

Abstract
Background and Purpose
Chloroquine is a traditional medicine to treat malaria. There is increasing evidence that chloroquine not only induces phagocytosis but regulates vascular tone. Few reports investigating the effect of chloroquine on vascular responsiveness of coronary arteries have been made. In this study, we examined how chloroquine affected endothelium‐dependent relaxation in coronary arteries under normal and diabetic conditions.
Experimental Approach
We isolated coronary arteries from mice and examined endothelium‐dependent relaxation (EDR). Human coronary endothelial cells and mouse coronary endothelial cells isolated from control and diabetic mouse (TALLYHO/Jng [TH] mice, a spontaneous type 2 diabetic mouse model) were used for the molecular biological or cytosolic NO and Ca2+ measurements.
Key Results
Chloroquine inhibited endothelium‐derived NO‐dependent relaxation but had negligible effect on endothelium‐derived hyperpolarization (EDH)‐dependent relaxation in coronary arteries of control mice. Chloroquine significantly decreased NO production in control human coronary endothelial cells partly by phosphorylating eNOSThr495 (an inhibitory phosphorylation site of eNOS) and attenuating the rise of cytosolic Ca2+ concentration after stimulation. EDR was significantly inhibited in diabetic mice in comparison to control mice. Interestingly, chloroquine enhanced EDR in diabetic coronary arteries by, specifically, increasing EDH‐dependent relaxation due partly to its augmenting effect on gap junction activity in diabetic mouse coronary endothelial cells.
Conclusions and Implications
These data indicate that chloroquine affects vascular relaxation differently under normal and diabetic conditions. Therefore, the patients' health condition such as coronary macrovascular or microvascular disease, with or without diabetes, must be taken account into the consideration when selecting chloroquine for the treatment of malaria.
Abbreviations
- CA
coronary artery
- CQ
chloroquine
- EC
endothelial cell
- EDR
endothelium‐dependent relaxation
- EDH
endothelium‐derived hyperpolarization
- EDNO
endothelium‐derived NO
- FPIX
ferriprotoporphyrin IX
- HCECs
human coronary endothelial cells
- MCECs
mouse coronary endothelial cells
- SMC
smooth muscle cell
- SOCE
store‐operated Ca2+ entry
- T2D
Type 2 diabetic
- TH mouse
TALLYHO/Jng mouse
- VDCC
voltage‐dependent L‐typee_k Ca2+ channel
- Wt
wild type
What is already known
Chloroquine is a useful anti‐malaria treatment but also serves as a regulator of vascular tone.
There is no reports examining the effect of chloroquine on vascular response in coronary arteries.
What this study adds
Chloroquine attenuates EDR by reducing NO but not EDH in normal mice CAs.
In diabetic mice, chloroquine enhances EDR via augmenting the activity of the gap junctions.
What is the clinical significance
Chloroquine should be used with caution in patients with non‐diabetic cardiovascular disease.
1. INTRODUCTION
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5535 (CQ), a 4‐aminoquinoline derivative, is an antimalarial drug that has also been used to treat other diseases including lupus erythematosus, rheumatoid arthritis, and amoebiasis (Egan, 2001; Rees & Maibach, 1963). Chloroquine exhibits weak base charge with pKa of 8.1 for the amino group at Position 7 and pK 2 = 10.1 for the amino group on the alkyl side chain (Larsson & Tjalve, 1979). This base character of chloroquine is responsible for killing malaria by accumulating chloroquine in the parasite's acidic digestive vacuole, where the parasites degrade haemoglobin a major nutrient source of parasites. During degradation of haemoglobin, the level of ferriprotoporphyrin IX (FPIX) is also increased in the digestive vacuole. In the meantime, parasites biocrystallize FPIX dimers into inert hemozoin crystals to avoid cell lysis. Chloroquine in the digestive vacuole inhibits hemozoin formation, which leads to FPIX accumulation, membrane permeabilization and cell lysis (Fong & Wright, 2013). The basic nature of chloroquine causes it be concentrated in lysosomers (lysosomotrophic action).. After the accumulation of chloroquine in lysosomes, the pH of the lysosome will increase from 4 to 6 (Kalia & Dutz, 2007; Kutner, Breuer, Ginsburg, & Cabantchik, 1987) and result in inactivation of many enzymes in the lysosome such as proteases. This is the main cause of chloroquine‐induced autophagy inhibition (Mauthe et al., 2018).
Recently, there is increasing evidence that chloroquine also regulates the function of vascular cells (Manson et al., 2014; Pestana, Oishi, Salistre‐Araujo, & Rodrigues, 2015; Sai et al., 2014; Wu et al., 2017) and cardiac myocytes (Sanchez‐Chapula, Salinas‐Stefanon, Torres‐Jacome, Benavides‐Haro, & Navarro‐Polanco, 2001; Tona, Ng, Akera, & Brody, 1990). Sai et al. (2014) demonstrated that chloroquine induces vasodilation in rat aorta at high concentration (>100 μmol·L−1) via inhibiting voltage‐dependent L‐type Ca2+ channel (VDCC). We have also found that pretreatment with chloroquine (200 μmol·L−1) attenuates vascular contraction in mouse pulmonary artery through inhibition of VDCC in smooth muscle cells (SMCs; Wu et al., 2017). Pestana et al. (2015) show that chloroquine relaxes vessels in a dose‐dependent manner by increasing NO production in rat aorta. In contrast, Manson et al. (2014) demonstrate that chloroquine induces vasodilation independent of endothelium, VDCC, and Ca2+‐activated K+ channel in guinea pig aorta. These data suggest that the effect of chloroquine on vascular response varies in different tissues and organs. In this study, we have aimed to investigate the effect of chloroquine on vascular relaxation in coronary arteries (CAs) isolated from control and diabetic mice.
Chronic hyperglycaemia leads to cardiovascular complications that are a leading cause of death in diabetic patients. Endothelial cell (EC) dysfunction is a major cause of vascular complications in diabetes, while coronary endothelial dysfunction is implicated in the increased morbidity of coronary artery disease and coronary microvascular disease in diabetes (Belin de Chantemele & Stepp, 2012; Labazi & Trask, 2017; Pant, Marok, & Klein, 2014; Pepine et al., 2015; Rocic, 2012). It is therefore scientifically important and clinically relevant to examine whether chloroquine has a maladaptive effect on coronary endothelial function in diabetes. To the best of our knowledge, there is no report examining chloroquine effect on coronary vascular function in diabetes, although chloroquine was examined on other organs of diabetics. Chloroquine appears to exert a beneficial effect in both insulin‐dependent and insulin‐independent diabetic patients (Blazar et al., 1984; Powrie et al., 1993; Smith, Amos, Mahler, & Peters, 1987) due partly to its inhibition of insulin degradation (Blazar et al., 1984). In diabetic animal models, chloroquine treatment significantly improves diastolic function of the left ventricle (Yuan et al., 2016), tubular function in the kidney (Jeong et al., 2018), and abnormal gluconeogenesis in the liver (Jarzyna, Kiersztan, Lisowa, & Bryla, 2001). Those data indicate that chloroquine might be a useful treatment to improve hyperglycaemia‐ and diabetes‐induced complications. In this study, we were the first to examine the effect of chloroquine on endothelium‐dependent relaxation (EDR) in CAs and compare the effect of chloroquine on coronary vasodilation in healthy and diabetic mice. We also determined the molecular mechanisms underlining these outcomes. The data show that chloroquine differentially affects coronary vascular function in healthy control and diabetic mice. The study provides important insights into the understanding of the adverse effect of chloroquine on coronary function in control patients or patients without diabetes.
2. METHODS
2.1. Animals
This study was conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at the University of Arizona (UA) and University of California, San Diego (UCSD). All animal use is in compliance with all current U.S. government regulations concerning the care and use of laboratory animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. The University has been certified by PHS with Animal Welfare Assurance numbers A3248‐01 (UA) and A3033‐01 (UCSD), and the approved IACUC protocol numbers for this study are 14‐520 (UA) and S18185 (UCSD). The laboratory personnel who conducted following experiments took all the training required for animal handling and were certified by the IACUC.
TALLYHO/Jng (TH) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA; RRID:IMSR_JAX:005314) and bred in our animal facility. The TH mouse is a polygenic type 2 diabetic (T2D) model, and we used C57BL/6 as a wild‐type (Wt) control (Kim & Saxton, 2012). Male C57BL/6 mice (ENVIGO, Placentia, California, USA; RRID:MGI:2161078) were used at the age of ≤16 weeks in Figure 1. TH male mice were used at the age of 16–25 weeks, and the age‐matched Wt mice were used as control. All mice were housed in a specific pathogen‐free facility with five mice per cage and maintained at 23–25°C with 12‐hr light–12‐hr dark cycle and standard rodent diet and water ad libitum.
Figure 1.
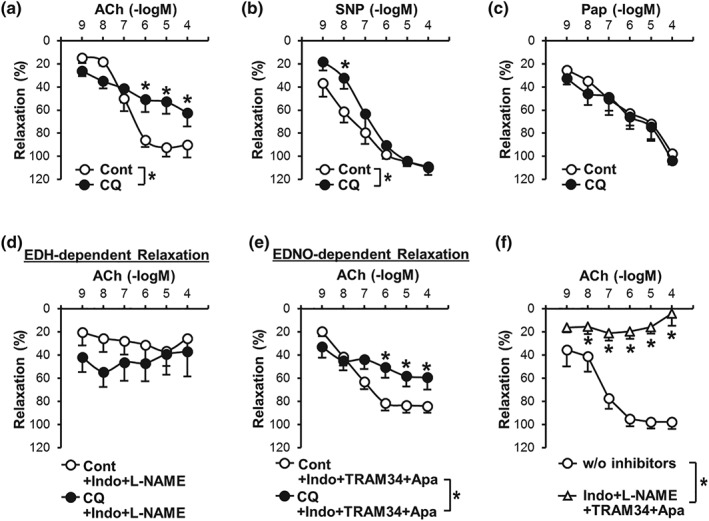
Endothelium‐dependent relaxation in coronary artery is attenuated by chloroquine. (a) Endothelium‐dependent relaxation (EDR) induced by ACh in the absence (Cont, open circle) or presence (CQ, closed circle) of chloroquine (CQ, 10 μM). n mice = 6 in each group. Data are mean ± SE. *P < .05 versus Cont. (b) Smooth muscle (SM)‐dependent relaxation induced by sodium nitroprusside (SNP, an NO donor) in the absence (Cont) or presence (CQ) of CQ. n mice = 7 in each group. (c) SM‐dependent relaxation induced by papaverine (Pap, a PDE inhibitor) in the absence (Cont) or presence (CQ) of CQ. n mice = 6 in each group. (d)Endothelium‐derived hyperpolarization (EDH)‐dependent relaxation in the absence (Cont) or presence (CQ) of CQ. EDH‐dependent relaxation was conducted by ACh administration in the presence of indomethacin (Indo, a COX inhibitor, 10 μM) and L‐NAME (an eNOS inhibitor, 100 μM). n mice = 8 in each group. (e) Endothelium‐derived NO (EDNO)‐dependent relaxation in the absence (Cont) or presence (CQ) of CQ. EDNO‐dependent relaxation was accessed by ACh administration in the presence of Indo, TRAM34 (an inhibitor of intermediate‐conductance Ca2+‐activated K+ channel, 100 nM), and apamin (an inhibitor of small conductance Ca2+ activated K+ channel, 100 nM). n mice = 13 in each group. (f) Pretreatment with Indo (to inhibit PGI2), L‐NAME (to inhibit NO), and TRAM34 and apamin (to inhibit EDH) abolishes ACh‐induced relaxation in coronary artery (CA). n mice = 8 in each group. *P < .05 compared to control (i.e., w/o inhibitors). Statistical comparison between dose–response curves was made by two‐way ANOVA with Bonferroni post hoc test
Mice were randomized to groups for each experiment. Oral glucose tolerance test (OGTT) and measurements of plasma cholesterol, HDL, triglyceride, and insulin levels were achieved as described in a previous paper (Cho, Basu, Dai, Heldak, & Makino, 2013). Heart dissection was performed under terminal anaesthesia with a mixture of ketamine (100 mg·kg−1, i.p.) and xylazine (5 mg·kg−1, i.p.). The body weight of mice used in this study was 29.2 ± 0.7 g in Wt mice and 35.2 ± 1.2 g in TH mice (P < .01), and blood glucose levels without fasting were 172.3 ± 9.3 mg·dl−1 in Wt mice and 422.9 ± 28.8 mg·dl−1 in TH mice (P < .01; Wt, n = 29; TH, n = 28).
2.2. Determination of chloroquine concentration for the experiment
The concentration of chloroquine (Cat# C6628, Sigma‐Aldrich Corp. St. Louis, MO, USA), which we used in this study, was determined based on the literature. It has been demonstrated that a plateau plasma concentration is 10 μM after administration of chloroquine at the dosage of 500 mg·day−1 and 1 μM at 250 mg·day−1 (Mackenzie, 1983). Chloroquine is commonly administered at 100–250 mg·kg−1, and 500 mg is the maximal dosage for daily administration (Mackenzie, 1983). The highest dosage is 1,500 mg·day−1 for 3 days for an acute episode of malaria (Reed & Campbell, 1962). Therefore, we used 10 μM of chloroquine for our experiments. Chloroquine was dissolved in saline and freshly prepared at 10 mM before further dilution for the experiment.
2.3. Isometric tension measurement in coronary arterial ring
Isometric tension was measured in isolated coronary arterial ring of mice to evaluate vascular function as previously described (Estrada et al., 2012; Makino, Platoshyn, Suarez, Yuan, & Dillmann, 2008). The heart was isolated from the mouse and put in modified Krebs–Henseleit solution (KHS) for dissection. The composition of KHS in mM: NaCl 118.0, KCl 4.7, NaHCO3 25.0, CaCl2 1.8, NaH2PO4 1.2, MgSO4 1.2, and glucose 11.0. Third‐order small coronary arteries were dissected, and adherent connective tissues and cardiac myocytes were removed. Arteries were cut into 1‐ to 1.5‐mm segments. The arterial rings were mounted in a 4‐channel wire myograph (620M, Danish Myo Technology A/S. Aarhus N, Denmark) over 20‐μm wires, set at a resting tension of 100 mg, and allowed to equilibrate for 45 min with intermittent washes every 15 min. After equilibration, each CA was contracted to achieve an active tension of about 50–100 mg (150–200 mg from the baseline) by treatment with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1884 2α. The concentration of PGF2α used for precontraction was 1–10 μM. The magnitude of contraction was 76.5 ± 6.1 mg in Wt and 66.2 ± 11.8 in TH. There was no statistical difference of PGF2α‐induced contraction between Wt and TH. The degree of relaxation was shown as a percent of PGF2α‐induced contraction.
The dose‐dependent curve was generated using the raw data; averaged data were shown for each dose in the figure. The maximal response shown in the tables was calculated using the value at the maximal response of relaxation (or the maximal relaxation value). EC50 was calculated using Sigma Plot 12.5 (Systat Software, Inc., San Jose, CA, USA).
2.4. Isolation of mouse coronary endothelial cell
Mouse coronary endothelial cell (MCECs) were isolated using a method previously described (Cho et al., 2013; Luo, Truong, & Makino, 2016; Makino et al., 2008; Makino et al., 2015).
2.5. Cytosolic NO measurement
Cytosolic NO concentration ([NO]cyto) was measured using a modified method described in our previous paper (Estrada et al., 2012). Briefly, human coronary endothelial cells (HCECs, Cat# CC‐2585, Lonza America, Inc., Alpharetta, GA, USA) were stained with DAF‐FM diacetate (5 μM) for 30 min in the dark at room temperature. The DAF‐FM‐loaded cells were then washed and treated with chloroquine (10 μM) for 30 min. The fluorescent images of cells were obtained using an EVOS FL Auto Imaging System (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The background intensity of DAF‐FM was subtracted from the cell intensity to precisely indicate the actual cell intensity of DAF‐FM. The actual cell intensity in treated cells was then normalized to the actual cell intensity in non‐treated cells to show changes of DAF‐FM intensity.
2.6. Cytosolic Ca2+ measurement
Cytosolic‐free Ca2+ concentration ([Ca2+]cyto) in HCECs was measured using a modified method previously described (Estrada et al., 2012). Briefly, cells were plated on glass slides coated with 5% gelatine and loaded with the membrane‐permeable acetoxymethyl ester form of fura 2 (fura2‐AM; 4 μM for 1 hr in the dark at room temperature). The fura2‐AM‐loaded cells were then superfused with physiological salt solution (PSS, pH 7.4, in mM: NaCl 141, KCl 4.7, CaCl2 1.8, MgCl2 1.2, HEPES 10, and glucose 10) for 30 min at 32°C. Fura‐2 fluorescence signals from the cells and background were imaged using a Nikon TE300 microscope (Nikon Instruments, Inc., Melville, New York, USA). The changes in [Ca2+]cyto are described as a normalized ratio F/F 0 (F = R 340/R 380, F 0 = averaged F measured for 5 min before application of extracellular agonist). The amplitude of peak increase in [Ca2+]cyto (∆F/F 0) and the AUC of the peak increase in [Ca2+]cyto per individual cells were measured and calculated. Data were normalized by the average of control data.
2.7. Western blot analysis
Protein expression was analysed using an SDS‐PAGE. Antibodies specifically against endothelial NOS (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249, 1:2000, sc‐654, Santa Cruz Biotechnology, Inc., Dallas, TX, USA; RRID:AB_631423), p‐eNOSSer1177 (1:1000, #9570, Cell Signaling Technology, Danvers, MA, USA. RRID:AB_823493), p‐eNOSThr495 (1:1000, #9574, Cell Signaling Technology; RRID:AB_2153176), and actin (1:4000, sc‐1616, Santa Cruz Biotechnology, Inc., RRID:AB_630836) were purchased from different companies as indicated above.
2.8. Dye transfer assay
After MCEC isolation, cells were plated in a 24‐well plate and cultured for 4 days to reach confluence. Cells were then incubated with chloroquine (10 μM) or vehicle (saline) for 30 min. Dye transfer assay was performed as described in our previous paper (Makino et al., 2015). Briefly, Lucifer yellow (0.5 mg·ml−1) was added to the well after chloroquine treatment, and the confluent cells were scratched by a scalpel in the middle of the well to allow dye to get into cells at the edge of the scrape. The dye solution was left for 20 min in the well, and then cells were washed and fixed. Images of dye transfer were captured using an EVOS FL Auto Imaging System. The distance of dye transfer from the scraping site to the farthest site of cells showing visual uptake of dye was measured using Image Pro‐Plus 7.0 software (Media Cybernetics, Inc., Rockville, MD, USA). The data were normalized to the averaged distance of dye transfer in Wt cells without chloroquine treatment.
2.9. Data analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. We conducted data analysis in a blinded fashion whenever possible and set proper controls for every experimental plan. The mouse numbers and independent experiment numbers are described in the figure legend.
Statistical analysis was performed using GraphPad Prism 8.1.2 (GraphPad Software, Inc., La Jolla, CA, USA; RRID:SCR_002798). Data are presented as mean ± SE. Student's t test was used for comparisons of two experimental groups (except Figure 2f,g), and one‐way ANOVA was used for multiple comparisons. Mann–Whitney test was used for Figure 2f,g. Statistical comparison between dose–response curves was made by a two‐way ANOVA and a post hoc test with the Bonferroni correction (if F achieved the necessary level of statistical significance (P < 0.05) and there is no significant variance in homogeneity). Differences were considered to be statistically significant when P value was less than .05 (*P < .05).
Figure 2.
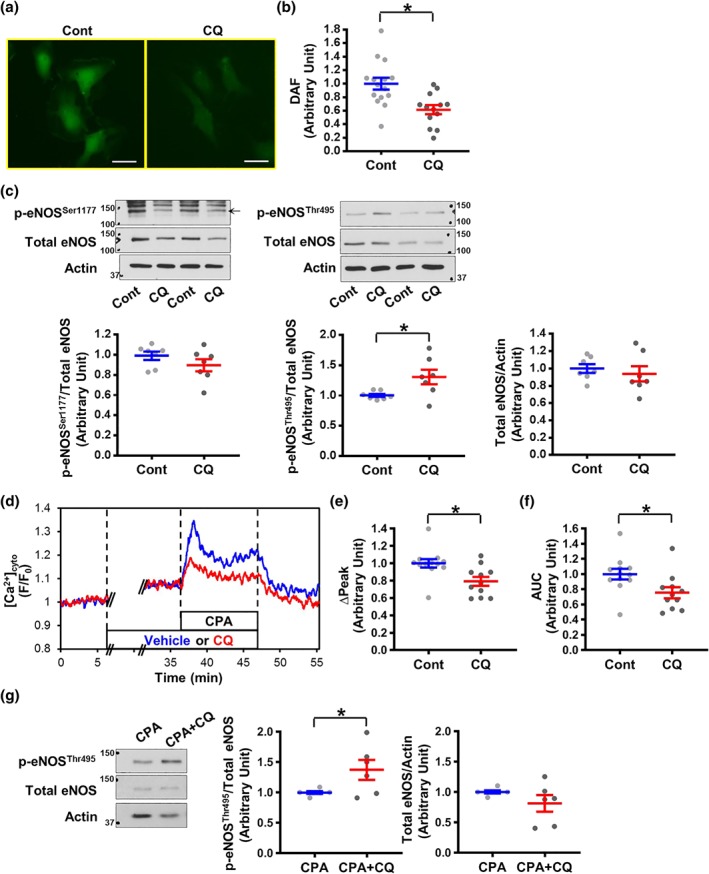
CQ decreases cytosolic NO level, increases phosphorylated eNOS495, and attenuates the rise in cytosolic Ca2+ concentration after stimulation in human coronary endothelial cells. (a) Representative images showing NO signals, determined by DAF, in human coronary endothelial cells (HCECs) with or without (w/o) CQ treatment for 30 min. Bar = 50 μm. (b) Summarized data showing DAF intensity in control cells (Cont, n experiment = 15, n cells = 127) and CQ‐treated cells (CQ, n experiment = 13, n cells = 118). Data are mean ± SE. *P < .05 compared to Cont. (c) WB analysis on total eNOS, phosphorylated eNOS at Ser1177 (p‐eNOSser1177), and Thr495 (p‐eNOSThr495) in control HCECs (Cont) and CQ‐treated HCECs (CQ). Actin was used as a loading control. The scattered plots show the level of p‐eNOS normalized to total eNOS (left panels) and the level of total eNOS normalized to actin (right panel). n experiment = 7. Data are mean ± SE. *P < .05 compared to Cont. (d) Representative records showing cytosolic Ca2+ concentration ([Ca2+]cyto) in HCECs before, during, and after stimulation with cyclopiazonic acid (CPA, an inhibitor of sarcoplasmic reticulum Ca2+‐ATPase to increase [Ca2+]cyto, 10 μM) in the absence (vehicle, blue tracing) or presence (CQ, red tracing) of CQ. (e,f) Summarized data showing the amplitude (∆Peak, e) and the AUC (f) of CPA‐induced increase in [Ca2+]cyto in control cells (Cont, n experiment = 12, n cells = 120) and CQ‐treated cells (CQ, n experiment = 11, n cells = 110). Data are mean ± SE. *P < .05 compared to Cont. (g) The level of p‐eNOSThr495 after the treatment of CPA with or without CQ. n experiment = 6. Data are mean ± SE. *P < .05 compared to CPA. Unpaired Student's t‐test was used for comparisons of two experimental groups in Figure 2b,c,g. Mann–Whitney test was used for Figures 2e,f
2.10. Materials
Ketamine and xylazine were purchased from Henry Schein Inc. (Melville, NY, USA). DAF‐FM diacetate (Cat# D23844), Fura2‐AM (Cat# F1221), and Lucifer yellow (Cat# L453) were purchased from Thermo Fisher Scientific Inc., Waltham, MA, USA. Other chemicals were obtained from Sigma‐Aldrich Corp. St. Louis, MO, USA.
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017; Alexander, Christopoulos et al., 2017).
3. RESULT
3.1. Chloroquine attenuates EDR in coronary artery
Acetylcholine (ACh) induces EDR via releasing NO and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915 and/or by hyperpolarizing cells. We first examined whether chloroquine affected endothelium‐dependent coronary vasodilation. ACh was administered to CAs in a dose‐dependent manner to evaluate the function of ECs. Pretreatment with chloroquine significantly attenuated ACh‐induced vasodilation in CAs (Figure 1a and Table 1). In contrast, chloroquine had negligible effect on coronary vasodilation induced by https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9533 (SNP, an NO donor; Figure 1b) and papaverine (Pap, a PDE inhibitor; Figure 1c). These data indicate that chloroquine significantly inhibits EDR but has no effect on endothelium‐independent and smooth muscle (SM)‐dependent vasodilation in CAs.
Table 1.
Maximal responses and EC50 values of agonist‐induced vasodilation in the absence or presence of chloroquine in mouse coronary arteries
| n | Vehicle | Chloroquine (CQ) | ||||
|---|---|---|---|---|---|---|
| Max. (%) | ‐log (EC50) | Max. (%) | ‐log (EC50) | |||
| ACh | Untreated | 6 | 93.9 ± 8.4 | 7.0 ± 0.1 | 64.7 ± 11.0* | 7.4 ± 0.7 |
| L‐NAME + Indo | 7 | 49.9 ± 11.0 | 6.2 ± 0.6 | 75.9 ± 10.3* | 7.1 ± 0.5 | |
| TRAM34 + Apa + Indo | 13 | 86.3 ± 6.2 | 7.8 ± 0.3 | 64.9 ± 9.1* | 7.7 ± 0.5 | |
| SNP | Untreated | 7 | 108.9 ± 3.7 | 9.2 ± 0.9 | 109.9 ± 6.1 | 7.0 ± 0.4 |
| Pap | Untreated | 6 | 98.0 ± 8.0 | 6.2 ± 0.8 | 103.7 ± 3.5 | 6.0 ± 0.5 |
Note. L‐NAME, 100 μM; Indo, 10 μM; TRAM34, 100 nM; Apa, 100 nM. Data are mean ± SE.
P < .05 compared to vehicle.
To examine the potential mechanisms involved in chloroquine‐mediated inhibition of EDR, we used different chemicals to block each signalling cascade. We pretreated vessels with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5213 (an eNOS inhibitor, 100 μM) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1909 (Indo, a COX inhibitor, 10 μM) to visualize endothelium‐derived hyperpolarization (EDH)‐dependent relaxation. We used a combination cocktail of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2311 (Apa, an inhibitor of small‐conductance Ca2+‐activated K+ [SKCa] channel, 100 nM) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2336 (an inhibitor of intermediate‐conductance Ca2+‐activated K+ [IKCa] channel, 100 nM) to inhibit EDH‐dependent relaxation. In the presence of Apa, TRAM34, and Indo, we were able to observe endothelium‐derived NO (EDNO)‐dependent relaxation in isolated CAs. Chloroquine treatment did not affect EDH‐dependent relaxation in CAs (Figure 1d), while chloroquine significantly attenuated EDNO‐dependent relaxation (Figure 1e and Table 1 ). Figure 1f demonstrates that the treatment of Indo, L‐NAME, Apa, and TRAM34 abolished EDR in CAs, confirming that ACh relaxes CAs through EDNO‐, EDH‐, and prostacyclin‐dependent relaxation. These data indicate that chloroquine attenuates EDR in CAs due primarily to its inhibitory effect on EDNO‐dependent relaxation, but not EDH‐dependent relaxation.
3.2. Chloroquine inhibits NO production in HCECs
To examine whether chloroquine‐mediated inhibition on EDNO‐dependent relaxation in CAs is, at least in part, due to its inhibitory effect on NO production in ECs, we measured and compared [NO]cyto in HCECs and chloroquine ‐treated HCECs. We stained HCECs with DAF‐FM to measure [NO]cyto. Cells were then treated with vehicle (saline) or chloroquine for 30 min, and the DAF‐FM image was taken and used to calculate [NO]cyto. Figure 2a,b shows that chloroquine significantly decreased [NO]cyto in HCECs. To examine the molecular mechanism by which chloroquine decreases [NO]cyto, we examined and compared the eNOS activity in vehicle and chloroquine ‐treated HCECs. The levels of total eNOS and phosphorylated (p)‐eNOSSer1177 (a positive regulatory site) were not altered by chloroquine, whereas the level of p‐eNOSThr495 (a negative regulatory site) was significantly increased by chloroquine (Figure 2c). These data suggest that chloroquine may reduce the resting level of NO production by phosphorylation of eNOS at the negative regulatory site (Thr495).
3.3. Chloroquine pretreatment attenuates the increase in [Ca2+]cyto after stimulation in HCECs
Since an increase in [Ca2+]cyto leads to eNOS activation and induces NO production, we examined the effect of chloroquine on the rise of [Ca2+]cyto after stimulation. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5350 (CPA, an inhibitor of sarcoplasmic reticulum Ca2+‐ATPase) was used to increase [Ca2+]cyto by inducing store‐operated Ca2+ entry (SOCE). Chloroquine treatment did not affect the resting level of [Ca2+]cyto (vehicle, 1.14 ± 0.06; chloroquine, 1.14 ± 0.02, P = .98) but significantly inhibited CPA‐induced increase in [Ca2+]cyto (Figure 2d–f). In addition, we found that chloroquine still increased the level of p‐eNOSThr495 in the presence of CPA (Figure 2g), implying that NO production was decreased by chloroquine due to attenuated Ca2+ influx and increased p‐eNOSThr495 level.
3.4. Chloroquine enhances EDH‐dependent relaxation in CAs of diabetic mice
To examine the effect of chloroquine on coronary vasodilation in diabetes, we used CAs dissected from control Wt mice and diabetic TH mice, a well‐established T2D mouse model with abnormal glucose tolerance (Figure 3a), hyperinsulinaemia, and dyslipidaemia (Table 2). EDR was significantly attenuated in CAs from diabetic mice compared to CAs from control mice (Figure 3b), whereas there was no difference of SM‐dependent relaxation in CAs dissected from control and diabetic mice (Figure 3e). Chloroquine significantly attenuated EDR in CAs from control mice (Figure 3c) but enhanced EDR in CAs from diabetic mice (Figure 3d). The chloroquine‐mediated inhibition of EDR, which we observed in CAs from control mice, was due obviously to the attenuated EDNO‐dependent relaxation (Figure 3h), but not to EDH‐dependent relaxation (Figure 3f). The chloroquine‐mediated augmentation of EDR, which we observed in CAs from diabetic mice, resulted from increased EDH‐dependent relaxation (Figure 3g). Chloroquine had a negligible effect on EDNO‐dependent relaxation in CAs from diabetic mice (Figure 3i). We also examined the effect of 18β‐glycyrrhetinic acid (18GA, a non‐specific gap junction inhibitor, 50 μM) on EDH‐dependent relaxation in the presence of chloroquine (Figure 3j,k). CAs were pre‐incubated with chloroquine and 18GA or vehicle (0.25% ethanol) for 30 min before preconstruction. Enhanced EDH‐dependent relaxation was significantly attenuated by 18GA treatment in control and diabetic mice. These data suggest that augmented EDH‐dependent relaxation by chloroquine was due partly to increased gap junction activity in diabetic mice (Table 3).
Figure 3.
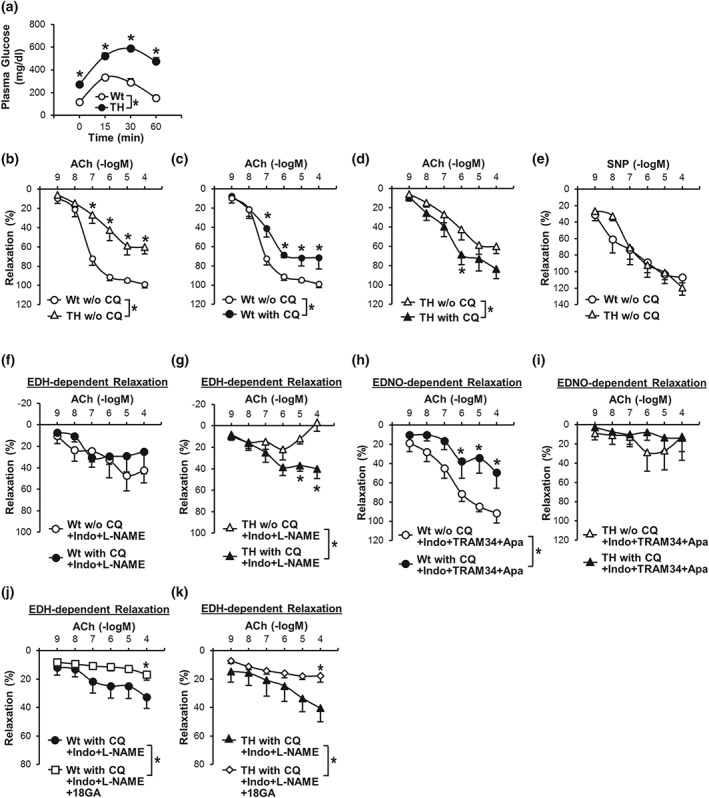
CQ enhances EDH‐dependent coronary vasodilation in diabetic mice. (a): Oral glucose tolerance test in Wt mice (n mice = 7) and TH mice (n mice = 7). *P < .05 compared to Wt mice. (b) EDR in CAs from Wt and TH mice without CQ treatment (w/o CQ). Wt w/o CQ, n mice = 13; TH w/o CQ, n mice = 12. Data are mean ± SE. *P < .05 compared to Wt w/o CQ. (c) Effect of CQ on EDR in CAs from Wt mice. Wt w/o CQ, n mice = 13; Wt with CQ, n mice = 9. Data are mean ± SE. *P < .05 compared to Wt w/o CQ. (d) Effect of CQ on EDR in CAs from TH mice. TH w/o CQ, n mice = 12; TH with CQ, n mice = 8. Data are mean ± SE. *P < .05 compared to TH w/o CQ. (e) SM‐dependent relaxation induced by SNP in CAs from Wt (n mice = 5) and TH mice (n mice = 5) in the absence of CQ treatment (w/o CQ). (f) EDH‐dependent relaxation in CAs isolated from Wt mice in the absence or presence of CQ. n mice = 7 in each group. (g) EDH‐dependent relaxation in CAs from TH mice in the absence or presence of CQ. n mice = 7 in each group. *P < .05 compared to w/o CQ. (h) EDNO‐dependent relaxation in CAs from Wt mice in the absence or presence of CQ. n mice = 6 in each group. *P < .05 versus w/o CQ. (i) EDNO‐dependent relaxation in CAs from TH mice in the absence or presence of CQ. n mice = 5 in each group. Indo and L‐NAME were used to inhibit PGI2 and NO in experiments shown in (f) and (g), while Indo, TRAM34, and apamin were used to inhibit PGI2 and EDH in experiments shown in (h) and (i). (j) EDH‐dependent relaxation in CAs isolated from Wt mice in the presence of CQ with vehicle (0.25% ethanol) or 18GA (a non‐specific Gap junction inhibitor, 50 μM). n mice = 8 in each group. (k) EDH‐dependent relaxation in CAs from TH mice in the presence of CQ with vehicle or 18GA. n mice = 8 in each group. *P < .05 versus w/o 18GA. Statistical comparison between dose–response curves were made by two‐way ANOVA with Bonferroni post hoc test
Table 2.
Pharmacological parameters of Wt and TH mice
| Wt (n = 8) | TH (n = 8) | |
|---|---|---|
| Body weight (g) | 27.9 ± 0.5 | 32.8 ± 1.8* |
| Blood glucose (mg·L−1) | 1633 ± 150 | 5388 ± 278* |
| Insulin (ng·mL−1) | 0.19 ± 0.03 | 0.48 ± 0.11* |
| TC (mg·L−1) | 806 ± 35 | 1536 ± 45* |
| HDL (mg·L−1) | 412 ± 40 | 719 ± 37* |
| TG (mg·L−1) | 306 ± 72 | 1614 ± 695* |
| LDL/VLDL (mg·L−1) | 333 ± 59 | 494 ± 102 |
Note. Data are mean ± SE.
Abbreviations: TC, plasma total cholesterol; TG, plasma triglyceride.
P < .05 compared to Wt.
Table 3.
Maximal responses and EC50 values of agonist‐induced vasodilation in the absence or presence of chloroquine (CQ) in mouse coronary arteries dissected from Wt or TH mice
| Wt | TH | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Vehicle | n | CQ | n | Vehicle | n | CQ | ||
| Max. response | |||||||||
| ACh | Untreated | 13 | 101.6 ± 2.8 | 9 | 84.4 ± 5.1* | 12 | 66.1 ± 8.1 | 8 | 85.8 ± 9.8 |
| L‐NAME + Indo | 7 | 55.3 ± 14.3 | 7 | 45.3 ± 16.1 | 7 | 27.5 ± 9.9 | 7 | 53.7 ± 5.0* | |
| TRAM34 + Apa + Indo | 6 | 93.0 ± 9.6 | 6 | 51.1 ± 17.0* | 5 | 35.4 ± 19.0 | 5 | 19.7 ± 13.2 | |
| SNP | Untreated | 5 | 107.1 ± 7.0 | 5 | 119.5 ± 9.1 | ||||
| ‐log (EC50) | |||||||||
| ACh | Untreated | 13 | 7.5 ± 0.3 | 9 | 7.0 ± 0.3 | 12 | 6.6 ± 0.3 | 8 | 7.1 ± 0.5 |
| L‐NAME + Indo | 7 | 7.4 ± 0.6 | 7 | 7.6 ± 0.4 | 7 | 8.2 ± 0.8 | 7 | 6.4 ± 0.5* | |
| TRAM34 + Apa + Indo | 6 | 7.6 ± 0.7 | 6 | 6.0 ± 0.5 | 5 | 6.0 ± 0.4 | 5 | 8.7 ± 0.1* | |
| SNP | Untreated | 5 | 7.7 ± 0.5 | 5 | 6.7 ± 0.4 | ||||
| n | Vehicle | n | 18GA | n | Vehicle | n | 18GA | ||
| Max. response | |||||||||
| ACh | L‐NAME + Indo + CQ | 8 | 37.1 ± 8.5 | 8 | 18.2 ± 3.6 | 8 | 41.9 ± 9.6 | 8 | 23.1 ± 3.2 |
| ‐log (EC50) | |||||||||
| ACh | L‐NAME + Indo + CQ | 8 | 5.4 ± 0.4 | 8 | 8.1 ± 0.8* | 8 | 6.1 ± 0.6 | 8 | 8.4 ± 0.8* |
Note. L‐NAME, 100 μM; Indo, 10 μM; TRAM34, 100 nM; Apa, 100 nM; vehicle for CQ, saline; 18GA, 50 μM; vehicle for 18GA, 0.25% ethanol. Data are mean ± SE.
P < .05 versus vehicle.
3.5. Chloroquine enhances gap junction activity in MCECs isolated from diabetic mice
To confirm that chloroquine regulates gap junction activity, we examined the effect of chloroquine on endothelial gap junctions in coronary ECs (MCECs) isolated from control and diabetic mice. Gap junction activity was assessed by the dye transfer experiment (Makino et al., 2015). Figure 4a,b demonstrates that the distance of dye transfer was significantly decreased in MCECs isolated from diabetic mice compared to MCECs from control mice and chloroquine significantly increased the distance of dye transfer in MCECs of diabetic mice. These data indicate that chloroquine enhances gap junction activity in diabetic MCECs and subsequently enhances EDH‐dependent relaxation in CAs of diabetic mice.
Figure 4.
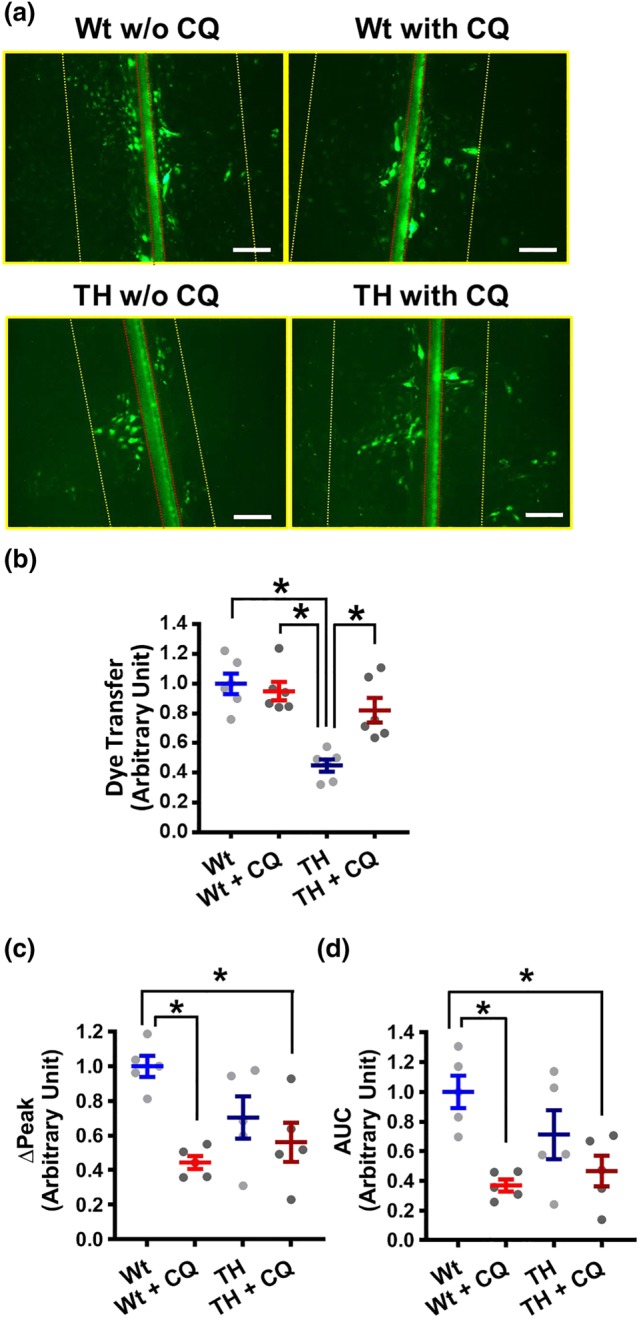
CQ increases gap junction activity but does not change the CPA‐induced [Ca2+]cyto increase, in MCECs isolated from TH mice. (a,b) Lucifer yellow dye transfer experiment in MCECs was conducted to examine the gap junction activity. (a) Representative photographs showing the images of LY dye transfer in ECs. The red line indicates the edge of the scrape. The yellow line indicates the farthest site of cells showing visual uptake of dye. The distance between yellow and red lines was calculated as a dye transfer. (b) The scattered dots show the normalized data of dye transfer by the distance in MCECs isolated from Wt without CQ (Wt‐MCECs w/o CQ). Bar = 100 μ. n mice = 6 in each group. (c,d) Summarized data showing ∆Peak (c) and the AUC (d) of CPA‐induced increase in [Ca2+]cyto in Wt MCECs w/o CQ (Wt, n mice = 5, n cells = 43), Wt MCECs with CQ (Wt + CQ, n mice = 5, n cells = 43), TH MCECs w/o CQ (TH, n mice = 5, n cells = 44), and TH MCECs with CQ (TH + CQ, n mice = 5, n cells = 44). *P < .05 between groups as indicated. Statistical comparison between the groups was made by one‐way ANOVA with Bonferroni post hoc test
3.6. Chloroquine pretreatment did not affect the increase in [Ca2+]cyto after stimulation in MCECs isolated from diabetic mice
To examine whether chloroquine differentially regulates Ca2+ influx between control and diabetes, we isolated MCECs from control and diabetic mice and conducted digital imaging fluorescence microscopy experiments to measure CPA‐induced [Ca2+]cyto changes in the presence or absence of chloroquine. CPA was used to induce SOCE as illustrated in Figure 2a. CPA‐induced [Ca2+]cyto increase was significantly decreased by chloroquine treatment in MCECs from control mice; however, there was no difference of CPA‐induced [Ca2+]cyto increase in the presence or absence of chloroquine in MCECs from diabetic mice (Figure 4c,d). These data suggest that enhanced EDH‐dependent relaxation in diabetic mice is due mainly to increased gap junction activity, but not to changing [Ca2+]cyto in MCECs.
4. DISCUSSION
The vascular response to the same agonist is not always the same among different organs and branches of vessels. Although there are several studies investigating the effect of chloroquine on vascular response (Manson et al., 2014; Pestana et al., 2015; Sai et al., 2014; Wu et al., 2017), there is no report so far that examines the effect of chloroquine on vasodilation in CAs isolated from healthy and diabetic mice. Muscarinic agonist (e.g. ACh) and https://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=4 induce EDR in all types of vessels; however, EDR in the large proximal artery is caused primarily by releasing NO from the endothelium, while EDH‐dependent vasodilation is prominent in small distal artery (Garland & Dora, 2017; Godo et al., 2016) because of more myoendothelial gap junctions in smaller vessels than in large vessels (Hill, Rummery, Hickey, & Sandow, 2002; Sandow & Hill, 2000; Straub, Zeigler, & Isakson, 2014). In this study, we used small CAs (third order), which enabled us to examine both EDH‐dependent and EDNO‐dependent vascular relaxation. Changes in EDH‐dependent relaxation in small distal arteries also play a critical role under pathophysiological conditions (i.e., coronary microvascular disease). In control mice, chloroquine attenuated EDR in CAs via inhibition of EDNO‐dependent relaxation (instead of EDH‐dependent relaxation; Figure 1). Both SNP and Pap lead to vascular relaxation by increasing cGMP in SMCs, and chloroquine did not affect SNP‐ and Pap‐induced vascular relaxation, suggesting that chloroquine attenuates NO‐dependent relaxation by altering EC function without changing SMC function. The chloroquine‐mediated inhibition of EDNO‐dependent relaxation could result from reduced NO production or decreased NO bioavailability in ECs. Figure 2b shows that NO concentration in HCECs is significantly decreased by chloroquine treatment. In our pilot study, we measured cytosolic ROS using DHE staining (Makino, Scott, & Dillmann, 2010) and found no change of DHE intensity after the treatment of chloroquine (Control, 9.97 ± 1.50 [n cell = 135, n experiment = 12]; chloroquine, 8.55 ± 1.46 [n cell = 122, n experiment = 12]; P > .05). These data suggest that the chloroquine ‐induced attenuation of EDNO‐dependent relaxation is more likely due to the decrease in NO production.
eNOS activation is regulated by phosphorylation and cofactors binding. Several phosphorylation sites in eNOS differentially regulate the activity of eNOS. Among those sites, Ser1177 and Thr495 are the two most studied phosphorylation sites in eNOS (Rafikov et al., 2011). Ser1177 is not phosphorylated under resting condition, and a rise of [Ca2+]cyto phosphorylates Ser1177 via CaMKII activation and increases NO production. On the other hand, Thr495 is a negative regulatory site for which phosphorylation leads to a decrease in eNOS activity. Chloroquine treatment in ECs significantly increased p‐eNOSThr495 without changing p‐eNOSSer1177 (Figure 2c), suggesting that NO production might be reduced by increasing p‐eNOSThr495 level. After binding of ACh to the muscarinic receptor, [Ca2+]cyto is increased through Ca2+ release from the endoplasmic reticulum, which activates eNOS via Ca2+/calmodulin binding. Therefore, inhibition of agonist‐induced increase in [Ca2+]cyto causes the reduction of NO production. In this study, we used CPA to increase [Ca2+]cyto by depleting intracellular Ca2+ stores and causing SOCE. The number of ACh receptor in ECs is often decreased each day after the isolation, and it is difficult to get homogenous response to ACh from isolated ECs (Tracey & Peach, 1992). However, isolated MCECs respond to CPA and lead to SOCE in all cells (Estrada et al., 2012). We found that chloroquine significantly inhibited CPA‐induced rise in [Ca2+]cyto (Figure 2d–f). The data in Figure 2 suggest that chloroquine attenuates NO production via reducing eNOS activity through increasing p‐eNOSThr495 and inhibiting the rise of [Ca2+]cyto. However, we do not know how chloroquine phosphorylates Thr495 in eNOS or how chloroquine decreases Ca2+ influx. Further studies are needed to define the mechanisms involved in chloroquine‐mediated phosphorylation of p‐eNOSThr495 and decrease in SOCE.
Change of [Ca2+]cyto may also affect EDH‐dependent vascular relaxation via reducing activation of SKCa channel and IKCa channels. However, we did not see a decrease in EDH‐dependent relaxation by chloroquine treatment; we actually observed a slight increase of relaxation in control mice (Figure 1d). In the absence of extracellular Ca2+ or in the presence of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2514 (a selective blocker of L‐type voltage‐dependent Ca2+ channels), ACh can still evoke membrane hyperpolarization in SMCs (Chen & Suzuki, 1990; Fukao, Hattori, Kanno, Sakuma, & Kitabatake, 1997). In addition, some endothelium‐derived hyperpolarization factors (EDHFs) can directly relax smooth muscle without activating SKCa and IKCa channels in ECs (Campbell & Fleming, 2010; O'Sullivan, Kendall, & Randall, 2004; Zhang et al., 2012). Therefore, it is possible that decreased CPA‐induced rise in [Ca2+]cyto after chloroquine treatment might not be sufficient to attenuate EDH‐dependent relaxation in control mice.
Diabetic patients exhibit a higher morbidity of malaria infection (Danquah, Bedu‐Addo, & Mockenhaupt, 2010); however, less information is available about the effect of the antimalarial drug on cardiovascular complications in diabetes. Major diabetic complications associated with morbidity are coronary artery and coronary microvascular disease. Ischaemia due to coronary artery disease is caused by occlusion or narrowing vessels as a result of lipid plaque formation, whereas the patient with coronary microvascular disease shows a clear angiograph with reduced coronary flow due to attenuated relaxation in the distal artery and reduced number of capillaries. Coronary endothelial dysfunction is implicated in the development of both coronary macrovascular and microvascular diseases (Belin de Chantemele & Stepp, 2012; Labazi & Trask, 2017; Pant et al., 2014; Pepine et al., 2015; Rocic, 2012). Therefore, we examined the effect of chloroquine on coronary endothelial function in control and diabetic mice. EDR, but not smooth muscle‐dependent relaxation, was significantly attenuated in CAs of diabetic mice compared to CAs of control mice (Figure 3b,e). Interestingly, while chloroquine attenuated EDR in CAs from control mice (Figure 3c), it enhanced EDR in CAs isolated from diabetic mice (Figure 3d). The enhanced EDR in CAs from diabetic mice might be due to augmented EDH‐dependent relaxation (Figure 3g).
EDR is usually caused by EDH and/or endothelium‐dependent NO release. EDH‐dependent relaxation is initiated by membrane hyperpolarization in ECs that can evoke SMC hyperpolarization by intercellular communication through gap junctions. EDH‐dependent relaxation can also be induced by releasing EDHFs (e.g., EETs, H2O2, and anandamide). We and other investigators demonstrate that the activity of endothelial gap junction is attenuated in diabetes (Makino et al., 2008; Makino et al., 2015; Roy, Jiang, Li, & Kim, 2017). Figure 3k demonstrates that pretreatment of 18GA (a gap junction inhibitor) significantly inhibits chloroquine‐induced augmentation of EDH‐dependent vascular relaxation in diabetic mice. These data suggest that increased gap junction activity is involved in enhanced EDH‐dependent relaxation by chloroquine in diabetes. Furthermore, we directly measured gap junction activity and found that chloroquine does increase gap junction activity in diabetic MCECs (Figure 4a,b). Gap junctions are cell membrane channels made of connexin (Cx) proteins. ECs predominantly express Cx40, Cx37, and Cx43 (along with Cx45 and Cx32 in some organs; Brisset, Isakson, & Kwak, 2009; Cai et al., 2001; Esseltine & Laird, 2016; Figueroa & Duling, 2009; Hill et al., 2002; Hong & Hill, 1998; Okamoto et al., 2009; Sohl & Willecke, 2004; Yeh, Dupont, Coppen, Rothery, & Severs, 1997). We have observed that Cx40 is highly expressed in coronary ECs in control mice but significantly down‐regulated in diabetic ECs and the down‐regulation of Cx40 is implicated in endothelial dysfunction in type 1 diabetes (Makino et al., 2008).
The t 1/2 of Cxs, which is much shorter (1–5 hr) than other plasma membrane proteins, can be shortened or delayed by chemical treatments (Falk, Kells, & Berthoud, 2014). The degradation process of Cxs involves in autophagosome, where chloroquine tends to accumulate and inhibits various enzyme activities (Mauthe et al., 2018). It is thus possible that chloroquine will change Cx levels in ECs by altering Cx degradation. Indeed, Chen, Guo, et al. (2017) demonstrate that chloroquine attenuates the degradation of Cx40 in rats with brain injury. In this study, we treated CAs and MCECs with chloroquine only for 30 min. The short‐term treatment with chloroquine may not be sufficient to affect the total amount of Cx proteins but may be enough to maintain Cxs in the plasma membrane (by reducing degradation and/or increasing trafficking) to increase the activity of gap junction. Further studies are required to examine the effects of chloroquine on Cx trafficking and degradation.
EDH‐dependent vascular relaxation can be enhanced if gap junction activity is augmented and/or if ECs are more hyperpolarized by increasing activity of SKCa and IKCa channels. In this study, we did not measure membrane potential and the activity of SKCa and IKCa channels in MCECs. CPA‐induced [Ca2+]cyto increase was significantly decreased by chloroquine treatment in control mice; however, chloroquine did not affect the response to CPA in diabetic MCECs (Figure 4c,d). These data imply that the activities of SKCa and IKCa channels were not altered by chloroquine treatment in diabetic MCECs. Even if chloroquine increases SKCa and IKCa channel activity in diabetic MCECs, this beneficial effect of chloroquine might not be translated well into vascular relaxation without improving gap junction activity by chloroquine. In Figure 4a,b, the activity of gap junction was significantly attenuated in MCECs of diabetic mice and chloroquine treatment restored gap junction activity. Therefore, increased EDH‐dependent vascular relaxation by chloroquine in diabetic mice is likely due to restored gap junction activity by chloroquine in diabetic MCECs. In type 1 diabetic mice, we also observed that gap junction activity was attenuated in MCECs and the recovery of gap junction activity restored attenuated vascular relaxation (Makino et al., 2015). Therefore, the increase in gap junction activity by chloroquine would be an efficient way to restore vascular relaxation in diabetic mice.
Chloroquine is also believed to stimulate bitter taste receptor (TAS2R) and leads to vascular relaxation (Chen, Ping, et al., 2017; Manson et al., 2014; Sai et al., 2014). Chloroquine is known as an agonist of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=117#660, TAS2R10, and TAS2R39 (Upadhyaya et al., 2014); therefore, it is natural to think that chloroquine regulates vascular tone through the activation of TAS2Rs. However, there is no study investigating whether the deletion of TAS2R inhibits chloroquine‐induced vascular response. In addition, chloroquine‐induced vascular relaxation is observed only at a high concentration (>10−4 M). This concentration is higher than the physiological concentration, which we could detect in the body after chloroquine treatment for malaria. Many chemicals could directly contract or relax vessels at the high concentration. Due to uncertain mechanisms of chloroquine‐mediated vascular response, we did not examine the direct effect of chloroquine, through this receptor, on coronary arterial response in current study.
In this study, we demonstrate that chloroquine attenuates EDR in CAs isolated from control mice but chloroquine enhances EDR in CAs from diabetic mice. The different outcome might result from the altered endothelial function in diabetic mice. The chloroquine ‐mediated attenuation of EDR, which was observed in CAs from control mice, is due partly to a decrease in endothelial NO production. The chloroquine‐mediated increase in EDR is mainly due to the enhancement of EDH‐dependent relaxation in diabetic CAs as a result of chloroquine‐increased gap junction activity. The observations from this study indicate that chloroquine should be used with caution in patients with non‐diabetic cardiovascular disease due to an adverse effect of chloroquine on coronary vascular function.
AUTHOR CONTRIBUTIONS
Q.Z. conducted the experiments, analysed the data and drafted and reviewed the manuscript. A.T., C.W., M.W., R.S., N.L., and Z.W. conducted the experiments and reviewed the manuscript. J.Y. and J.W. reviewed and edited the manuscript. A.M. conceived the project, designed the experiments, analysed the data, and reviewed and edited the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENT
This work was supported by a grant from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL142214 and HL146764 to A. Makino).
Zhang Q, Tsuji‐Hosokawa A, Willson C, et al. Chloroquine differentially modulates coronary vasodilation in control and diabetic mice. Br J Pharmacol. 2020;177:314–327. 10.1111/bph.14864
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin de Chantemele, E. J. , & Stepp, D. W. (2012). Influence of obesity and metabolic dysfunction on the endothelial control in the coronary circulation. Journal of Molecular and Cellular Cardiology, 52, 840–847. 10.1016/j.yjmcc.2011.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazar, B. R. , Whitley, C. B. , Kitabchi, A. E. , Tsai, M. Y. , Santiago, J. , White, N. , … Brown, D. M. (1984). In vivo chloroquine‐induced inhibition of insulin degradation in a diabetic patient with severe insulin resistance. Diabetes, 33, 1133–1137. 10.2337/diab.33.12.1133 [DOI] [PubMed] [Google Scholar]
- Brisset, A. C. , Isakson, B. E. , & Kwak, B. R. (2009). Connexins in vascular physiology and pathology. Antioxidants & Redox Signaling, 11, 267–282. 10.1089/ars.2008.2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, W. J. , Koltai, S. , Kocsis, E. , Scholz, D. , Schaper, W. , & Schaper, J. (2001). Connexin37, not Cx40 and Cx43, is induced in vascular smooth muscle cells during coronary arteriogenesis. Journal of Molecular and Cellular Cardiology, 33, 957–967. 10.1006/jmcc.2001.1360 [DOI] [PubMed] [Google Scholar]
- Campbell, W. B. , & Fleming, I. (2010). Epoxyeicosatrienoic acids and endothelium‐dependent responses. Pflügers Archiv, 459, 881–895. 10.1007/s00424-010-0804-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G. F. , & Suzuki, H. (1990). Calcium dependency of the endothelium‐dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. The Journal of Physiology, 421, 521–534. 10.1113/jphysiol.1990.sp017959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. G. , Ping, N. N. , Liang, D. , Li, M. Y. , Mi, Y. N. , Li, S. , … Cao, Y. X. (2017). The expression of bitter taste receptors in mesenteric, cerebral and omental arteries. Life Sciences, 170, 16–24. 10.1016/j.lfs.2016.11.010 [DOI] [PubMed] [Google Scholar]
- Chen, W. , Guo, Y. , Yang, W. , Zheng, P. , Zeng, J. , & Tong, W. (2017). Involvement of autophagy in connexin 40 reduction in the late phase of traumatic brain injury in rats. Brain Research Bulletin, 131, 100–106. 10.1016/j.brainresbull.2017.03.014 [DOI] [PubMed] [Google Scholar]
- Cho, Y. E. , Basu, A. , Dai, A. , Heldak, M. , & Makino, A. (2013). Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. American Journal of Physiology. Cell Physiology, 305, C1033–C1040. 10.1152/ajpcell.00234.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danquah, I. , Bedu‐Addo, G. , & Mockenhaupt, F. P. (2010). Type 2 diabetes mellitus and increased risk for malaria infection. Emerging Infectious Diseases, 16, 1601–1604. 10.3201/eid1610.100399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, T. J. (2001). Structure‐function relationships in chloroquine and related 4‐aminoquinoline antimalarials. Mini Reviews in Medicinal Chemistry, 1, 113–123. 10.2174/1389557013407188 [DOI] [PubMed] [Google Scholar]
- Esseltine, J. L. , & Laird, D. W. (2016). Next‐generation connexin and pannexin cell biology. Trends in Cell Biology, 26, 944–955. 10.1016/j.tcb.2016.06.003 [DOI] [PubMed] [Google Scholar]
- Estrada, I. A. , Donthamsetty, R. , Debski, P. , Zhou, M. H. , Zhang, S. L. , Yuan, J. X. , … Makino, A. (2012). STIM1 restores coronary endothelial function in type 1 diabetic mice. Circulation Research, 111, 1166–1175. 10.1161/CIRCRESAHA.112.275743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, M. M. , Kells, R. M. , & Berthoud, V. M. (2014). Degradation of connexins and gap junctions. FEBS Letters, 588, 1221–1229. 10.1016/j.febslet.2014.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa, X. F. , & Duling, B. R. (2009). Gap junctions in the control of vascular function. Antioxidants & Redox Signaling, 11, 251–266. 10.1089/ars.2008.2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, K. Y. , & Wright, D. W. (2013). Hemozoin and antimalarial drug discovery. Future Medicinal Chemistry, 5, 1437–1450. 10.4155/fmc.13.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao, M. , Hattori, Y. , Kanno, M. , Sakuma, I. , & Kitabatake, A. (1997). Sources of Ca2+ in relation to generation of acetylcholine‐induced endothelium‐dependent hyperpolarization in rat mesenteric artery. British Journal of Pharmacology, 120, 1328–1334. 10.1038/sj.bjp.0701027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland, C. J. , & Dora, K. A. (2017). EDH: Endothelium‐dependent hyperpolarization and microvascular signalling. Acta Physiologica (Oxford, England), 219, 152–161. 10.1111/apha.12649 [DOI] [PubMed] [Google Scholar]
- Godo, S. , Sawada, A. , Saito, H. , Ikeda, S. , Enkhjargal, B. , Suzuki, K. , … Shimokawa, H. (2016). Disruption of physiological balance between nitric oxide and endothelium‐dependent hyperpolarization impairs cardiovascular homeostasis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 36, 97–107. 10.1161/ATVBAHA.115.306499 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, C. E. , Rummery, N. , Hickey, H. , & Sandow, S. L. (2002). Heterogeneity in the distribution of vascular gap junctions and connexins: Implications for function. Clinical and Experimental Pharmacology & Physiology, 29, 620–625. 10.1046/j.1440-1681.2002.03699.x [DOI] [PubMed] [Google Scholar]
- Hong, T. , & Hill, C. E. (1998). Restricted expression of the gap junctional protein connexin 43 in the arterial system of the rat. Journal of Anatomy, 192(Pt 4), 583–593. 10.1046/j.1469-7580.1998.19240583.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarzyna, R. , Kiersztan, A. , Lisowa, O. , & Bryla, J. (2001). The inhibition of gluconeogenesis by chloroquine contributes to its hypoglycaemic action. European Journal of Pharmacology, 428, 381–388. 10.1016/S0014-2999(01)01221-3 [DOI] [PubMed] [Google Scholar]
- Jeong, H. Y. , Kang, J. M. , Jun, H. H. , Kim, D. J. , Park, S. H. , Sung, M. J. , … Lee, S. Y. (2018). Chloroquine and amodiaquine enhance AMPK phosphorylation and improve mitochondrial fragmentation in diabetic tubulopathy. Scientific Reports, 8, 8774 10.1038/s41598-018-26858-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia, S. , & Dutz, J. P. (2007). New concepts in antimalarial use and mode of action in dermatology. Dermatologic Therapy, 20, 160–174. 10.1111/j.1529-8019.2007.00131.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , & Saxton, A. M. (2012). The TALLYHO mouse as a model of human type 2 diabetes. Methods in Molecular Biology, 933, 75–87. 10.1007/978-1-62703-068-7_6 [DOI] [PubMed] [Google Scholar]
- Kutner, S. , Breuer, W. V. , Ginsburg, H. , & Cabantchik, Z. I. (1987). On the mode of action of phlorizin as an antimalarial agent in in vitro cultures of Plasmodium falciparum. Biochemical Pharmacology, 36, 123–129. 10.1016/0006-2952(87)90389-3 [DOI] [PubMed] [Google Scholar]
- Labazi, H. , & Trask, A. J. (2017). Coronary microvascular disease as an early culprit in the pathophysiology of diabetes and metabolic syndrome. Pharmacological Research, 123, 114–121. 10.1016/j.phrs.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, B. , & Tjalve, H. (1979). Studies on the mechanism of drug‐binding to melanin. Biochemical Pharmacology, 28, 1181–1187. 10.1016/0006-2952(79)90326-5 [DOI] [PubMed] [Google Scholar]
- Luo, S. , Truong, A. H. , & Makino, A. (2016). Isolation of mouse coronary endothelial cells. Journal of Visualized Experiments, 113, e53985 10.3791/53985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie, A. H. (1983). Dose refinements in long‐term therapy of rheumatoid arthritis with antimalarials. The American Journal of Medicine, 75, 40–45. 10.1016/0002-9343(83)91269-X [DOI] [PubMed] [Google Scholar]
- Makino, A. , Dai, A. , Han, Y. , Youssef, K. D. , Wang, W. , Donthamsetty, R. , … Dillmann, W. H. (2015). O‐GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. American Journal of Physiology. Cell Physiology, 309, C593–C599. 10.1152/ajpcell.00069.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino, A. , Platoshyn, O. , Suarez, J. , Yuan, J. X. , & Dillmann, W. H. (2008). Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin‐induced diabetic mice. American Journal of Physiology. Cell Physiology, 295, C221–C230. 10.1152/ajpcell.00433.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino, A. , Scott, B. T. , & Dillmann, W. H. (2010). Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia, 53, 1783–1794. 10.1007/s00125-010-1770-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson, M. L. , Safholm, J. , Al‐Ameri, M. , Bergman, P. , Orre, A. C. , Sward, K. , … Adner, M. (2014). Bitter taste receptor agonists mediate relaxation of human and rodent vascular smooth muscle. European Journal of Pharmacology, 740, 302–311. 10.1016/j.ejphar.2014.07.005 [DOI] [PubMed] [Google Scholar]
- Mauthe, M. , Orhon, I. , Rocchi, C. , Zhou, X. , Luhr, M. , Hijlkema, K. J. , … Reggiori, F. (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome‐lysosome fusion. Autophagy, 14, 1435–1455. 10.1080/15548627.2018.1474314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, T. , Akiyama, M. , Takeda, M. , Gabazza, E. C. , Hayashi, T. , & Suzuki, K. (2009). Connexin32 is expressed in vascular endothelial cells and participates in gap‐junction intercellular communication. Biochemical and Biophysical Research Communications, 382, 264–268. 10.1016/j.bbrc.2009.02.148 [DOI] [PubMed] [Google Scholar]
- O'Sullivan, S. E. , Kendall, D. A. , & Randall, M. D. (2004). Heterogeneity in the mechanisms of vasorelaxation to anandamide in resistance and conduit rat mesenteric arteries. British Journal of Pharmacology, 142, 435–442. 10.1038/sj.bjp.0705810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant, R. , Marok, R. , & Klein, L. W. (2014). Pathophysiology of coronary vascular remodeling: Relationship with traditional risk factors for coronary artery disease. Cardiology in Review, 22, 13–16. 10.1097/CRD.0b013e31829dea90 [DOI] [PubMed] [Google Scholar]
- Pepine, C. J. , Ferdinand, K. C. , Shaw, L. J. , Light‐McGroary, K. A. , Shah, R. U. , Gulati, M. , … ACC CVD in Women Committee (2015). Emergence of nonobstructive coronary artery disease: A woman's problem and need for change in definition on angiography. Journal of the American College of Cardiology, 66, 1918–1933. 10.1016/j.jacc.2015.08.876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestana, C. R. , Oishi, J. C. , Salistre‐Araujo, H. S. , & Rodrigues, G. J. (2015). Inhibition of autophagy by chloroquine stimulates nitric oxide production and protects endothelial function during serum deprivation. Cellular Physiology and Biochemistry, 37, 1168–1177. 10.1159/000430240 [DOI] [PubMed] [Google Scholar]
- Powrie, J. K. , Shojaee‐Moradie, F. , Watts, G. F. , Smith, G. D. , Sonksen, P. H. , & Jones, R. H. (1993). Effects of chloroquine on the dyslipidemia of non‐insulin‐dependent diabetes mellitus. Metabolism, 42, 415–419. 10.1016/0026-0495(93)90096-7 [DOI] [PubMed] [Google Scholar]
- Rafikov, R. , Fonseca, F. V. , Kumar, S. , Pardo, D. , Darragh, C. , Elms, S. , … Black, S. M. (2011). eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. The Journal of Endocrinology, 210, 271–284. 10.1530/JOE-11-0083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, H. , & Campbell, A. A. (1962). Central scotomata following chloroquine therapy. Canadian Medical Association Journal, 86, 176–178. [PMC free article] [PubMed] [Google Scholar]
- Rees, R. B. , & Maibach, H. I. (1963). Chloroquine: A review of reactions and dermatologic indications. Archives of Dermatology, 88, 280–289. 10.1001/archderm.1963.01590210038006 [DOI] [PubMed] [Google Scholar]
- Rocic, P. (2012). Why is coronary collateral growth impaired in type II diabetes and the metabolic syndrome? Vascular Pharmacology, 57, 179–186. 10.1016/j.vph.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S. , Jiang, J. X. , Li, A. F. , & Kim, D. (2017). Connexin channel and its role in diabetic retinopathy. Progress in Retinal and Eye Research, 61, 35–59. 10.1016/j.preteyeres.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai, W. B. , Yu, M. F. , Wei, M. Y. , Lu, Z. , Zheng, Y. M. , Wang, Y. X. , … Liu, Q. H. (2014). Bitter tastants induce relaxation of rat thoracic aorta precontracted with high K+ . Clinical and Experimental Pharmacology & Physiology, 41, 301–308. 10.1111/1440-1681.12217 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Chapula, J. A. , Salinas‐Stefanon, E. , Torres‐Jacome, J. , Benavides‐Haro, D. E. , & Navarro‐Polanco, R. A. (2001). Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. The Journal of Pharmacology and Experimental Therapeutics, 297, 437–445. [PubMed] [Google Scholar]
- Sandow, S. L. , & Hill, C. E. (2000). Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium‐derived hyperpolarizing factor‐mediated responses. Circulation Research, 86, 341–346. 10.1161/01.RES.86.3.341 [DOI] [PubMed] [Google Scholar]
- Smith, G. D. , Amos, T. A. , Mahler, R. , & Peters, T. J. (1987). Effect of chloroquine on insulin and glucose homoeostasis in normal subjects and patients with non‐insulin‐dependent diabetes mellitus. British Medical Journal (Clinical Research Ed.), 294, 465–467. 10.1136/bmj.294.6570.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohl, G. , & Willecke, K. (2004). Gap junctions and the connexin protein family. Cardiovascular Research, 62, 228–232. 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Straub, A. C. , Zeigler, A. C. , & Isakson, B. E. (2014). The myoendothelial junction: Connections that deliver the message. Physiology (Bethesda), 29, 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tona, L. , Ng, Y. C. , Akera, T. , & Brody, T. M. (1990). Depressant effects of chloroquine on the isolated guinea‐pig heart. European Journal of Pharmacology, 178, 293–301. 10.1016/0014-2999(90)90108-I [DOI] [PubMed] [Google Scholar]
- Tracey, W. R. , & Peach, M. J. (1992). Differential muscarinic receptor mRNA expression by freshly isolated and cultured bovine aortic endothelial cells. Circulation Research, 70, 234–240. 10.1161/01.RES.70.2.234 [DOI] [PubMed] [Google Scholar]
- Upadhyaya, J. D. , Singh, N. , Sikarwar, A. S. , Chakraborty, R. , Pydi, S. P. , Bhullar, R. P. , … Chelikani, P. (2014). Dextromethorphan mediated bitter taste receptor activation in the pulmonary circuit causes vasoconstriction. PLoS ONE, 9, e110373 10.1371/journal.pone.0110373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, K. , Zhang, Q. , Wu, X. , Lu, W. , Tang, H. , Liang, Z. , … Wang, J. (2017). Chloroquine is a potent pulmonary vasodilator that attenuates hypoxia‐induced pulmonary hypertension. British Journal of Pharmacology, 174, 4155–4172. 10.1111/bph.13990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, H. I. , Dupont, E. , Coppen, S. , Rothery, S. , & Severs, N. J. (1997). Gap junction localization and connexin expression in cytochemically identified endothelial cells of arterial tissue. The Journal of Histochemistry and Cytochemistry, 45, 539–550. 10.1177/002215549704500406 [DOI] [PubMed] [Google Scholar]
- Yuan, X. , Xiao, Y. C. , Zhang, G. P. , Hou, N. , Wu, X. Q. , Chen, W. L. , … Zhang, G. S. (2016). Chloroquine improves left ventricle diastolic function in streptozotocin‐induced diabetic mice. Drug Design, Development and Therapy, 10, 2729–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. X. , Borbouse, L. , Gebremedhin, D. , Mendoza, S. A. , Zinkevich, N. S. , Li, R. , & Gutterman, D. D. (2012). H2O2‐induced dilation in human coronary arterioles: Role of protein kinase G dimerization and large‐conductance Ca2+‐activated K+ channel activation. Circulation Research, 110, 471–480. 10.1161/CIRCRESAHA.111.258871 [DOI] [PMC free article] [PubMed] [Google Scholar]