

Abstract
Background and Purpose
The 5‐lipoxygenase product 5‐oxo‐6E,8Z,11Z,14Z‐eicosatetraenoic acid (5‐oxo‐ETE), acting through the OXE receptor, is a potent eosinophil chemoattractant that may be an important proinflammatory mediator in eosinophilic diseases such as asthma. We previously identified a series of indole‐based OXE receptor antagonists that rapidly appear in the blood following oral administration but have limited lifetimes. The objective of this study was to increase the potency and plasma half‐lives of these compounds and thereby identify the optimal candidate for future preclinical studies in monkeys, as rodents do not have an OXE receptor orthologue.
Experimental Approach
We synthesized a series of substituted phenylalkyl indoles and compared their antagonist potencies, pharmacokinetics, and metabolism to those of our earlier compounds. The potencies of some of their metabolites were also investigated.
Key Results
Among the compounds tested, the S‐enantiomer of the m‐chlorophenyl compound (S‐Y048) was the most potent, with an pIC50 of about 10.8 for inhibition of 5‐oxo‐ETE‐induced calcium mobilization in human neutrophils. When administered orally to cynomolgus monkeys, S‐Y048 rapidly appeared in the blood and had a half‐life in plasma of over 7 hr, considerably longer than any of the other OXE analogues tested. A major hydroxylated metabolite, with a potency close to that of its precursor, was identified in plasma.
Conclusion and Implications
Because of its highly potent antagonist activity and its long lifetime in vivo, S‐Y048 may be a useful anti‐inflammatory agent for the treatment of eosinophilic diseases such as asthma, allergic rhinitis, and atopic dermatitis.
What is already known
The eicosanoid 5‐oxo‐ETE is a potent eosinophil chemoattractant that acts via the OXE receptor.
OXE receptor activation can be blocked by indole‐based antagonists mimicking the structure of 5‐oxo‐ETE.
What does this study add
We have structurally modified our OXE antagonists to substantially increase both potency and plasma half‐life.
S‐Y048 and its active hydroxylated metabolite have potencies in the picomolar range.
What is the clinical significance
S‐Y048 is a good candidate for in vivo efficacy studies in monkeys.
This OXE receptor antagonist may be a novel therapeutic agent in eosinophilic diseases in humans.
Abbreviations
- 230
5‐(5‐chloro‐2‐hexyl‐1‐methyl‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoate
- 5‐LO
5‐lipoxygenase
- 5‐oxo‐ETE
5‐oxo‐6E,8Z,11Z,14Z‐eicosatetraenoic acid
- C025
5‐(5‐chloro‐1‐methyl‐2‐(6‐phenylhexyl)‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoic acid
- NP‐HPLC
normal‐phase HPLC
- RP‐HPLC
reversed‐phase HPLC
- S‐230
(S)‐5‐(5‐chloro‐2‐hexyl‐1‐methyl‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoate
- S‐C025
(S)‐5‐(5‐chloro‐1‐methyl‐2‐(6‐phenylhexyl)‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoic acid
- S‐Y048
(S)‐5‐(5‐chloro‐2‐(6‐(3‐chlorophenyl)hexyl)‐1‐methyl‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoate
- S‐Y048M
(S)‐5‐(5‐chloro‐2‐((S)‐6‐(3‐chlorophenyl)‐1‐hydroxyhexyl)‐1‐methyl‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoate
- Y048
5‐(5‐chloro‐2‐(6‐(3‐chlorophenyl)hexyl)‐1‐methyl‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoate
1. INTRODUCTION
Arachidonic acid metabolites formed by the 5‐lipoxygenase (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1385) pathway are important mediators in asthma, and drugs targeting their biosynthesis reduce disease symptoms in humans (Haeggstrom, 2018; Peters‐Golden & Henderson, 2007). While the focus has principally been on LTD4, acting through the selective https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=269 (Peters‐Golden & Henderson, 2007; Yokomizo, Nakamura, & Shimizu, 2018), other 5‐LO products may also play a role. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2487, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3353, and other CysLTs are only very weak chemoattractants for human eosinophils (Morita, Schroder, & Christophers, 1989; Powell, Chung, & Gravel, 1995; Sun et al., 1991), which play a major role in asthma and other allergic diseases (McBrien & Menzies‐Gow, 2017). In contrast, another 5‐LO product, 5‐oxo‐6E,8Z,11Z,14Z‐eicosatetraenoic acid (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3391), is a potent eosinophil chemoattractant, both in vitro (Powell et al., 1995) and in vivo (Muro et al., 2003). Its actions are mediated by the G protein‐coupled oxoeicosanoid (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=271) receptor (Bäck et al., 2014; Hosoi et al., 2002; Jones et al., 2003; Takeda, Yamamoto, & Haga, 2002), which is encoded by the OXER1 gene. The OXE receptor is highly expressed on eosinophils and basophils and to a lesser extent on neutrophils, monocytes, and macrophages (Iikura et al., 2005; Jones et al., 2003; Sturm et al., 2005).
The pathophysiological role of 5‐oxo‐ETE has been difficult to establish, in part due to the lack of an orthologue of the OXE receptor in rodents. Nevertheless, its potent effects on human eosinophils suggest that it might be involved in eosinophilic diseases. In addition to its direct chemoattractant effects on these cells, 5‐oxo‐ETE has been shown to induce the transendothelial migration of eosinophils (Dallaire et al., 2003). This is due not only to its effect on cell migration but also to its ability to increase the expression and release of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1633 as well as the expression of the urokinase‐type plasminogen activator receptor, resulting in degradation of components of the extracellular matrix, thereby facilitating the passage of eosinophils into the tissues (Guilbert et al., 1999; Langlois, Ferland, Tremblay, & Laviolette, 2006). Our prior experiments showing that 5‐oxo‐ETE can elicit the infiltration of eosinophils into the skin in humans, especially in asthmatic subjects (Muro et al., 2003), would be consistent with a role for this mediator in human inflammatory diseases. We have detected 5‐oxo‐ETE in feline bronchoalveolar lavage fluid (Cossette et al., 2015), and appreciable levels of this substance have also recently been detected in exhaled breath condensate from humans (Kowal, Gielicz, & Sanak, 2017). Furthermore, allergen challenge of human subjects, sensitive to house dust mite, resulted in significant increases in 5‐oxo‐ETE, suggesting that it may play a role in asthma (Kowal et al., 2017). Consistent with this, anti‐IgE was shown to elicit the release of 5‐oxo‐ETE from human bronchial segments (Kolmert et al., 2018). 5‐Oxo‐ETE may also play a role in the development of nasal polyps, as it is formed by epithelial cells from nasal polyps and was shown to increase the levels of eosinophil cationic protein in organ cultures of nasal polyps (Lin et al., 2018).
As the potent effects of 5‐oxo‐ETE are due to its interaction with a selective receptor, rather than to its electrophilic properties (cf. Schopfer, Cipollina, & Freeman, 2011), a selective OXE receptor antagonist that could block 5‐oxo‐ETE signalling would be an important asset in determining the pathophysiological role of this lipid mediator and could offer a novel therapeutic approach to alleviate the symptoms of eosinophilic diseases such as asthma, allergic rhinitis, and atopic dermatitis. We therefore sought to develop synthetic OXE antagonists with high potency and resistance to metabolism. Using 5‐oxo‐ETE‐induced calcium mobilization in human neutrophils to screen potential antagonists, we identified indoles containing adjacent hexyl and 3S‐methyl‐5‐oxovalerate side chains (e.g., S‐230, Figure 1) as selective OXE antagonists (pIC50 ~ 8; Gore et al., 2014). Because of the absence of the OXE receptor in rodents, we looked for an alternative animal model to test the effects of our OXE antagonists in vivo. We first considered cats, which are prone to develop asthma, but, although feline leukocytes respond well to 5‐oxo‐ETE, our antagonists were not very potent in this species (Cossette et al., 2015), presumably due to differences between the feline and human receptors, which are ~75% identical. We finally settled on cynomolgus monkeys, which have an OXE receptor that is 95% identical to the human receptor. In contrast to its modest effects on feline granulocytes, compound 230 was a potent inhibitor of 5‐oxo‐ETE‐induced activation of monkey granulocytes (Cossette et al., 2016). High levels of 230 could be detected in the blood shortly after oral administration to monkeys at a dose of 30 mg·kg−1, but its concentration declined rapidly due to extensive ω‐oxidation of the hexyl side chain. In an attempt to reduce ω‐oxidation, we replaced the hexyl side chain of S‐230 with a phenyl group, resulting in an antagonist (S‐C025) that is considerably more potent than S‐230 and, despite being cleared fairly rapidly from the blood, displays improved pharmacokinetic properties (Chourey et al., 2018).
Figure 1.
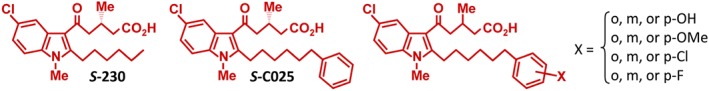
Structures of OXE receptor antagonists
Although the monkey is an excellent animal model for investigation of human diseases because of its similarity to humans, the cost of these experiments is very high, and the experimental design is consequently limited due to the relatively small numbers of animals that can be used. Before initiating studies using monkey models of allergic disease, we wished to optimize the properties of the OXE antagonist to be tested in these costly experiments. We therefore sought to optimize our lead compound, S‐C025, with respect to both potency and pharmacokinetic properties by investigating the effects of a variety of substituents on the phenyl ring. This resulted in the identification of S‐Y048, which has enhanced potency and an increased lifetime in blood, and is converted to an abundant long‐lasting plasma metabolite with nearly equivalent potency.
2. METHODS
2.1. Measurement of intracellular calcium levels in human neutrophils
The potencies of OXE receptor antagonists were evaluated by determining their effects on 5‐oxo‐ETE‐induced calcium mobilization in human neutrophils (Gore et al., 2014). Neutrophils were prepared from healthy human subjects after removal of red cells by dextran sedimentation and mononuclear cells by centrifugation over Ficoll‐Paque. After labelling the cells with indo‐1, fluorescence was measured at 37°C using a Cary Eclipse spectrofluorometer (Agilent Technologies, Santa Clara, CA, USA) equipped with a temperature‐controlled cuvette holder and a magnetic stirrer. After stabilization of the fluorescence, antagonists were added, followed by addition of 5‐oxo‐ETE (10 nM) 2 min later and digitonin (final concentration of 0.1%) after a further 1 min to determine maximal fluorescence.
2.2. Measurement of polymerized actin
F‐Actin was measured in unfractionated leukocytes from whole blood obtained from healthy human subjects, following removal of red blood cells using dextran and hypotonic lysis as described previously (Gore et al., 2014), with some minor modifications. The leukocytes were initially incubated with anti‐CD16‐Pe/Cy5 (1.5 μl per 106 cells; mouse IgG1, clone 3G8; BioLegend, San Diego, CA, USA; catalogue number 302010; lot no. B15039; https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=854 https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=874) for 30 min on ice. After centrifugation and washing once with PBS, the cells were suspended in PBS containing Ca++ (1.8 mM) and Mg++ (1 mM) at a concentration of 5.5 × 106 cells·ml−1. Aliquots (90 μl) of the labelled cells were preincubated for 5 min at 37°C with vehicle (1‐μl DMSO) or S‐Y048 (10 or 1,000 nM), followed by addition of vehicle (10‐μl PBS containing Ca++, Mg++, and 0.1% BSA), 5‐oxo‐ETE (final concentration, 10 nM), PGD2 (10 nM), LTB4 (10 nM), or eotaxin‐1 (10 nM). After 20 s, the incubations were terminated by addition of formaldehyde (37%) to give a final concentration of 8.5% and kept on ice for 30 min. A mixture of lysophosphatidylcholine (30 μg in 23.8‐μl PBS) and N‐(7‐nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)phallacidin (NBD‐phallacidin; Molecular Probes; 49 pmol in 6.2‐μl MeOH; final concentration, 0.3 μM) was added to each sample, followed by incubation overnight in the dark at 4°C. Immediately prior to data acquisition by flow cytometry (BD LSRFortessa X‐20, BD Biosciences, San Jose, CA, USA), 300 μl of PBS was added to each sample. Eosinophils were identified by high side scatter and low expression of CD16, whereas neutrophils also displayed high side scatter but high CD16 expression.
2.3. Pharmacokinetic experiments in cynomolgus monkeys
All animal care and experimental procedures were performed in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the local institutional animal care committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Pharmacokinetic experiments were performed using six female cynomolgus monkeys (2.5 to 5 kg), housed at INRS‐Institut Armand‐Frappier, Laval, Quebec. Animals were housed in pairs in cages with a 12‐hr light/12‐hr dark schedule. They received a diet consisting of Teklad Global chow for non‐human primates supplemented with fresh fruit and vegetables and breakfast cereal and had unlimited access to water. Monkeys were first habituated to the oral gavage procedure on three successive days and then weighed to determine the correct dose. Three days later, all food was withdrawn at the end of the afternoon, and a blood sample was taken. The following morning, various doses of S‐Y048, S‐70, or S‐71 were delivered by oral gavage, and blood samples (1 ml) were taken from the femoral vein 0.5, 1, 2, 4, 8, 12, and 24 hr later. Feeding was resumed after the first blood sample was obtained at 30 min.
To provide a dose of 5 mg·kg−1, OXE receptor antagonists (S‐Y048, S‐70, and S‐71) were dissolved in ethanol (25 mg·ml−1) and stored at −80°C before use. On the morning of the experiment, the ethanolic solution was thawed and vortexed, and the required amount was added to 10 volumes of 20 mM NaHCO3 (pH 8.0). The resulting suspension (2.2 ml·kg−1; 9.1% EtOH) was immediately vortexed and administered by oral gavage to a monkey. An identical procedure was followed for doses of 2 mg·kg−1, except that the antagonist (i.e., S‐Y048) was dissolved in ethanol at a concentration of 10 mg·ml−1. In some cases, a second identical dose of antagonist was administered 8 hr after the first dose, immediately after obtaining an 8‐hr blood sample. Blood samples (1 to 2 ml) were collected in heparinized tubes 1 hr prior to gavage and 0.5, 1, 2, 4, 8, 12, and 24 hr after gavage. If a second dose was administered, an additional sample was collected 1 hr later. The blood samples were centrifuged, and the resulting plasma was frozen and stored at −80°C prior to extraction and analysis.
2.4. Measurement of OXE antagonists and their metabolites in plasma
Plasma samples were thawed and diluted with 2 volumes of MeOH, prior to the addition of internal standards. For analysis of S‐Y048, the tetramethylene analogue of C‐025 (i.e., 5‐(5‐chloro‐2‐(4‐phenylbutyl)‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoic acid; 1.3 μg) was used as the internal standard, whereas for S‐70 and S‐71, the corresponding pentamethylene analogue (i.e., 5‐(5‐chloro‐2‐(5‐phenylpentyl)‐1H‐indol‐3‐yl)‐3‐methyl‐5‐oxopentanoic; 1 μg) was used. The samples were stored overnight at −80°C, and the precipitated material was removed by centrifugation. After adjusting the concentration of MeOH in the supernatant to 30% by the addition of water, the sample was loaded onto a C18 Sep‐Pak cartridge (Waters Corp), which was washed with 30% MeOH, followed by the elution of the antagonist and its metabolites with 100% MeOH. The solvent was then removed in vacuo using a rotary evaporator, and the residue dissolved in 30% MeOH containing 2.5‐mM H3PO4 and analysed by precolumn extraction‐RP‐HPLC using a modified Waters 2695 Alliance system (Waters Corporation, Mississauga, Ontario, Canada) equipped with a Waters model 2996 photodiode array detector. Automated precolumn extraction was performed as described previously (Powell, 1987) using a C18 SecurityGuard cartridge (4 × 3 mm; Phenomenex, Torrance, CA, USA) coupled with a Kinetex C18 column (Phenomenex; see figure legends for precise chromatographic conditions). All solvents used for extraction and chromatographic analysis were purchased from Fisher Scientific, Markham, ON, Canada.
2.5. Normal‐phase HPLC
Normal‐phase (NP) HPLC of α‐hydroxy‐Y048 stereoisomers was performed using an Econosphere column (250 × 4.6 mm; 5‐μm particle size; Alltech Associates, Deerfield, IL, USA). The mobile phase was hexane/isopropanol/acetic acid (98.4:1.5:0.1) at a flow rate of 1.5 ml·min−1 and a temperature of 30°C.
2.6. Chiral HPLC
The S‐ and R‐enantiomers of compounds 69–72 and Y048 were separated by chiral HPLC with a Cellulose‐1 column (5‐μm particle size; 250 × 4.6 mm; Phenomenex) as the stationary phase. Except for compound 70, isocratic elution was used with a mobile phase consisting of hexane/methanol/acetic acid (98.7:1.2:0.1) with a flow rate of 1.2 ml·min−1 and a column temperature of 30°C. For compound 70, the mobile phase was hexane/methanol/acetic acid (98.4:1.5:0.1), and the flow rate was 1.5 ml·min−1.
Stereoisomers of synthetic α‐hydroxy‐Y048 as well as S‐Y048M from plasma were analysed by chiral HPLC using a Cellulose‐2 column (4.6 × 250 mm; 5‐μm particle size; Phenomenex) with hexane/ethanol/acetic acid (93:7:0.1) as the mobile phase. The flow rate was 1 ml·min−1, and the column temperature was 45°C.
2.7. Identification of the major plasma metabolite of S‐Y048 by LC‐MS/MS
To identify S‐Y048M, column fractions corresponding to the peak at 23.5 min in Figure 5f were collected during the analysis of a plasma extract from blood obtained 12 hr after administration of S‐Y048 (5 mg·kg−1; see legend to Figure 5f for further details). The combined fractions were concentrated to dryness in vacuo and dissolved in methanol (100 μl) prior to analysis by LC‐MS/MS, which was performed using a model 1100 HPLC system (Agilent Technologies) connected to an LTQ Velos Orbitrap high‐resolution mass spectrometer by a heated electrospray ionization source (Thermo Scientific, San Jose, CA, USA). The stationary phase was a Phenomenex Kinetex C18 column (2.6‐μm particle size; 50 × 2.1 mm), whereas the mobile phase was a linear gradient between Solvents A (0.02% HOAc in water) and B (0.02% HOAc in MeCN) as follows: 0.0 min, 30% B; 1.0 min, 30% B; 25.0 min, 55% B; 32.0 min, 55% B; 32.1 min, 90% B; and 37.0 min, 90% B. The flow rate was 0.3 ml·min−1, the column temperature was 25°C, and the injection volume was 10 μl. Analyses were performed in negative electrospray ionization mode as follows: capillary temperature of 350°C, source heater temperature of 300°C, sheath gas flow of 20, auxiliary gas flow of 10, and source voltage of −3.0 kV. The MS settings were S lens RF level of 60%, automatic gain control target of 1 × 106 ions, mass range of m/z 250 to m/z 700, and resolution of 100,000. Multiple levels of MSn analysis in data‐dependent acquisition mode were used to identify S‐Y048M. In data‐dependent acquisition mode, the selection of the precursor ion for MS2 analysis was based on the chlorine isotope pattern and/or isolation of the top three most intense ions from the full MS scan. MS2 settings were collision‐induced dissociation, signal threshold of 5,000, normalized collision energy of 35, isolation width of 2 Da, and activation time of 50 ms. MS3 was performed using parent and product mass lists to trigger MS3 for selected ions and was performed with the same settings as MS2 except that a normalized collision energy of 45 was used.
Figure 5.
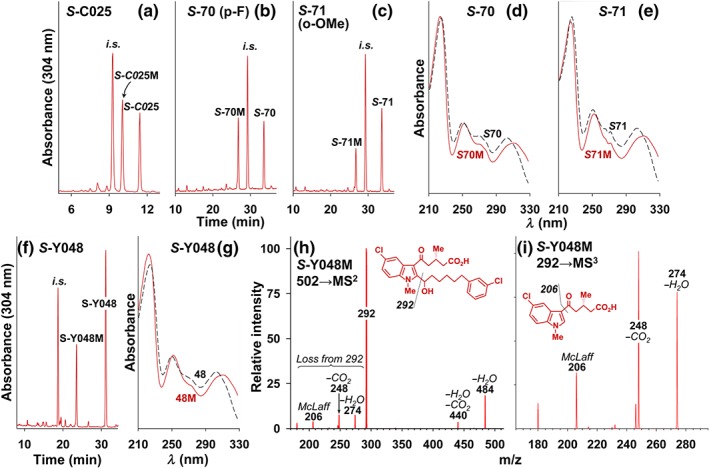
Identification of major plasma metabolites of OXE receptor antagonists. (a) S‐C025, (b, d) S‐70 (p‐fluoro‐S‐C025), (c, e) S‐71 (o‐methoxy‐S‐C025), and (f, g) S‐Y048 (m‐chloro‐S‐C025), all at doses of 5 mg·kg−1, were administered to monkeys by oral gavage, and blood samples (1 ml) were taken after 12 hr. Internal standards (tetramethylene analogue of C025 for S‐C025 and S‐Y048 and pentamethylene analogue of C025 for compounds S‐70 and S‐71) were added to the plasma samples, which were extracted and analysed by RP‐HPLC. For (a) S‐C025, the data were taken from a previous study (Chourey et al., 2018) employing a Novapak C18 column and a gradient of 70% to 100% MeCN over 15 min. For panels (b) and (c), a Kinetex C18 column (2.6‐μm particle size; 100 × 4.6 mm) was used with a gradient between 38% and 65% MeCN over 35 min, followed by 5 min at 65% MeCN. For panel (f), a Kinetex C18 column (5‐μm particle size; 250 × 4.6 mm) was used with a gradient between 55% and 75% MeCN over 30 min, followed by 5 min at 75% MeCN. All solvents contained 0.02% HOAc. The flow rates were 1 ml·min−1, and the column temperature was maintained at 30°C. The UV spectra of S‐70, S‐71, and S‐Y048 (broken lines) along with their major plasma metabolites (red solid lines) are shown in panels (d), (e), and (g), respectively. (h) MS2 fragmentation of the [M − H]− ion at m/z 502 for S‐Y048M, isolated from plasma as shown in panel (f). (i) MS3 fragmentation of the ion at m/z 292 shown in panel (h). The ion at m/z 206 is formed as the result of a McLafferty rearrangement
2.8. Data analysis
. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. Data are represented either as individual values or as means ± SEM. The statistical significance of differences among multiple groups was evaluated using one‐way ANOVA with the Bonferroni test as a multiple comparison method. Differences between two groups were evaluated using t tests. Differences with a P value of less than .05 were considered to be statistically significant
2.9. Materials
5‐Oxo‐ETE (Khanapure, Shi, Powell, & Rokach, 1998) and LTB4 (Zamboni & Rokach, 1982) were synthesized as previously described. 5‐Oxo‐ETE was purified by reversed‐phase (RP) HPLC prior to use. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=769 (CCL11) was obtained from Cedarlane, Burlington, ON, Canada, whereas https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1881 was purchased from Cayman Chemical, Ann Arbor, MI, USA. The procedures for the synthesis of all the OXE receptor antagonists evaluated in this study have been provided as Supporting Information.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. Effects of phenyl ring substituents on OXE antagonist potency
We first examined the abilities of the hydroxyphenyl compounds 74–76 to inhibit 5‐oxo‐ETE‐induced calcium mobilization in human neutrophils, but none was as potent as racemic C025 (pIC50, 9.61 ± 0.07; Figure 2a). The most potent of these was the p‐hydroxy compound 76, which had a pIC50 about 0.3 units lower than that of C025, whereas those of the meta and ortho OH‐substituted phenyl analogues were about 0.9 and 1.4 units lower (Figure 2a,f). A different pattern emerged when we examined the methoxy‐substituted analogues 71–73 (Figure 2b). In this case, the p‐methoxy compound (73) was the least potent with a pIC50 about 0.4 units lower than that of C025, whereas the ortho‐methoxy compound (71) was equipotent, and the meta‐methoxy compound (72) was more potent. A similar pattern was observed for the fluoro‐substituted analogues, with the m‐fluoro compound (69) having a pIC50 about 0.5 units higher than C025 and the ortho and para compounds being less potent (Figure 2c). However, the most potent among this series of compounds proved to be the m‐chloro analogue Y048, which had a pIC50 of 10.47, 0.86 units higher than that of C025 (Figure 2d). In contrast, the ortho‐ and para‐ chloro compounds 66 and 67 were slightly less potent than C025.
Figure 2.
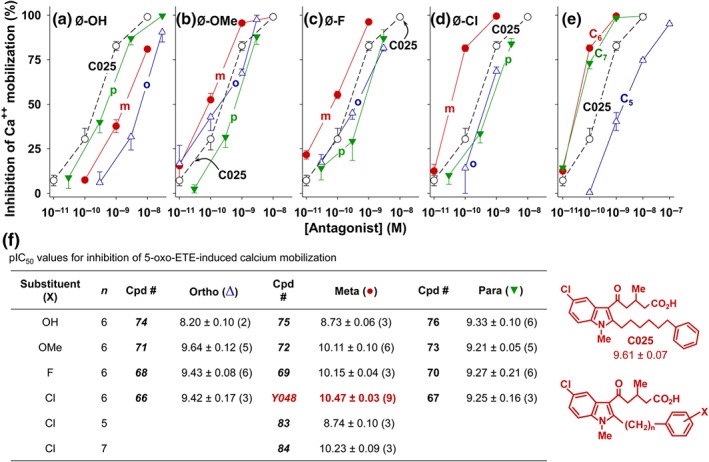
Effects of ortho‐, meta‐, and para‐ substituents on antagonist potency. The effects of analogues of racemic 025 containing hydroxyl (a), methoxyl (b), fluoro (c), and chloro (d) substituents in the ortho (o), meta (m), and para (p) positions on 5‐oxo‐ETE‐induced calcium mobilization were determined. (e) Effect of the length of the polymethylene spacer (C5,; C6 [i.e., Y048]; C7) on the potency of m‐Cl‐substituted antagonists. (f) The pIC50 values for all of the compounds depicted in panels (a) to (e). The values are means ± SE with the numbers of independent experiments indicated in brackets. All compounds are racemic mixtures. Data for C025 (○; pIC50, 9.61 ± 0.07 [n = 10]), which was included in this experiment for comparison, are also shown
3.2. Effect of the length of the methylene chain of Y048 on antagonist potency
To examine the effect of the length of the polymethylene chain connecting the m‐chlorophenyl group to the indole moiety, we prepared the pentamethylene and heptamethylene analogues of Y048. Reducing the polymethylene spacer by a single methylene group (compound 83) had a large impact on antagonist potency, reducing it by over 50‐fold (Figure 2e). In contrast, addition of a methylene group, as in the heptamethylene analogue 84, had relatively little impact on potency.
3.3. Potencies of S‐ and R‐enantiomers of OXE receptor antagonists
All of the data shown in Figure 2 are for racemic compounds, whereas we have previously shown that most of the antagonist activity of 5‐oxo‐3‐methylvalerate‐substituted indoles resides in the S‐enantiomers (Gore et al., 2014; Reddy et al., 2015). We therefore separated the S‐ and R‐enantiomers of some of the above antagonists by chiral HPLC and examined the potencies of the purified enantiomers. We initially investigated the p‐fluoro and o‐methoxy compounds, since they were among the first members of this series to be synthesized, and then investigated the more potent meta‐substituted compounds. Although the S/R potency ratios differed from one substituent to another, in all cases, the S‐enantiomer was considerably more potent than the corresponding R‐enantiomer (Table 1). The greatest difference was seen with the methoxy derivatives (ortho and meta), where the S‐enantiomers had pIC50 values approximately 2.5 to 3 units higher than the R‐enantiomers (Table 1 and Figure 3b,c). The difference between enantiomers was less with the fluoro compounds (Figure 3a,d), for which the S‐enantiomers had pIC50 values about 1.1 to 1.7 units higher than the R‐enantiomers. Of all the compounds tested, the S‐enantiomer of the m‐chloro compound S‐Y048 (pIC50, 10.81) was the most potent, with the corresponding R‐enantiomer having a pIC50 1.6 units lower (Figure 3e).
Table 1.
Potencies of S‐ and R‐enantiomers of some OXE antagonists and metabolites
| Compound | pIC50 | |
|---|---|---|
| S‐enantiomer | R‐enantiomer | |
| C025 | 10.06 ± 0.04 (46) | 7.64 ± 0.04 (3) |
| 70 (p‐fluoro) | 9.61 ± 0.14 (9) | 8.53 ± 0.01 (2) |
| 71 (o‐methoxy) | 10.01 ± 0.09 (11) | 7.41 ± 0.15 (6) |
| 72 (m‐methoxy) | 10.30 ± 0.04 (5) | 7.35 ± 0.11 (5) |
| 72 (m‐fluoro) | 10.45 ± 0.06 (5) | 8.78 ± 0.08 (5) |
| Y048 (m‐chloro) | 10.81 ± 0.08 (14)* | 9.21 ± 0.06 (5) |
| S‐70M | 8.87 ± 0.11 (3) | |
| S‐71M | 9.29 ± 0.14 (3) | |
| S‐Y048M | 10.15 ± 0.10 (6) | |
Significantly lower than all other S‐enantiomers (P < .05).
Figure 3.
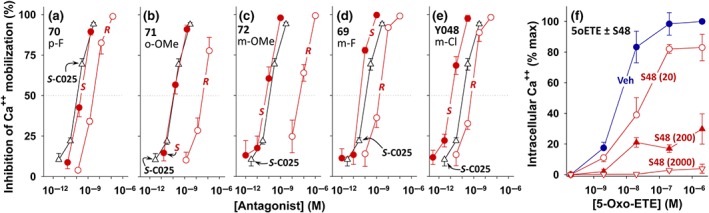
Effects of the S‐ and R‐enantiomers of OXE‐R antagonists on 5‐oxo‐ETE‐induced calcium mobilization. The effects of the S‐enantiomers (●) and R‐enantiomers (○) of (a) p‐fluoro (70), (b) o‐methoxy (71), (c) m‐methoxy (72), (d) m‐fluoro (69), and (e) m‐chloro (Y048) substituted phenyl antagonists on calcium mobilization induced in human neutrophils by 10 nM 5‐oxo‐ETE are shown. The response to S‐C025 (Δ; pIC50, 10.06 ± 0.01) is included for comparison. The values are means ± SE (see Table 1 for numbers of experiments). (f) Effects of increasing concentrations of S‐Y048 (S48) on the concentration–response curve for 5‐oxo‐ETE‐induced calcium mobilization in human neutrophils. Either vehicle (Veh) or different concentrations (20 pM; 200 pM or 2,000 pM) of S‐Y048 were added to indo‐1‐loaded neutrophils, followed 2 min later by the addition of different concentrations of 5‐oxo‐ETE (see Section 2 for further details)
We also investigated the effects of increasing concentrations of S‐Y048 on the concentration–response curve to 5‐oxo‐ETE (Figure 3f). Indo‐1‐loaded neutrophils were preincubated with S‐Y048 (20, 200, and 2,000 pM) for 2 min prior to the addition of increasing concentrations (2, 20, 200, and 2,000 nM) of 5‐oxo‐ETE, and changes in intracellular calcium levels were measured. The lowest concentration of S‐Y048 tested (20 pM) resulted in a rightward shift in the concentration–response curve for 5‐oxo‐ETE and appeared to reduce the maximal response by about 20%. Higher concentrations of S‐Y048 further reduced the maximal response to 5‐oxo‐ETE, which was nearly undetectable at a concentration of 2 nM.
3.4. S‐Y048 is selective for the OXE receptor
To ensure that S‐Y048 is selective for the OXE receptor, we compared its effects to other mediators that activate eosinophils and/or neutrophils. S‐Y048 (10 nM) completely blocked actin polymerization induced in human eosinophils by 5‐oxo‐ETE (10 nM; Figure 4a). In contrast, a 100‐fold higher concentration of S‐Y048 (i.e., 1 μM) had no effect on actin polymerization in eosinophils in response to identical concentrations of PGD2, LTB4, or human eotaxin‐1. Similarly, S‐Y048 blocked 5‐oxo‐ETE‐induced actin polymerization in neutrophils but had no effect on the response to LTB4 (Figure 4b). As expected, neutrophils did not respond to either PGD2 or eotaxin‐1.
Figure 4.
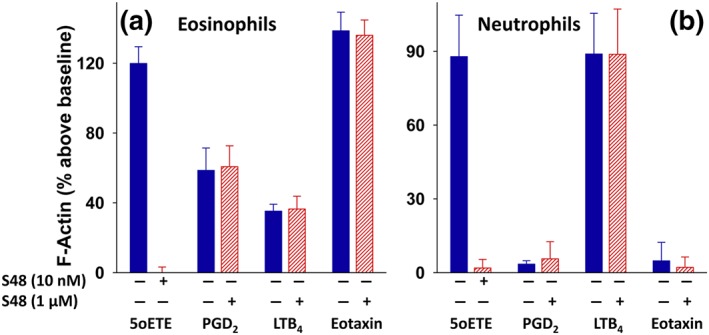
Selectivity of S‐Y048 for the OXE receptor. Unfractionated human leukocytes labelled with anti‐CD16‐PE/Cy5 were incubated for 5 min with either vehicle or concentrations of S‐Y048 (S48) of either 10 nM (5‐oxo‐ETE) or 1 μM (all other agonists). 5‐Oxo‐ETE, PGD2, LTB4, and eotaxin‐1 (all 10 nM) were then added, and incubations were terminated after a further 20 s by the addition of formaldehyde. Polymerized F‐actin was measured in eosinophils and neutrophils by flow cytometry as described in Section 2
3.5. In vivo metabolism of OXE receptor antagonists
Because of their much higher potency, we decided to synthesize the S‐enantiomers of some of the above compounds for further in vivo pharmacokinetic and metabolic studies. We initially prepared the S‐enantiomers of some of the first compounds that we synthesized for the above in vitro studies, including the p‐fluoro compound 70 and the o‐methoxy compound 71. Once we realized the high potency of S‐Y048, purified from racemic Y048, we also prepared this compound by total synthesis. Our initial pharmacokinetic experiments were done with the S‐enantiomers of compounds 70 and 71. The antagonists were dissolved in ethanol, which was added to bicarbonate, and the resulting suspension was administered by oral gavage at a dose of 5 mg·kg−1. Analysis of plasma samples taken 12 hr later revealed that, as with the unsubstituted phenyl compound S‐C025 (Figure 5a), substantial levels of S‐70 (Figure 5b) and S‐71 (Figure 5c) were still present. At this time point, for each antagonist, we observed a major metabolite with a tR lower than that of the parent compound. Small amounts of other unidentified metabolites were also present, but we did not observe any other major metabolites with similar or different UV spectra. It is likely that glucuronide conjugates were also formed but did not reach high concentrations in plasma due to their rapid clearance. We have previously detected glucuronides in monkey plasma after administration of the related compound S‐230 (Chourey et al., 2017).
The UV spectrum for each of the above major metabolites and their parent compounds are shown in Figure 5d,e. For each of these metabolites, the peak at 303 nm for the parent compound underwent a bathochromic shift to 311 nm, similar to what we previously observed for the major plasma metabolite of S‐C025, which contains a hydroxyl group on the hexamethylene chain on the methylene group adjacent (i.e., α) to the indole (Chourey et al., 2018).
We subsequently investigated the in vivo metabolism of the more potent m‐chloro antagonist S‐Y048, which was also converted to a major plasma metabolite (S‐Y048M; Figure 5f) with similar UV properties (Figure 5g). To conclusively identify this metabolite, we isolated it from plasma by RP‐HPLC following oral administration of S‐Y048 (5 mg·kg−1) and analysed it by LC‐MS/MS. S‐Y048M had an [M − H] − ion at m/z 502.1598, compared to the theoretical value of m/z 502.1557 expected for a hydroxy metabolite of S‐Y048 (mass accuracy, 8 ppm). MS2 fragmentation of this ion (Figure 5h) resulted in intense ions at m/z 484 (loss of H2O), 440 (loss of H2O + CO2), and 292 (base peak, loss of the chlorophenylhydroxyalkyl side chain), consistent with the presence of a hydroxyl group α to the indole. Fragmentation of the ion at m/z 292 gave an MS3 spectrum (Figure 5i) with intense ions at m/z 274 (loss of H2O), 248 (loss of CO2), 206 (loss of CH2 = CH–CH2–CO2H due to a McLafferty rearrangement), and 180. The latter four ions were also observed in the MS2 spectrum shown in Figure 5h. We previously observed an intense ion at m/z 292 in the MS2 spectrum of the α‐hydroxy metabolites of S‐230 (hexyl group in the 2‐position of the indole; Chourey et al., 2017) and S‐C025 (phenylhexyl group in the 2‐position of the indole; Chourey et al., 2018), suggesting that S‐C048M also has a hydroxyl group in this position and is therefore identical to α‐hydroxy‐S‐Y048.
3.6. Pharmacokinetics of OXE receptor antagonists
Figure 6 shows the plasma levels of the above three compounds as well as S‐C025 (Figure 6a) and their major plasma metabolites over 24 hr following oral administration. Preliminary experiments with S‐70 (Figure 6b; n = 1) and S‐71 (Figure 6c; n = 2) suggest that these compounds have similar pharmacokinetic profiles to that of S‐C025 (Table 2). In contrast, S‐Y048 appears to have a slightly lower but more prolonged maximal plasma concentration (Figure 6d). Furthermore, the t½ for S‐Y048 is about 4 times longer than that for S‐C025 (P < .05), and the AUC is about 70% greater (P < .05; Table 2). Thus, although the Cmax for S‐Y048 tended to be a little lower than that for S‐C025 (not statistically significant), its concentration dropped more slowly over the full 24‐hr period, at which time the plasma concentration of S‐Y048 was about 2.7 times higher than that of S‐C025. However, the latter difference did not quite reach statistical significance, probably due to the limited number of animals (n = 3) that we could study due to the high cost of experiments with monkeys. Another difference between these two antagonists is that the concentrations of the major plasma metabolite of S‐C025 (i.e., S‐025M) exceeded those of S‐C025 at times longer than 8 hr (Figure 6a), whereas the plasma concentrations of S‐Y048 exceeded those of S‐Y048M at 24 hr in all three animals in this group (Figure 6d).
Figure 6.
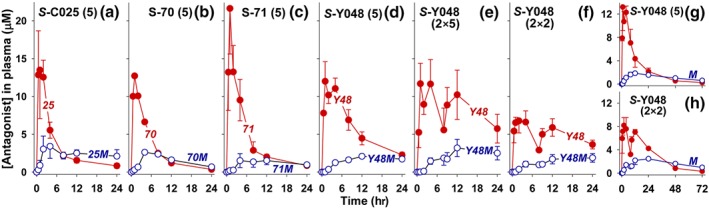
Levels of OXE receptor antagonists and their major metabolites in plasma following oral administration. OXE receptor antagonists were administered to cynomolgus monkeys by oral gavage as described in Section 2. Blood samples were collected after various times, and the plasma was subjected to solid‐phase extraction, and the amounts of unmetabolized antagonist (●) along with their major plasma metabolite (○) were measured by RP‐HPLC, using appropriate internal standards as shown in Figure 5. Data for S‐C025 (5 mg·kg−1; n = 3), taken from a previous study (Chourey et al., 2018), are shown in panel (a). (b) S‐70 (p‐fluoro; 5 mg·kg−1; n = 1); (c) S‐71 (o‐methoxy; 5 mg·kg−1; n = 2); (d) S‐Y048 (m‐chloro; 5 mg·kg−1; n = 3); (e) S‐Y048 (5 mg·kg−1 followed 8 hr later by a second dose of 5 mg·kg−1); (f) S‐Y048 (2 mg·kg−1 followed 8 hr later by a second dose of 2 mg·kg−1). In two animals, measurements of S‐Y048 and S‐Y048M (M) were made up to 72 hr for doses of S‐Y048 of 5 mg·kg−1 (g) and 2 × 2 mg·kg−1 (h)
Table 2.
Pharmacokinetic data for OXE receptor antagonists in monkeys
| Antagonist | n | Dose (mg·kg−1) | tmax (hr) | Cmax (μM) | C24 hr (μM) | t½ (hr) | AUCb (Ant) | AUCb (Met) |
|---|---|---|---|---|---|---|---|---|
| S‐C025 a | 3 | 5 | 1.2 ± 0.4 | 18.0 ± 3.8 | 0.83 ± 0.34 | 1.9 ± 0.2 | 78 ± 12 | 57 ± 18 |
| S‐70 (p‐F) | 1 | 5 | 1.0 | 12.7 | 0.36 | 3.3 | 71 | 36 |
| S‐71 (o‐OMe) | 2 | 5 | 1.0 ± 0.0 | 21.6 ± 6.0 | 0.86 ± 0.08 | 2.3 ± 0.0 | 104 ± 28 | 37 ± 20 |
| S‐Y048 | 3 | 5 | 2.0 ± 1.0 | 13.4 ± 2.2 | 2.27 ± 0.37 | 7.5 ± 1.9* | 138 ± 13* | 38 ± 4 |
| S‐Y048 | 3 | 2 | 1.2 ± 0.4 | 7.3 ± 1.3 | 5.8 ± 0.4 |
The data for S‐C025 are taken from Chourey et al. (2018).
The unit for the AUCs for the antagonists (Ant) and its major plasma metabolite (Met) is nmol·hr·ml−1.
Significantly greater than the corresponding value for S‐C025.
With a single dose of 5 mg·kg−1, the plasma levels of S‐Y048 were maintained between about 2 and 12 μM over a period of 24 hr (Figure 6d). When a second dose was administered 8 hr after the first, the plasma levels of S‐Y048 remained between about 5 and 12 μM over 24 hr (Figure 6e). We also examined a lower dose of S‐Y048 (2 × 2 mg·kg−1, given 8 hr apart), which resulted in plasma levels between about 3 and 7 μM over a period of 24 hr (Figure 6f). In a limited number of animals (n = 2), we measured the plasma concentrations of S‐Y048 and S‐Y048M over 72 hr following administration of S‐Y048 at doses of 5 (Figure 6g) and 2 × 2 (Figure 6h) mg·kg−1. The concentrations of S‐Y048M declined much more slowly than those of its precursor, and between 48 and 72 hr were about 2 to 3 times higher with both dosing regimens.
3.7. Potencies of major plasma metabolites of OXE receptor antagonists
To determine whether they possess antagonist activity, the metabolites of the o‐methoxy, p‐fluoro, and m‐chloro compounds were purified from plasma by RP‐HPLC, and their abilities to block 5‐oxo‐ETE‐induced calcium mobilization in neutrophils were compared to those of their parent compounds (Figure 7a–c). In all cases, the plasma metabolites were potent OXE receptor antagonists, their pIC50 values being only about 0.7 units lower than their respective precursors (Table 1). S‐Y048M was the most potent of the metabolites examined, with a pIC50 of 10.15.
Figure 7.
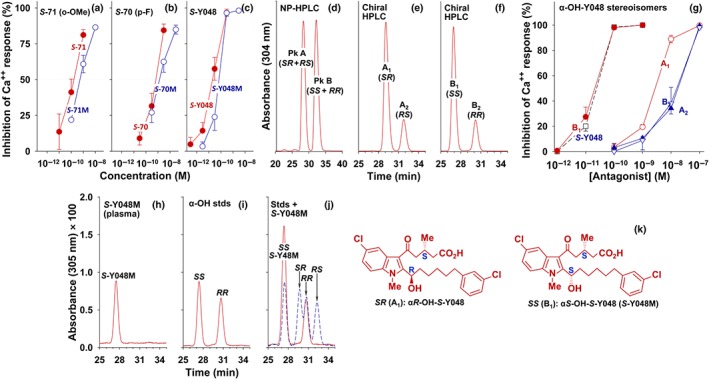
Antagonist potencies of the major plasma metabolites and their synthetic stereoisomers of some OXE receptor antagonists. The major plasma metabolites (shown as M) of (a) S‐71 (o‐methoxy‐S‐C025), (b) S‐70 (p‐fluoro‐S‐C025), and (c) S‐Y048 (m‐chloro‐S‐C025) were purified by RP‐HPLC, as shown in Figure 5, from extracts of plasma from monkeys that had received the corresponding antagonist. Metabolites purified from samples taken between 4 and 24 hr were pooled and tested for their abilities to inhibit 5‐oxo‐ETE‐induced calcium mobilization in neutrophils. The values shown for antagonists and their metabolites are means ± SEM (n = 3), except for the data shown in panel (c); n = 6. (d–f) αRS‐Hydroxy‐RS‐Y048 was prepared using the chiral synthon methyl (R)‐5‐chloro‐3‐methyl‐5‐oxopentanoate (R:S = 9:1), which in this preparation resulted in mixture of stereoisomers of α‐OH‐Y048 in which the S:R ratio for the methyl group in the acyl side chain of the final product was about 4:1. (d) Separation of RR/SS‐enantiomer pair from RS/SR‐enantiomer pair by NP‐HPLC. (e) Separation of the SR‐ and RS‐enantiomers from Peak A in panel (d) by chiral HPLC. (f) Separation of the SS‐ and RR‐enantiomers from Peak B in panel (d) by chiral HPLC. (g) Effects of synthetic S‐Y048 (□), A1 (αR‐hydroxy‐S‐Y048), A2 (αS‐hydroxy‐R‐Y048), B1 (αS‐hydroxy‐S‐Y048), and B2 (αR‐hydroxy‐R‐Y048) on 5‐oxo‐ETE‐induced calcium mobilization in human neutrophils. (h) Chiral HPLC of S‐Y048M after isolation from plasma by RP‐HPLC. (i) Chiral HPLC of a mixture of synthetic αS‐OH‐S‐Y048 and αR‐OH‐R‐Y048; (j) Co‐chromatography of S‐Y048M with the standards shown in panel (i). The dashed blue curve shows a mixture of all four synthetic stereoisomers, run separately. (k) Structures of αS‐hydroxy‐S‐Y048 (SS) and αR‐hydroxy‐S‐Y048 (SR). See Section 2 for the NP‐HPLC and chiral HPLC conditions
3.8. Chirality of the α‐hydroxyl group in S‐Y048M
To determine the chirality of the hydroxyl group α to the indole in S‐Y048M and to further investigate the antagonist properties of α‐hydroxy‐Y048 stereoisomers, we synthesized a mixture of stereoisomers of this compound, using a procedure similar to that used to prepare the corresponding α‐hydroxy derivatives of S‐230 (Chourey et al., 2017), and separated them by a combination of NP‐HPLC and chiral HPLC. Although we had hoped to synthesize principally the αS‐OH and αR‐OH stereoisomers of S‐Y048, in our initial synthesis, appreciable amounts of the R‐methyl stereoisomers were also formed (see Supporting Information for further details), which enabled us to purify all four stereoisomers and examine their antagonist potencies.
Synthetic α‐OH‐Y048 was separated into two peaks by NP‐HPLC (Figure 7d). The material in Peak A was then separated into two peaks, A1 (αR‐OH‐S‐Y048) and A2 (αS‐OH‐R‐Y048) by chiral HPLC (Figure 7e). Similarly, the material in Peak B was separated into B1 (αS‐OH‐S‐Y048) and B2 (αR‐OH‐R‐Y048) using identical chiral chromatography conditions (Figure 7f). The above assignments of the chirality of the methyl and hydroxyl groups are based on a combination of chiral synthesis and by analogy between the elution order of α‐OH‐Y048 stereoisomers and that of the corresponding stereoisomers of α‐OH‐230 (containing a hexyl side chain instead of a chlorophenylhexyl side chain), which we have characterized using single crystal X‐ray diffractometry (Chourey et al., 2017).
The effects of the four stereoisomers of α‐OH‐Y048 on 5‐oxo‐ETE‐induced calcium mobilization are shown in Figure 7g. One of the four isomers (B1, αS‐OH‐S‐Y048) was extremely potent, with a pIC50 approximately equivalent to that of S‐Y048, whereas the other three stereoisomers had pIC50 values approximately 2 to 3 units lower (Table 3).
Table 3.
Antagonist potencies of synthetic α‐hydroxy derivatives of S‐Y048
| Compound | Identity | pIC50 |
|---|---|---|
| S‐Y048 | 10.62 ± 0.3 (4) | |
| A 1 | αR‐OH‐S‐Y048 (SR) | 8.55 ± 0.02 (3) |
| A 2 | αS‐OH‐R‐Y048 (RS) | 7.79 ± 0.07 (4) |
| B 1 | αS‐OH‐S‐Y048 (SS) | 10.71 ± 0.09 (4) |
| B 2 | αR‐OH‐R‐Y048 (RR) | 7.86 ± 0.16 (4) |
A co‐chromatography experiment was performed to determine the chirality of the α‐hydroxyl group in S‐Y048M. S‐Y048M alone gave a single peak when examined by chiral HPLC (Figure 7h). A chromatogram showing the elution positions of a mixture of synthetic αS‐OH‐S‐Y048 and αR‐OH‐R‐Y048 is shown in Figure 7i, whereas Figure 7j shows a mixture of these two standards with S‐Y048M. A chromatogram of all four of the synthetic stereoisomers, run separately, is also shown in Figure 7j (dashed lines). S‐Y048 is clearly identical to αS‐OH‐S‐Y048 (Figure 7k), based on both its antagonist potency and its chromatographic behaviour.
4. DISCUSSION
The primary objective of the current study was to improve the pharmacokinetic properties and the potency of S‐C025 by modification of the phenyl ring. We initially tested the effects of the ortho, meta, and para hydroxy derivatives of C025, which we had previously synthesized as potential plasma metabolites of C025. However, we were unable to detect significant amounts of any of these compounds in plasma, even with the highest dose (30 mg·kg−1) of C025 tested (data not shown). None of these hydroxy derivatives was as potent as C025 in blocking the effects of 5‐oxo‐ETE, with pIC50 values between 0.3 and 1.4 units lower than that of the racemic parent compound. Because of their reduced potency and the fact that addition of a hydroxyl group might promote electrophilic substitution of the phenyl ring, which could potentially lead to increased metabolism, we did not investigate the hydroxyphenyl compounds further.
We next examined the antagonist potencies of a series of substituted phenyl compounds containing methoxy, fluoro, and chloro substituents in different position of the phenyl ring. Interestingly, in contrast to the hydroxyphenyl derivatives, among which the p‐hydroxy isomer was the most potent, among the halogen‐ and methoxy‐substituted compounds that we investigated, those with a meta substituent were consistently the most potent and, in all cases, were more potent than C025. This difference could possibly be related to the more polar nature of the hydroxyl group and its ability to form hydrogen bonds, either with substituents on the receptor or with water molecules.
Of all the compounds we investigated, the m‐chloro derivative Y048 was the most potent. In our previous study, which focused on S‐C025, we examined a series of phenylalkyl compounds containing between three and six methylene groups and found the hexamethylene analogue (i.e., S‐C025) to be the most potent. This also appears to be true for the m‐chlorophenyl series, in which case the hexamethylene compound (i.e., S‐Y048) has a pIC50 over 1.7 units higher than the pentamethylene analogue 83. In contrast, addition of another methylene group (heptamethylene compound 84) did not have an appreciable effect on potency.
To examine the relative potencies of the R‐ and S‐enantiomers of some of the above compounds, we initially used chiral chromatography to separate the enantiomers from the racemic mixtures. As expected, in all cases, the S‐enantiomers were far more potent than the R‐enantiomers. We therefore decided to focus on the S‐enantiomers for further in vivo pharmacokinetic studies and synthesized several of these compounds by chiral synthesis using a procedure similar to the one we developed for the synthesis of S‐230 (Reddy et al., 2015). Because of the far greater quantities of antagonists required for studies in larger animals, as well as the substantial cost of performing in vivo experiments with monkeys, we were able to perform only a limited number of experiments with selected compounds. We initially prepared the S‐enantiomer of the p‐fluoro derivative 70, as the racemic form of this compound was the first in this series to be synthesized. However, because its pharmacokinetic properties did not appear to be substantially different from those of S‐C025, and since it had somewhat reduced potency, we did not pursue this compound further. We next prepared the S‐enantiomer of another of our early compounds, the o‐methoxy derivative 71, which was approximately equipotent with S‐C025 in blocking the OXE receptor, but neither did this compound offer any advantage over S‐C025 with respect to its pharmacokinetic properties. The last series of compounds that we synthesized and tested were those containing meta substituents, of which the m‐chloro derivative was the most potent, with the racemic compound and the S‐enantiomer having pIC50 values approximately 0.8 units higher than that those of C025 and its S‐enantiomer. We therefore prepared S‐Y048 by total chemical synthesis and found it to have a much longer half‐life in blood as well as a higher AUC compared to an equivalent dose of S‐C025. Furthermore, the blood levels of S‐Y048 could be maintained at a relatively constant level over 24 hr by administration of two doses 8 hr apart.
We previously showed that one of our earlier related antagonists (racemic 230) selectively blocks 5‐oxo‐ETE‐induced calcium mobilization in neutrophils, without affecting the responses to the neutrophil agonists https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1831, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1022, and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=821 (Gore et al., 2014). Because our ongoing in vivo studies are focused on eosinophils, we wanted to ensure that S‐Y048 is selective for the OXE receptor compared to other receptors involved in the activation of these cells. Taking advantage of flow cytometry to distinguish between eosinophils and neutrophils, we found that S‐Y048 has no effect on responses of eosinophils mediated by the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=339, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=267, or https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=60 receptors or responses of neutrophils mediated by the BLT1 receptor.
S‐Y048 is a lipophilic molecule with a calculated Log P value of 5.50, and it is possible that this may contribute to its long lifetime in the blood, as it will be bound to plasma proteins. Although this could theoretically compromise its in vivo efficacy, we have recently shown that this antagonist can block 5‐oxo‐ETE‐induced eosinophil infiltration into the skin of rhesus monkeys and significantly inhibit dermal eosinophilia in response to intradermal injection of house dust mite antigen (Miller et al., 2019). These results clearly demonstrate that the lipophilic nature of S‐Y048 does not prevent its in vivo activity in primates, possibly due to its strong binding to the OXE receptor, as it appears to be an insurmountable antagonist, as shown in Figure 3f. Prolonged receptor residence time due to slow dissociation from a receptor has been shown to be an important determinant of in vivo drug efficacy (Seow et al., 2016). Furthermore, its main plasma metabolite αS‐hydroxy‐S‐Y048M has nearly equivalent potency, a long lifetime in the circulation, and is less lipophilic due to its additional hydroxyl group (calculated Log P of 3.75), and is therefore likely to contribute to the effects of S‐Y048, especially at later time points.
The high potencies of αS‐hydroxy‐S‐Y048 and the corresponding metabolite of S‐C025, αS‐hydroxy‐S‐C025, are very interesting. We identified a similar αS‐hydroxy metabolite of S‐230 (Chourey et al., 2017), but there was a much greater loss of potency (over 2 log units) compared to the corresponding metabolites of the phenyl antagonists. It is clear from our earlier studies that the terminal hydrophobic portion of 5‐oxo‐ETE is required for its biological activity (Patel et al., 2008), and the addition of a hydrophilic group to this part of the molecule might be expected to interfere with its interaction with the OXE receptor. However, the presence of a hydroxyl group in the equivalent position of 5‐oxo‐ETE in the 15‐lipoxygenase‐generated metabolite https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6167 has a relatively modest inhibitory effect on agonist activity (O'Flaherty et al., 1996; Powell et al., 1995; Schwenk & Schröder, 1995), suggesting that a hydroxyl group may be tolerated in this position. The αR‐hydroxy derivatives of S‐Y048, S‐C025, and S‐230 were all far less potent than the metabolically formed αS‐hydroxy derivatives. This might possibly be due to differences in the degree of hydrogen bonding between the α‐hydroxyl group and the oxo group of the 5‐oxovalerate side chain, which could possibly result in a less favourable conformation for the αR‐hydroxy compounds, thereby impeding their interaction with the OXE receptor.
Because of its potency and prolonged lifetime in the blood, αS‐hydroxy‐S‐C048 (S‐C048M) might in itself be an interesting drug candidate, especially if it is well absorbed from the GI tract. The slow rate of decline in its plasma concentration over 3 days following administration of S‐Y048 suggests that once‐daily administration of a relatively low dose might be feasible. However, the two chiral centres of αS‐hydroxy‐S‐Y048 make its large‐scale synthesis more challenging, and we have not yet been able to test this hypothesis.
In conclusion, we have identified a potent OXE receptor antagonist with a pIC50 of about 10.8 (~20 pM) in inhibiting calcium mobilization induced by a 500‐fold higher concentration of 5‐oxo‐ETE. S‐Y048 appears rapidly in the blood after oral administration and remains at relatively high levels over 24 hr, especially when administered twice daily. In the present study, S‐Y048 was administered as a suspension in bicarbonate buffer, but with improved formulation, it should be possible to extend its lifetime in the blood even further. Our initial data from in vivo studies in rhesus monkeys demonstrate that S‐Y048 has in vivo efficacy in inhibiting allergen‐induced infiltration of eosinophils into the skin (Miller et al., 2019) and lungs (unpublished data). The high potency of S‐Y048 together with its favourable pharmacokinetic properties suggest that this OXE receptor antagonist may be a credible drug candidate for the treatment of eosinophilic diseases in humans, such as atopic dermatitis, asthma, and allergic rhinitis.
CONFLICT OF INTEREST
W.S.P. and J.R. have been granted a patent covering 230 and have applied for a patent covering S‐Y048.
AUTHOR CONTRIBUTIONS
Q.Y., S.C., C.N.R., and R.W. performed the chemical syntheses. S.G. evaluated antagonist potencies. C.C. conducted HPLC analyses. I.S. and D.V. performed mass spectrometric analyses. W.S.P. and J.R. designed the study. W.S.P., J.R., Q.Y., S.C., and D.V. wrote or contributed to the writing of the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Data S1
Supporting Information
ACKNOWLEDGEMENTS
This work was supported by the Canadian Institutes of Health Research (W.S.P.: Grants MOP‐6254 and PP2‐133388), the American Asthma Foundation (J.R.: Grant 12‐0049), and the National Heart, Lung, and Blood Institute (J.R.: Grant R01HL081873) and by AmorChem (Montreal, QC). The Meakins‐Christie Laboratories‐MUHC‐RI are supported in part by a Centre grant from Le Fond de la Recherche en Santé du Québec as well as by the J. T. Costello Memorial Research Fund. J.R. also wishes to acknowledge the National Science Foundation for the AMX‐360 (Grant CHE‐90‐13145) and Bruker 400 MHz (Grant CHE‐03‐42251) NMR instruments. D.V. and I.S. were supported by the Natural Sciences and Engineering Research Council of Canada (Grant RGPIN/435814‐2103). I.S. also wishes to acknowledge the Centre for Biological Applications of Mass Spectrometry at Concordia University for PhD scholarship funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Ye Q, Chourey S, Reddy CN, et al. Novel highly potent OXE receptor antagonists with prolonged plasma lifetimes that are converted to active metabolites in vivo in monkeys. Br J Pharmacol. 2020;177:388–401. 10.1111/bph.14874
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … Collaborators C (2017). The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäck, M. , Powell, W. S. , Dahlén, S. E. , Drazen, J. M. , Evans, J. F. , Serhan, C. N. , … Rovati, G. E. (2014). Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. British Journal of Pharmacology, 171, 3551–3574. 10.1111/bph.12665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourey, S. , Ye, Q. , Reddy, C. N. , Cossette, C. , Gravel, S. , Zeller, M. , … Powell, W. S. (2017). In vivo a‐hydroxylation of a 2‐alkylindole antagonist of the OXE receptor for the eosinophil chemoattractant 5‐oxo‐6,8,11,14‐eicosatetraenoic acid in monkeys. Biochemical Pharmacology, 138, 107–118. 10.1016/j.bcp.2017.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourey, S. , Ye, Q. , Reddy, C. N. , Wang, R. , Cossette, C. , Gravel, S. , … Powell, W. S. (2018). Novel highly potent and metabolically resistant oxoeicosanoid (OXE) receptor antagonists that block the actions of the granulocyte chemoattractant 5‐oxo‐6,8,11,14‐eicosatetraenoic acid (5‐oxo‐ETE). Journal of Medicinal Chemistry, 61, 5934–5948. 10.1021/acs.jmedchem.8b00154 [DOI] [PubMed] [Google Scholar]
- Cossette, C. , Chourey, S. , Ye, Q. , Nagendra Reddy, C. , Gore, V. , Gravel, S. , … Powell, W. S. (2016). Pharmacokinetics and metabolism of selective oxoeicosanoid (OXE) receptor antagonists and their effects on 5‐oxo‐6,8,11,14‐eicosatetraenoic acid (5‐oxo‐ETE)‐induced granulocyte activation in monkeys. Journal of Medicinal Chemistry, 59, 10127–10146. 10.1021/acs.jmedchem.6b00895 [DOI] [PubMed] [Google Scholar]
- Cossette, C. , Gravel, S. , Reddy, C. N. , Gore, V. , Chourey, S. , Ye, Q. , … Powell, W. S. (2015). Biosynthesis and actions of 5‐oxoeicosatetraenoic acid (5‐oxo‐ETE) on feline granulocytes. Biochemical Pharmacology, 96, 247–255. 10.1016/j.bcp.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallaire, M. J. , Ferland, C. , Page, N. , Lavigne, S. , Davoine, F. , & Laviolette, M. (2003). Endothelial cells modulate eosinophil surface markers and mediator release. The European Respiratory Journal, 21, 918–924. 10.1183/09031936.03.00102002 [DOI] [PubMed] [Google Scholar]
- Gore, V. , Gravel, S. , Cossette, C. , Patel, P. , Chourey, S. , Ye, Q. , … Powell, W. S. (2014). Inhibition of 5‐oxo‐6,8,11,14‐eicosatetraenoic acid‐induced activation of neutrophils and eosinophils by novel indole OXE receptor antagonists. Journal of Medicinal Chemistry, 57, 364–377. 10.1021/jm401292m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbert, M. , Ferland, C. , Bosse, M. , Flamand, N. , Lavigne, S. , & Laviolette, M. (1999). 5‐oxo‐6,8,11,14‐eicosatetraenoic acid induces important eosinophil transmigration through basement membrane components—Comparison of normal and asthmatic eosinophils. American Journal of Respiratory Cell and Molecular Biology, 21, 97–104. 10.1165/ajrcmb.21.1.3517 [DOI] [PubMed] [Google Scholar]
- Haeggstrom, J. Z. (2018). Leukotriene biosynthetic enzymes as therapeutic targets. The Journal of Clinical Investigation, 128, 2680–2690. 10.1172/Jci97945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … Nc‐Iuphar (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi, T. , Koguchi, Y. , Sugikawa, E. , Chikada, A. , Ogawa, K. , Tsuda, N. , … Ohnuki, T. (2002). Identification of a novel eicosanoid receptor coupled to Gi/o . The Journal of Biological Chemistry, 277, 31459–31465. 10.1074/jbc.M203194200 [DOI] [PubMed] [Google Scholar]
- Iikura, M. , Suzukawa, M. , Yamaguchi, M. , Sekiya, T. , Komiya, A. , Yoshimura‐Uchiyama, C. , … Hirai, K. (2005). 5‐Lipoxygenase products regulate basophil functions: 5‐Oxo‐ETE elicits migration, and leukotriene B4 induces degranulation. The Journal of Allergy and Clinical Immunology, 116, 578–585. 10.1016/j.jaci.2005.04.029 [DOI] [PubMed] [Google Scholar]
- Jones, C. E. , Holden, S. , Tenaillon, L. , Bhatia, U. , Seuwen, K. , Tranter, P. , … Finan, P. (2003). Expression and characterization of a 5‐oxo‐6E,8Z,11Z,14Z‐eicosatetraenoic acid receptor highly expressed on human eosinophils and neutrophils. Molecular Pharmacology, 63, 471–477. 10.1124/mol.63.3.471 [DOI] [PubMed] [Google Scholar]
- Khanapure, S. P. , Shi, X. X. , Powell, W. S. , & Rokach, J. (1998). Total synthesis of a potent proinflammatory 5‐oxo‐ETE and its 6,7‐dihydro biotransformation product. The Journal of Organic Chemistry, 63, 337–342. https://doi.org/DOI 10.1021/jo9716993 [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmert, J. , Fauland, A. , Fuchs, D. , Safholm, J. , Gomez, C. , Adner, M. , … Wheelock, C. E. (2018). Lipid mediator quantification in isolated human and guinea pig airways: An expanded approach for respiratory research. Analytical Chemistry, 90, 10239–10248. 10.1021/acs.analchem.8b01651 [DOI] [PubMed] [Google Scholar]
- Kowal, K. , Gielicz, A. , & Sanak, M. (2017). The effect of allergen‐induced bronchoconstriction on concentration of 5‐oxo‐ETE in exhaled breath condensate of house dust mite‐allergic patients. Clinical and Experimental Allergy, 47, 1253–1262. 10.1111/cea.12990 [DOI] [PubMed] [Google Scholar]
- Langlois, A. , Ferland, C. , Tremblay, G. M. , & Laviolette, M. (2006). Montelukast regulates eosinophil protease activity through a leukotriene‐independent mechanism. The Journal of Allergy and Clinical Immunology, 118, 113–119. 10.1016/j.jaci.2006.03.010 [DOI] [PubMed] [Google Scholar]
- Lin, L. , Chen, Z. , Tang, X. Y. , Dai, F. , Wei, J. J. , & Sun, G. B. (2018). 5‐Oxo‐ETE from nasal epithelial cells upregulates eosinophil cation protein by eosinophils in nasal polyps in vitro. International Archives of Allergy and Immunology, 177, 107–115. 10.1159/000489819 [DOI] [PubMed] [Google Scholar]
- McBrien, C. N. , & Menzies‐Gow, A. (2017). The biology of eosinophils and their role in asthma. Frontiers in medicine (Lausanne), 4(93), 1–14. 10.3389/fmed.2017.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, L. A. , Cossette, C. , Chourey, S. , Ye, Q. , Reddy, C. N. , Rokach, J. , & Powell, W. S. (2019). Inhibition of allergen‐induced dermal eosinophilia by an oxoeicosanoid (OXE) receptor antagonist in nonhuman primates. British Journal of Pharmacology. 10.1111/bph.14872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, E. , Schroder, J. M. , & Christophers, E. (1989). Differential sensitivities of purified human eosinophils and neutrophils to defined chemotaxins. Scandinavian Journal of Immunology, 29, 709–716. 10.1111/j.1365-3083.1989.tb01175.x [DOI] [PubMed] [Google Scholar]
- Muro, S. , Hamid, Q. , Olivenstein, R. , Taha, R. , Rokach, J. , & Powell, W. S. (2003). 5‐Oxo‐6,8,11,14‐eicosatetraenoic acid induces the infiltration of granulocytes into human skin. The Journal of Allergy and Clinical Immunology, 112, 768–774. 10.1067/mai.2003.1736 [DOI] [PubMed] [Google Scholar]
- O'Flaherty, J. T. , Kuroki, M. , Nixon, A. B. , Wijkander, J. , Yee, E. , Lee, S. L. , … Daniel, L. W. (1996). 5‐Oxo‐eicosatetraenoate is a broadly active, eosinophil‐selective stimulus for human granulocytes. Journal of Immunology, 157, 336–342. [PubMed] [Google Scholar]
- Patel, P. , Cossette, C. , Anumolu, J. R. , Gravel, S. , Lesimple, A. , Mamer, O. A. , … Powell, W. S. (2008). Structural requirements for activation of the 5‐oxo‐6E,8Z, 11Z,14Z‐eicosatetraenoic acid (5‐oxo‐ETE) receptor: Identification of a mead acid metabolite with potent agonist activity. The Journal of Pharmacology and Experimental Therapeutics, 325, 698–707. 10.1124/jpet.107.134908 [DOI] [PubMed] [Google Scholar]
- Peters‐Golden, M. , & Henderson, W. R. Jr. (2007). Leukotrienes. The New England Journal of Medicine, 357, 1841–1854. 10.1056/NEJMra071371 [DOI] [PubMed] [Google Scholar]
- Powell, W. S. (1987). Precolumn extraction and reversed‐phase high‐pressure liquid chromatography of prostaglandins and leukotrienes. Analytical Biochemistry, 164, 117–131. 10.1016/0003-2697(87)90375-7 [DOI] [PubMed] [Google Scholar]
- Powell, W. S. , Chung, D. , & Gravel, S. (1995). 5‐Oxo‐6,8,11,14‐eicosatetraenoic acid is a potent stimulator of human eosinophil migration. Journal of Immunology, 154, 4123–4132. [PubMed] [Google Scholar]
- Reddy, C. N. , Ye, Q. J. , Chourey, S. , Gravel, S. , Powell, W. S. , & Rokach, J. (2015). Stereoselective synthesis of two highly potent 5‐oxo‐ETE receptor antagonists. Tetrahedron Letters, 56, 6896–6899. 10.1016/j.tetlet.2015.10.097 [DOI] [Google Scholar]
- Schopfer, F. J. , Cipollina, C. , & Freeman, B. A. (2011). Formation and signaling actions of electrophilic lipids. Chemical Reviews, 111, 5997–6021. 10.1021/cr200131e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk, U. , & Schröder, J. M. (1995). 5‐Oxo‐eicosanoids are potent eosinophil chemotactic factors—Functional characterization and structural requirements. The Journal of Biological Chemistry, 270, 15029–15036. [DOI] [PubMed] [Google Scholar]
- Seow, V. , Lim, J. , Cotterell, A. J. , Yau, M. K. , Xu, W. , Lohman, R. J. , … Fairlie, D. P. (2016). Receptor residence time trumps drug‐likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Scientific reports‐Uk, 6, 24575 10.1038/srep24575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm, G. J. , Schuligoi, R. , Sturm, E. M. , Royer, J. F. , Lang‐Loidolt, D. , Stammberger, H. , … Heinemann, A. (2005). 5‐oxo‐6,8,11,14‐eicosatetraenoic acid is a potent chemoattractant for human basophils. The Journal of Allergy and Clinical Immunology, 116, 1014–1019. 10.1016/j.jaci.2005.08.001 [DOI] [PubMed] [Google Scholar]
- Sun, F. F. , Crittenden, N. J. , Czuk, C. I. , Taylor, B. M. , Stout, B. K. , & Johnson, H. G. (1991). Biochemical and functional differences between eosinophils from animal species and man. Journal of Leukocyte Biology, 50, 140–150. [DOI] [PubMed] [Google Scholar]
- Takeda, S. , Yamamoto, A. , & Haga, T. (2002). Identification of a G protein‐coupled receptor for 5‐oxo‐eicosatetraenoic acid. Biomedical Research‐Tokyo, 23, 101–108. 10.2220/biomedres.23.101 [DOI] [Google Scholar]
- Yokomizo, T. , Nakamura, M. , & Shimizu, T. (2018). Leukotriene receptors as potential therapeutic targets. The Journal of Clinical Investigation, 128, 2691–2701. 10.1172/JCI97946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni, R. , & Rokach, J. (1982). Simple efficient synthesis of LTB4 and 12‐epi‐LTB4. Tetrahedron Letters, 23, 2631–2634. 10.1016/S0040-4039(00)87415-8 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Supporting Information