

Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy with a dismal prognosis in patients with resistant or relapsed disease. Although NOTCH is a known driver in T-ALL, its clinical inhibition has significant limitations. Our previous studies suggested that NRARP, a negative regulator of Notch signaling, could have a suppressive role in T-ALL. Here, we report that NRARP levels are significantly increased in primary T-ALL cells suggesting that NRARP is not sufficient to block NOTCH oncogenic signals. Interestingly, although NRARP overexpression blocks NOTCH1 signaling and delays the proliferation of T-ALL cells that display high levels of Notch1 signaling, it promotes the expansion of T-ALL cells with lower levels of Notch1 activity. We found that NRARP interacts with lymphoid enhancer-binding factor 1 (LEF1) and potentiates Wnt signaling in T-ALL cells with low levels of Notch. Together these results indicate that NRARP plays a dual role in T-ALL pathogenesis, regulating both Notch and Wnt pathways, with opposite functional effects depending on Notch activity. Consistent with this hypothesis, mice transplanted with T-cells co-expressing NOTCH1 and NRARP develop leukemia later than mice transplanted with T-NOTCH1 cells. Importantly, mice transplanted with T-cells overexpressing NRARP alone developed leukemia with similar kinetics to those transplanted with T-NOTCH1 cells. Our findings uncover a role for NRARP in T-ALL pathogenesis and indicate that Notch inhibition may be detrimental for patients with low levels of Notch signaling, which would likely benefit from the use of Wnt signaling inhibitors. Importantly, our findings may extend to other cancers where Notch and Wnt play a role.
Subject terms: Acute lymphocytic leukaemia, Cell signalling, Cancer genetics, Oncogenes
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy that accounts for 15 and 25% of pediatric and adult acute lymphoblastic leukemia, respectively [1]. Although the outcome of T-ALL patients has improved significantly, the prognosis of patients with resistant or relapsed disease remains dismal [2, 3]. This and the high toxicity of the existing therapies underline the need to develop more specific and effective therapeutic strategies [3, 4].
Notch signaling plays a fundamental role in the pathogenesis of T-ALL, with more than 50% of human T-ALL patients displaying NOTCH activating mutations [5]. The prominent role of Notch in the pathogenesis of T-ALL led to the clinical inhibition of this pathway [6, 7]. However, the strategies developed so far lack sufficient antileukemia effects and have been associated with high toxicity [6, 7]. Thus, a better understanding of the oncogenic mechanisms associated to Notch function and regulation in T-ALL is necessary.
NOTCH regulated ankyrin repeat protein (NRARP) is a transcriptional target of NOTCH [8, 9] and a negative regulator of the Notch signaling [9, 10]. NRARP is expressed throughout human T cell development and its expression inversely correlates with NOTCH1 [11, 12]. In the mouse, overexpression of Nrarp in hematopoietic stem cells inhibits T-cell lineage commitment and early thymocyte development [12]. Our previous studies revealed that loss of mir-181ab1 inhibits leukemia development at least in part by derepressing the expression of Nrarp [13]. These results suggested that deregulation of NRARP may contribute to the pathogenesis of T-ALL. Here, we uncover a dual role for NRARP dependent on NOTCH1 intracellular domain (NICD) levels, with opposite functional outcomes in T-ALL pathogenesis. Importantly, our findings establish a new paradigm in what regards the outcomes of the cross talk between Notch and Wnt signaling pathways in T-ALL, with important therapeutic implications.
Results
NRARP is upregulated in T-ALL cells but it is insufficient to block Notch signaling
To understand if NRARP plays a role in T-ALL pathogenesis we started by characterizing NRARP expression in T-ALL primary cells and cell lines. NRARP protein levels were upregulated in T-ALL cells in comparison with normal thymocytes (Fig. 1a). In addition, we observed a positive correlation between NRARP and NICD levels (Fig. 1b). These observations are consistent with the fact that NRARP is a direct transcriptional target of NOTCH1. Nonetheless, they also suggest that either the NRARP protein expressed in T-ALL cells is not functional or that its levels, although increased, are not sufficient to block NOTCH1 oncogenic signals. To address these questions, we used shRNAs to silence NRARP expression in the T-ALL cell lines DND4.1 and MOLT-4. Although we achieved a knockdown of only 40–50% at the mRNA level (Supplementary Fig. S1 A) that was sufficient to increase NICD levels in both cell lines (Fig. 1c). Consistent with the increase in NICD1 levels, DND4.1 and MOLT-4 cells knocked down for NRARP proliferated more than their parental counterparts (Fig. 1d). Rescue of NRARP expression in MOLT-4 shNRARP cells (Supplementary Fig. S1B) significantly decreased their proliferative capacity (Supplementary Fig. S1 C). Importantly, these results showed that NRARP is functional in T-ALL cells and that, as in normal T-cells, it negatively regulates the Notch pathway.
Fig. 1.

NRARP expression is increased in T-ALL cells but it is insufficient to block Notch signaling. a NRARP protein levels in T-ALL primary cells (n = 5) and cell lines (n = 10) determined by western blot (WB) and normalized to NRARP levels in total thymocytes (n = 8). b Correlation between NRARP and NICD protein levels in total thymocytes and T-ALL primary cell lines. Protein levels were determined by WB. c WB analysis of NICD expression in DND4.1 and MOLT-4 T-ALL cell lines upon NRARP knockdown using shRNAs. d Effects of NRARP knockdown in DND4.1 and MOLT-4 cell proliferation. Cells were transduced with an shRNA against NRARP (shNRARP) or a scramble sequence as control (shSCR). Representative assay of three biological experiments, each performed in triplicate. e Effects of NRARP overexpression in T-ALL cell lines NICD levels (determined by WB). To better visualize the changes induced by NRARP overexpression, T-ALL cells transduced with an Empty vector (control condition) or an NRARP vector were treated with the proteasome inhibitor MG132. f Relative expression of Notch transcriptional targets in T-ALL cells overexpressing NRARP. mRNA levels were normalized to control condition (Empty cells). In d and f data represent the mean ± SEM. Statistical values were obtained using either the Student’s t test (a), 2way ANOVA (d), or Pearson correlation (b). **p < 0.01, ***p < 0.001
We next overexpressed NRARP in human T-ALL cell lines (Supplementary Fig. S1D, E), which led to NICD downregulation (Fig. 1e). NRARP has been shown to regulate NICD degradation through the proteasome [10]. Curiously, treatment of DND4.1 cells with the proteasome inhibitor MG132 did not reverse NRARP overexpression effects on NICD1 levels, suggesting that NRARP may also induce the degradation of NICD in a proteasome-independent way. Furthermore, NRARP overexpression blocked NOTCH transcriptional activity as shown by the overall decreased expression of NOTCH1 downstream targets HES1 and DTX1 in NRARP-overexpressing cells (Fig. 1f). Together, these results are consistent with the reported function of NRARP as a negative regulator of Notch signaling [9, 10], and suggest that in T-ALL NRARP levels, although elevated, are not sufficient to completely block NOTCH signals.
NRARP can not only inhibit but also promote the proliferation of T-ALL cells
Next, we investigated the functional effects of NRARP-induced inhibition of Notch signaling in T-ALL cell proliferation and survival. As expected, NRARP overexpression delayed proliferation and expansion of CEM and DND4.1 cell lines (Fig. 2a, b). However, to our surprise, NRARP had the opposite effect on Loucy and TALL-1 cells, promoting their proliferation and expansion (Fig. 2a, b).
Fig. 2.

NRARP can not only inhibit but also promote the proliferation of T-ALL cells. a Effects of NRARP overexpression in T-ALL cell proliferation and b expansion. In a one of three independent assays (each performed in triplicate) is displayed, whereas in b the fold change induced by NRARP is shown in comparison to control (Empty condition). c Effects of NRARP overexpression in T-ALL cell death as determined by annexinV staining and flow cytometry analysis. One of three independent assays performed in triplicate is displayed. d Effects of NRARP overexpression in cell cycle progression. Cells were sorted in G1 and cultured for 8 h. n = 3. e Analysis of the protein levels of the cell cycle regulator pRB. In a, b, c, and d data represent the mean ± SEM. Statistical values were obtained using the Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001
With the exception of DND4.1 cells, we did not observe significant differences in leukemia cell viability upon NRARP overexpression (Fig. 2c). Thus, we next characterized NRARP effects on cell cycle progression. We sorted cells in G1 and analyzed cell cycle progression after 8 h in culture. Compatible with the proliferation data, we observed a delay in cell cycle progression in both CEM and DND4.1 NRARP-overexpressing cells (Fig. 2d). In contrast, Loucy and TALL-1 cells progressed faster in the cycle upon NRARP overexpression (Fig. 2d). In line with these results, we observed increased levels of phosphorylated retinoblastoma protein in NRARP-overexpressing Loucy and TALL-1 cells (Fig. 2e). When phosphorylated, this cell cycle regulator becomes inactivated, allowing for G1-to-S phase cell cycle transition.
NRARP promotes Wnt signaling in T-ALL cells
Interestingly, we noticed that cMYC was not downregulated in Loucy and TALL-1 cells overexpressing NRARP. On the contrary cMYC transcript (Fig. S2A) and protein levels (Fig. S2B) were considerably upregulated in Loucy cells in particular. Since cMYC is a well-known transcriptional target of NOTCH, these data were in apparent contradiction with the inhibition of NOTCH we observed in the T-ALL cell lines, upon NRARP overexpression (Fig. 1f, e). On the other hand, these observations were consistent with the increased proliferation observed in Loucy and TALL-1 NRARP cells (Fig. 2) and suggested that NRARP may lead (directly or indirectly) to the activation of another pathway that maintains (or elevates) cMYC levels. Because in endothelial and neuro crest cells, NRARP has been shown to positively regulate canonical Wnt signaling [14, 15], which in turn it is known to regulate cMYC [16], we sought to investigate if NRARP could impact cMYC levels through the activation of the Wnt pathway. We started by evaluating β-catenin and phospho-β-catenin Ser675 levels as measures of Wnt signaling activity (phosphorylation of serine 675 has been shown to increase β-catenin stability and its transcriptional activity) [17] and found that both were upregulated in Loucy and TALL-1 cells overexpressing NRARP (Fig. 3a). These results suggested that NRARP overexpression positively regulates Wnt signaling in these cell lines. In contrast, NRARP overexpression in CEM and DND4.1 cells, did not affect or downregulated somewhat Wnt signaling pathway (Fig. 3a). Thus, to evaluate the impact of Wnt signaling in NRARP-overexpressing cells, we exposed T-ALL cells to the Wnt inhibitor PRI-724. This inhibitor binds to CPB blocking its interaction with β-catenin, inhibiting the transcription of Wnt downstream targets (Supplementary Fig. S3A) [18, 19]. Although Wnt inhibition initially affected the proliferation of both DND4.1 and Loucy NRARP cells, DND4.1 cells recovered their proliferative capacity while Loucy NRARP cells did not (Fig. 3b). Analysis of cell proliferation in parental cells further shows that Loucy NRARP cells are more sensitive to Wnt inhibition than Loucy Empty cells (Fig. 3b). Together, these results demonstrate that in Loucy cells overexpression of NRARP promotes proliferation through Wnt signaling. We also observed that Wnt inhibition decreased cell viability, in particular of Loucy NRARP cells (Fig. S3B). Curiously, blockade of Wnt signaling revert the loss in viability induced by NRARP in DND4.1 cells (Fig. 2c, Supplementary Fig. S3B), an effect that was partially maintained up to 144 h (Supplementary Fig. S3C). Supporting the importance of Wnt signaling pathway in the proliferative capacity of Loucy cells, we observed that activation of this pathway using a GSK3 inhibitor (Supplementary Fig. S3D) promotes Loucy Empty cells proliferation (Supplementary Fig. S3E). The activation of this pathway by NRARP in Loucy cells may explain the fact that, in Loucy NRARP cells, treatment with GSK3 inhibitor does not promote proliferation (Supplementary Fig. S3E). In what concerns DND4.1 Empty and NRARP cells we observed a decrease in proliferation, which is consistent with the role described for GSK3 in the positive regulation of Notch1 signaling and NICD1 stability (Supplementary Fig, S3E) [20]. As shown in the Supplementary Fig. S3D treatment with the GSK3 inhibitor leads to a downregulation of NICD1 levels.
Fig. 3.

NRARP promotes Wnt signaling in T-ALL cells. a Analysis of WNT signaling activity by measuring β-catenin and pβ-catenin Ser675 protein levels by WB in T-ALL cell lines with and without NRARP overexpression. b Effects of Wnt inhibition in the proliferation of T-ALL cells overexpressing or not NRARP using the inhibitor PRI-724. One of three independent assays performed in triplicate is shown. c Analysis of LEF1 protein levels in T-ALL cell lines upon NRARP overexpression. LEF1 has several isoforms due to an alternative promoter and alternative splicing. d Effects of LEF1 knockdown in β-catenin, pβ-catenin Ser675, and cMYC protein levels in T-ALL cell lines with and without NRARP overexpression. e Effects of LEF1 knockdown in the proliferation of T-ALL cells overexpressing or not NRARP. One of three independent assays performed in triplicate is shown. Quantitative analysis of f NRARP:LEF1 interactions and g LEF1-β-catenin interactions performed by the quantification of cells with and without PLA signals per cell per microscope field. The combination of three independent assays and the analysis of five microscope fields per assay are shown. In b, e, f, and g data represent the mean ± SEM. Statistical values were obtained using the Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Of note, while NRARP negatively modulates Notch signaling by promoting NICD degradation, it is known to positively regulate Wnt signaling by promoting LEF1 protein stability [14]. LEF1 is a DNA-binding transcription factor of the T-cell factor (TCF) family, acting downstream of the Wnt signaling pathway by interacting with nuclear β-catenin [21]. Analysis of LEF1 protein levels in NRARP-overexpressing cells showed an increase in Loucy and TALL-1 cells of 27% and 13%, respectively (Fig. 3c). LEF1 has several isoforms with apparently different, sometimes opposing, functions [22–24]. Thus, we next dissected the impact of LEF1 downstream from NRARP by silencing LEF1 in DND4.1 and Loucy cells with and without NRARP overexpression (Supplementary Fig. S4A, B). LEF1 knockdown blocked Wnt signaling in both Empty and NRARP cells (Fig. 3d). We then evaluated the functional effects of LEF1 knockdown in NRARP expressing cells and found that LEF1 silencing reversed NRARP-induced proliferation in Loucy cells, whereas it did not impact DND4.1 NRARP cells (Fig. 3e). These changes in Loucy NRARP cells were paralleled by cMYC downregulation (Fig. 3d). This set of results provide evidence that NRARP, aside from having a suppressive role in T-ALL by inhibiting Notch signaling (e.g., in CEM and DND4.1 cells) can also promote T-ALL proliferation by potentiating Wnt signaling through LEF1 (e.g., in Loucy and TALL-1). Moreover, the fact that LEF1 knockdown in DND4.1 control cells augmented cell proliferation, suggests that Wnt signaling may has an inhibitory effect in some T-ALL cases (Fig. 3e). This inhibitory effect of LEF1 knockdown was further confirmed in the MOLT-4 cells (Supplementary Fig. S4C, D). In both cell lines, although not statistically significant, LEF1 knockdown results in minor increments in NOTCH1 downstream targets (Supplementary Fig. S4E).
To understand if NRARP regulates LEF1 in a direct fashion we carried out in situ PLA assays to evaluate the interaction between NRARP and LEF1 (Supplementary Fig. S5A). We found that, in both DND4.1 and Loucy cell lines, NRARP overexpression increased the percentage of cells with NRARP:LEF1 interactions (Fig. 3f) and, in the case of Loucy cells, the number of interactions per cell (Supplementary Fig. S5B). However, this increase in NRARP:LEF1 interactions only translated into increased Wnt signaling activity in Loucy cells as determined by the increase in LEF1:β-catenin interactions in these cells upon NRARP overexpression (Fig. 3g and Supplementary Fig. S5 C, D). These data are consistent with the functional results obtained by the chemical inhibition of Wnt signaling and LEF1 knockdown, and show that although NRARP interacts with LEF1 in both cell lines, it leads to Wnt signaling activity only in Loucy cells.
NRARP anti- or pro-tumoral role in T-ALL depends on NICD1 levels
CEM and DND4.1 cells have NOTCH1-activating mutations while Loucy and TALL-1 cells do not [25] (of note TALL-1 cells have a NOTCH3 mutation [26]). Consequently, CEM and DND4.1 cells have higher levels of NICD1 than Loucy and TALL-1 cells (Fig. 4a). Therefore, we investigated whether the dual role of NRARP related to NICD1 levels. First we confirmed that the dual effect of NRARP correlated with NICD1 levels also in MOLT-4 and Jurkat T-ALL cells (Supplementary Fig. S1F, G). Although these cell lines have NOTCH1-activating mutations [25], they have very different levels of NICD1 (Fig. 4a). Taken together, our data suggest that NRARP may prevent the expansion of cells with high levels of NICD1 (such as the NOTCH1-mutant cell lines CEM, DND4.1, and MOLT-4) and promote the growth of cells with normal (lower) levels of NICD1 (e.g. Loucy, TALL-1, and Jurkat cell lines).
Fig. 4.

NRARP anti- or pro-tumoral role in T-ALL depends on NICD1 levels. a Flow cytometry analysis of NICD1 levels in T-ALL cell lines (n = 3). b Kaplan–Meier survival curve of immunocompromised (NSG) mice transplanted with p53-null CD4-CD8- precursor T-cells (D1 cells) overexpressing NICD1, NRARP or both. Whereas NRARP alone induced leukemia development with a similar kinetic of that of NICD1 (median survival of 72.5 and 74 days, respectively), its co-expression with NICD1 had a suppressive effect, increasing significantly the median survival to 86 days (p < 0.05). c Kaplan–Meier survival curve of NSG mice transplanted with D1 cells isolated from mice primarily transplanted with D1 cells overexpressing NICD1, NRARP, or both. The secondary transfer of these cells confirmed their leukemogenic potential and NRARP suppressive role when co-expressed with NICD1 (p < 0.05). d NICD protein levels in D1 cells isolated from spleens of mice transplanted with D1 NICD1, D1 NRARP, or D1 NICD1 NRARP-overexpressing cells. e Western blot analysis of cMYC, β-catenin, and pβ-catenin Ser675 protein levels in cells collected from spleens of mice transplanted D1 NRARP cells and D1 Empty cells (control condition)
To further test this possibility we overexpressed NICD1 in the p53-null CD4−CD8− precursor T-cell line D1 [27], with or without co-expression of NRARP (Supplementary Fig. S6). D1 cells were then transplanted into immunocompromised mice. As expected, mice transplanted with NICD1-expressing D1 cells developed leukemia rapidly, whereas mice transferred with D1 cells co-expressing NICD1 and NRARP developed leukemia significantly later (Fig. 4b). By contrast, mice transplanted with D1 cells overexpressing NRARP alone developed leukemia with similar kinetics to those transplanted with D1 NICD1 cells (Fig. 4b), confirming the oncogenic potential of NRARP in the context of low levels of Notch1 signaling. Transfer of these cells into secondary recipient mice confirmed their leukemogenic nature (Fig. 4c). In agreement with the in vitro data using human T-ALL cell lines, NRARP NICD1 D1 cells, collected from the spleen, showed decreased NICD1 levels as compared with D1 overexpressing NICD1 alone (Fig. 4d). Moreover, D1 cells expressing NRARP alone displayed higher levels of β-catenin and cMYC than control, empty vector cells (Fig. 4e).
NRARP negative impact on primary T-ALL cells associates with Notch pathway activation status
To validate the dual role of NRARP in human primary T-ALL cells we exposed primary or patient-derived xenograft (PDX) T-ALL cells, classified according to their NOTCH1 mutational status/levels of NICD1 (Supplementary Fig. S7A, B) [28], to NRARP recombinant protein (rNRARP) (Supplementary Fig. S7C, D). Consistent with our observations in cell lines, NRARP forced expression in NOTCH1 mutant/NICD1 high T-ALL samples negatively impacted cell number (Fig. 5a) and viability (Fig. 5b), whereas it had the opposite effect on NOTCH1 wild type/NICD low cases (Fig. 5a, b). Also in agreement with our observations in T-ALL cell lines, rNRARP led to the downregulation of NOTCH targets in NOTCH1-mutant samples and upregulated WNT targets in NOTCH1-WT cells (Fig. 5c). Overall, the data from primary T-ALL samples and PDXs support our hypothesis that NRARP plays a dual role in T-ALL, with opposite functional outcomes that depend on NICD1 levels.
Fig. 5.

NRARP negative impact on primary T-ALL cells associates with Notch pathway activation status. Analysis of primary and patient-derived xenograft (PDX) T-ALL cell proliferation a and b viability upon delivery of NRARP recombinant protein (rNRARP). Samples with NOTCH1 mutations/high levels of NICD1 are depicted in orange and samples NOTCH1-WT/low levels of NICD1 in blue. c mRNA analysis of Notch1 and Wnt signaling transcriptional targets by quantitative-PCR, in primary and PDX T-ALL cells treated with rNRARP. In a and b data represent the mean ± SEM. Statistical values were obtained using the Student’s t test. *p < 0.05, **p < 0.01
Discussion
NRARP is a transcriptional target of NOTCH1 that promotes the degradation of NICD, originating a negative feedback loop that downregulates Notch signaling. Although it is known that the constitutive activation of this signaling pathway plays a pivotal role in T-ALL, a putative role for NRARP in this disease context has not been investigated. Our studies on the role of mir-181ab1 in T-ALL suggested that NRARP could have a suppressive role in T-ALL [13]. Thus, in the current work we set out to evaluate the contribution of NRARP to T-ALL pathogenesis. Interestingly, our work demonstrates that NRARP has a dual role in T-ALL, dependent of Notch1 activity levels. On one hand, NRARP has a ‘tumor suppressor’-like role in T-ALL cells with high levels of NICD by inhibiting Notch1 signaling. On the other hand, in T-ALL cells with normal levels of NICD, NRARP unexpectedly promotes leukemogenesis, by activating the Wnt signaling pathway (Fig. 6).
Fig. 6.
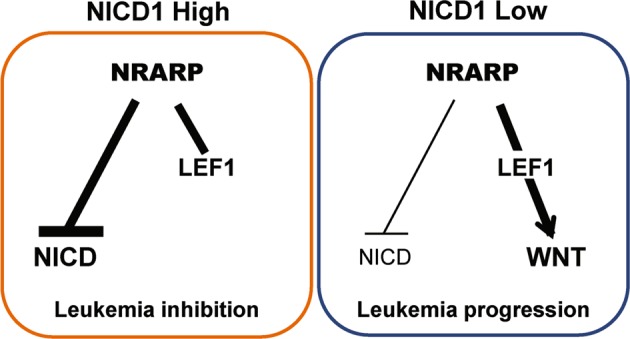
Schematic representation of NRARP dual role in T-ALL. In cells with high levels of NICD1, NRARP overexpression blocks Notch signaling and suppresses leukemia development. In cells with low/normal levels of NICD1, NRARP activates the Wnt pathway promoting leukemia progression
NRARP is expressed in hematopoietic progenitor cells [11], as well as in thymocyte subsets, with highest levels in CD4+CD8+ T-cells [12]. Its overexpression in murine bone marrow stem/progenitor cells blocks T-cell lineage commitment and the progression of T-cell development through DN stages [12]. Notably, we now show that NRARP overexpression leads to the malignant transformation of CD4−CD8− T-cell precursors, inducing T-ALL as efficiently as NOTCH1 but through Wnt signaling activation. Our work identifies an unexpected role for NRARP in T-ALL pathogenesis and warrants the search for genomic lesions (or any other alteration—–e.g., transcriptional, epigenetic, or post transcriptional) that may lead to the deregulation of NRARP levels and/or function. According to our findings, an alteration leading to the upregulation of NRARP levels in NOTCH WT cells can promote leukemogenesis through the activation of the Wnt signaling. On the opposite, alterations that lead to the downregulation of NRARP levels in a NOTCH1 high context, can promote leukemogenesis by derepressing Notch oncogenic signaling. Thus, if identified, it would be interesting to understand how these alterations correlate with the activation of Notch and Wnt pathways.
Importantly, our results highlight the therapeutic potential of NRARP in a context of high levels of NOTCH or NOTCH1 mutations. We show that, although increased in T-ALL cells with higher levels of NOTCH, NRARP is not sufficient to block those oncogenic signals. However, delivery of NRARP recombinant protein to T-ALL cells with high levels of NOTCH negatively impacts their proliferation and viability, as demonstrated by our studies. This implicates that therapeutic strategies leading to NRARP activation will be potentially useful for the treatment of a majority of T-ALL cases (with NOTCH1 hyper activation). Moreover, our findings also demonstrate that such strategies may be detrimental in the context of some NOTCH1-WT/low NICD1 T-ALL cases and/or Wnt signaling dependent T-ALL cases.
NRARP has been shown to independently regulate canonical Wnt and Notch signaling [14, 15]. Curiously, NRARP negatively modulates the Notch pathway by promoting NICD degradation, but positively regulates Wnt signaling by promoting LEF1 protein stability [14]. LEF1 is a DNA-binding transcription factor of the TCF family, acting downstream of the Wnt signaling pathway by interacting with nuclear β-catenin [21]. Our data demonstrates that in T-ALL, NRARP also positively regulates Wnt signaling through LEF1. Overexpression of NRARP increases LEF1 protein levels and knockdown of LEF1 reverses NRARP-induced proliferation in T-ALL cells with low levels of NICD1. In addition, our data shows that NRARP overexpression increases the number of NRARP:LEF1 interactions which in T-ALL cells with low levels of NICD1 translates into more LEF1:β-catenin interactions. Because NRARP binds to NICD1 and LEF1, it is possible that in cells with high levels of NICD1, by targeting NICD to degradation, NRARP drags LEF1 along, impairing Wnt signaling activation.
Given their essential roles throughout T-cell development, the cross talk between Notch and Wnt pathways has been postulated to also occur in T-ALL. Nonetheless, this has not been clearly demonstrated and the existing data are not consensual. Some studies point for a positive correlation between these pathways. For example, in adult T-ALL, LEF1 activating mutations were associated with NOTCH1 mutations [29]. In addition, Lef1 has been reported as a transcriptional target of NOTCH1 in T-cell lymphomas and to accelerate Notch1-induced lymphomagenesis in mice [30]. In contrast, other evidences point to the inactivation of Wnt signaling when Notch signaling is active [31–33]. Particularly, in pediatric T-ALL, LEF1 inactivation has been associated with NOTCH1-activating mutations [34]. The fact that proliferation is potentiated by the chemical inhibition of Wnt signaling and by LEF1 silencing in NOTCH1-mutant DND4.1 and MOLT-4 cells is in agreement with the latter studies, and suggests that Wnt signaling may be detrimental for T-ALL cells with NOTCH1 constitutive activation.
Overall, our results identify a role for NRARP in T-cell leukemogenesis and indicate that Wnt signaling has an oncogenic role in human T-ALL pathogenesis, independent of Notch signals. Our findings open new therapeutic perspectives and underline the need to consider NOTCH1 and WNT mutational status into T-ALL therapeutic decisions. While the therapeutic inhibition of Notch signaling may be beneficial in patients with the constitutive activation of this pathway, in patients with low levels of Notch signaling this approach may be therapeutically disadvantageous. In these cases, the inhibition of Wnt signaling may prove to be beneficial. In contrast, our data further suggests that Wnt signaling inhibition may be disadvantageous for T-ALL patients with high levels of Notch1 activity. Importantly, our findings may extend to other cancer types where Notch and Wnt are known to play a role, such as breast, lung, and colon cancer.
Materials and methods
Primary leukemia cells
Primary leukemia cells were obtained from bone marrow and/or peripheral blood of diagnostic pediatric T-ALL. Informed consent was obtained in accordance with the Declaration of Helsinki and under the ethical review board approval of Instituto Português de Oncologia (Lisbon, Portugal) and Centro Infantil Boldrini (Campinas, São Paulo, Brazil). When available, T-ALL cells from PDXs were used.
Cell lines
T-ALL cell lines CEM, MOLT-4, DND4.1, Jurkat, Loucy, and TALL-1 were cultured in RPMI 1640 (Gibco, Thermo Fisher Scientific, Gaithersburg, MD, USA) supplemented with 10% FBS (Biowest, Nuaillé, France), 1% penicillin/streptomycin (Gibco) and 1% HEPES (Gibco), at 37 °C in a 5% CO2 environment. Cells were kept at an optimal concentration of 0.5 × 106 cells/mL. D1 cells were cultured in the same conditions plus 25 ng/mL rmIL-7 (PeproTech EC, London, UK). HEK-293T cells were maintained in DMEM (Gibco) supplemented with 10% FBS, 1% l-glutamine, and 1% penicillin/streptomycin. All cell lines were periodically checked for mycoplasma by PCR and were found to be negative.
RNA extraction, cDNA synthesis, and quantitative-PCR
Total RNA was extracted using TRIzol® reagent (Ambion™, Thermo Fisher Scientific). Synthesis of cDNA was performed with up to 1 μg of total RNA, using the reverse transcriptase SuperScript™ III kit (Invitrogen, Thermo Fisher Scientific) and random hexamers. Quantitative-PCR was performed using Taqman and SyberGreen methods. NRARP and 18S expression was quantified using Taqman Gene Expression Assays (Applied Biosystems, Foster City, CA, USA). The expression of the remaining genes was quantified using Power SYBR Green PCR Master Mix (Applied Biosystems) and the specific primers are indicated in Supplementary Table S1. The PCR reactions were performed using a ViiA™ 7 Real-Time PCR System (Thermo Fisher Scientific). The expression of each gene was normalized to the expression level of the ribosomal RNA 18S using the dCt method and fold difference comparisons between samples evaluated using the ddCT method.
Immunoblot
Lysates were prepared as described [35], resolved by SDS-PAGE and immunoblotted with antibodies against NRARP (E-13 and C-12), β-Catenin (E-5), β-Actin (Santa Cruz Biotechnology, Dallas, TX, USA), phospho-β-catenin (Ser675), LEF1 (C18A7), Cleaved Notch1 (Val1744), cMyc (D3N8F) (Cell Signaling Technology, Danvers, MA, USA), Rb (G3-245), and phospho-Rb (S807/S811, J112-906) (BD PharmingenTM, San Jose, CA, USA). Densitometry analysis was performed using Adobe Photoshop CS5 Extended software (Adobe Systems). Results were normalized to the loading control.
Proximity ligation assay (PLA)
We utilized the Duolink® PLA technology for the in situ detection of NRARP:LEF1 and LEF1:β- catenin interaction in T-ALL cell lines with and without NRARP overexpression. We used the Duolink® In Situ Red Starter Kit Goat/Rabbit and In Situ Red Starter Kit Mouse/Rabbit (Sigma-Aldrich, St. Louis, MO, USA) for the detection of NRARP:LEF1 and LEF1:β-catenin interactions, respectively. We followed manufacturer’s instructions and used the primary antibodies anti-LEF1 Rabbit mAb (HPA002087) (Sigma-Aldrich), anti-β-catenin mouse mAb (E-5), and anti-NRARP goat mAb (E-13) (Santa Cruz Biotechnology). Samples were analyzed in a confocal laser point-scanning microscope (Zeiss LSM 710), using the integrated software ZEN 2012. Cell counting and image configuration were performed using Fiji/ImageJ software.
Proteasome inhibitor treatment
Cells were treated with 1 µL/mL of MG132 inhibitor (10 mM in DMSO) (Calbiochem), at a cell density of 1 × 106 cells/mL and incubated for 8 h at 37 °C. Cells were then harvested and lysed as described above.
Production of retro and lentiviruses
Retroviruses were produced using the pCLeco packaging vector and vesicular-stomatitis-virus-pseudotyped lentiviruses using a third-generation system. Both viruses were produced by co-transfection into HEK-293T cells and using Lipofectamine 2000 (Life technologies, Carlsbad, CA, USA) as previously describe [13, 35].
Transduction of cell lines for gene overexpression or knockdown
NRARP-overexpressing cells (referred to as NRARP) were established by transduction with lentiviruses produced using the lentiviral vector pCDH-CMV-MCS-T2AcopGFP (SBI, Palo Alto, CA, USA) cloned with NRARP ORF [13]. Cells transduced with the pCDH vector without an insert are referred to as “Empty”. For NICD overexpression, cells were transduced with retroviruses produced using the retroviral construct MigR1-NICD1 [13]. Cell lines knocked down for NRARP or LEF1 was established using OmicsLink™ shRNA vectors from GeneCopoeia (Rockville, MD, USA). The control vector (named SCR) expresses a scramble shRNA sequence. Briefly, 1 × 106 cells were incubated with 1 mL of viral supernatant and 8 ng/mL of polybrene (Sigma-Aldrich). Cells were spun down for 120 min at room temperature at 2300 rpm. The resulting transduced cell lines were sorted for an equivalent reporter expression.
Flow cytometry analysis
Standard procedures were used to stain the cells with fluorochrome conjugated antibodies or to verify reporter protein expression. In this study we used a PE anti-NOTCH1 (mN1A) antibody (Biolegend, San Diego, CA, USA). Samples were acquired in an LSRFortessa cell analyzer (BD Biosciences). Data analyses were performed using the FlowJo software.
Cell growth assays
To assess cell growth potential of T-ALL cell lines, expansion and proliferation assays were carried out. Expansion assays were performed by culturing a fixed number of cells at optimal density and counting cells in a hemocytometer using trypan blue for dead cells exclusion, at days 3, 6, 9, and 12 of culture. For proliferation experiments 1.5 × 105 cells were plated at optimal density and harvested at 24, 48, 72, and 96 h. Cell number was determined by flow cytometry (LSRFortessa cell analyzer, BD Biosciences) using counting beads (Beckmon Coulter). Data analyses were performed using FlowJo™ software.
Viability assays
To assess cell viability 1.5 × 105 cells were plated at optimal density and harvested at 24, 48, 72 and 96 h. Viability was determined by flow cytometry (LSRFortessa cell analyzer, BD Biosciences) using annexinV-APC (eBioScience, Thermo Fisher Scientific) and 7AAD dye (BD Pharmingen), according to manufacturer’s protocol. Data were analyzed using FlowJo™ software.
Cell cycle analysis
To analyze cell cycle profiles of NRARP-overexpressing T-ALL cell lines (as comparing with empty control vectors), 1 × 106 cells were incubated for 1 h at 4 °C and plated afterwards into 12-well plates at optimal density.
To determine the effects of NRARP in T-ALL cell cycle progression we sorted T-ALL cells in the G1 phase of cell cycle and compared the cell cycle profile of those cells upon 8 h in culture. We used the Vybrant® DyeCycle™ Violet Stain dye (Thermo Fisher Scientific) to sort cells in G1 phase using a BD FACSAria™ Cell Sorter (containing a violet laser, 407 nm). Briefly, for cell cycle analysis, cells were collected, washed, resuspended in cold PBS, and fixed with equal volume of 80% ethanol in PBS. Before staining, cells were washed twice with cold PBS and incubated with RNase A solution of 50 µg/mL in PBS, for 30 min at 37 °C. Propidium iodide was added at a concentration of 2.5 × 10−3 µg/µL and incubated overnight. Cell cycle profile was assessed next day by flow cytometry using a FACSCalibur. Data analyses were performed using FlowJo™ or ModFit LT™ software, as appropriate.
NRARP recombinant protein production
Plasmid encoding recombinant NRARP protein was used to transform BL21 bacteria to facilitate isopropyl-b-D-thiogalactoside induction for protein expression in LB medium cultures following standard methods. Eluents containing protein were measured using Bradford reagent, combined and dialyzed overnight at 4 °C and stored at appropriate temperature until further use.
D1 cells transfer in vivo model
Age and sex-matched (8–14 weeks) NSG mice were transferred with D1 cells overexpressing NRARP and/or NICD not blinding. Cells (5 × 106) were injected intravenously in the tail. For the analysis of mouse overall survival humane endpoints were established. In particular, mice were euthanized when presenting a 20% weight loss or signs of lethargy. These endpoints were used to build the survival curve. The Kaplan–Meier estimator was used to determine the median rate of survival. The p-value was determined using the log-rank (Mantel–Cox) test. At the time of the humane endpoints mice were sacrificed with anesthesia overdose (Isoflurane). Experimental procedures were approved by the institutional Animal Ethics Committee from Instituto de Medicina Molecular João Lobo Antunes and followed the recommendations for the care and use of laboratory animals from the European Commission and Portuguese authorities. The minimal number of animals sufficient for the generation of statistically significant data (n = 5) was used.
Statistical analysis
GraphPad Prism software was used for statistical analysis. Unless stated otherwise functional assays were evaluated using the unpaired Student’s two-tailed t-test. Significance was set for p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001: ****p < 0.001).
Supplementary information
Acknowledgements
The authors thank the members of the JBarata laboratory for comments and discussions, and Sérgio Almeida e João Sabino for the valuable help with PLA assays. The authors especially thank the generosity of patients and their families for providing primary samples, and the health care professionals (Service directors, physicians, and nurses) from IPO Lisboa and Centro Infantil Boldrini involved in the collection of those samples. This work was supported by the grant IF/00788/2013 from Fundação para a Ciência e a Tecnologia (FCT) awarded to RF and by the consolidator grant ERC CoG-648455 from the European Research Council awarded to JTB. PA is a Marie Curie ITN PhD student, supported by Marie Curie Protein Conjugates ITNand. MLO is a LisbonBioMed student supported by an FCT PhD fellowship (PD/BD/114102/2015). RF and JTB are FCT starting and consolidator investigators, respectively. Publication costs of this work were supported by UID/BIM/50005/2019, project funded by Fundação para a Ciência e a Tecnologia (FCT)/Ministério da Ciência, Tecnologia e Ensino Superior (MCTES) through Fundos do Orçamento de Estado.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Inês Pinto, Mafalda Duque
Supplementary information
The online version of this article (10.1038/s41388-019-1042-9) contains supplementary material, which is available to authorized users.
References
- 1.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. New Engl J Med. 2004;350:1535–48. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 2.Smith MA. Update on developmental therapeutics for acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2009;4:175–82. doi: 10.1007/s11899-009-0024-3. [DOI] [PubMed] [Google Scholar]
- 3.Fullmer A, O’Brien S, Kantarjian H, Jabbour E. Novel therapies for relapsed acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2009;4:148–56. doi: 10.1007/s11899-009-0021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roti G, Stegmaier K. New Approaches to Target T-ALL. Front Oncol. 2014;4:170. doi: 10.3389/fonc.2014.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 6.Papayannidis C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015;5:e350. doi: 10.1038/bcj.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling-are we there yet? Nat Rev Drug Discov. 2014;13:357–78. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 8.Krebs LT, Deftos ML, Bevan MJ, Gridley T. The Nrarp gene encodes an ankyrin-repeat protein that is transcriptionally regulated by the notch signaling pathway. Dev Biol. 2001;238:110–9. doi: 10.1006/dbio.2001.0408. [DOI] [PubMed] [Google Scholar]
- 9.Lahaye K, Kricha S, Bellefroid EJ. XNAP, a conserved ankyrin repeat-containing protein with a role in the Notch pathway during Xenopus primary neurogenesis. Mech Dev. 2002;110:113–24. doi: 10.1016/s0925-4773(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 10.Lamar E, Deblandre G, Wettstein D, Gawantka V, Pollet N, Niehrs C, et al. Nrarp is a novel intracellular component of the Notch signaling pathway. Genes Dev. 2001;15:1885–99. doi: 10.1101/gad.908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van de Walle I, De Smet G, De Smedt M, Vandekerckhove B, Leclercq G, Plum J, et al. An early decrease in Notch activation is required for human TCR-alphabeta lineage differentiation at the expense of TCR-gammadelta T cells. Blood. 2009;113:2988–98. doi: 10.1182/blood-2008-06-164871. [DOI] [PubMed] [Google Scholar]
- 12.Yun TJ, Bevan MJ. Notch-regulated ankyrin-repeat protein inhibits Notch1 signaling: multiple Notch1 signaling pathways involved in T cell development. J Immunol. 2003;170:5834–41. doi: 10.4049/jimmunol.170.12.5834. [DOI] [PubMed] [Google Scholar]
- 13.Fragoso R, Mao T, Wang S, Schaffert S, Gong X, Yue S, et al. Modulating the strength and threshold of NOTCH oncogenic signals by mir-181a-1/b-1. PLoS Genet. 2012;8:e1002855. doi: 10.1371/journal.pgen.1002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishitani T, Matsumoto K, Chitnis AB, Itoh M. Nrarp functions to modulate neural-crest-cell differentiation by regulating LEF1 protein stability. Nat Cell Biol. 2005;7:1106–12. doi: 10.1038/ncb1311. [DOI] [PubMed] [Google Scholar]
- 15.Phng LK, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell. 2009;16:70–82. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 17.Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8:33972–89. doi: 10.18632/oncotarget.15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arensman MD, Telesca D, Lay AR, Kershaw KM, Wu N, Donahue TR, et al. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol Cancer Ther. 2014;13:2303–14. doi: 10.1158/1535-7163.MCT-13-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] PNAS. 2004;101:12682–7. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin YH, Kim H, Oh M, Ki H, Kim K. Regulation of Notch1/NICD and Hes1 expressions by GSK-3alpha/beta. Mol cells. 2009;27:15–9. doi: 10.1007/s10059-009-0001-7. [DOI] [PubMed] [Google Scholar]
- 21.van Noort M, Clevers H. TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev Biol. 2002;244:1–8. doi: 10.1006/dbio.2001.0566. [DOI] [PubMed] [Google Scholar]
- 22.Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, et al. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53–7. doi: 10.1038/ng0501-53. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Ji P, Steffen B, Metzger R, Schneider PM, Halfter H, et al. Alterations of lymphoid enhancer factor-1 isoform expression in solid tumors and acute leukemias. Acta Biochim Biophy Sin. 2005;37:173–80. [PubMed] [Google Scholar]
- 24.Kobielak A, Kobielak K, Trzeciak WH. A novel isoform of human lymphoid enhancer-binding factor-1 (LEF-1) gene transcript encodes a protein devoid of HMG domain and nuclear localization signal. Acta Biochim Pol. 2001;48:221–6. [PubMed] [Google Scholar]
- 25.Squiban B, Ahmed ST, Frazer JK. Creation of a human T-ALL cell line online database. Leuk Lymphoma. 2017;58:2728–30. doi: 10.1080/10428194.2017.1300896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernasconi-Elias P, Hu T, Jenkins D, Firestone B, Gans S, Kurth E, et al. Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene. 2016;35:6077–86. doi: 10.1038/onc.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43:932–9. doi: 10.1038/ng.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silva A, Jotta PY, Silveira AB, Ribeiro D, Brandalise SR, Yunes JA, et al. Regulation of PTEN by CK2 and Notch1 in primary T-cell acute lymphoblastic leukemia: rationale for combined use of CK2- and gamma-secretase inhibitors. Haematologica. 2010;95:674–8. doi: 10.3324/haematol.2009.011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo X, Zhang R, Liu J, Li M, Song C, Dovat S, et al. Characterization of LEF1 High Expression and Novel Mutations in Adult Acute Lymphoblastic Leukemia. PLoS One. 2015;10:e0125429. doi: 10.1371/journal.pone.0125429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spaulding C, Reschly EJ, Zagort DE, Yashiro-Ohtani Y, Beverly LJ, Capobianco A, et al. Notch1 co-opts lymphoid enhancer factor 1 for survival of murine T-cell lymphomas. Blood. 2007;110:2650–8. doi: 10.1182/blood-2007-04-084202. [DOI] [PubMed] [Google Scholar]
- 31.Kwon C, Cheng P, King IN, Andersen P, Shenje L, Nigam V, et al. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat Cell Biol. 2011;13:1244–51. doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin YH, Kim H, Ki H, Yang I, Yang N, Lee KY, et al. Beta-catenin modulates the level and transcriptional activity of Notch1/NICD through its direct interaction. Biochim Biophys Acta. 2009;1793:290–9. [DOI] [PubMed]
- 33.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 34.Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. 2010;115:2845–51. doi: 10.1182/blood-2009-07-234377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Correia NC, Fragoso R, Carvalho T, Enguita FJ, Barata JT. MiR-146b negatively regulates migration and delays progression of T-cell acute lymphoblastic leukemia. Sci Rep. 2016;6:31894. doi: 10.1038/srep31894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.