

Abstract
Background
The current study is aimed to examine the impact of pharmacokinetics and gene polymorphisms of enzymes involving in absorption, distribution, metabolism and excretion (ADME) on the efficacy of gefitinib in non-small cell lung cancer (NSCLC) patients.
Methods
Eligible patients with indication of gefitinib treatment were prospectively enrolled in this study. Two peripheral blood samples at baseline and before cycle 2 day 1 were collected for the detection of single nucleotide polymorphisms (SNPs) of drug ADME enzymes and trough drug concentration (Ctrough) at steady state. Thirteen SNPs were genotyped using the Sequenom Massarray system. Ctrough was determined by validated high-performance liquid chromatographic method with tandem mass spectrometric (LC-MS/MS).
Results
Fifty-eight NSCLC patients were enrolled in this study. The median of Ctrough was 175ng/mL (range from 47.8 to 470 ng/mL). The trough concentration was not associated with either objective response or progression free survival (PFS). Ctrough was significantly lower in CYP3A4 rs2242480 CC + CT genotype than in TT genotype (P=0.019) and in ABCG2 rs2231142 AA genotype than in AC + CC genotype (P=0.031). ABCB1 rs2032582 dominant model was significantly correlated with overall response rate (ORR) and patients with GG phenotype respond better than patients with GT + TT phenotypes (84.6% vs. 51.2%, P=0.032). ABCB1 rs10256836 recessive model was significantly correlated with PFS and patients with GG phenotype achieved longer PFS than patients with GC + CC phenotypes (17.40 vs. 10.33 months, P=0.040).
Conclusions
The Ctrough of gefitinib was significantly different between CYP3A4 and ABCG2 genotypes, but not with the efficacy of gefitinib treatment. ABCB1 rs2032582 and rs10256836 polymorphisms were correlated treatment outcome. Polymorphisms analysis of ABCB1 could be a predictive biomarker for gefitinib treatment.
Keywords: Non-small cell lung cancer (NSCLC), gefitinib, pharmacokinetic, ABCB1, single nucleotide polymorphisms (SNPs)
Introduction
Non-small cell lung cancer (NSCLC) patients containing sensitive somatic mutations of the epidermal growth factor receptor (EGFR) gene, such as the small deletions [747–750] and point mutations at codon 858 (L858R), are highly responsive to gefitinib (1,2). However, there are still 30% patients with EGFR mutations could not benefit from gefitinib treatment. Thus, it’s important to identify biomarkers that allow clear separation of patients with or without a relevant chance of clinical benefit from this drug (2,3). Recently findings suggest that concomitant oncogenes strongly correlated with treatment outcome of gefitinib, and indicate poor clinical benefit, such as TP53, KRAS, MYC, APC, etc. (4,5). Some studies focus on the genes involved in drug targets or signaling pathways (6,7). The deletion polymorphism of BIM, a pro-apoptotic member of the BCL2 family, could lead to intrinsic resistance to gefitinib (8,9).
However, very few studies focus on the pharmacogenomics impact of gefitinib on its treatment outcome. A different approach is to identify germline gene polymorphisms of enzymes involving in gefitinib absorption, distribution, metabolism and excretion (ADME) processes. The ADME of a drug is largely a host-mediated process. As a tyrosine kinase inhibitor, the efficacy of gefitinib in vivo could be significantly affected by genetic variability of these potential pharmacokinetics or pharmacogenomics linked enzymes (10,11). ATP-binding cassette (ABC) transporters function as substrate enzymes for multiple compounds and play a leading role in transporting drugs through the membrane between or out of body compartments (12-14). Cytochrome P450 proteins including CYP3A5, CYP2D6, CYP3A4, CYP1A1, CYP1A2 and POR are the main metabolizing enzymes involved in gefitinib metabolism (15-17). A variety of single-nucleotide polymorphisms (SNPs) in these enzymes have been described to predict toxicities and efficacy of chemotherapeutic agents (18,19).
However, few studies have determined the impact of gene polymorphisms of enzymes in ADME processes on the efficacy of gefitinib in advanced NSCLC patients with EGFR mutations. Therefore, this study is aimed to investigate whether gene polymorphisms of enzymes in ADME processes could be used as predictive biomarkers for gefitinib.
Methods
Patients and study design
Patients were enrolled in this study prospectively. Key eligible criteria included histologically confirmed NSCLC, indication of gefitinib treatment (e.g., EGFR active mutation, unknown EGFR status failed standard treatment), evaluable disease according to the Response Evaluation Criteria in Solid Tumor (RECIST) v1.1, ≥18 years old and Eastern Cooperative Oncology Group (ECOG) performance score (PS) ≤2. Gefitinib was administrated 250 mg daily between 8 am to 9 am and consecutive treatment for one month was counted for one cycle. Anti-tumor activities were evaluated by investigators per RECIST v1.1 using image examination (computed tomography or magnetic resonance imaging scan) at baseline, first cycle (1 month), and every 2 cycles after. Overall response rate (ORR) included complete response (CR) and partial response (PR). Disease control rate (DCR) included CR, PR and stable disease (SD). Progression free survival (PFS) was defined as the time from the first dose to progression disease (PD), death and the start of second systemic treatment at the nearest evaluation before cut-off or the earliest event that shall prevail. This study was in accordance with the International Standards of Good Clinical Practice and approved by Sun Yat-sen University Cancer Center Independent Review Board (B2013-038-01) and registered on clinicaltrial.gov (NCT01994057). Written informed consents were provided by all patients.
Samples collection and detection
Two peripheral blood samples (2 mL) were collected for all patients using EDTA vacuum tubes. The first one was drawn before first gefitinib dose and the second one was drawn within 5 min before gefitinib administration at day 1 of cycle 2. Samples were stored at 2–8 °C and centrifuged within 4 h to separate plasma for somatic mutation detection. White blood cells were used for germline mutation detection. Both plasma and white blood cells were stored at −80 °C until analysis. The plasma of second blood was collected to measure the trough concentration of gefitinib at steady state using validated high-performance liquid chromatography with tandem mass spectrometry (LC-MS/MS) (20). EGFR mutations in primary tumors, metastatic lymph nodes or pleural effusion were detected using direct sequencing or real-time PCR (RT-PCR).
Genotype analysis
DNAs were extracted using the Genome TIANGEN Blood DNA Extraction Kit (AP348, China, Beijing). The quantity and quality of the extracted DNA were measured by calculating their optical density using ultraviolet spectrophotometer. SNPs were genotyped by matrix-assisted laser desorption/ionization time-of-flight platform using the Sequenom Massarray system (San Diego, CA, USA). Thirteen SNPs with allele frequency >5% in Chinese population were used for genotyping including CYP1A1 (rs2606345, rs1048943), CYP1A2 (rs762551), CYP3A4 (rs2242480), cytochrome P450 reductase (POR) (rs1057868, rs17685), UGT1A7 (rs6759892), ABCB1 (rs10256836, rs1045642, rs1128503, rs2032582) and ABCG2 (rs2231137, rs2231142). Since multiple genetic models have been adapted to explore the biological rationales behind the preference of these genetic models and there is no concrete evidence for each SNP, in this research, the allele with higher frequency was considered as the dominant allele and its homozygote and heterozygote genotypes were categorized as the dominant genotypes. Meanwhile, the homozygote genotype of the allele with lower frequency was considered as recessive genotype (21,22).
Statistical analysis
Clinical and experimental data were imported using EpiData 3.0. Statistical analyses were performed using SPSS v13 statistical software (USA). All descriptive data were expressed as mean ± standard deviation and categorical variables were presented as counts and percent. The distribution of genotypes was tested for Hardy-Weinberg equilibrium (HWE). Fisher’s exact test and log-rank test were used to compare ORR and PFS in different genotypes and clinical characteristics. Univariate analysis was applied in SNPs and ORR correlation study as unadjusted results. With consideration of clinical confounding factors, multivariate logistic regression was used to analyze the correlation as adjusted results. Multivariate logistic regression was used to reanalyze the statistical significance of each marked SNP with ORR. Odds ratios (OR) and 95% confidence interval (95% CI) were estimated in all univariate analysis and multivariate analysis. The Wilcoxon Mann-Whitney U test and Kruskal-Wallis H test was adopted to test the difference in gefitinib trough concentration between different genotype groups. A two-tailed P value of 0.05 indicated statistical significance. The figures were drawn using the GraphPad Prism5.0.
Results
Patients
A total of 58 NSCLC patients from Sun Yat-sen University Cancer Center from July 2011 to January 2014 were consecutively enrolled into this study. Data cutoff was set on April 30, 2015. Patients’ demographic and clinical characteristics are shown in Table 1. The population was made up of 48% male and 52% female, with a median age of 56 years at diagnosis. Most of them (90%) had adenocarcinoma. Twenty-two (38%) patients harbored EGFR exon 19 deletion and 27 (47%) harbored EGFR exon 21 L858R point mutation. Thirty-five (60%) patients achieved PR with ORR of 60% and DCR of 95%. The datasets with detailed disease description were listed in (http://cdn.amegroups.cn/static/application/05c49f1f89637a6bb3769bc81ba85e62/atm.2019.12.60-1.xlsx) with the private information blinded. The correlation of baseline clinical characteristics with ORR and PFS were analyzed and no significant clinical confounding characters had significant effect on gefitinib efficacy (Figure 1).
Table 1. Patients’ demographics and baseline clinical characteristics.
| Characters | No. of patients [%] |
|---|---|
| Gender | |
| Male | 28 [48] |
| Female | 30 [52] |
| Age, median (range), years | 56 [31–77] |
| BSA, median (range), m2 | 1.64 (1.26–2.06) |
| ECOG PS | |
| 0–1 | 45 [78] |
| >2 | 10 [17] |
| Unknown | 3 [5] |
| Smoking history | |
| Smoker | 36 [62] |
| Non-smoker | 18 [31] |
| Unknown | 4 [7] |
| Disease staging | |
| Recurrent | 21 [36] |
| IIIB or IV | 37 [64] |
| Pathology | |
| Adenocarcinoma | 52 [90] |
| Adenosquamous carcinoma | 2 [4] |
| Squamous carcinoma | 4 [7] |
| EGFR mutation status | |
| Exon19 deletions | 22 [38] |
| Exon21 L858R | 27 [47] |
| Other sensitive mutations* | 6 [10] |
| Unknown | 3 [5] |
| Treatment line of gefitinib | |
| 1st | 23 [40] |
| 2nd or later | 35 [60] |
| Platinum based chemotherapy | |
| With | 18 [31] |
| Without | 40 [69] |
| Best response to gefitinib | |
| Partial response | 35 [60] |
| Stable disease | 20 [34] |
| Progressive disease | 3 [5] |
| Progression free survival | |
| ≤10 months | 24 [41] |
| >10 months | 34 [59] |
*, 3 patients with exon 21L861Q, 2 with exon 20S768I and 1 with exon 18G719X. Staging: according to IASLC 2007 staging method; BSA, body surface area; ECOG PS, Eastern Cooperative Oncology Group-performance status.
Figure 1.

The impact of patients’ demographics and clinical characteristics on Gefitinib efficacy (overall response rate and progression free survival). ORR, overall response rate; 95% CI, 95% confidence interval.
Genotypes and pharmacokinetics
The distributions of genotypes of all 13 SNPs were consistent with Hardy-Weinberg equilibrium (Table S1). Population distribution analysis indicated that the frequencies of SNPs in Chinese were highly consistent with those in Japanese NSCLC patients, but significantly different those in Caucasians and African Americans (Table S2).
Table S1. Genotype frequency and Hardy-Weinberg equilibrium test for SNPs.
| Gene | Rs | N | Genotype frequency | Minor allelic frequency | HWE P value | ||
|---|---|---|---|---|---|---|---|
| wt/wt, n [%] | wt/m, n [%] | m/m, n [%] | |||||
| CYP1A1 | rs1048943 (CYP1A1*2C) | 53 | 29 [54] | 19 [37] | 5 [9] | 0.27 | 0.47 |
| rs2606345 | 58 | 50 [86] | 7 [12] | 1 [2] | 0.08 | 0.23 | |
| CYP1A2 | rs762551 (CYP1A2*1F) | 56 | 26 [47] | 26 [46] | 4 [7] | 0.30 | 0.46 |
| CYP3A4 | rs2242480 (CYP3A4*1G) | 57 | 25 [45] | 29 [50] | 3 [5] | 0.30 | 0.14 |
| POR | rs1057868 (POR*37) | 55 | 18 [33] | 28 [51] | 9 [16] | 0.43 | 0.73 |
| rs17685 (POR*11) | 53 | 17 [33] | 29 [54] | 7 [13] | 0.40 | 0.32 | |
| UGT1A7 | rs6759892 | 55 | 28 [52] | 25 [45] | 2 [3] | 0.26 | 0.20 |
| ABCB1 | rs1045642 | 54 | 19 [35] | 28 [53] | 7 [12] | 0.39 | 0.50 |
| rs10256836 | 58 | 39 [67] | 18 [31] | 1 [2] | 0.17 | 0.49 | |
| rs1128503 | 58 | 24 [42] | 26 [44] | 8 [14] | 0.35 | 0.82 | |
| rs2032582 | 56 | 12 [21] | 32 [58] | 12 [21] | 0.50 | 0.28 | |
| ABCG2 | rs2231137 | 56 | 25 [44] | 27 [49] | 4 [7] | 0.32 | 0.36 |
| rs2231142 | 55 | 26 [46] | 24 [45] | 5 [9] | 0.31 | 0.87 | |
HWE, Hardy-Weinberg equilibrium.
Table S2. Comparison of genotypes distributions in different populations.
| Gene | Rs | Minor allelic frequency | Han Chinese | Japanese | Caucasian | African American |
|---|---|---|---|---|---|---|
| CYP1A1 | rs1048943 (CYP1A1*2C) | 0.27 | 0.26 | 0.25 | 0.03** | 0.01** |
| rs2606345 | 0.08 | 0.03 | 0.03 | 0.45** | 0.17 | |
| CYP1A2 | rs762551 (CYP1A2*1F) | 0.3 | 0.31 | 0.36 | 0.32 | 0.44 |
| CYP3A4 | rs2242480 (CYP3A4*1G) | 0.3 | 0.28 | 0.30 | 0.20 | 0.12** |
| POR | rs1057868 (POR*37) | 0.43 | 0.42 | 0.40 | 0.29 | 0.08** |
| rs17685 (POR*11) | 0.4 | 0.28 | 0.36 | 0.29 | 0.29 | |
| UGT1A7 | rs6759892 | 0.26 | 0.23 | 0.21 | 0.45** | 0.67** |
| ABCB1 | rs1045642 | 0.39 | 0.42 | 0.46 | 0.45 | 0.07** |
| rs10256836 | 0.17 | 0.11 | 0.14 | 0.30** | 0.09 | |
| rs1128503 | 0.35 | 0.29 | 0.41 | 0.42 | 0.14** | |
| rs2032582 | 0.50 | 0.38 | 0.48 | 0.44 | 0.13** | |
| ABCG2 | rs2231137 | 0.32 | 0.29 | 0.19 | 0.06** | 0.06** |
| rs2231142 | 0.31 | 0.29 | 0.31 | 0.09** | 0.04** |
**, the distribution of different alleles in different populations (Japanese/Caucasian/African American) compared with Han Chinese population by Chi-square test.
Pharmacokinetic results are showed in Figure S1. The Ctrough of gefitinib was in the range of 47.8–470 ng/mL with median value of 175 ng/mL and interquartile range of 130.5–236.6 ng/mL. Nonparametric tests were adopted to test the difference of Ctrough of gefitinib among different genotype groups (Table S3). In CYP3A4 rs2242480 dominant model, the Ctrough of gefitinib was significantly lower in patients with CC + CT genotypes than patients with TT genotype (Wilcoxon Mann-Whitney U test, P=0.019, Figure S2). For ABCG2 rs2231142, the Ctrough of gefitinib was significantly different among patients with different genotypes (Kruskal-Wallis H test, P=0.045). The Ctrough of gefitinib was significantly lower in patients with AA genotype than in patients with AC + CC genotype (Wilcoxon Mann-Whitney U test, P=0.031, Figure S3).
Figure S1.
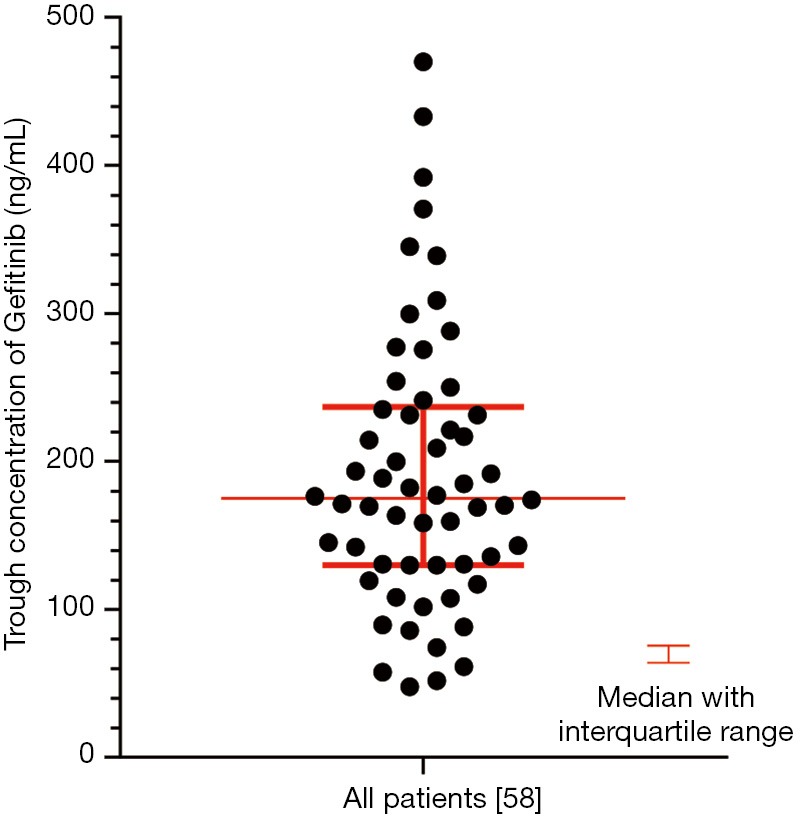
The distribution of gefitinib trough concentration in all 58 patients.
Table S3. The Wilcoxon Mann-Whitney U and Kruskal-Wallis H test for the gefitinib blood concentration among different genotype groups.
| Gene | Polymorphisms | Gene model | P# | P* |
|---|---|---|---|---|
| CYP1A1 | rs1048943 (CYP1A1*2C) | DOM | 0.203 | 0.317 |
| REC | 0.802 | |||
| rs2606345 | DOM | 0.260 | ||
| CYP1A2 | rs762551 | REC | 0.954 | |
| CYP3A4 | rs2242480 (CYP3A4*1G) | DOM | 0.019 | 0.078 |
| REC | 0.531 | |||
| POR | rs1057868 (POR*37) | DOM | 0.302 | 0.546 |
| REC | 0.963 | |||
| rs17685 (POR*11) | DOM | 0.120 | 0.242 | |
| REC | 0.879 | |||
| UGT1A7 | rs6759892 | DOM | 0.755 | |
| ABCB1 | rs1045642 | DOM | 0.479 | 0.743 |
| REC | 0.986 | |||
| rs10256836 | REC | 0.565 | ||
| rs1128503 | DOM | 0.130 | 0.316 | |
| REC | 0.572 | |||
| rs2032582 | DOM | 1.000 | 0.168 | |
| REC | 0.069 | |||
| ABCG2 | rs2231137 | DOM | 0.585 | 0.239 |
| REC | 0.154 | |||
| rs2231142 | DOM | 0.064 | 0.045 | |
| REC | 0.031 |
#, Wilcoxon Mann-Whitney U test for the gefitinib trough concentration among different genotype model groups; *, Krustal-Wallis H test for the gefitinib blood concentration among different genotype groups. DOM, dominant model; REC, recessive model.
Figure S2.

The distribution of gefitinib trough concentration in CYP3A4 rs2242480 genotype groups. *, P value <0.05.
Figure S3.

The distribution of gefitinib trough concentration in ABCG2 rs2231142 genotype groups. *, P value <0.05.
Association between gefitinib pharmacokinetics and efficacy
Nonparametric tests were adopted to test the difference in Ctrough of gefitinib between patients with different gefitinib responses. The median Ctrough of gefitinib was 173.9 ng/mL with interquartile range of 108–231 ng/mL in PR group and 191.5 ng/mL with interquartile range of 135.5–275.5 ng/mL in SD + PD group, respectively (P=0.236). X-Tile was used to determine the cutoff value of Ctrough based on ORR (Ctrough cutoff at 200 ng/mL) (23). Chi-square test showed that the ORR was not significantly different in patients with Ctrough >200 ng/mL and patients with Ctrough <200 ng/mL [64% (23/36) vs. 55% (12/22), P=0.480].
In addition, no significant correlation was found between patients’ PFS and Ctrough of gefitinib either. The median PFS of patients with Ctrough >200 and <200 ng/mL was 11.80 and 14.00 months, respectively (P=0.78, Table S4, Figure S4).
Table S4. Log-rank test for PFS between patients with Ctrough <200 ng/mL and Ctrough >200 ng/mL.
| Group | Median | Standard deviation | 95% confidence intervals | |
|---|---|---|---|---|
| Lower | Upper | |||
| Ctrough <200 ng/mL | 14.000 | 2.880 | 8.355 | 19.645 |
| Ctrough >200 ng/mL | 11.800 | 4.591 | 2.802 | 20.798 |
| Total | 13.533 | 3.073 | 7.510 | 19.556 |
Figure S4.
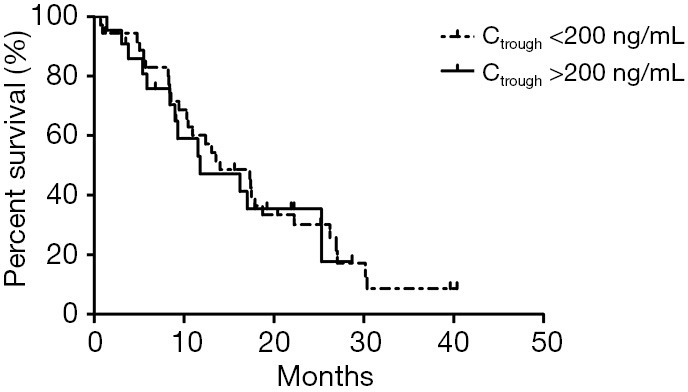
The Kaplan-Meier curve of the PFS in high trough concentration (>200 ng/mL) group compared with that in low trough concentration (<200 ng/mL) group. PFS, progression free survival.
Association between genotypes and efficacy
We analyzed the impact of gene polymorphisms on the efficacy of gefitinib treatment in all 58 patients. With consideration of clinical confounding factors, multivariate logistic regression was used to re-analyze each SNP and those factors with ORR as an adjusted result. The results indicated that 3 SNPs (CYP1A2 rs762551 recessive model, POR rs1057868 dominant model and ABCB1 rs2032582 dominant model) were significant high-risk determinants for ORR as shown in Table 2. Further multivariate logistic regression (stepwise method) was applied with considering 2 SNPs and clinical confounders as independent covariate factors. The results suggested only ABCB1 rs2032582 dominant model was a significant high-risk determinant for ORR (GT + TT/GG: HR =0.111, 95% CI: 0.013 to 0.965). The response rate was significantly lower in patients with ABCB1 rs2032582 (GT + TT) genotypes than in patients with ABCB1 rs2032582 (GG) genotype, with ORR of 51.2% and 84.6%, respectively (P=0.032, Tables 3,S5).
Table 2. Analysis of Association of SNPs gene models with ORR with/without consideration of potential confounders.
| Gene | Polymorphisms | Gene model | ORR | ORR | |||
|---|---|---|---|---|---|---|---|
| P-SV# | OR-SV# (95% CI) | P-MV* | OR-MV* (95% CI) | ||||
| CYP1A1 | rs1048943 (CYP1A1*2C) | DOM | 0.449 | 2.40 (0.248–23.181) | 0.229 | 5.223 (0.354–77.086) | |
| REC | 0.358 | 1.714 (0.543–5.410) | 0.813 | 1.201 (0.264–5.465) | |||
| rs2606345 | DOM | 0.168 | 2.963 (0.633–13.869) | 0.292 | 3.300 (0.358–30.398) | ||
| CYP1A2 | rs762551 | REC | 0.082 | 0.368 (0.120–1.134) | 0.048 | 0.196 (0.039–0.983) | |
| CYP3A4 | rs2242480 (CYP3A4*1G) | DOM | 0.330 | 0.294 (0.025–3.454) | 0.486 | 0.387 (0.027–5.589) | |
| REC | 0.722 | 0.822 (0.279–2.419) | 0.942 | 1.060 (0.220–5.101) | |||
| POR | rs1057868 (POR*37) | DOM | 0.273 | 2.588 (0.477–13.711) | 0.039 | 14.729 (1.146–189.37) | |
| REC | 0.336 | 0.547 (0.160–1.870) | 0.682 | 0.726 (0.158–3.347 | |||
| rs17685 (POR*11) | DOM | 0.594 | 1.607 (0.281–9.188) | 0.142 | 6.654 (0.529–83.747) | ||
| REC | 0.801 | 0.857 (0.258–2.844) | 0.873 | 1.126 (0.263–4.821) | |||
| UGT1A7 | rs6759892 | DOM | 0.468 | 0.667 (0.223–1.994) | 0.907 | 1.089 (0.261–4.551) | |
| ABCB1 | rs10256836 | REC | 0.847 | 1.118 (0.359–3.484) | 0.772 | 1.260 (0.263–6.029) | |
| rs1045642 | DOM | 0.298 | 0.425 (0.085–2.128) | 0.679 | 0.674 (0.104–4.383) | ||
| REC | 0.419 | 0.615 (0.190–1.995) | 0.187 | 0.277 (0.041–1.861) | |||
| rs1128503 | DOM | 0.778 | 1.167 (0.399–3.408) | 0.936 | 1.059 (0.263–4.254) | ||
| REC | 0.893 | 0.900 (0.193–4.194) | 0.782 | 1.346 (0.164–11.043) | |||
| rs2032582 | DOM | 0.068 | 0.219 (0.043–1.117) | 0.020 | 0.033 (0.002–0.585) | ||
| REC | 0.050 | 0.259 (0.067–1.000) | 0.136 | 0.286 (0.055–1.480) | |||
| ABCG2 | rs2231137 | DOM | 0.431 | 1.556 (0.518–4.669) | 0.399 | 1.764 (0.472–6.600) | |
| REC | 0.829 | 0.762 (0.065–8.943) | 0.742 | 1.757 (0.061–50.617) | |||
| rs2231142 | DOM | 0.968 | 0.978 (0.329–2.907) | 0.891 | 1.102 (0.275–4.411) | ||
| REC | 0.396 | 0.375 (0.039–3.605) | 0.546 | 0.376 (0.016–8.967) | |||
#, single variant logistic analysis for the contribution of the SNPs gene models for the ORR; *, adjusted P value by multivariate logistic regression analysis (Enter method) for potential clinical confounders, including gender, BSA, age, PS, smoking history, staging, pathology, EGFR status, treatment lines and platinum chemotherapy. SV, single variant logistic analysis; MV, multivariate logistic regression analysis; ORR, overall response rate; OR, odds ratio; 95% CI, 95% confidence interval; DOM, dominant model; REC, recessive model; SNPs, single nucleotide polymorphisms.
Table 3. Response rate of gefitinib in ABCB1 rs2032582 dominant model.
| Objective response | rs2032582 (GG) | rs2032582 (TT + GT) | P value |
|---|---|---|---|
| CR + PR | 11 (84.6%) | 22 (51.2%) | – |
| SD + PD | 2 (15.4%) | 21 (48.8%) | – |
| Total | 13 | 43 | 0.032 |
CR, complete response; PR, partial response; SD, stable disease; PD, progression disease.
Table S5. Multivariable logistic regression analysis of ORR.
| Parameter | P | OR | 95% CI |
|---|---|---|---|
| Intercept | 0.037 | 9.000 | |
| rs2032582 (TT + GT/GG) | 0.047 | 0.111 | 0.013 to 0.965 |
OR, odds ratio; 95% CI, confidence interval. ORR, overall response rate.
All 58 patients were analyzed to identify the determinant of PFS in the 13 SNPs. Kaplan-Meier curves and log-rank test were applied to compare the PFS of patients with different gene polymorphisms in gefitinib treatment. The results indicated that ABCB1 rs10256836 recessive model was significantly associated with PFS of patients subjected to gefitinib treatment, as shown in Table 4 and Figure 2. CYP3A4 rs2242480 dominant model was excluded due to unbalanced proportion of CC + CT (54/57) and TT genotype (3/57). For ABCB1 rs10256836 recessive model, the median PFS was 10.33 months (95% CI: 7.58–13.09) for patients with GC + CC genotypes, significantly shorter than that of 17.40 months (95% CI: 12.19–22.61) for patients with GG genotype (P=0.04). The Cox regression with stepwise methods was adopted to select the significant determinant of PFS in gefitinib treatment (Table S6). The results showed that ABCB1 rs10256836 recessive models were significantly associated with PFS of patients subjected to gefitinib treatment.
Table 4. Log-rank test for the association of SNPs gene models with PFS.
| Gene | Polymorphisms | Gene model | P | Median PFS (95% CI) |
|---|---|---|---|---|
| CYP1A1 | rs1048943 (CYP1A1*2C) | DOM | 0.656 | 16.23 (9.44–23.02) vs. 14.00 (6.41–21.59) |
| REC | 0.443 | 17.03 (10.68–23.29) vs. 14.0 (4.60–23.40) | ||
| rs2606345 | DOM | 0.868 | 16.23 (4.27–28.20) vs. 13.07 (6.90–19.24) | |
| CYP1A2 | rs762551 | REC | 0.926 | 13.53 (11.14–15.92) vs. 16.23 (8.50–23.97) |
| CYP3A4 | rs2242480 (CYP3A4*1G) | DOM | 0.002 | 14.00 (8.58–19.42) vs. 5.37 (1.63–9.10) |
| REC | 0.327 | 17.90 (14.91–20.89) vs. 10.33 (8.42–12.24) | ||
| POR | rs1057868 (POR*37) | DOM | 0.874 | 12.40 (5.98–18.82) vs. 14.00 (6.67–21.33) |
| REC | 0.253 | 11.53 (9.56–13.50) vs. 17.03 (12.05–22.02) | ||
| rs17685 (POR*11) | DOM | 0.423 | 12.40 (6.03–18.77) vs. 13.53 (0.36–26.71) | |
| REC | 0.181 | 11.53 (9.56–13.50) vs. 16.23 (11.25–21.22) | ||
| UGT1A7 | rs6759892 | DOM | 0.995 | 13.53 (8.52–18.55) vs. 14.00 (4.20–23.80) |
| ABCB1 | rs1045642 | REC | 0.797 | 12.40 (9.46–15.34) vs. 14.00 (8.12–19.88) |
| rs10256836 | REC | 0.040 | 17.40 (12.19–22.61) vs. 10.33 (7.58–13.09) | |
| rs1128503 | DOM | 0.721 | 16.23 (8.12–24.35) vs. 13.53 (7.51–19.56) | |
| REC | 0.975 | 10.97 (0–24.27) vs. 14.00 (7.67–20.33) | ||
| rs2032582 | DOM | 0.159 | 11.80 (6.86–16.74) vs. 17.03 (9.92–24.15) | |
| REC | 0.322 | 11.80 (8.47–15.13) vs. 18.73 (16.52–20.54) | ||
| ABCG2 | rs2231137 | DOM | 0.390 | 11.53 (8.22–4.85) vs. 17.50 (11.29–23.71) |
| REC | 0.652 | 8.23 (4.18–12.29) vs. 14.00 (7.94–20.06) | ||
| rs2231142 | DOM | 0.554 | 12.40 (8.47–16.33) vs. 16.23 (9.32–23.15) | |
| REC | 0.484 | 12.40 (7.96–16.84) vs. 13.53 (7.36–19.71) |
SNPs, single nucleotide polymorphisms; PFS, progression free survival; 95% CI, 95% confident interval; DOM, dominant model; REC, recessive model.
Figure 2.
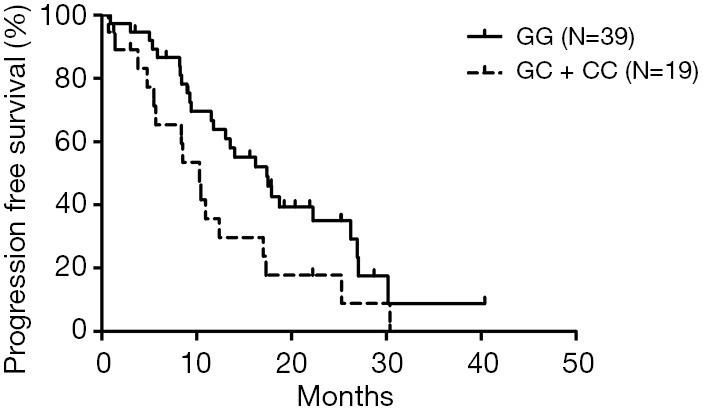
Kaplan-Meier curve and log-rank test for progression-free survival in advanced NSCLC patients treated with gefitinib: association with ABCB1 rs10256836 recessive model (GG vs. GC + CC). NSCLC, non-small cell lung cancer.
Table S6. Multivariable Cox regression of PFS.
| Parameter | Odds ratio | 95% CI of OR | P |
|---|---|---|---|
| Rs2242480DOM | 7.542 | 2.082 to 27.319 | 0.002 |
| Rs10256836REC | 2.170 | 1.136 to 4.146 | 0.019 |
OR, odds ratio; DOM, dominant model; REC, recessive model. PFS, progression free survival.
Discussion
Current study explored the impacts of blood Ctrough of gefitinib and ADME enzymes on the outcomes of gefitinib treatment. The results from 58 patients indicated that the Ctrough of gefitinib was associated with neither patient’ response to gefitinib nor immediate survival, while ATP-binding cassette gene polymorphisms were significantly related with the outcome of gefitinib treatment.
In this research, the Ctrough of gefitinib was significantly different between patients with CYP3A4 rs2242480 and ABCG2 rs2231142, implying that the blood Ctrough of gefitinib might be influenced by the polymorphisms of metabolic enzymes in the liver and transporters in the kidney. Second, both preclinical and clinical researches revealed that the concentration of gefitinib in tumor and skin was much higher than its plasma concentration in xenograft mice and patients (24-26). Using positron emission tomography, 11C and 14C gefitinib was accumulated in tumor tissues and normal tissues such as skin and intestine rather than evenly distributed in blood, both in rat and human (25,27,28). Haura’s study indicated that gefitinib level in tumor was approximately 40-fold higher than its plasma level (29). These findings indicated that the blood Ctrough of gefitinib is not a good substitute indicator for its concentration in tumor, insistent with the previous research (20). Third, the ratio of gefitinib concentration in tumor to that in plasma is ranged from 1.12–250 to 1 (median 40 to 1), which demonstrated tremendous inter-individual variability (30). A previous research also indicated that the trough concentration at d8 and d15 was not associated with response rates or survival times with gefitinib (11). The polymorphisms of transporters (ABCB1) and metabolic enzymes (CYP1A1) in lung might contribute to the variability of tumor concentration (31). Therefore, it is reasonable to speculate that SNPs of these enzymes could cause variability of gefitinib concentration in tumor and affect tumor response.
ABC transporters distributed as membrane proteins in various organs, where they functioned as substrate enzymes for multiple compounds, and played a leading role in transporting drugs through the membrane between or out of body compartments (32,33). Thus, the genes polymorphisms of specific transporters that involved in these processes might have not negligible impact on the variation in drug response or toxicities. Vlaming’s research indicated that human ABCB1 and ABCG2 transporters were over-expressed in NSCLC tumor cell lines (31). Based on that, our hypothesis is that SNPs affect the activities of ABCB1 or ABCG2 in tumor tissues, causing abnormal variability of gefitinib concentration in tumor and eventually leading to different response to gefitinib. Our findings in this research indicated that ABCB1 rs2032582 and rs10256836 gene polymorphisms were highly correlated with gefitinib efficacy. Gefitinib is a substrate of ABCB1 (32,33). ABCB1 rs2032582 G>T mutation G2677T/A is a nonsynonymous mutation in exon 21. Three nucleotides (G/T/A) were in rs2032582, encoding 3 amino acids (Ala893Ser/Thr). However, the function of this SNP is controversial. Some studies concluded that rs2032582 G>T mutation will weaken the drug efflux function of ABCB1 (34,35). Based on that, GG phenotype in rs2032582 is probably the risk factor of gefitinib response. Clinical data indicated that rs10256836 G>C mutation increased the expression or activity of transporters proteins (36-38). Our results also indicated patients with rs10256836 GG genotype had longer PFS, consistent with the previous research. Previous research also suggested that ABCB1 rs1128503 TT genotype was a significant high-risk determinant of both skin rash and diarrhea (39). In this research, neither ABCB1 rs1128503 nor rs1045642 was significantly associated with ORR and PFS. However, only polymorphisms have been detected in this cohort, the gene expression data and concentration data from target tissue would be helpful to give a better illustration to the complete pharmacokinetics and pharmacogenomics inter-individual variability. In vivo and in vitro experiments were needed to confirm our findings.
Conclusions and limitation
Our study indicated that gefitinib trough concentration was significantly different between CYP3A4 and ABCG2 genotypes; blood gefitinib trough concentration was not associated with its efficacy. Moreover, pharmacogenomic analysis revealed that patients with GT + TT genotypes in ABCB1 rs2032582 dominant model respond better to gefitinib and patients carried with GC + CC genotypes in ABCB1 rs10256836 dominant recessive model had longer PFS after gefitinib treatment. However, our findings were based on a small sample prospective cohort study. Therefore, further randomized clinical trials must be conducted to confirm that correlation and to elucidate the biomarker function of ABC transporters family gene polymorphisms (ABCB1 rs2032582 and rs10256836) in clinical practice.
Acknowledgments
We thank the patients and their families for contributing to this study.
Funding: This study was supported by the National Nature Science Foundation of China (81973398 to X Wang, 81730103 to M Huang, 81872499 to L Zhang, 81473283 to X Wang), the National Key Research and Development Program of China (2016YFC0905000 to M Huang, 2016YFC0905503 to L Zhang), the Science, Technology and Innovation Commission of Shenzhen Municipality (JCYJ20170817145454378 to S Xin) and the 111 project (B16047 to M Huang).
Ethical Statement: The authors are accountable for all aspects of the work, and the questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was approved by the Ethical Committee of Sun Yat-sen University Cancer Center (B2013-08-01). Informed consent written informed consent was obtained from all participating subjects. This trial was registered at ClinicalTrials.gov. (NCT01994057, date of registration: 2013/11/23).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin - paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57. 10.1056/NEJMoa0810699 [DOI] [PubMed] [Google Scholar]
- 2.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8. 10.1056/NEJMoa0909530 [DOI] [PubMed] [Google Scholar]
- 3.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121-8. 10.1016/S1470-2045(09)70364-X [DOI] [PubMed] [Google Scholar]
- 4.Hong S, Gao F, Fu S, et al. Concomitant Genetic Alterations With Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With EGFR-Mutant Advanced Non-Small Cell Lung Cancer. JAMA Oncol 2018;4:739-42. 10.1001/jamaoncol.2018.0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Cheng Y, An T, et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single-arm, multicentre clinical trial. Lancet Respir Med 2018;6:681-90. 10.1016/S2213-2600(18)30264-9 [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Zhang Y, Tang H, et al. EGFR gene copy number as a predictive/biomarker for patients with non-small-cell lung cancer receiving tyrosine kinase inhibitor treatment: a systematic review and meta-analysis. J Investig Med 2017;65:72-81. 10.1136/jim-2016-000252 [DOI] [PubMed] [Google Scholar]
- 7.Dahabreh IJ, Linardou H, Kosmidis P, et al. EGFR gene copy number as a predictive biomarker for patients receiving tyrosine kinase inhibitor treatment: a systematic review and meta-analysis in non-small-cell lung cancer. Ann Oncol 2011;22:545-52. 10.1093/annonc/mdq432 [DOI] [PubMed] [Google Scholar]
- 8.Douillard JY, Shepherd FA, Hirsh V, et al. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin Oncol 2010;28:744-52. 10.1200/JCO.2009.24.3030 [DOI] [PubMed] [Google Scholar]
- 9.Zhao M, Zhang Y, Cai W, et al. The Bim deletion polymorphism clinical profile and its relation with tyrosine kinase inhibitor resistance in Chinese patients with non-small cell lung cancer. Cancer 2014;120:2299-307. 10.1002/cncr.28725 [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi H, Sato K, Niioka T, et al. Relationship Among Gefitinib Exposure, Polymorphisms of Its Metabolizing Enzymes and Transporters, and Side Effects in Japanese Patients With Non-Small-Cell Lung Cancer. Clin Lung Cancer 2015;16:274-81. 10.1016/j.cllc.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 11.Hirose T, Fujita K, Kusumoto S, et al. Association of pharmacokinetics and pharmacogenomics with safety and efficacy of gefitinib in patients with EGFR mutation positive advanced non-small cell lung cancer. Lung Cancer 2016;93:69-76. 10.1016/j.lungcan.2016.01.005 [DOI] [PubMed] [Google Scholar]
- 12.Beretta GL, Cassinelli G, Pennati M, et al. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur J Med Chem 2017;142:271-89. 10.1016/j.ejmech.2017.07.062 [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Ramirez J, House L, et al. Comparison of the drug-drug interactions potential of erlotinib and gefitinib via inhibition of UDP-glucuronosyltransferases. Drug Metab Dispos 2010;38:32-9. 10.1124/dmd.109.029660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal S, Sane R, Gallardo JL, et al. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 2010;334:147-55. 10.1124/jpet.110.167601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Cusatis G, Brahmer J, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther 2007;6:432-8. 10.4161/cbt.6.3.3763 [DOI] [PubMed] [Google Scholar]
- 16.McKillop D, McCormick AD, Millar A, et al. Cytochrome P450-dependent metabolism of Gefitinib. Xenobiotica 2005;35:39-50. 10.1080/00498250400026464 [DOI] [PubMed] [Google Scholar]
- 17.Li J, Zhao M, He P, et al. Differential metabolism of Gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res 2007;13:3731-7. 10.1158/1078-0432.CCR-07-0088 [DOI] [PubMed] [Google Scholar]
- 18.Goldstein DB, Tate SK, Sisodiya SM. Pharmacogenetics goes genomic. Nat Rev Genet 2003;4:937-47. 10.1038/nrg1229 [DOI] [PubMed] [Google Scholar]
- 19.Ma Q, Lu AY. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol Rev 2011;63:437-59. 10.1124/pr.110.003533 [DOI] [PubMed] [Google Scholar]
- 20.Xin S, Zhao Y, Wang X, et al. The Dissociation of Gefitinib Trough Concentration and Clinical Outcome in NSCLC Patients with EGFR Sensitive Mutations. Sci Rep 2015;5:12675. 10.1038/srep12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke GM, Anderson CA, Pettersson FH, et al. Basic statistical analysis in genetic case-control studies. Nat Protoc 2011;6:121-33. 10.1038/nprot.2010.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao F, Song M, Wang Y, et al. Genetic model. J Cell Mol Med 2016;20:765. 10.1111/jcmm.12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res 2004;10:7252-9. 10.1158/1078-0432.CCR-04-0713 [DOI] [PubMed] [Google Scholar]
- 24.Marko-Varga G, Fehniger TE, Rezeli M, et al. Drug localization in different lung cancer phenotypes by MALDI mass spectrometry imaging. J Proteomics 2011;74:982-92. 10.1016/j.jprot.2011.03.019 [DOI] [PubMed] [Google Scholar]
- 25.McKillop D, Partridge EA, Kemp JV, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther 2005;4:641-9. 10.1158/1535-7163.MCT-04-0329 [DOI] [PubMed] [Google Scholar]
- 26.Marko-Varga G, Fehniger TE, Rezeli M, et al. Drug localization in different lung cancer phenotypes by MALDI mass spectrometry imaging. J Proteomics 2011;74:982-92. 10.1016/j.jprot.2011.03.019 [DOI] [PubMed] [Google Scholar]
- 27.Ballard P, Yates JW, Yang Z, et al. Preclinical Comparison of Osimertinib with Other EGFR-TKIs in EGFR-Mutant NSCLC Brain Metastases Models, and Early Evidence of Clinical Brain Metastases Activity. Clin Cancer Res 2016;22:5130-40. 10.1158/1078-0432.CCR-16-0399 [DOI] [PubMed] [Google Scholar]
- 28.Zhang MR, Kumata K, Hatori A, et al. (11C) Gefitinib ((11C) Iressa): radiosynthesis, in vitro uptake, and in vivo imaging of intact murine fibrosarcoma. Mol Imaging Biol 2010;12:181-91. 10.1007/s11307-009-0265-5 [DOI] [PubMed] [Google Scholar]
- 29.Haura EB, Sommers E, Song L, et al. A pilot study of preoperative gefitinib for early-stage lung cancer to assess intratumor drug concentration and pathways mediating primary resistance. J Thorac Oncol 2010;5:1806-14. 10.1097/JTO.0b013e3181f38f70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cusatis G, Gregorc V, Li J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 2006;98:1739-42. 10.1093/jnci/djj469 [DOI] [PubMed] [Google Scholar]
- 31.Vlaming ML, Läppchen T, Jansen HT, et al. PET-CT imaging with ((18)F)-gefitinib to measure Abcb1a/1b (P-gp) and Abcg2 (Bcrp1) mediated drug-drug interactions at the murine blood-brain barrier. Nucl Med Biol 2015;42:833-41. 10.1016/j.nucmedbio.2015.07.004 [DOI] [PubMed] [Google Scholar]
- 32.Tamura M, Kondo M, Horio M, et al. Genetic polymorphisms of the adenosine triphosphate-binding cassette transporters (ABCG2, ABCB1) and gefitinib toxicity. Nagoya J Med Sci 2012;74:133-40. [PMC free article] [PubMed] [Google Scholar]
- 33.Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science 2007;315:525-8. 10.1126/science.1135308 [DOI] [PubMed] [Google Scholar]
- 34.Salama NN, Yang Z, Bui T, et al. MDR1 haplotypes significantly minimize intracellular uptake and transcellular P-gp substrate transport in recombinant LLC-PK1 cells. J Pharm Sci 2006;95:2293-308. 10.1002/jps.20717 [DOI] [PubMed] [Google Scholar]
- 35.Lin KM, Chiu YF, Tsai IJ, et al. ABCB1 gene polymorphisms are associated with the severity of major depressive disorder and its response to escitalopram treatment. Pharmacogenet Genomics 2011;21:163-70. [DOI] [PubMed] [Google Scholar]
- 36.Daud AN, Bergman JE, Kerstjens-Frederikse WS, et al. The Risk of Congenital Heart Anomalies Following Prenatal Exposure to Serotonin Reuptake Inhibitors-Is Pharmacogenetics the Key? Int J Mol Sci 2016. doi: . 10.3390/ijms17081333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Foulkes AS. Latent variable modeling paradigms for genotype-trait association studies. Biom J 2011;53:838-54. 10.1002/bimj.201000218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paule B, Castagne V, Picard V, et al. MDR1 polymorphism role in patients treated with cetuximab and irinotecan in irinotecan refractory colorectal cancer. Med Oncol 2010;27:1066-72. 10.1007/s12032-009-9336-3 [DOI] [PubMed] [Google Scholar]
- 39.Rudin CM, Liu W, Desai A, et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol 2008;26:1119-27. 10.1200/JCO.2007.13.1128 [DOI] [PMC free article] [PubMed] [Google Scholar]