that abdominal fat can be developmentally programmed during early-to-mid gestation (Poissonnet et al., 1988) (Figure 1).
Maternal-fetal interactions
Altered endocrine-metabolic interactions in pregnancy also can influence fetal growth. In this regard, prenatally T-treated female sheep (Steckler et al., 2005, Manikkam et al., 2004, Crespi et al., 2006) and rats (Slob et al., 1983, McGivern, 1989) exhibit intrauterine growth retardation and low birth weight near term (Padmanabhan et al., 2006, McGivern, 1989, Manikkam et al., 2004). In prenatally T-treated fetal sheep, these events accompany advanced placental differentiation (Beckett et al., 2014) that is insufficient to maintain placental function during late gestation, leading to intrauterine growth retardation, mainly in female offspring (Beckett et al., 2014), followed by postnatal weight gain (or catch-up growth) (Manikkam et al., 2004) and insulin resistance in adulthood (Cardoso et al., 2016). Moreover, precocious puberty occurs in prenatally T-treated sheep independent of body weight and is prevented by prenatal co-treatment with flutamide or rosiglitazone, confirming pubertal timing as a programmed androgen-insulin mediated event (Padmanabhan et al., 2015). Therefore, prenatally T-treated sheep and rats may be suitable models for placental insufficiency, particularly since these sheep also have hypertension and myocardial changes associated with left ventricular hypertrophy (King et al., 2007, Vyas et al., 2016), while the rodents have increased mortality (Wolf et al., 2004).
In contrast, prenatally-T treated rats have a normal birth weight, with exaggerated weight gain by 30 days of age (Tehrani et al., 2014). Similarly, early prenatally T-treated female monkeys have a normal birth weight, with an increase in body weight beginning in early infancy (Herman et al., 2000, Abbott et al., 2007, Abbott et al., 2008, Abbott et al., 2010) followed by delayed puberty as seen in males and an adult PCOS-like phenotype. Mechanisms beyond T-induced developmental programming likely exist since exposure of monkeys to prenatal T excess impairs glucose-insulin homeostasis without affecting body weight in both adult sexes (Abbott et al., 2007).
From a human perspective, macrosomic infants of obese glucose-intolerant mothers represent successful fetal adaptation to maternal nutrient overabundance (Mane et al., 2017). On the other hand, low infant birthweight in women with impaired placental aromatization and diminished uteroplacental perfusion represents impaired fetal nutrient availability from placental insufficiency (Tanguy et al., 1981, Thoumsin et al., 1982). Both large and small for gestational age infants have been reported in pregnant women with PCOS (Palomba et al., 2014a, Palomba et al., 2014b), with birth weight and maternal midgestational BMI positively correlated with newborn adiposity as determined by cord blood leptin levels (Maliqueo et al., 2009).
Poor intrauterine growth and low birth weight accompany precocious puberty and PCOS in northern Spanish women (Ibanez et al., 1998, Ibanez et al., 1996) and in PCOS pregnancies in Chilean and Iranian women (Mehrabian and Kelishadi, 2012, Sir-Petermann et al., 2005), but not in larger groups of Finnish (Laitinen et al., 2003) and Dutch individuals (Sadrzadeh et al., 2003, Koster et al., 2015). Low or normal birth weight in some infants of pregnant women with PCOS can accompany placental abnormalities, including chronic villitis/intervillositis, impaired decidual trophoblast invasion and reduced placental size, that limit nutrient delivery to the fetus and may (Palomba et al., 2014c) or may not (Koster et al., 2015) reduce the ratio of fetal to placental weight.
Since infants of PCOS mothers also can have normal birth weights (Dumesic et al., 2014, Laitinen et al., 2003), developmental programming of adiposity can occur in the human fetus (Poissonnet et al., 1988) without altering infant birth weight, yet still increase the risk of excessive postnatal weight gain and onset of PCOS in susceptible offspring. Mechanisms independent of T-induced developmental programming are supported by findings that male relatives of PCOS women exhibit metabolic dysfunction similar to that of their female relatives (Dumesic et al., 2007, Yilmaz et al., 2018). Additional transgenerational transmission of PCOS through the male line is also supported by a strong association of genetic variants previously linked to male-pattern balding with PCOS phenotypes (Day et al., 2018).
Endocrine antecedents of PCOS
Endocrine antecedents of PCOS occur in female infants born to PCOS mothers and further emphasize that interactions between genes of the fetus and the endocrine-metabolic environment of pregnancy may program offspring after birth. Infant girls born to PCOS versus non-PCOS mothers exhibit increased sebum production by the pilosebaceous unit as a marker of prenatal androgen excess (Homburg et al., 2017). They also exhibit AMH overproduction as a marker of growing follicles, which persists in prepubertal life and improves, along with exaggerated ovarian responsiveness to GnRH analog administration, when PCOS mothers receive metformin in pregnancy, beginning at or before conception (Sir-Petermann et al., 2006, Crisosto et al., 2012). Consistent with these findings, female children of PCOS women have enlarged ovaries and hyperinsulinemia that precede onset of LH hypersecretion and androgen excess during puberty in some (Crisosto et al., 2007, Kent et al., 2008, Sir-Petermann et al., 2009), but not all (Legro et al., 2017) studies.
From a metabolic perspective, adiposity of infants from PCOS women positively correlates with birth weight and maternal midgestational BMI (Maliqueo et al., 2009), the latter of which increases disproportionally in normal- and over-weight PCOS women compared to other pregnant women (Kent et al., 2018). Infant males born to PCOS mothers exhibit higher body weight and develop insulin resistance in adulthood as a risk factor for acquiring type 2 diabetes (Recabarren et al., 2008). Although metformin use in pregnancy appears to improve some ovarian characteristics in female offspring of PCOS mothers (Sir-Petermann et al., 2006, Crisosto et al., 2012), increased childhood adiposity, insulin resistance and increased newborn head size following gestational treatment of PCOS women with metformin demonstrates the need to minimize gestational interventions in humans (Hanem et al., 2019). In addition, interactions between genes of the susceptible fetus and the maternal PCOS endocrine-metabolic environment are likely further altered by postnatal environmental inputs, since widespread epigenomic remodeling occurs throughout human aging, being inherited across generations through non-genetic mechanisms (Benayoun et al., 2015, Simpkin et al., 2016).
Conclusion
The collective data suggest that epigenetic changes in fetal life may impact the developmental origins of PCOS, assuming a critical time interval of fetal susceptibility beginning in early- to mid-gestation when developmental programming occurs (Dumesic et al., 2007, Abbott et al., 2019). By inducing permanent PCOS–like phenotypes characterized by LH hypersecretion from reduced steroid negative feedback, hyperandrogenism, ovulatory dysfunction and impaired glucose-insulin homeostasis, animal models of prenatal T-treatment support clinical studies of PCOS women in providing insights into when developmental programming occurs during human development, how placental function alters the maternal-fetal relationship to affect fetal growth, and perhaps why birth weights of PCOS women differ throughout the world. Improved maternal endocrine-metabolic health of PCOS women to lower their risk of pregnancy-related complications could potentially reduce intergenerational susceptibility to PCOS and its metabolic derangements in offspring.
Acknowledgements:
We thank Karla Largaespada for subject recruitment strategies and administrative responsibilities that were crucial for the successful studies of the normal-weight PCOS subjects.
Funding: This study was supported by a grant from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health (NIH) under awards P50HD071836 and P51 ODO11092 for the Endocrine Technologies Support Core (ETSC) through the Oregon National Primate Research Center, and P50HD028934 through the Wisconsin National Primate Research Center and the University of Virginia and HD 44232 through the University of Michigan; statistical analyses by the NIH National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR001881; and the Santa Monica Bay Woman’s Club. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Declaration of interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- ABBOTT DH, BARNETT DK, BRUNS CM & DUMESIC DA 2005. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update, 11, 357–74. [DOI] [PubMed] [Google Scholar]
- ABBOTT DH, BARNETT DK, LEVINE JE, PADMANABHAN V, DUMESIC DA, JACORIS S & TARANTAL AF 2008. Endocrine antecedents of polycystic ovary syndrome in fetal and infant prenatally androgenized female rhesus monkeys. Biol Reprod, 79, 154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABBOTT DH, BRUNS CM, BARNETT DK & DUMESIC DA 2007. Fetal programming of polycystic ovary syndrome In: KOVACS G & NORMAN R (eds.) Polycystic Ovary Syndrome. 2nd ed Cambridge: Cambridge University Press. [Google Scholar]
- ABBOTT DH, BRUNS CR, BARNETT DK, DUNAIF A, GOODFRIEND TL, DUMESIC DA & TARANTAL AF 2010. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am J Physiol Endocrinol Metab, 299, E741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABBOTT DH, DUMESIC DA, EISNER JE, KEMNITZ JW & GOY RW 1997. The Prenatally Androgenized Female Rhesus Monkey as a Model for PCOS In: NESTLER JE, DEWAILLY D & AZZIZ R (eds.) Androgen Excess Disorders in Women. Phyladelphia, Pennsylvannia: Lippincott-Raven Press. [Google Scholar]
- ABBOTT DH, DUMESIC DA & LEVINE JE 2019. Hyperandrogenic origins of polycystic ovary syndrome - implications for pathophysiology and therapy. Expert Rev Endocrinol Metab, 14, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABBOTT DH, EISNER JR, COLMAN RJ, KEMNITZ JW & DUMESIC DA 2002. In: CHANG RJ, DUNAIF A & HIENDEL J (eds.) Polycystic Ovary Syndrome. New York, New York: Marcel Dekker, Inc. [Google Scholar]
- ABBOTT DH, VEPRASKAS SH, HORTON TH, TERASAWA E & LEVINE JE 2018. Accelerated Episodic Luteinizing Hormone Release Accompanies Blunted Progesterone Regulation in PCOS-like Female Rhesus Monkeys (Macaca Mulatta) Exposed to Testosterone during Early-to-Mid Gestation. Neuroendocrinology, 107, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABI SALLOUM B, VEIGA-LOPEZ A, ABBOTT DH, BURANT CF & PADMANABHAN V 2015. Developmental programming: exposure to testosterone excess disrupts steroidal and metabolic environment in pregnant sheep. Endocrinology, 156, 2323–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AHMAD AK, KAO CN, QUINN M, LENHART N, ROSEN M, CEDARS MI & HUDDLESTON H 2018. Differential rate in decline in ovarian reserve markers in women with polycystic ovary syndrome compared with control subjects: results of a longitudinal study. Fertil Steril, 109, 526–531. [DOI] [PubMed] [Google Scholar]
- ANDERSON H, FOGEL N, GREBE SK, SINGH RJ, TAYLOR RL & DUNAIF A 2010. Infants of women with polycystic ovary syndrome have lower cord blood androstenedione and estradiol levels. J Clin Endocrinol Metab, 95, 2180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNER P 2005. Effects of testosterone on fat cell lipolysis. Species differences and possible role in polycystic ovarian syndrome. Biochimie, 87, 39–43. [DOI] [PubMed] [Google Scholar]
- ASRM 2015. Obesity and reproduction: a committee opinion. Fertil Steril, 104, 1116–26. [DOI] [PubMed] [Google Scholar]
- BARBIERI RL, SALTZMAN DH, TORDAY JS, RANDALL RW, FRIGOLETTO FD & RYAN KJ 1986. Elevated concentrations of the beta-subunit of human chorionic gonadotropin and testosterone in the amniotic fluid of gestations of diabetic mothers. Am J Obstet Gynecol, 154, 1039–43. [DOI] [PubMed] [Google Scholar]
- BARKER DJ 2002. Fetal programming of coronary heart disease. Trends Endocrinol Metab, 13, 364–8. [DOI] [PubMed] [Google Scholar]
- BARKER DJ & OSMOND C 1986. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet, 1, 1077–81. [DOI] [PubMed] [Google Scholar]
- BARNES RB, ROSENFIELD RL, EHRMANN DA, CARA JF, CUTTLER L, LEVITSKY LL & ROSENTHAL IM 1994. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab, 79, 1328–33. [DOI] [PubMed] [Google Scholar]
- BARRETT ES, HOEGER KM, SATHYANARAYANA S, ABBOTT DH, REDMON JB, NGUYEN RHN & SWAN SH 2018. Anogenital distance in newborn daughters of women with polycystic ovary syndrome indicates fetal testosterone exposure. J Dev Orig Health Dis, 9, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRY JA, KAY AR, NAVARATNARAJAH R, IQBAL S, BAMFO JE, DAVID AL, HINES M & HARDIMAN PJ 2010. Umbilical vein testosterone in female infants born to mothers with polycystic ovary syndrome is elevated to male levels. J Obstet Gynaecol, 30, 444–6. [DOI] [PubMed] [Google Scholar]
- BECK-PECCOZ P, PADMANABHAN V, BAGGIANI AM, CORTELAZZI D, BUSCAGLIA M, MEDRI G, MARCONI AM, PARDI G & BEITINS IZ 1991. Maturation of hypothalamic-pituitary-gonadal function in normal human fetuses: circulating levels of gonadotropins, their common alpha-subunit and free testosterone, and discrepancy between immunological and biological activities of circulating follicle-stimulating hormone. J Clin Endocrinol Metab, 73, 525–32. [DOI] [PubMed] [Google Scholar]
- BECKETT EM, ASTAPOVA O, STECKLER TL, VEIGA-LOPEZ A & PADMANABHAN V 2014. Developmental programing: impact of testosterone on placental differentiation. Reproduction, 148, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENAYOUN BA, POLLINA EA & BRUNET A 2015. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol, 16, 593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUNS CM, BAUM ST, COLMAN RJ, DUMESIC DA, EISNER JR, JENSEN MD, WHIGHAM LD & ABBOTT DH 2007. Prenatal androgen excess negatively impacts body fat distribution in a nonhuman primate model of polycystic ovary syndrome. Int J Obes (Lond), 31, 1579–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDOSO RC, VEIGA-LOPEZ A, MOELLER J, BECKETT E, PEASE A, KELLER E, MADRIGAL V, CHAZENBALK G, DUMESIC D & PADMANABHAN V 2016. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Adiposity and Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology, 157, 522–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARMINA E, NAPOLI N, LONGO RA, RINI GB & LOBO RA 2006. Metabolic syndrome in polycystic ovary syndrome (PCOS): lower prevalence in southern Italy than in the USA and the influence of criteria for the diagnosis of PCOS. Eur J Endocrinol, 154, 141–5. [DOI] [PubMed] [Google Scholar]
- CHANG R & DUMESIC D 2018. Polycystic Ovary Syndrome and Hyperandrogenic States In: STRAUSS JI & BARBIERI R (eds.) Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology and Clinical Management. Eighth ed Philadelphia: Elsevier Saunders. [Google Scholar]
- CHAZENBALK G, CHEN YH, HENEIDI S, LEE JM, PALL M, CHEN YD & AZZIZ R 2012. Abnormal expression of genes involved in inflammation, lipid metabolism, and Wnt signaling in the adipose tissue of polycystic ovary syndrome. J Clin Endocrinol Metab, 97, E765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAZENBALK G, SINGH P, IRGE D, SHAH A, ABBOTT DH & DUMESIC DA 2013. Androgens inhibit adipogenesis during human adipose stem cell commitment to preadipocyte formation. Steroids, 78, 920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHHABRA S, MCCARTNEY CR, YOO RY, EAGLESON CA, CHANG RJ & MARSHALL JC 2005. Progesterone inhibition of the hypothalamic gonadotropin-releasing hormone pulse generator: evidence for varied effects in hyperandrogenemic adolescent girls. J Clin Endocrinol Metab, 90, 2810–5. [DOI] [PubMed] [Google Scholar]
- CHRIST JP, GUNNING MN, MEUN C, EIJKEMANS MJC, VAN RIJN BB, BONSEL GJ, LAVEN JSE & FAUSER B 2019. Pre-Conception Characteristics Predict Obstetrical and Neonatal Outcomes in Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab, 104, 809–818. [DOI] [PubMed] [Google Scholar]
- COLE B, HENSINGER K, MACIEL GA, CHANG RJ & ERICKSON GF 2006. Human fetal ovary development involves the spatiotemporal expression of p450c17 protein. J Clin Endocrinol Metab, 91, 3654–61. [DOI] [PubMed] [Google Scholar]
- CORBOULD A 2007. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol, 192, 585–94. [DOI] [PubMed] [Google Scholar]
- CRESPI EJ, STECKLER TL, MOHANKUMAR PS & PADMANABHAN V 2006. Prenatal exposure to excess testosterone modifies the developmental trajectory of the insulin-like growth factor system in female sheep. J Physiol, 572, 119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRISOSTO N, CODNER E, MALIQUEO M, ECHIBURU B, SANCHEZ F, CASSORLA F & SIR-PETERMANN T 2007. Anti-Mullerian hormone levels in peripubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab, 92, 2739–43. [DOI] [PubMed] [Google Scholar]
- CRISOSTO N, ECHIBURU B, MALIQUEO M, PEREZ V, LADRON DE GUEVARA A, PREISLER J, SANCHEZ F & SIR-PETERMANN T 2012. Improvement of hyperandrogenism and hyperinsulinemia during pregnancy in women with polycystic ovary syndrome: possible effect in the ovarian follicular mass of their daughters. Fertil Steril, 97, 218–24. [DOI] [PubMed] [Google Scholar]
- CRISOSTO N, LADRON DE GUEVARA A, ECHIBURU B, MALIQUEO M, CAVADA G, CODNER E, PAEZ F & SIR-PETERMANN T 2019. Higher luteinizing hormone levels associated with antimullerian hormone in postmenarchal daughters of women with polycystic ovary syndrome. Fertil Steril, 111, 381–388. [DOI] [PubMed] [Google Scholar]
- DAY F, KARADERI T, JONES MR, MEUN C, HE C, DRONG A, KRAFT P, LIN N, HUANG H, BROER L, et al. 2018. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet, 14, e1007813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DESAI M, BEALL M & ROSS MG 2013. Developmental origins of obesity: programmed adipogenesis. Curr Diab Rep, 13, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DICKER A, RYDEN M, NASLUND E, MUEHLEN IE, WIREN M, LAFONTAN M & ARNER P 2004. Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia, 47, 420–428. [DOI] [PubMed] [Google Scholar]
- DRISCOLL SG, BENIRSCHKE K & CURTIS GW 1960. Neonatal deaths among infants of diabetic mothers. Postmortem findings in ninety-five infants. Am J Dis Child, 100, 818–35. [DOI] [PubMed] [Google Scholar]
- DUMESIC DA, ABBOTT DH & PADMANABHAN V 2007. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord, 8, 127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMESIC DA, AKOPIANS AL, MADRIGAL VK, RAMIREZ E, MARGOLIS DJ, SARMA MK, THOMAS AM, GROGAN TR, HAYKAL R, SCHOOLER TA, et al. 2016. Hyperandrogenism Accompanies Increased Intra-Abdominal Fat Storage in Normal Weight Polycystic Ovary Syndrome Women. J Clin Endocrinol Metab, 101, 4178–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMESIC DA, GOODARZI MO, CHAZENBALK GD & ABBOTT DH 2014. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med, 32, 159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMESIC DA, PATANKAR MS, BARNETT DK, LESNICK TG, HUTCHERSON BA & ABBOTT DH 2009. Early prenatal androgenization results in diminished ovarian reserve in adult female rhesus monkeys. Hum Reprod, 24, 3188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMESIC DA, PHAN JD, LEUNG KL, GROGAN TR, DING X, LI X, HOYOS LR, ABBOTT DH & CHAZENBALK GD 2019. Adipose Insulin Resistance in Normal-Weight Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab, 104, 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMESIC DA, SCHRAMM RD, PETERSON E, PAPROCKI AM, ZHOU R & ABBOTT DH 2002. Impaired developmental competence of oocytes in adult prenatally androgenized female rhesus monkeys undergoing gonadotropin stimulation for in vitro fertilization. J Clin Endocrinol Metab, 87, 1111–9. [DOI] [PubMed] [Google Scholar]
- EAGLESON CA, GINGRICH MB, PASTOR CL, ARORA TK, BURT CM, EVANS WS & MARSHALL JC 2000. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab, 85, 4047–52. [DOI] [PubMed] [Google Scholar]
- EISNER JR, DUMESIC DA, KEMNITZ JW & ABBOTT DH 2000. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J Clin Endocrinol Metab, 85, 1206–10. [DOI] [PubMed] [Google Scholar]
- EISNER JR, DUMESIC DA, KEMNITZ JW, COLMAN RJ & ABBOTT DH 2003. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res, 11, 279–86. [DOI] [PubMed] [Google Scholar]
- EK I, ARNER P, BERGQVIST A, CARLSTROM K & WAHRENBERG H 1997. Impaired adipocyte lipolysis in nonobese women with the polycystic ovary syndrome: a possible link to insulin resistance? J Clin Endocrinol Metab, 82, 1147–53. [DOI] [PubMed] [Google Scholar]
- EK I, ARNER P, RYDEN M, HOLM C, THORNE A, HOFFSTEDT J & WAHRENBERG H 2002. A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes, 51, 484–92. [DOI] [PubMed] [Google Scholar]
- FAULDS G, RYDEN M, EK I, WAHRENBERG H & ARNER P 2003. Mechanisms behind lipolytic catecholamine resistance of subcutaneous fat cells in the polycystic ovarian syndrome. J Clin Endocrinol Metab, 88, 2269–73. [DOI] [PubMed] [Google Scholar]
- FISCH SC, NIKOU AF, WRIGHT EA, PHAN JD, LEUNG KL, GROGAN TR, ABBOTT DH, CHAZENBALK GD & DUMESIC DA 2018. Precocious subcutaneous abdominal stem cell development to adipocytes in normal-weight women with polycystic ovary syndrome. Fertil Steril, 110, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUEDIKIAN AA, LEE AY, GROGAN TR, ABBOTT DH, LARGAESPADA K, CHAZENBALK GD & DUMESIC DA 2018. Reproductive and metabolic determinants of granulosa cell dysfunction in normal-weight women with polycystic ovary syndrome. Fertil Steril, 109, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANEM LGE, SALVESEN O, JULIUSSON PB, CARLSEN SM, NOSSUM MCF, VAAGE MO, ODEGARD R & VANKY E 2019. Intrauterine metformin exposure and offspring cardiometabolic risk factors (PedMet study): a 5–10 year follow-up of the PregMet randomised controlled trial. Lancet Child Adolesc Health, 3, 166–174. [DOI] [PubMed] [Google Scholar]
- HANSEN NS, STRASKO KS, HJORT L, KELSTRUP L, HOUSHMAND-OREGAARD A, SCHROLKAMP M, SCHULTZ HS, SCHEELE C, PEDERSEN BK, LING C, et al. 2017. Fetal Hyperglycemia Changes Human Preadipocyte Function in Adult Life. J Clin Endocrinol Metab, 102, 1141–1150. [DOI] [PubMed] [Google Scholar]
- HART R, SLOBODA DM, DOHERTY DA, NORMAN RJ, ATKINSON HC, NEWNHAM JP, DICKINSON JE & HICKEY M 2010. Circulating maternal testosterone concentrations at 18 weeks of gestation predict circulating levels of antimullerian hormone in adolescence: a prospective cohort study. Fertil Steril, 94, 1544–7. [DOI] [PubMed] [Google Scholar]
- HERMAN RA, JONES B, MANN DR & WALLEN K 2000. Timing of prenatal androgen exposure: anatomical and endocrine effects on juvenile male and female rhesus monkeys. Horm Behav, 38, 52–66. [DOI] [PubMed] [Google Scholar]
- HICKEY M, SLOBODA DM, ATKINSON HC, DOHERTY DA, FRANKS S, NORMAN RJ, NEWNHAM JP & HART R 2009. The relationship between maternal and umbilical cord androgen levels and polycystic ovary syndrome in adolescence: a prospective cohort study. J Clin Endocrinol Metab, 94, 3714–20. [DOI] [PubMed] [Google Scholar]
- HOGG K, MCNEILLY AS & DUNCAN WC 2011. Prenatal androgen exposure leads to alterations in gene and protein expression in the ovine fetal ovary. Endocrinology, 152, 2048–59. [DOI] [PubMed] [Google Scholar]
- HOMBURG R, GUDI A, SHAH A & M. L. A 2017. A novel method to demonstrate that pregnant women with polycystic ovary syndrome hyper-expose their fetus to androgens as a possible stepping stone for the developmental theory of PCOS. A pilot study. Reprod Biol Endocrinol, 15, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HULTQUIST GT & OLDING LB 1981. Endocrine pathology of infants of diabetic mothers. A quantitative morphological analysis including a comparison with infants of iso-immunized and of non-diabetic mothers. Acta Endocrinol Suppl (Copenh), 241, 1–202. [PubMed] [Google Scholar]
- IBANEZ L, POTAU N, FRANCOIS I & DE ZEGHER F 1998. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism in girls: relation to reduced fetal growth. J Clin Endocrinol Metab, 83, 3558–62. [DOI] [PubMed] [Google Scholar]
- IBANEZ L, POTAU N, ZAMPOLLI M, PRAT N, VIRDIS R, VICENS-CALVET E & CARRASCOSA A 1996. Hyperinsulinemia in postpubertal girls with a history of premature pubarche and functional ovarian hyperandrogenism. J Clin Endocrinol Metab, 81, 1237–43. [DOI] [PubMed] [Google Scholar]
- KELLER E, CHAZENBALK GD, AGUILERA P, MADRIGAL V, GROGAN T, ELASHOFF D, DUMESIC DA & ABBOTT DH 2014. Impaired preadipocyte differentiation into adipocytes in subcutaneous abdominal adipose of PCOS-like female rhesus monkeys. Endocrinology, 155, 2696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENT J, DODSON WC, KUNSELMAN A, PAULI J, STONE A, DIAMOND MP, COUTIFARIS C, SCHLAFF WD, ALVERO R, CASSON P, et al. 2018. Gestational Weight Gain in Women With Polycystic Ovary Syndrome: A Controlled Study. J Clin Endocrinol Metab, 103, 4315–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENT SC, GNATUK CL, KUNSELMAN AR, DEMERS LM, LEE PA & LEGRO RS 2008. Hyperandrogenism and hyperinsulinism in children of women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab, 93, 1662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KING AJ, OLIVIER NB, MOHANKUMAR PS, LEE JS, PADMANABHAN V & FINK GD 2007. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab, 292, E1837–41. [DOI] [PubMed] [Google Scholar]
- KOSTER MP, DE WILDE MA, VELTMAN-VERHULST SM, HOUBEN ML, NIKKELS PG, VAN RIJN BB & FAUSER BC 2015. Placental characteristics in women with polycystic ovary syndrome. Hum Reprod, 30, 2829–37. [DOI] [PubMed] [Google Scholar]
- KUMAR S, GORDON GH, ABBOTT DH & MISHRA JS 2018. Androgens in maternal vascular and placental function: implications for preeclampsia pathogenesis. Reproduction, 156, R155–R167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAITINEN J, TAPONEN S, MARTIKAINEN H, POUTA A, MILLWOOD I, HARTIKAINEN AL, RUOKONEN A, SOVIO U, MCCARTHY MI, FRANKS S & JARVELIN MR 2003. Body size from birth to adulthood as a predictor of self-reported polycystic ovary syndrome symptoms. Int J Obes Relat Metab Disord, 27, 710–5. [DOI] [PubMed] [Google Scholar]
- LEGRO RS, KUNSELMAN AR, DODSON WC & DUNAIF A 1999. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab, 84, 165–9. [DOI] [PubMed] [Google Scholar]
- LEGRO RS, KUNSELMAN AR, STETTER CM, GNATUK CL, ESTES SJ, BRINDLE E, VESPER HW, BOTELHO JC, LEE PA & DODSON WC 2017. Normal Pubertal Development in Daughters of Women With PCOS: A Controlled Study. J Clin Endocrinol Metab, 102, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LONGO M, RACITI GA, ZATTERALE F, PARRILLO L, DESIDERIO A, SPINELLI R, HAMMARSTEDT A, HEDJAZIFAR S, HOFFMANN JM, NIGRO C, et al. 2018. Epigenetic modifications of the Zfp/ZNF423 gene control murine adipogenic commitment and are dysregulated in human hypertrophic obesity. Diabetologia, 61, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUNDGREN JA, KIM SH, BURT SOLORZANO CM, MCCARTNEY CR & MARSHALL JC 2018. Progesterone Suppression of Luteinizing Hormone Pulse Frequency in Adolescent Girls With Hyperandrogenism: Effects of Metformin. J Clin Endocrinol Metab, 103, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALIQUEO M, ECHIBURU B, CRISOSTO N, AMIGO P, ARANDA P, SANCHEZ F & SIR-PETERMANN T 2009. Metabolic parameters in cord blood of newborns of women with polycystic ovary syndrome. Fertil Steril, 92, 277–82. [DOI] [PubMed] [Google Scholar]
- MALIQUEO M, LARA HE, SANCHEZ F, ECHIBURU B, CRISOSTO N & SIR-PETERMANN T 2013. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol, 166, 151–5. [DOI] [PubMed] [Google Scholar]
- MANE L, FLORES-LE ROUX JA, BENAIGES D, RODRIGUEZ M, MARCELO I, CHILLARON JJ, PEDRO-BOTET J, LLAURADO G, GORTAZAR L, CARRERAS R, et al. 2017. Role of First-Trimester HbA1c as a Predictor of Adverse Obstetric Outcomes in a Multiethnic Cohort. J Clin Endocrinol Metab, 102, 390–397. [DOI] [PubMed] [Google Scholar]
- MANIKKAM M, CRESPI EJ, DOOP DD, HERKIMER C, LEE JS, YU S, BROWN MB, FOSTER DL & PADMANABHAN V 2004. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology, 145, 790–8. [DOI] [PubMed] [Google Scholar]
- MANNERAS-HOLM L, LEONHARDT H, KULLBERG J, JENNISCHE E, ODEN A, HOLM G, HELLSTROM M, LONN L, OLIVECRONA G, STENER-VICTORIN E, et al. 2011. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab, 96, E304–11. [DOI] [PubMed] [Google Scholar]
- MCGIVERN RF 1989. Low birthweight in rats induced by prenatal exposure to testosterone combined with alcohol, pair-feeding, or stress. Teratology, 40, 335–8. [DOI] [PubMed] [Google Scholar]
- MEHRABIAN F & KELISHADI R 2012. Comparison of the metabolic parameters and androgen level of umbilical cord blood in newborns of mothers with polycystic ovary syndrome and controls. J Res Med Sci, 17, 207–11. [PMC free article] [PubMed] [Google Scholar]
- MORAN LJ, MISSO ML, WILD RA & NORMAN RJ 2010. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update, 16, 347–63. [DOI] [PubMed] [Google Scholar]
- NEEL JV 1962. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet, 14, 353–62. [PMC free article] [PubMed] [Google Scholar]
- NOROOZZADEH M, RAMEZANI TEHRANI F, SEDAGHAT K, GODINI A & AZIZI F 2015. The impact of prenatal exposure to a single dose of testosterone on insulin resistance, glucose tolerance and lipid profile of female rat’s offspring in adulthood. J Endocrinol Invest, 38, 489–95. [DOI] [PubMed] [Google Scholar]
- O’REILLY MW, KEMPEGOWDA P, WALSH M, TAYLOR AE, MANOLOPOULOS KN, ALLWOOD JW, SEMPLE RK, HEBENSTREIT D, DUNN WB, TOMLINSON JW, et al. 2017. AKR1C3-Mediated Adipose Androgen Generation Drives Lipotoxicity in Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab, 102, 3327–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORTEGA HH, SALVETTI NR & PADMANABHAN V 2009. Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction, 137, 865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, MANIKKAM M, RECABARREN S & FOSTER D 2006. Prenatal testosterone excess programs reproductive and metabolic dysfunction in the female. Mol Cell Endocrinol, 246, 165–74. [DOI] [PubMed] [Google Scholar]
- PADMANABHAN V, SMITH P & VEIGA-LOPEZ A 2012. Developmental programming: Impact of prenatal testosterone treatment and postnatal obesity on ovarian follicular dynamics. J Dev Orig Health Dis, 3, 276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V & VEIGA-LOPEZ A 2013. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol, 373, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, VEIGA-LOPEZ A, ABBOTT DH & DUMESIC DA 2007. Developmental programming of ovarian dysfunction In: GONZALEZ-BULNES A (ed.) Novel concepts in Ovarian Endocrinology. Kerala, India: Research Signpost. [Google Scholar]
- PADMANABHAN V, VEIGA-LOPEZ A, ABBOTT DH, RECABARREN SE & HERKIMER C 2010. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology, 151, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, VEIGA-LOPEZ A, HERKIMER C, ABI SALLOUM B, MOELLER J, BECKETT E & SREEDHARAN R 2015. Developmental Programming: Prenatal and Postnatal Androgen Antagonist and Insulin Sensitizer Interventions Prevent Advancement of Puberty and Improve LH Surge Dynamics in Prenatal Testosterone-Treated Sheep. Endocrinology, 156, 2678–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALOMBA S, DE WILDE MA, FALBO A, KOSTER MP, LA SALA GB & FAUSER BC 2015. Pregnancy complications in women with polycystic ovary syndrome. Hum Reprod Update, 21, 575–92. [DOI] [PubMed] [Google Scholar]
- PALOMBA S, FALBO A, CHIOSSI G, MUSCOGIURI G, FORNACIARI E, ORIO F, TOLINO A, COLAO A, LA SALA GB & ZULLO F 2014a. Lipid profile in nonobese pregnant women with polycystic ovary syndrome: a prospective controlled clinical study. Steroids, 88, 36–43. [DOI] [PubMed] [Google Scholar]
- PALOMBA S, FALBO A, CHIOSSI G, ORIO F, TOLINO A, COLAO A, LA SALA GB & ZULLO F 2014b. Low-grade chronic inflammation in pregnant women with polycystic ovary syndrome: a prospective controlled clinical study. J Clin Endocrinol Metab, 99, 2942–51. [DOI] [PubMed] [Google Scholar]
- PALOMBA S, FALBO A, CHIOSSI G, TOLINO A, TUCCI L, LA SALA GB & ZULLO F 2014c. Early trophoblast invasion and placentation in women with different PCOS phenotypes. Reprod Biomed Online, 29, 370–81. [DOI] [PubMed] [Google Scholar]
- PALOMBA S, MAROTTA R, DI CELLO A, RUSSO T, FALBO A, ORIO F, TOLINO A, ZULLO F, ESPOSITO R & LA SALA GB 2012. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-control study. Clin Endocrinol (Oxf), 77, 898–904. [DOI] [PubMed] [Google Scholar]
- POISSONNET CM, LAVELLE M & BURDI AR 1988. Growth and development of adipose tissue. J Pediatr, 113, 1–9. [DOI] [PubMed] [Google Scholar]
- PUTTABYATAPPA M, LU C, MARTIN JD, CHAZENBALK G, DUMESIC D & PADMANABHAN V 2018. Developmental Programming: Impact of Prenatal Testosterone Excess on Steroidal Machinery and Cell Differentiation Markers in Visceral Adipocytes of Female Sheep. Reprod Sci, 25, 1010–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RECABARREN SE, PADMANABHAN V, CODNER E, LOBOS A, DURAN C, VIDAL M, FOSTER DL & SIR-PETERMANN T 2005. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am J Physiol Endocrinol Metab, 289, E801–6. [DOI] [PubMed] [Google Scholar]
- RECABARREN SE, SMITH R, RIOS R, MALIQUEO M, ECHIBURU B, CODNER E, CASSORLA F, ROJAS P & SIR-PETERMANN T 2008. Metabolic profile in sons of women with polycystic ovary syndrome. J Clin Endocrinol Metab, 93, 1820–6. [DOI] [PubMed] [Google Scholar]
- REYNOLDS RM, ALLAN KM, RAJA EA, BHATTACHARYA S, MCNEILL G, HANNAFORD PC, SARWAR N, LEE AJ, BHATTACHARYA S & NORMAN JE 2013. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ, 347, f4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON JE, BIRCH RA, FOSTER DL & PADMANABHAN V 2002. Prenatal exposure of the ovine fetus to androgens sexually differentiates the steroid feedback mechanisms that control gonadotropin releasing hormone secretion and disrupts ovarian cycles. Arch Sex Behav, 31, 35–41. [DOI] [PubMed] [Google Scholar]
- ROBINSON JE, FORSDIKE RA & TAYLOR JA 1999. In utero exposure of female lambs to testosterone reduces the sensitivity of the gonadotropin-releasing hormone neuronal network to inhibition by progesterone. Endocrinology, 140, 5797–805. [DOI] [PubMed] [Google Scholar]
- ROMACHO T, ELSEN M, ROHRBORN D & ECKEL J 2014. Adipose tissue and its role in organ crosstalk. Acta Physiol (Oxf), 210, 733–53. [DOI] [PubMed] [Google Scholar]
- ROSENFIELD RL, BARNES RB, CARA JF & LUCKY AW 1990. Dysregulation of cytochrome P450c 17 alpha as the cause of polycystic ovarian syndrome. Fertil Steril, 53, 785–91. [PubMed] [Google Scholar]
- SADRZADEH S, KLIP WA, BROEKMANS FJ, SCHATS R, WILLEMSEN WN, BURGER CW, VAN LEEUWEN FE, LAMBALK CB & GROUP OP 2003. Birth weight and age at menarche in patients with polycystic ovary syndrome or diminished ovarian reserve, in a retrospective cohort. Hum Reprod, 18, 2225–30. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-FERRER ML, MENDIOLA J, HERNANDEZ-PENALVER AI, CORBALAN-BIYANG S, CARMONA-BARNOSI A, PRIETO-SANCHEZ MT, NIETO A & TORRES-CANTERO AM 2017. Presence of polycystic ovary syndrome is associated with longer anogenital distance in adult Mediterranean women. Hum Reprod, 32, 2315–2323. [DOI] [PubMed] [Google Scholar]
- SARMA HN, MANIKKAM M, HERKIMER C, DELL’ORCO J, WELCH KB, FOSTER DL & PADMANABHAN V 2005. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology, 146, 4281–91. [DOI] [PubMed] [Google Scholar]
- SHAO M, HEPLER C, VISHVANATH L, MACPHERSON KA, BUSBUSO NC & GUPTA RK 2017. Fetal development of subcutaneous white adipose tissue is dependent on Zfp423. Mol Metab, 6, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMPKIN AJ, HEMANI G, SUDERMAN M, GAUNT TR, LYTTLETON O, MCARDLE WL, RING SM, SHARP GC, TILLING K, HORVATH S, et al. 2016. Prenatal and early life influences on epigenetic age in children: a study of mother-offspring pairs from two cohort studies. Hum Mol Genet, 25, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIR-PETERMANN T, ANGEL B, MALIQUEO M, CARVAJAL F, SANTOS JL & PEREZ-BRAVO F 2002a. Prevalence of Type II diabetes mellitus and insulin resistance in parents of women with polycystic ovary syndrome. Diabetologia, 45, 959–64. [DOI] [PubMed] [Google Scholar]
- SIR-PETERMANN T, CODNER E, MALIQUEO M, ECHIBURU B, HITSCHFELD C, CRISOSTO N, PEREZ-BRAVO F, RECABARREN SE & CASSORLA F 2006. Increased anti-Mullerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab, 91, 3105–9. [DOI] [PubMed] [Google Scholar]
- SIR-PETERMANN T, CODNER E, PEREZ V, ECHIBURU B, MALIQUEO M, LADRON DE GUEVARA A, PREISLER J, CRISOSTO N, SANCHEZ F, CASSORLA F, et al. 2009. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab, 94, 1923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIR-PETERMANN T, HITCHSFELD C, MALIQUEO M, CODNER E, ECHIBURU B, GAZITUA R, RECABARREN S & CASSORLA F 2005. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum Reprod, 20, 2122–6. [DOI] [PubMed] [Google Scholar]
- SIR-PETERMANN T, MALIQUEO M, ANGEL B, LARA HE, PEREZ-BRAVO F & RECABARREN SE 2002b. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod, 17, 2573–9. [DOI] [PubMed] [Google Scholar]
- SLOB AK, DEN HAMER R, WOUTERSEN PJ & VAN DER WERFF TEN BOSCH JJ 1983. Prenatal testosterone propionate and postnatal ovarian activity in the rat. Acta Endocrinol (Copenh), 103, 420–7. [DOI] [PubMed] [Google Scholar]
- STECKLER T, MANIKKAM M, INSKEEP EK & PADMANABHAN V 2007a. Developmental programming: follicular persistence in prenatal testosterone-treated sheep is not programmed by androgenic actions of testosterone. Endocrinology, 148, 3532–40. [DOI] [PubMed] [Google Scholar]
- STECKLER T, WANG J, BARTOL FF, ROY SK & PADMANABHAN V 2005. Fetal programming: prenatal testosterone treatment causes intrauterine growth retardation, reduces ovarian reserve and increases ovarian follicular recruitment. Endocrinology, 146, 3185–93. [DOI] [PubMed] [Google Scholar]
- STECKLER TL, ROBERTS EK, DOOP DD, LEE TM & PADMANABHAN V 2007b. Developmental programming in sheep: administration of testosterone during 60–90 days of pregnancy reduces breeding success and pregnancy outcome. Theriogenology, 67, 459–67. [DOI] [PubMed] [Google Scholar]
- SULLIVAN SD & MOENTER SM 2004. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci U S A, 101, 7129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SWINBURN BA, NYOMBA BL, SAAD MF, ZURLO F, RAZ I, KNOWLER WC, LILLIOJA S, BOGARDUS C & RAVUSSIN E 1991. Insulin resistance associated with lower rates of weight gain in Pima Indians. J Clin Invest, 88, 168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANGUY G, THOUMSIN HJ, ZORN JR & CEDARD L 1981. DHEA-S-loading test in cases of intrauterine growth retardation: relationship between the pattern of the maternal plasma metabolites and the fetoplacental dysfunction. Gynecol Obstet Invest, 12, 305–16. [DOI] [PubMed] [Google Scholar]
- TATA B, MIMOUNI NEH, BARBOTIN AL, MALONE SA, LOYENS A, PIGNY P, DEWAILLY D, CATTEAU-JONARD S, SUNDSTROM-POROMAA I, PILTONEN TT, et al. 2018. Elevated prenatal anti-Mullerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med, 24, 834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEEDE HJ, MISSO ML, COSTELLO MF, DOKRAS A, LAVEN J, MORAN L, PILTONEN T, NORMAN RJ & INTERNATIONAL PN 2018. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum Reprod, 33, 1602–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEHRANI FR, NOROOZZADEH M, ZAHEDIASL S, PIRYAEI A & AZIZI F 2014. Introducing a rat model of prenatal androgen-induced polycystic ovary syndrome in adulthood. Exp Physiol, 99, 792–801. [DOI] [PubMed] [Google Scholar]
- THOUMSIN HJ, ALSAT E & CEDARD L 1982. In vitro aromatization of androgens into estrogens in placental insufficiency. Gynecol Obstet Invest, 13, 37–43. [DOI] [PubMed] [Google Scholar]
- VEIGA-LOPEZ A, MOELLER J, PATEL D, YE W, PEASE A, KINNS J & PADMANABHAN V 2013. Developmental programming: impact of prenatal testosterone excess on insulin sensitivity, adiposity, and free fatty acid profile in postpubertal female sheep. Endocrinology, 154, 1731–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VYAS AK, HOANG V, PADMANABHAN V, GILBREATH E & MIETELKA KA 2016. Prenatal programming: adverse cardiac programming by gestational testosterone excess. Sci Rep, 6, 28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISS PA, SCHOLZ HS, HAAS J, TAMUSSINO KF, SEISSLER J & BORKENSTEIN MH 2000. Long-term follow-up of infants of mothers with type 1 diabetes: evidence for hereditary and nonhereditary transmission of diabetes and precursors. Diabetes Care, 23, 905–11. [DOI] [PubMed] [Google Scholar]
- WILSON EA & JAWAD MJ 1979. The effect of trophic agents on fetal ovarian steroidogenesis in organ culture. Fertil Steril, 32, 73–9. [DOI] [PubMed] [Google Scholar]
- WOLF CJ, LEBLANC GA & GRAY LE JR. 2004. Interactive effects of vinclozolin and testosterone propionate on pregnancy and sexual differentiation of the male and female SD rat. Toxicol Sci, 78, 135–43. [DOI] [PubMed] [Google Scholar]
- WU Y, ZHONG G, CHEN S, ZHENG C, LIAO D & XIE M 2017. Polycystic ovary syndrome is associated with anogenital distance, a marker of prenatal androgen exposure. Hum Reprod, 32, 937–943. [DOI] [PubMed] [Google Scholar]
- XU N, KWON S, ABBOTT DH, GELLER DH, DUMESIC DA, AZZIZ R, GUO X & GOODARZI MO 2011. Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS One, 6, e27286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YILMAZ B, VELLANKI P, ATA B & YILDIZ BO 2018. Diabetes mellitus and insulin resistance in mothers, fathers, sisters, and brothers of women with polycystic ovary syndrome: a systematic review and meta-analysis. Fertil Steril, 110, 523–533 e14. [DOI] [PubMed] [Google Scholar]
- ZHOU R, BRUNS CM, BIRD IM, KEMNITZ JW, GOODFRIEND TL, DUMESIC DA & ABBOTT DH 2007. Pioglitazone improves insulin action and normalizes menstrual cycles in a majority of prenatally androgenized female rhesus monkeys. Reprod Toxicol, 23, 438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]