

Abstract
Background and Purpose
Chronic inflammation may play a role in the pathogenesis of Parkinson's disease (PD). Noradrenaline is an endogenous neurotransmitter with anti‐inflammatory properties. In the present investigation, we assessed the immunomodulatory and neuroprotective efficacy of pharmacologically targeting the CNS noradrenergic system in a rat model of PD.
Experimental Approach
The impact of treatment with the β2‐adrenoceptor agonists clenbuterol and formoterol was assessed in the intranigral LPS rat model of PD. The immunomodulatory potential of formoterol to influence the CNS response to systemic inflammation was also assessed.
Key Results
LPS‐induced deficits in motor function (akinesia and forelimb‐use asymmetry) and nigrostriatal dopamine loss were rescued by both agents. Treatment with the noradrenaline reuptake inhibitor atomoxetine reduced striatal dopamine loss and motor deficits following intranigral LPS injection. Co‐treatment with the β2‐adrenoceptor antagonist ICI 118,551 attenuated the protective effects of atomoxetine. Systemic LPS challenge exacerbated reactive microgliosis, IL‐1β production, dopamine cell loss in the substantia nigra, nerve terminal degeneration in the striatum, and associated motor impairments in animals that previously received intranigral LPS. This exacerbation was attenuated by formoterol treatment.
Conclusion and Implications
The results indicate that pharmacologically targeting β2‐adrenoceptors has the propensity to regulate the neuroinflammatory phenotype in vivo and may be a potential neuroprotective strategy where inflammation contributes to the progression of dopaminergic neurodegeneration. In accordance with this, clinical agents such as β2‐adrenoceptor agonists may prove useful as immunomodulatory agents in the treatment of neurodegenerative conditions associated with brain inflammation.
Abbreviations
- CD68
cluster of differentiation 68
- DSP4
N‐(2‐chloroethyl)‐N‐ethyl‐2‐bromobenzylamine
- Iba1
ionised calcium binding adaptor molecule 1
- IL‐1RA
IL‐1 receptor antagonist
- LC
locus coeruleus
- PD
Parkinson's disease
- SN
substantia nigra
- 6‐OHDA
6‐hydroxydopamine
What is already known
Noradrenaline exerts putative anti‐inflammatory effects in the CNS via activating β2‐adrenoceptors.
The selective β2‐adrenoceptor agonist formoterol is currently prescribed for the treatment of asthma in humans.
What this study adds
Treatment with formoterol inhibits microglial activation and attenuates the loss of nigrostriatal dopamine neurons.
Formoterol prevents the progression of dopamine loss and motor dysfunction in response to systemic inflammation.
What is the clinical significance
Pharmacologically targeting β2‐adrenoceptors for immunomodulation may confer disease‐modifying effects in the treatment of Parkinson's disease.
1. INTRODUCTION
Progressive degeneration of dopaminergic neurons projecting from the substantia nigra (SN) is a pathological hallmark of Parkinson's disease (PD) associated with motor impairment (Dickson, 2012). Microglial activation and release of pro‐inflammatory cytokines promote oxidative stress and toxicity are widely considered to contribute to the degeneration of the nigrostriatal tract (McGeer & McGeer, 2004; Tansey & Goldberg, 2010). Inflammatory‐based models of PD such as intranigral administration of the bacterial endotoxin and immune stimulus LPS simulate this process and provoke degeneration of dopamine neurons in the SN (Hoban et al., 2013; Yssel, O'Neill, Nolan, Connor, & Harkin, 2018).
Previous reports have indicated that the endogenous catecholaminergic transmitter noradrenaline suppresses the inflammatory response and production of neurotoxic substances including https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974, and https://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=nitric+oxide&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&submit=Search+Database in the brain (Feinstein et al., 2002; McNamee, Ryan, Kilroy, & Connor, 2010; Ryan et al., 2013). Moreover, noradrenaline negatively regulates the IL‐1 system in glial cells via up‐regulation of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5878 (IL‐1RA) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2321 decoy receptor in vitro (McNamee, Ryan, Kilroy, & Connor, 2010) and in vivo (McNamee, Griffin, Ryan, et al., 2010) and raises CNS expression of the broad‐spectrum anti‐inflammatory cytokine https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4975 and its downstream signalling molecule suppressor of cytokine signalling 3 (McNamee, Ryan, Griffin, et al., 2010). Noradrenaline reuptake inhibitors inhibit microglial activation and production of inflammatory mediators TNF‐α and NO and inhibit activation of the inflammatory transcription factor https://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=NF-κB&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&submit=Search+Database in rat brain (O'Sullivan, Ryan, Curtin, Harkin, & Connor, 2009). More recently, we have determined that treatment of rats with the noradrenaline reuptake inhibitor https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7118 yields a robust anti‐inflammatory effect, suppressing microglial activation and TNF‐α expression, whilst preventing loss in TH positive (TH+) dopamine neurons and resulting in functional improvement in motor deficits in the LPS inflammatory model of PD in rats (Yssel et al., 2018). These results have confirmed that pharmacological enhancement of noradrenergic tone has the propensity to regulate the neuroinflammatory phenotype in vivo and act as an endogenous neuroprotective mechanism where inflammation contributes to the progression of dopamine loss.
The β2 https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29&familyId=4&familyType=GPCR is a G protein‐coupled noradrenaline receptor subtype expressed post‐synaptically on neurons and also on microglial and astrocytic cells (Tanaka, Kashima, Suzuki, Ono, & Sawada, 2002). In the periphery, activation of β2‐adrenoceptors leads to relaxation of smooth muscles in the lungs, bronchodilation and vasodilation of blood vessels (Barisione, Baroffio, Crimi & Brusasco, 2010; Guimaraes and Moura, 2001). Stimulation of β2‐adrencptors (a) activates adenylyl cyclase which raises intracellular cAMP leading to (b) activation of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=284 which in turn phosphorylates cAMP‐response element binding protein and (c) induces de novo synthesis of the NF‐κB inhibitory protein IκBα (and possibly preventing its phosphorylation), thus stabilising cytosolic levels of IκBα which (d) inhibits transcriptional activity of NF‐κB by preventing its translocation into the nucleus, ultimately (e) decreasing pro‐inflammatory gene expression. Downstream signalling through β2‐adrenoceptors inhibits microglial activation and reduces pro‐inflammatory cytokine production (Farmer & Pugin, 2000; Kin & Sanders, 2006; van der Poll, Jansen, Endert, Sauerwein, & van Deventer, 1994). The β2‐adrenoceptor agonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=559 elicits anti‐inflammatory and neuroprotective effects in the CNS and inhibits dopamine neuron loss induced by LPS in vitro and in vivo (Qian et al., 2011). Clenbuterol is a brain penetrant β2‐adrenoceptor agonist used in the treatment of respiratory disorders including asthma and chronic obstructive pulmonary disease (Baronti, Grieco, & Vibelli, 1980; Boner et al., 1988) and has been shown to have neuroprotective properties both in vitro and in vivo by reducing apoptosis induced by the excitotoxin, kainic acid (Gleeson, Ryan, Griffin, Connor, & Harkin, 2010; Semkova, Schilling, Henrich‐Noack, Rami, & Krieglstein, 1996), in rodent models of cerebral ischaemia (Culmsee et al., 2004; Culmsee, Stumm, Schäfer, Weihe, & Krieglstein, 1999; Junker et al., 2002; Semkova et al., 1996; Zhu, Culmsee, Semkova, & Krieglstein, 1998) and in a murine model of motor neurone disease (Teng et al., 2006). Pretreatment with clenbuterol attenuates expression of the pro‐inflammatory molecules IL‐1β, IFN‐γ, and inducible NOS in the kainic acid model of excitotoxicity (Gleeson et al., 2010) and suppresses NF‐κB activity and expression of the NF‐κB‐inducible genes TNF‐α and ICAM‐1 in response to bacterial LPS (i.c.v.) in rats, whilst concurrently elevating expression of the NF‐κB‐inhibitory protein IκBα (Ryan et al., 2013). https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3465 is a long‐acting, highly selective β2‐adrenoceptor agonist which, in conjunction with the anti‐inflammatory corticosteroid https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7434, is in clinical use as an FDA‐approved metred dose inhaler for the treatment of asthma and airflow obstruction in patients with chronic obstructive pulmonary disease (Rosenhall et al., 2002). Formoterol has a rapid onset of action and acts locally as a long‐acting bronchodilator with anti‐inflammatory effects on mast cells in the lungs. It has a minimal adverse effects profile and is safe to use for prolonged periods of time in humans (Rosenhall et al., 2003). Due to its lipophilicity and high selectivity as a β2‐adrenoceptor agonist, formoterol is blood–brain barrier permeant and can enter the CNS parenchyma to exert potent immunomodulatory effects on glial cells with potential for disease modifying outcomes where inflammation is implicated.
In the present investigation, the immunomodulatory and neuroprotective efficacy of pharmacologically targeting β2‐adrenoceptors directly with both clenbuterol and formoterol, to inhibit microglial activation and protect against the onset and development of dopamine loss and motor deficits in response to intranigral LPS, was investigated. Microglia are susceptible to react more vigorously to a subsequent immune stimulus of systemic origin with indelible neurotoxic consequences (Perry & Holmes, 2014), and as such, the ability of formoterol to influence the CNS response to a subsequent systemic LPS stimulus was also investigated.
2. METHODS
2.1. Animals
All animal experiments were conducted in compliance with the European directive 2010/63/EU on the protection of animals used for scientific purposes, approved by the Animal Research Ethics Committee in Trinity College Dublin and performed under licence granted by the Health Products Regulatory Authority. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Male Wistar rats aged 6–8 weeks (220–250 g) were obtained from the Comparative Medicine Unit (TCD) and housed two per cage in hard‐bottomed polypropylene cages with stainless steel wire tops and wood shaving used as bedding. Animals were kept in climate‐controlled rooms set to 21°C (±2°C) with relative humidity levels of 50%, on a 12:12‐hr light–dark cycle and were allowed food and water ad libitum.
2.2. Experimental design
2.2.1. Experiment 1: Anti‐inflammatory and neuroprotective effects of clenbuterol or formoterol following intranigral LPS administration in rats
Adult male Wistar rats (n = 60) were allocated randomly into the following six treatment groups: (a) vehicle + vehicle, (b) vehicle + clenbuterol, (c) vehicle + formoterol, (d) LPS + vehicle, (e) LPS + clenbuterol, and (f) LPS + formoterol n = 10 per group. All experiments were designed to generate treatment groups of equal size using randomisation and blinded analysis. Sample size estimation was conducted via power analysis (GPower 3.1) from previous data sets. Rats received a unilateral stereotaxic injection of LPS (10 μg/2 μl) or vehicle (sterile PBS) into the SN. Four hours following surgery, rats received an i.p. injection of the β2‐adrenoceptor agonist clenbuterol (100 μg·kg−1), formoterol (100 μg·kg−1), or saline vehicle (0.89% [w/v] NaCl) and then once daily (q.d.) thereafter for 7 days. Behavioural testing in the staircase, stepping, and cylinder tests were conducted at 7 and 13 days post‐LPS. On the following day, rats were killed either by transcardial perfusion fixation for post mortem immunohistochemical analysis of the brain or decapitated followed by brain retrieval, dissection, and subsequent analysis of midbrain and striatal tissue dopamine concentrations.
Drugs are administered for 7 days after intranigral LPS administration to test their effect on microglial activation, the onset of dopaminergic neuropathology, and the development of motor deficits in the model. This is consistent with the treatment regimen described in previous published work from the group to date in respect of targeting the noradrenergic system in the LPS rat model of PD (Yssel et al., 2018). Furthermore, cessation of formoterol treatment after 7 days allows for the assessment of long‐withstanding anti‐inflammatory and neuroprotective action in the absence of acutely administered drug. This, in turn, further confirms the anti‐inflammatory and neuroprotective potential of treatment in rats exhibiting PD‐related neuropathology and motor deficits.
2.2.2. Experiment 2: Neuroprotective effects of atomoxetine following intranigral LPS administration in rats; impact of pretreatment with the β2‐AR antagonist ICI 118,551
Previously, we reported anti‐inflammatory and neuroprotective properties of atomoxetine in the LPS rat model of PD (Yssel et al., 2018). A role for the β2‐adrenoceptor in mediating these effects is assessed in support of specifically targeting this receptor in the model. Adult male Wistar rats (n = 50) were allocated randomly into the following five treatment groups: (a) vehicle + vehicle, (b) LPS + vehicle, (c) LPS + atomoxetine, (d) LPS + ICI 118,551, and (e) LPS + atomoxetine/ICI 118,551 n = 10 per group. Sample size estimation was conducted via power analysis (GPower 3.1) from previous data sets. Rats received a unilateral stereotaxic injection of LPS (10 μg/2 μl) or vehicle (sterile PBS) into the SN. Four hours following surgery, rats received an i.p. injection of atomoxetine (3 mg·kg−1), the β2‐AR antagonist ICI 118,551 (5 mg·kg−1), a combination of atomoxetine + ICI 118,551, or saline vehicle (0.89% [w/v] NaCl) and then twice a day (b.i.d.) thereafter for 7 days. ICI 118,551/vehicle was administered 30 min before atomoxetine. Two weeks following LPS administration, animals were killed by decapitation and brain retrieved and dissected for subsequent analysis of tissue dopamine concentrations.
2.2.3. Experiment 3: Effect of formoterol on intranigral LPS‐induced neuropathology and motor dysfunction following subsequent systemic LPS administration
Adult male Wistar rats (n = 48) were randomly assorted into eight treatment groups via block randomisation to prevent bias and achieve balance in the allocation of rats to their respective treatment groups: (a) Sham + vehicle + saline, (b) Sham + LPS + saline, (c) LPS + vehicle + saline, (d) LPS + LPS + saline, (e) Sham + vehicle + formoterol, (f) Sham + LPS + formoterol, (g) LPS + vehicle + formoterol, and (h) LPS + LPS + formoterol n = 8 per group. Sample size estimation was conducted via power analysis (GPower 3.1) from previous data sets. Rats received a unilateral intranigral injection of LPS (10 μg/2 μl) or vehicle (2‐μl 0.89% sterile saline). Rats were subsequently challenged with bacterial LPS (250 μg·kg−1 i.p.) or vehicle control (sterile saline) 4 weeks post‐intranigral LPS administration (Day 28). Treatment with formoterol (100 μg·kg−1 i.p.) or saline commenced 4 hr following systemic LPS challenge and continued once daily (i.p.) for 7 days. Behavioural testing in the staircase, stepping, and cylinder tests were conducted 6 weeks post‐intranigral LPS administration (Day 42). Rats were killed by transcardial perfusion fixation in preparation for immunohistochemical analysis or decapitated followed by brain retrieval and dissection for analysis of tissue dopamine concentrations.
2.3. Stereotaxic surgery
Rats were anaesthetised with the gaseous anaesthetic isoflurane (induction at 5% and maintenance at 2% in 1.5 L·min−1 O2) and positioned in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). LPS (10 μg in 2 μl of 0.89% sterile NaCl solution [saline]; Escherichia coli: serotype 0111:B4; Sigma‐Aldrich, Ireland) or vehicle (2‐μl sterile saline) was injected unilaterally into the SN at coordinates AP 5.3 mm, ML ± 2.0 mm, and DV 8.5 mm from bregma according to The Rat Brain in Stereotaxic Coordinates (Paxinos & Watson, 1986). For all injections, a Hamilton® Neuros syringe (7002 KH SYR; Hamilton, Switzerland) was used. Following surgery, the incision was closed using surgical adhesive, and rats were individually housed for a few hours to aid recovery.
2.4. Staircase test of skilled motor function
The staircase test was conducted to assess skilled motor function as previously described (Montoya, Campbell‐Hope, Pemberton, & Dunnett, 1991; Yssel et al., 2018). Briefly, rats were placed inside the staircase apparatus containing food pellets on an ascending stairwell, and the number of ipsilateral and contralateral forelimb retrievals of food pellets were recorded on video over a 10 min period for analysis of skilled motor function. These recordings were analysed to determine three parameters: (a) the number of successful reaching attempts made, (b) the number of unsuccessful attempts made, and (c) the number of pellets eaten using the tongue. Following this data collection, the success rate was calculated for each limb by experimenters blinded to the experimental treatment groups, as the number of successful reaching attempts expressed as a percentage of total attempts made.
2.5. Stepping test of forelimb akinesia
The stepping test was conducted as a measurement of forelimb akinesia and was performed as previously described (Olsson, Nikkhah, Bentlage, & Bjorklund, 1995; Yssel et al., 2018). Rats were held by the experimenter, blinded to the experimental treatment groups, with both hands so that three limbs (two hind limbs and one forelimb) were immobilised leaving the remaining forelimb unrestrained. Holding the rat perpendicular to a table's edge, with the free paw touching the table, the rat was directed lengthways (90 cm in 5 s) forward and backward, and the number of adjusting steps that contacted the table top was recorded in the forehand and backhand direction for each limb. The number of steps made by the contralateral limb in both the forehand and backhand direction was expressed as a percentage of the total number of steps made by both limbs in each direction.
2.6. Cylinder test of asymmetric limb use
The cylinder test was conducted as a measurement of forelimb‐use asymmetry as previously described (Schallert, Fleming, Leasure, Tillerson, & Bland, 2000). Briefly, rats were placed in an empty, transparent plexiglass cylinder and allowed to freely explore for 5 min. The number of forelimb wall placements made upon rearing was recorded by two independent observers blinded to the experimental treatment groups. Wall placements were recorded as “left limb,” “right limb,” or “simultaneous” (if the rat placed both paws against the wall of the cylinder at the same time) upon rearing. The number of wall placements made with the contralateral forelimb was expressed as a percentage of the total number of wall placements made.
2.7. HPLC coupled to electrochemical detection
Following decapitation, brains were quickly removed and the striatum and midbrain (containing the SN) were hand‐dissected on wet ice using Palkovits and Brownstein brain atlas (1988) for reference. Dopamine concentrations in the midbrain and striatum were measured as previously described (Yssel et al., 2018). In brief, peak heights were identified by retention times set to known biogenic amine standards, and data were normalised for total wet weight of tissue sample. Dopamine concentrations were quantified by electrochemical detection and analysis of the resulting chromatograms generated using a Merck Hitachi D‐2000 integrator. Inclusion of the internal standard (N‐methyl serotonin) in each sample allowed for correction of processing losses. These data, together with the brain tissue weights, were used to calculate the concentration of neurotransmitter in each sample. Results are expressed in terms of neurotransmitter (ng) per wet weight of tissue (g).
2.8. Brain tissue processing and immunohistochemistry
Rats were anaesthetised with urethane and transcardially perfused with ice‐cold PBS followed by 4% paraformaldehyde. Brains were then removed and stored in 4% paraformaldehyde at 4°C for 2 days followed by 30% sucrose solution until they sank; 30‐μm coronal sections through the SN and striatum were sliced using a cryostat (Leica, Germany) and stored in an anti‐freeze solution. For analysis of TH+ neurons and activated microglia, sections were incubated in 1% hydrogen peroxide solution in 20% methanol for 20 min to block endogenous peroxide activity, washed (3× PBS rinses), and incubated with blocking buffer (10% normal horse serum, 0.05% Triton X‐100; PBS for TH‐immunolabelling or 10% normal rabbit serum, 0.05% Triton X‐100; PBS for ionised calcium binding adaptor molecule 1 [Iba1]‐immunolabelling) for 45 min. Slices were incubated overnight with primary anti‐TH antibody (mouse monoclonal from Millipore, UK; MAB318, RRID:AB_2201528 used at 1:2,000) to label dopaminergic neurons in the SN and dopaminergic nerve fibres in the striatum, or anti‐Iba1 (polyclonal goat from Abcam, UK; ab5076, RRID:AB_2224402 used at 1:2,000) to label nigral microglia or anti‐cluster of differentiation 68 (CD68; monoclonal mouse from Santa Cruz Biotechnology [KP1]: sc‐20060, RRID:AB_627158 used at 1:500) to label microglia/invading macrophage activation. The following day, sections were washed (3× PBS rinses) and incubated with a biotinylated horse anti‐mouse secondary antibody (1:200 in 3% normal horse serum; PBS) for TH‐labelling or CD68 immunostaining, or a rabbit anti‐goat secondary antibody (1:200 in 3% normal rabbit serum; PBS) for Iba1‐immunolabelling for 90 min, followed by another 3 × 5 min PBS rinses and then incubated with an avidin‐biotinylated peroxidase complex formulated in PBS for 90 min, as described by the manufacturer (Vector Laboratories, UK). Following this, brain sections were washed again (3 × 5 min in PBS) and incubated with a 10% 3,3′‐diaminobenzidine solution (in 0.4% hydrogen peroxide; PBS). The sections were rinsed in PBS and mounted onto gelatin‐coated slides. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.9. Confocal immunofluorescence microscopy
For immunofluorescence staining of IL‐1β, 30‐μm sections of the SN were permeabilised in Triton‐X 100 (0.1%) for 20 min, rinsed in PBS (3 × 5 min), and incubated with blocking buffer (20% NGS, 0.05% Triton‐X 100; PBS) for 45 min. Sections were then incubated overnight with primary anti‐IL‐1β (polyclonal rabbit, ab9722 Abcam, UK, RRID:AB_308765 diluted 1:200 in 1% BSA; PBS). Sections were rinsed in PBS (3 × 5 min) and incubated with the secondary antibody (Alexa Fluor™ 488 goat anti‐rabbit from Invitrogen, A11008 RRID:AB_143165 diluted 1:1,000 in 1% BSA; PBS) for 2 hr in the dark. Sections were subsequently rinsed in PBS (3 × 5 min) and mounted onto gelatin‐coated slides with Vectashield mounting medium. Coverslips were applied and sealed with nail varnish. Nigral sections were visualised by fluorescent confocal microscopy (Zeiss LSM 510‐META). The primary antibody was omitted as a control for nonspecific binding of the secondary antibody. No immunoreactivity was detected under these conditions.
2.10. Histological quantification
For analysis of nigral TH+ cells and nerve fibres, Iba1+ cells, and CD68 immunoreactivity, images were taken using an Olympus DRP72 camera mounted on an Olympus BX51 normal light microscope and analysed using Image J software (Image J v1.38x; NIH, USA, RRID:SCR_003070). TH+ and Iba1+ dopamine and microglial cells respectively along with nigral CD68 immunoreactivity were assessed in 10 30‐μm‐thick serial sections of the SN per animal (AP −4.80 to −6.04 mm from bregma) before the injection site, at the injection site, and after the injection site to best represent extent of the lesion. The area of TH immunoreactivity in the striatum was evaluated in 20 30‐μm‐thick serial sections of the SN per animal (AP 1.60 to −0.92 mm from bregma) to assess dopaminergic nerve fibre integrity. Images were converted to 8‐bit, thresholded, and the area of the striatum was delineated. Data were expressed as a percentage of the intact side (contralateral hemisphere). The same images used to assess nigral Iba1 immunoreactivity were also used for the quantification of Iba1+ amoeboid cells in the SN. Amoeboid microglia were verified via perimeter analysis on Image J. In brief, images were converted to 8‐bit, a threshold was set, and the average Iba1+ cell perimeter length (μm) of each cell per image was calculated. Images containing amoeboid cells with an enlarged cell soma and lacking branching (unramified cells) were used for our analysis; Iba1+ cells that met these criteria were counted for each nigral image per treatment group by an unbiased experimenter blind to the experimental objectives. For analysis of nigral IL‐1β expression, images were taken using a fluorescent confocal microscope (Zeiss LSM 510‐META) and analysed as before using Image J software. All immunohistochemical data were expressed as a percentage of the intact (un‐lesioned) hemisphere. Tissue sections needed to be structurally intact and sliced evenly so both hemispheres are at the same AP position. Post‐immunohistochemical staining, any tissue sections that did not meet these requirements were excluded from the analysis. Immunohistochemical analysis was performed by two experimenters blinded to the experimental treatment groups to reduce any perceived bias.
2.11. Data and statistical analysis
The optimum sample sizes and animal numbers were determined by power analysis of pre‐existing data. All data were analysed using GraphPad Prism 8 (GraphPad Prism, San Diego, CA, RRID:SCR_002798). Data distributions were tested for normality using a Schapiro–Wilk test, and outliers were assessed using z‐scores (any z‐score greater than 3 or less than −3 SDs from the mean were considered an outlier). All data were normally distributed. Immunohistochemical data was expressed as a percentage of the intact side to normalise the data and minimise unwanted sources of variation that may arise with hemispheric staining procedures/intensity between subjects. Statistical analysis was undertaken only for studies where each group size was at least n = 5. The declared group size is the number of independent values, and statistical comparisons were performed using these independent values via ANOVA. The data were analysed either by one‐way ANOVA, two‐way ANOVA, or three‐way ANOVA depending on the experiment. If significant changes were observed following ANOVA (e.g., P < .05), the data were further analysed using a Newman–Keuls post hoc test. For determining whether groups differ, the level of probability (P) was set at P < .05 for constituting the threshold for statistical significance. Results are expressed as mean ± SEM. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
2.12. Materials
All drugs were injected via the i.p. route in an injection volume of 1 ml·kg−1 apart from clenbuterol which was pre‐dissolved at a concentration of 30 μg·ml−1. Doses were as follows: Atomoxetine (3 mg·kg−1; Eli Lilly and Co.), clenbuterol hydrochloride (100 μg·kg−1), and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=543 (5 mg·kg−1; MedchemExpress) were dissolved in 0.89% sterile saline. Formoterol hemifumarate (100 μg·kg−1; Tocris Bioscience, UK) was prepared in 1% DMSO. Saline or 1% DMSO were used as vehicle control where appropriate.
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. Clenbuterol and formoterol protect against intranigral LPS‐induced motor impairments
In the stepping test of forelimb akinesia, intranigral LPS injection decreased the number of contralateral adjusting steps made in the backhand and forehand direction at both 7 and 13 days post‐lesioning relative to vehicle‐injected controls. Treatment with clenbuterol or formoterol attenuated the LPS‐induced deficits in forelimb kinesis at 13 days post‐lesioning (Figure 1a+ b). In the cylinder test of forelimb‐use asymmetry, intranigral LPS decreased contralateral wall placements made at 7 and 14 days post‐lesioning relative to vehicle‐injected controls. Treatment with clenbuterol or formoterol attenuated LPS‐induced forelimb‐use asymmetry at 13 days post‐lesioning (Figure 1c). In the staircase test of skilled motor function, intranigral LPS decreased the contralateral success rate at 13 days post‐lesioning relative to vehicle‐injected controls. Treatment with formoterol attenuated LPS‐induced skilled motor deficits at 13 days post‐lesioning (Figure 1d).
Figure 1.
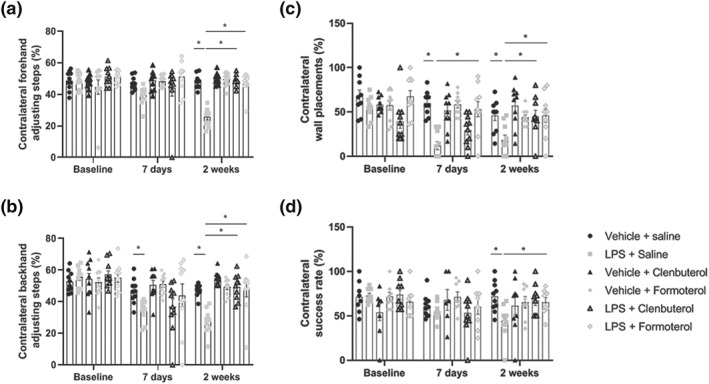
β2‐adrenoceptor activation attenuates LPS‐induced motor dysfunction. (a) Stepping test of forelimb akinesia (forehand direction). (b) Stepping test of forelimb akinesia (backhand direction). (c) Cylinder test of asymmetric limb use. (d) Staircase test of skilled motor function. The effect of clenbuterol and formoterol treatment on motor function was assessed at baseline (pre‐lesion), 7 days, and 2 weeks following intranigral LPS administration. Treatment with clenbuterol (100 μg·kg−1 i.p.) or formoterol (100 μg·kg−1 i.p.) began 4 hr following LPS and continued for 7 days (q.d.). Intranigral LPS induced bidirectional forelimb akinesia in the stepping test, forelimb‐use asymmetry in the cylinder test, and skilled motor deficits in the staircase test. Treatment with clenbuterol and formoterol attenuated motor deficits in response to intranigral lesioning with LPS. Data expressed as mean ± SEM (n = 10), * P < .05 compared to control via two‐way repeated measures ANOVA with post hoc Newman–Keuls
3.2. Clenbuterol and formoterol protect against nigrostriatal dopaminergic neurodegeneration
Exploratory analyses of anti‐TH immunohistochemistry in the post mortem SN and striatum revealed that intranigral LPS injection decreased the number of TH+ cells and the area of TH immunoreactivity in the SN and striatum respectively relative to sham‐lesioned controls. Treatment with clenbuterol or formoterol attenuated the LPS‐induced loss of TH+ dopamine cells in the SN (Figure 2a+ c). Treatment with formoterol, but not clenbuterol, attenuated the LPS‐induced loss of TH+ dopaminergic nerve fibres in the striatum (Figure 2b+ c).
Figure 2.
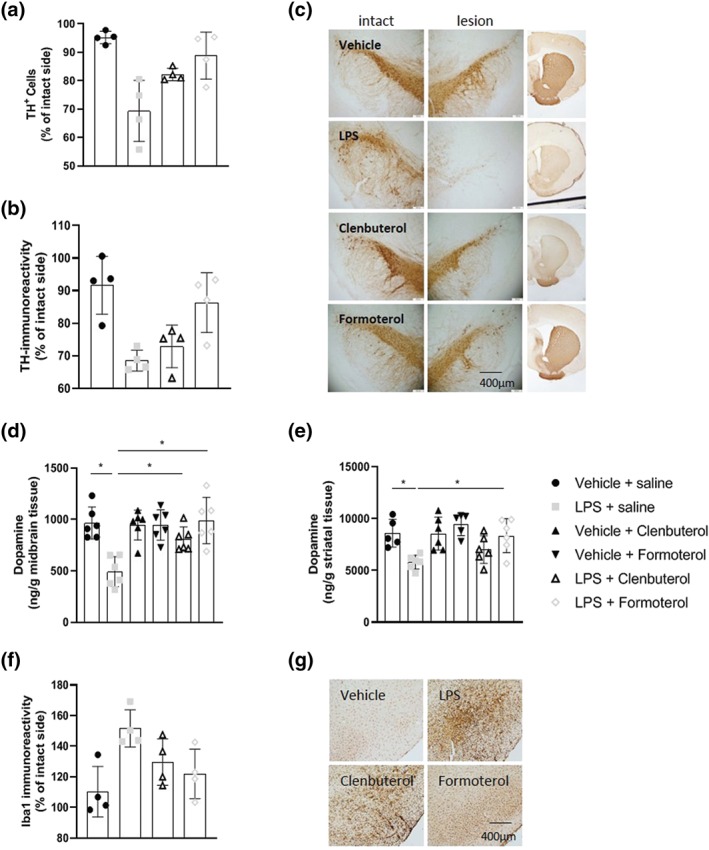
Clenbuterol and formoterol attenuate intranigral LPS‐induced loss of nigrostriatal dopamine neurons. (a) TH+ cell counts in the substantia nigra. (b) Representative images of anti‐TH immunostaining in the substantia nigra and ipsilateral striatum. (c) Area of TH immunoreactivity in the striatum. (d) Midbrain dopamine concentrations measured via HPLC. (e) Striatal dopamine concentrations measured via HPLC. The effect of clenbuterol and formoterol treatment was assessed on nigrostriatal dopamine neurons and midbrain and striatal dopamine concentrations 2 weeks 14 days following intranigral LPS administration. Treatment with clenbuterol (100 μg·kg−1 i.p.) or formoterol (100 μg·kg−1 i.p.) began 4 hr following LPS and continued for 7 days (q.d.). Intranigral LPS induced dopaminergic neurodegeneration along the nigrostriatal tract. Treatment with clenbuterol and formoterol attenuated the loss of dopamine neurons and suppressed reductions in nigrostriatal dopamine content. Data expressed as mean ± SEM (n = 4 for immunoreactivity; n = 6 for dopamine concentrations), * P < .05 via one‐way ANOVA with post 14 days following intranigral LPS administration. hoc Newman–Keuls
HPLC analysis of midbrain and striatal dopamine concentrations revealed that intranigral LPS injection reduced nigrostriatal dopamine concentrations relative to sham‐lesioned controls. Treatment with clenbuterol and formoterol attenuated the LPS‐induced loss of midbrain dopamine concentrations (Figure 2d). Treatment with formoterol, but not clenbuterol, attenuated LPS‐induced striatal dopamine depletion (Figure 2e).
Preliminary analyses showed that intranigral LPS induced microglial activation relative to vehicle controls as indicated by increased Iba1 immunoreactivity ipsilateral to the injection site. Treatment with formoterol, but not clenbuterol, ameliorated LPS‐induced microglial activation (Figure 2f+ g) 14 days following intranigral LPS administration.
3.3. Atomoxetine protects against intranigral LPS‐induced dopamine loss and motor dysfunction in a β2‐adrenoceptor dependent manner
In the stepping test of forelimb akinesia, intranigral LPS injection decreased the number of contralateral adjusting steps made in both the forehand and backhand direction at 7 and 13 days post‐lesioning relative to vehicle‐injected controls. Treatment with atomoxetine attenuated LPS‐induced bidirectional deficits in forelimb kinesis at both 7 and 13 days post‐lesioning. Co‐treatment with the β2‐adrenoceptor antagonist ICI 118,551 abrogated the protective effects of atomoxetine against LPS‐induced forelimb akinesia in both the forehand and backhand direction at 7 and 13 days post‐lesioning (Figure 3a+ b). In the cylinder test of asymmetric limb use, intranigral LPS decreased the number of contralateral wall placements made at both 7 and 13 days post‐lesioning relative to vehicle‐injected controls. Treatment with atomoxetine attenuated LPS‐induced forelimb‐use asymmetry at 7 and 13 days post‐lesioning. Co‐treatment with the β2‐adrenoceptor antagonist ICI 118,551 abrogated the protective effects of atomoxetine against LPS‐induced forelimb‐use asymmetry at 7 days post‐lesioning (Figure 3c). HPLC analysis of midbrain dopamine concentrations revealed that intranigral LPS decreased dopamine levels when assessed 13 days post‐lesioning relative to vehicle‐injected controls. Treatment with atomoxetine attenuated the loss of midbrain dopamine content. Co‐treatment with the β2‐adrenoceptor antagonist ICI 118,551 abrogated the protective effects of atomoxetine against LPS‐induced midbrain dopamine depletion (Figure 3d).
Figure 3.
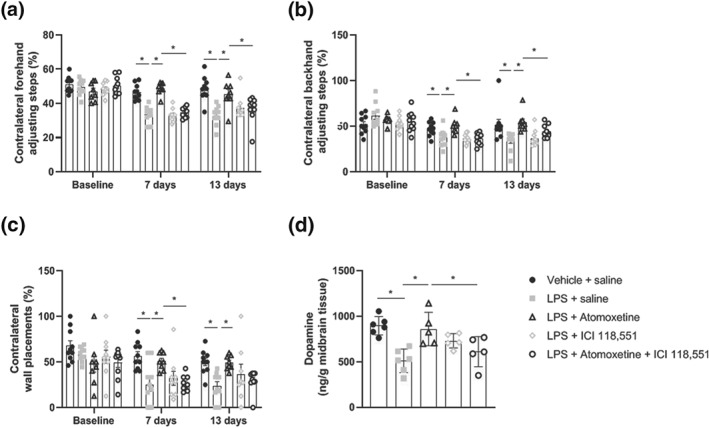
Co‐treatment with ICI 118,551 attenuates atomoxetine‐mediated protection against LPS‐induced motor deficits and reductions in midbrain dopamine concentration. (a) Stepping test of forelimb akinesia (forehand direction). (b) Stepping test of forelimb akinesia (backhand direction). (c) Cylinder test of asymmetric limb use. (d) Midbrain dopamine concentrations measured via HPLC. Treatment with atomoxetine (3 mg·kg−1; i.p.) following intranigral LPS (10 μg) began 4 hr following surgery and continued for 7 days. ICI 118,551 (5 mg·kg−1; i.p.) was administered 30 min prior to atomoxetine. Pharmacological antagonism of the β2‐adrenoceptor with ICI118,551 abrogated the protection against intranigral LPS‐induced dopamine loss and motor impairments afforded by treatment with atomoxetine. Data expressed as mean ± SEM (n = 10 in behaviour; n = 6 dopamine concentrations), * P < .05 versus control via one‐way (d) and two‐way repeated measures ANOVA (a–c) with post hoc Newman–Keuls
3.4. Formoterol protects against the progression of motor impairments in response to systemic LPS challenge
In the staircase test of skilled motor function, intranigral LPS injection decreased the contralateral success rate relative to sham‐lesioned controls. Systemic LPS challenge exacerbated skilled motor deficits in animals previously lesioned with intranigral LPS. Treatment with formoterol did not prevent exacerbations in skilled motor dysfunction in response to systemic LPS challenge (Figure 4a).
Figure 4.
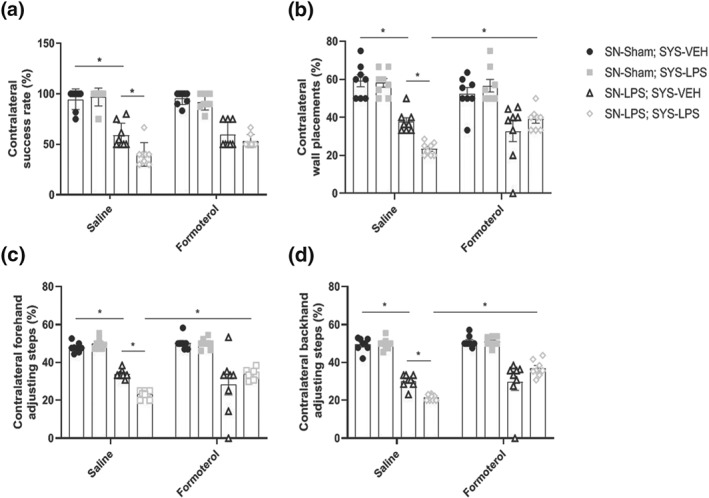
Formoterol protects against progressive motor impairment in response to systemic LPS challenge. (a) Staircase test of skilled motor function. (b) Cylinder test of forelimb‐use asymmetry. (c) Stepping test of forelimb akinesia (forehand direction). (d) Stepping test of forelimb akinesia (backhand direction). Intranigral LPS induced skilled motor deficits in the staircase test, forelimb‐use asymmetry in the cylinder test, and bidirectional forelimb akinesia in the stepping test at 6 weeks post‐lesioning. Systemic LPS challenge exacerbated motor deficits in the staircase test, cylinder test, and stepping test. Treatment with formoterol protected against the exacerbated decline in forelimb‐use asymmetry and forelimb akinesia. Data expressed as mean ± SEM (n = 8) * P < .05 compared to control via three‐way ANOVA with post hoc Newman–Keuls
In the cylinder test of asymmetric limb use, intranigral LPS injection reduced the number of contralateral wall placements made relative to sham‐lesioned controls. Systemic LPS challenge exacerbated forelimb‐use asymmetry in animals previously lesioned with intranigral LPS. Treatment with formoterol prevented advancements in forelimb‐use asymmetry in response to systemic LPS challenge (Figure 4b).
In the stepping test of forelimb akinesia, intranigral LPS injection reduced the number of contralateral adjusting steps made in both the forehand and backhand direction relative to sham‐lesioned controls. Systemic LPS challenge exacerbated bidirectional forelimb akinesia in animals previously lesioned with intranigral LPS. Treatment with formoterol prevented exacerbations in forelimb akinesia in both the forehand and backhand direction in response to systemic LPS challenge (Figure 4c + d).
3.5. Formoterol protects against exacerbations in dopamine cell loss and nigrostriatal dopamine depletion in response to systemic LPS challenge
Intranigral LPS alone and in combination with systemic LPS challenge reduced the number of TH+ dopamine cells in the SN by 61% and 83% respectively relative to vehicle‐treated controls. Systemic administration of LPS alone had no effect on nigral TH+ cell counts. Systemic administration of LPS exacerbated the TH+ dopamine cell loss within the SN by 22% in animals previously treated with LPS centrally. Treatment with formoterol did not restore the loss of TH+ dopamine neurons within the SN in response to intranigral LPS. Treatment with formoterol prevented exacerbations in TH+ cell loss within the SN in intranigral LPS‐lesioned rats subsequently exposed to systemic LPS challenge relative to intranigral LPS + systemic LPS rats treated with saline control (Figure 5a + e).
Figure 5.
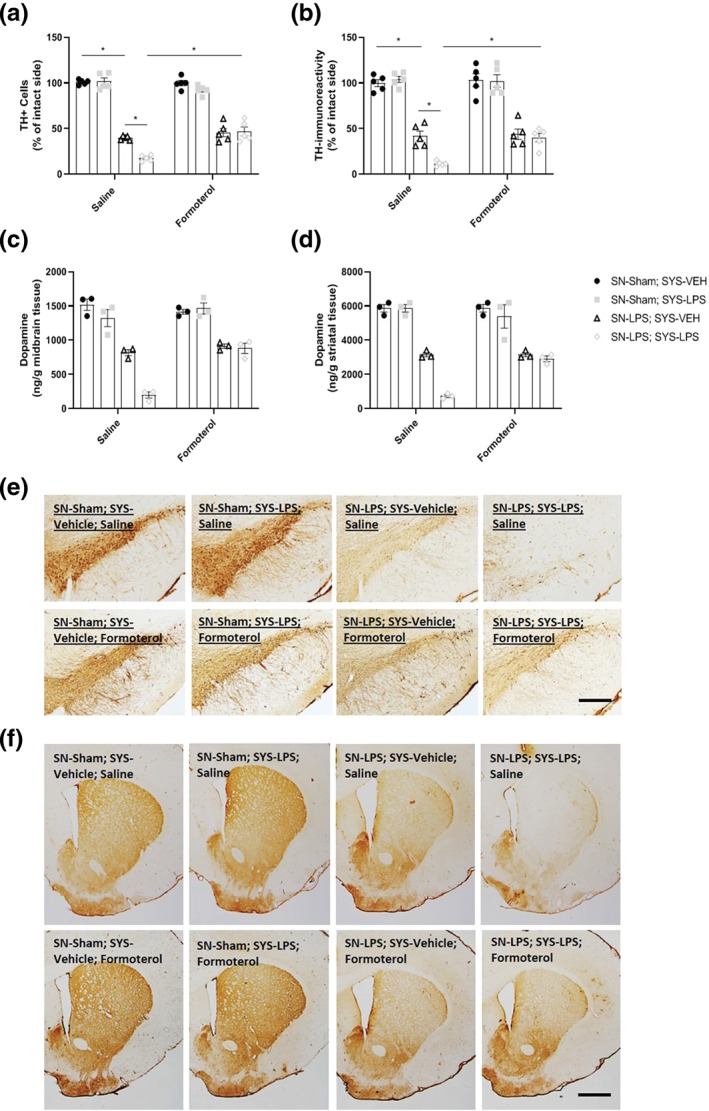
Formoterol protects against exacerbations in nigrostriatal dopaminergic neurodegeneration and dopamine depletion in response to systemic LPS challenge. (a) TH+ cell counts in the substantia nigra. (b) TH immunoreactivity in the striatum. (c) Midbrain dopamine concentrations measured via HPLC. (d) Striatal dopamine concentrations measured via HPLC. (e) Representative images for anti‐TH immunostaining in the substantia nigra. (f) Representative images of anti‐TH immunostaining in the striatum intranigral administration of LPS induced marked reductions in TH+ dopaminergic cells in the SN, TH+ dopaminergic neuronal fibre loss in the striatum, and depleted nigrostriatal dopamine concentrations. Systemic LPS exacerbated dopamine neuronal loss in the SN, nerve terminal degeneration in the striatum, and reductions in nigrostriatal dopamine concentrations in animals previously lesioned with intranigral LPS. Treatment with formoterol attenuated systemic LPS‐induced exacerbations in nigrostriatal dopaminergic neuronal loss and dopamine depletion observed in intranigral LPS‐lesioned animals. Scale bar = 200 μm (s.nigra) and 800 μm (striatum). Data expressed as mean ± SEM (n = 5 immunoreactivity; n = 3 dopamine concentrations), * P < .05 versus control via three‐way ANOVA with post hoc Newman–Keuls
Intranigral LPS alone or in combination with a subsequent systemic LPS challenge reduced TH+ immunoreactivity in the striatum by 58% and 88% respectively compared to sham‐lesioned controls challenged with saline. Systemic LPS alone had no effect on striatal TH+ immunoreactivity but exacerbated the loss of TH+ immunoreactivity observed in the striatum by approximately 30% following intranigral LPS. Treatment with formoterol did not restore intranigral LPS‐induced loss of striatal TH immunoreactivity but prevented exacerbations of intranigral LPS‐induced TH+ striatal denervation in response to subsequent systemic LPS challenge (Figure 5b + f).
Further exploratory analyses showed a reduction in midbrain dopamine concentration in response to intranigral LPS injection. Systemic LPS administration exacerbated midbrain dopamine depletion in intranigral LPS‐lesioned rats. Treatment with formoterol prevented exacerbations in midbrain dopamine loss (Figure 5c). Similarly, there was a reduction in striatal dopamine concentration in the intranigral LPS group relative to sham‐controls. Systemic LPS administration exacerbated dopamine depletion in intranigral LPS‐lesioned rats. Treatment with formoterol prevented exacerbations in striatal dopamine loss in intranigral LPS‐lesioned rats subsequently challenged with systemic LPS (Figure 5d).
3.6. Formoterol inhibits microglial activation and prevents enhanced microgliosis in response to systemic LPS challenge
Intranigral LPS injection induced robust microglial activation as indicated by increases in the area of Iba1 immunoreactivity and increased Iba1+ amoeboid cell numbers relative to sham‐lesioned controls. Systemic LPS challenge aggravated microglial activation in animals previously lesioned with intranigral LPS, as indicated by expansion in the area of Iba1 immunoreactivity and exacerbations in the number of Iba1+ amoeboid cells. Treatment with formoterol inhibited intranigral LPS‐induced microglial activation and prevented exacerbations in the area of Iba1 immunoreactivity and the number of Iba1+ amoeboid cells in response to a subsequent systemic LPS challenge (Figure 6a–c).
Figure 6.
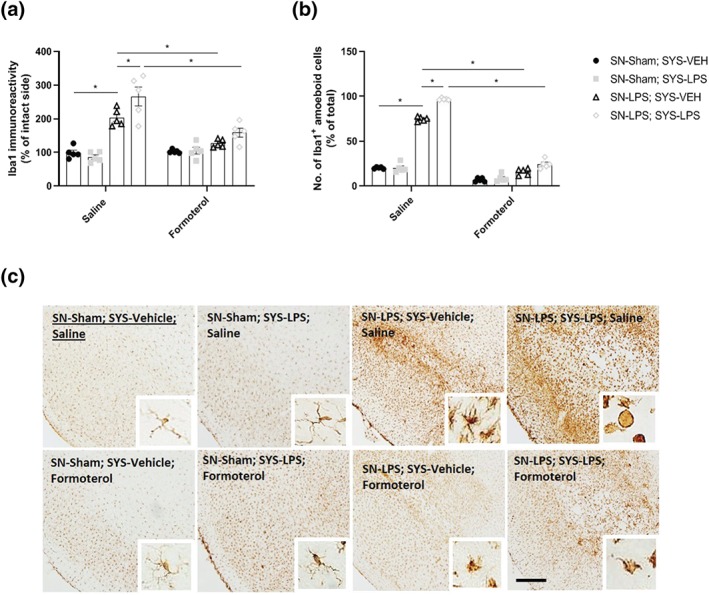
Formoterol inhibits intranigral LPS‐induced microglial activation and restrains aggravated microglial activation in response to systemic LPS challenge. (a) Area of Iba1 immunoreactivity in the substantia nigra. (b) Number of Iba1+ amoeboid cells in the substantia nigra. (c) Representative images of anti‐Iba1 immunostaining in the substantia nigra. Intranigral LPS induced robust increases in Iba1+ immunoreactivity and clear morphological alterations in Iba1+ cells indicative of microgliosis 6 weeks following intranigral LPS administration. Subsequent systemic LPS administration exacerbated nigral microgliosis and increased the number of Iba1+ amoeboid cells in intranigral LPS‐lesioned rats. Formoterol reduced Iba1 immunoreactivity in intranigral LPS‐lesioned rats and suppressed the exacerbation of nigral microgliosis and increased number of Iba1+ amoeboid cells following subsequent systemic LPS challenge. Scale bar = 200 μm (10×) and 50 μm (20×). Data expressed as mean ± SEM (n = 5), *(P < .05) compared to control via three‐way ANOVA with post hoc Newman–Keul
3.7. Formoterol ameliorates intranigral LPS‐induced increases in CD68 expression and attenuates the IL‐1β response to systemic LPS
Intranigral LPS injection increased the area of CD68 expression relative to sham‐lesioned controls. Systemic LPS challenge exacerbated CD68 expression in animals previously lesioned with intranigral LPS. Treatment with formoterol attenuated intranigral LPS‐induced increases in CD68 expression and prevented exacerbations in CD68 expression levels in response to systemic immune challenge with LPS (Figure 7a + c). Intranigral LPS injection increased the area of IL‐1β immunoreactivity relative to sham‐lesioned controls. Systemic LPS challenge exacerbated IL‐1β expression in animals previously lesioned with intranigral LPS. Treatment with formoterol attenuated the exacerbated IL‐1β response to systemic LPS challenge in animals previously lesioned with intranigral LPS (Figure 7b + d).
Figure 7.
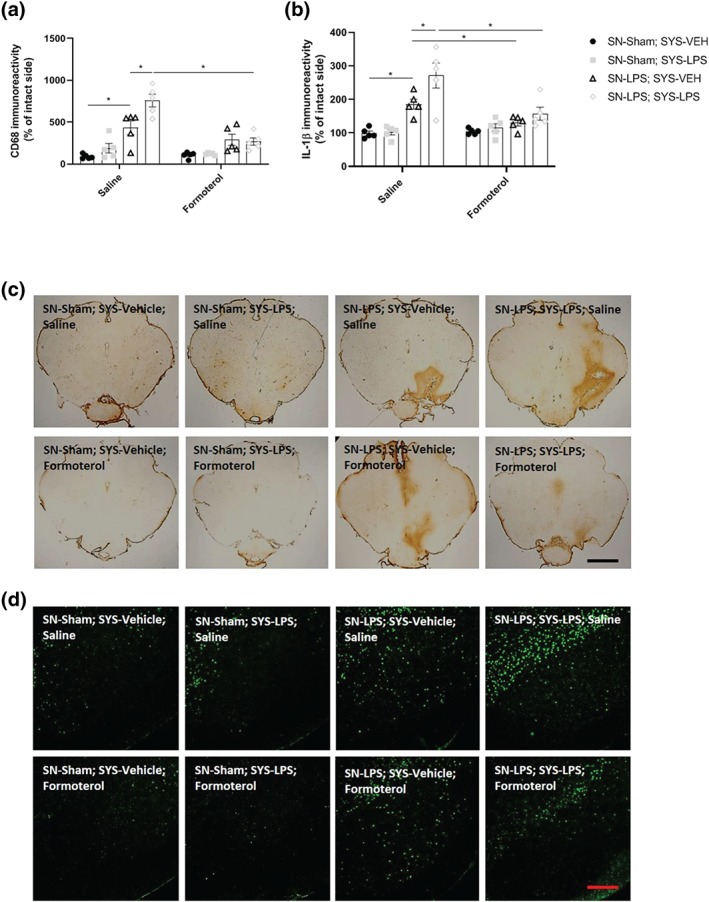
Formoterol attenuates exacerbations in intranigral LPS‐induced CD68 and IL‐1β expression in response to subsequent systemic LPS challenge. (a) Area of CD68 immunoreactivity in the substantia nigra. (b) IL‐1β protein expression in the SN. (c) Representative images of anti‐CD68 immunostaining in the substantia nigra. (d) Representative images of IL‐1β immunofluorescence in the substantia nigra. Intranigral LPS increases CD68 and IL‐1β immunoreactivity in the SN. Subsequent systemic LPS challenge exacerbates CD68 and IL‐1β immunoreactivity in intranigral LPS‐lesioned animals. Formoterol attenuates nigral CD68 expression and mitigates the enhanced IL‐1β response to systemic LPS challenge in animals previously lesioned with intranigral LPS. Scale bar = 800 μm (CD68) and 200 μm (IL‐1β). Data expressed as mean ± SEM (n = 5), *(P < .05) compared to control via three‐way ANOVA with post hoc Newman–Keuls
4. DISCUSSION
The results of this investigation provide evidence in support of the use of β2‐adrenoceptor agonists to restrict microglial activation and protect against the onset and progression of dopamine neuronal cell loss and related motor deficits provoked by central or systemic inflammation. Treatment with formoterol attenuates the loss of TH+ dopamine neurons in the substantia nigra (SN) and striatal denervation, the concordant depletion in midbrain and striatal dopamine concentrations, and associated motor deficits in response to intranigral LPS in rats. Formoterol also reduced LPS‐induced microgliosis within the SN. Clenbuterol produced similar albeit less robust effects and failed to significantly attenuate TH+ striatal denervation, dopamine depletion, deficits in skilled motor function in the staircase test, and increased microgliosis in response to intranigral LPS. Thus, formoterol conferred a more consummate level of protection against LPS‐induced degeneration of the nigrostriatal dopaminergic system and ensuing motor impairments, which in turn was likely to be incumbent on significantly ameliorating nigral microgliosis. Formoterol reportedly has a much greater affinity for the β2—versus the β1‐adrenoceptor (Anderson, 1993), and as β2‐adrenoceptors are highly expressed on microglia (Tanaka et al., 2002), this may explain the superior neuroprotective efficacy of formoterol relative to that observed with clenbuterol. Formoterol was therefore employed in subsequent experiments to assess its effects on the progression of dopamine loss and motor impairments induced by intranigral LPS and a subsequent systemic LPS challenge.
Previously, treatment with the noradrenaline reuptake inhibitor atomoxetine inhibits intranigral LPS‐induced microglial activation and ameliorates the loss of dopamine neurons in the SN, restricts dopaminergic nerve fibre degeneration in the striatum, and suppresses nigrostriatal dopamine depletion, thus providing partial protection against motor impairments (Yssel et al., 2018). In the current investigation, co‐treatment of atomoxetine with the β2‐adrenoceptor antagonist ICI 118,551 blocks the protective effects of atomoxetine, which is consistent with the aforementioned protective effects of formoterol and demonstrates a proposed β2‐adrenoceptor dependent mechanism underlying the neuroprotection observed.
Furthermore, prior intranigral LPS‐induced degeneration of the nigrostriatal tract predisposed to a heightened degree of reactive microgliosis and IL‐1β production in response to subsequent immune challenge with systemic LPS. These findings are accompanied by an exacerbated loss of TH+ nigrostriatal dopamine neurons and deficits in motor function. Systemic LPS alone however did not induce dopamine neuronal loss, nigrostriatal dopamine depletion, or motor dysfunction, as previously reported in mice (Byler et al., 2009). Other groups have demonstrated that inflammatory priming of the SN with the viral mimetic and toll‐like receptor 3 agonist polyinosinic‐polycytidylic acid sensitises midbrain dopamine neurons and striatal nerve fibres to oxidative stress‐induced degeneration in response to subsequent intra‐striatal delivery of a low dose of 6‐hydroxydopamine (6‐OHDA) 12 days later, findings which were ameliorated by systemic administration of IL1‐RA (Deleidi, Hallett, Koprich, Chung, & Isacson, 2010), highlighting a role for IL‐1 in exacerbating neuronal loss in response to a subsequent neurotoxic insult. Indeed, a subtoxic, intranigral LPS injection shifts primed microglia to a pro‐inflammatory state, elevates nigral IL‐1β production and exacerbates prior intra‐striatal 6‐OHDA‐induced degeneration of dopamine neurons, and increases the severity of motor deficits in the stepping test of forelimb akinesia (Pott Godoy, Tarelli, Ferrari, Sarchi, & Pitossi, 2008). Moreover, chronic systemic (i.v.) adenoviral‐mediated IL‐1β expression commencing 7 days following intra‐striatal 6‐OHDA delivery promotes end‐stage major histocompatibility complex class ІІ+ microglial activation and exacerbates dopaminergic neuronal loss in the SN, findings which were reversed upon administration of IL‐1RA. Thus, midbrain microglia are likely to be a major cellular source of IL‐1β production in the inflammatory‐primed midbrain in response to a subsequent systemic LPS challenge, which contributes to the exacerbated degeneration of proximal dopaminergic neurons and associated motor impairments.
Treatment with formoterol restricted microgliosis in the SN, attenuated intranigral LPS‐induced increases in IL‐1β expression, and, moreover, reduced the subsequent IL‐1β response to systemic LPS in intranigral LPS‐lesioned rats. Furthermore, formoterol attenuated the progression of TH+ dopamine cell loss in the SN, striatal denervation, and the further deterioration of motor function associated with systemic LPS challenge. Intranigral LPS‐induced microglial activation, IL‐1β production, and dopaminergic neurodegeneration more extensively pervade throughout the SN in response to systemic LPS challenge. Targeting β2‐adrenoceptors with formoterol imbues an intervening prophylactic mechanism to protect against the progression of neurodegeneration and exacerbated decline in motor function associated with systemic and central inflammation. Thus, it appears that treatment with formoterol could be prophylactic against the progression of PD‐related neuropathology and motor impairments induced by inflammation, provided that it is administered in a timely fashion relative to presentation of the inflammatory stimulus.
Degeneration of locus coeruleus (LC) noradrenergic neurons (up to 80%) and proceeding CNS noradrenaline depletion, as occurs in PD even before the onset of dopaminergic neuronal loss and motor abnormalities (Baloyannis, Costa, & Baloyannis, 2006; Zarow, Lyness, Mortimer, & Chui, 2003), is a salient yet overlooked pathological feature of this disease pathogenesis. Selective lesions of the LC‐noradrenergic system with N‐(2‐chloroethyl)‐N‐ethyl‐2‐bromobenzylamine (DSP4) exacerbate the loss of TH+ dopaminergic neurons in the rodent SN in response to systemic LPS (5 mg·kg−1 i.p.) administration (Jiang et al., 2015). Reports (Li et al., 2018) have shown that administration of DSP4 (50 mg·kg−1 i.p.) induced a 34% loss of dopamine β‐hydroxylase‐positive noradrenergic cells in the LC, leading to spatial learning and memory deficits, aggravates striatal dopamine depletion, and potentiates hypokinesia in response to a subsequent 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine regimen. Interestingly, DSP4‐mediated degeneration of LC‐noradrenergic neurons and ensuing brain noradrealine depletion is sufficient to initiate robust microglial activation and oxidative stress in vulnerable LC‐innervated brain regions, driving the sequential, progressive loss of TH+ dopamine cells in the SN followed by neuronal loss in the primary motor cortex and hippocampus, akin to the discrete pattern of caudo‐rostral neurodegeneration observed in PD patients (Song et al., 2019). Thus, the early loss of LC‐noradrenergic cell bodies and ensuing noradrenergic loss may be a crucial process initiating chronic neuroinflammation and the ascending pattern of neurodegeneration underlying PD progression. Noradrenaline, and indeed β2‐adrenoceptor stimulation, may exert crucial immunomodulatory and neuroprotective effects in PD by restraining microglial activation and ameliorating neuroinflammation (O'Neill & Harkin, 2018). Indeed, a 7‐day treatment regimen with atomoxetine inhibits intranigral LPS‐induced microglial activation and midbrain TNF‐α, IL‐6, and IL‐1β mRNA expression whilst up‐regulating brain‐derived neurotrophic factor, cerebral dopamine neurotrophic factor, and glial‐derived neurotrophic factor gene transcripts and attenuates the loss of TH+ dopamine neurons in the SN, preserving nerve terminal degeneration in the striatum and abrogating nigrostriatal dopamine depletion, leading to partial improvements in motor function (Yssel et al., 2018).
More recently, the β2‐adrenoceptor has been reported to regulate the α‐synuclein gene and relative α‐synuclein protein abundance, a major protein constituent of Lewy Body pathology in PD brains. Salbutamol (also a brain‐penetrant asthma medication) is associated with a decreased risk of developing PD, and, conversely, blockade of the β2‐AR with the antagonist propranolol is associated with an increased risk of developing PD (Mittal et al., 2017). Data from the same study revealed that the β2‐AR regulates the transcription of the human α‐synuclein gene SNCA through H3K27 acetylation (H3K27ac) of promoters and enhancers in the human SNCA locus and that treatment with selective β2‐adrenoceptor agonist clenbuterol is correlated with a decrease in H3K27ac levels and relative expression of SNCA mRNA levels. Thus, β2‐adrenoceptor agonists may also be particularly useful pharmacological agents for alleviating the burdening manifestation of aberrant α‐synuclein‐enriched Lewy Body pathology in PD patients.
Accumulation of α‐syn in the brain is a pathological feature of PD and leads to microglial activation, inflammatory cytokine production, and neurodegeneration (Kirik et al., 2002; Theodore, Cao, McLean, & Standaert, 2008; Xu et al., 2002). Given the importance of the β2‐adrenoceptor in regulating microglia and the α‐SYN gene in driving risk of PD, pharmacotherapies aimed at increasing noradrenergic tone to ameliorate inflammation are likely to have therapeutic potential. Long‐term treatment with noradrenaline reuptake inhibitors represents a clinically feasible neuroprotective strategy in PD, as these agents are currently used in the treatment of depression and attention deficit hyperactivity disorder and are safe when taken for prolonged periods (Michelson et al., 2003; Nelson, Mazure, Jatlow, Bowers, & Price, 2004). Similarly, our current findings support the use of a β2‐adrenoceptor agonist (particularly formoterol) to slow/halt PD progression in instances where an inflammatory component is driving disease progression. What makes this potentially prospective PD therapeutic even more promising is that formoterol is a widely used FDA‐approved treatment (marketed under brand name: Symbicort® as a metered dose inhaler) for asthmatics and chronic obstructive airway disease patients, and thus, it is also safe to use for prolonged periods of time in humans. In clinical practice, such agents may produce shakiness (tremor) in humans but this is considered unrelated to “Parkinsonian tremor” due to degeneration of the nigrostriatal dopaminergic system. Indeed, formoterol could be a viable prophylactic medication to slow disease onset and/or progression. In support of this, it has recently been reported that individuals who take long‐acting β2‐adrenoceptor agonists for asthma or chronic obstructive airway disease have a decreased risk of developing PD later in life, whereas people who take a β2‐adrenoceptor antagonist have an increased risk of developing PD later in life (Mittal et al., 2017).
AUTHOR CONTRIBUTIONS
E.O'N. performed most of the experiments and analyses involved and wrote the paper. J.Y. performed some of the experiments and analysis. C.M. analysed the behavioural data. A.H. conceived, designed, and supervised the study, performed the analyses, and wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
We would like to dedicate this publication to the memory of the late Professor Thomas J. Connor (1971–2013) whose pioneering research into the anti‐inflammatory properties of noradrenaline inspired the current investigation. The authors gratefully acknowledge the support of the Higher Education Authority of Ireland and the Programme for Research in Third Level Institutions MolCellBio programme. E.O'N. was supported by a Trinity College postgraduate research award. The authors also wish to acknowledge the support of the Trinity Foundation in memory of Thomas J. Connor.
O'Neill E, Yssel JD, McNamara C, Harkin A. Pharmacological targeting of β2‐adrenoceptors is neuroprotective in the LPS inflammatory rat model of Parkinson's disease. Br J Pharmacol. 2020;177:282–297. 10.1111/bph.14862
REFERENCES
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Roberts, R. E. , Broughton, B. R. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, G. P. (1993). Formoterol: Pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2‐adrenoceptor agonist bronchodilator. Life Sciences, 52(26), 2145–2160. 10.1016/0024-3205(93)90729-M [DOI] [PubMed] [Google Scholar]
- Baloyannis, S. J. , Costa, V. , & Baloyannis, I. S. (2006). Morphological alterations of the synapses in the locus coeruleus in Parkinson's disease. Journal of the Neurological Sciences, 248(1–2), 35–41. 10.1016/j.jns.2006.05.006 [DOI] [PubMed] [Google Scholar]
- Barisione, G. , Baroffio, M. , Crimi, E. & Brusasco, V. (2010). Beta‐Adrenergic Agonists. Pharmaceuticals (Basel), 3(4), 1016–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronti, A. , Grieco, A. , & Vibelli, C. (1980). Oral NAB 365 (clenbuterol) and terbutaline in chronic obstructive lung disease: A double‐blind, two‐week study. International Journal of Clinical Pharmacology, Therapy, and Toxicology, 18(1), 21–25. [PubMed] [Google Scholar]
- Boner, A. L. , Vallone, G. , Brighenti, C. , Schiassi, M. , Miglioranzi, P. , & Richelli, C. (1988). Comparison of the protective effect and duration of action of orally administered clenbuterol and salbutamol on exercise‐induced asthma in children. Pediatric Pulmonology, 4(4), 197–200. 10.1002/ppul.1950040402 [DOI] [PubMed] [Google Scholar]
- Byler, S. L. , Boehm, G. W. , Karp, J. D. , Kohman, R. A. , Tarr, A. J. , Schallert, T. , & Barth, T. M. (2009). Systemic lipopolysaccharide plus MPTP as a model of dopamine loss and gait instability in C57Bl/6J mice. Behavioural Brain Research, 198(2), 434–439. 10.1016/j.bbr.2008.11.027 [DOI] [PubMed] [Google Scholar]
- Culmsee, C. , Junker, V. , Kremers, W. , Thal, S. , Plesnila, N. , & Krieglstein, J. (2004). Combination therapy in ischemic stroke: Synergistic neuroprotective effects of memantine and clenbuterol. Stroke, 35(5), 1197–1202. 10.1161/01.STR.0000125855.17686.6d [DOI] [PubMed] [Google Scholar]
- Culmsee, C. , Stumm, R. K. , Schäfer, M. K. H. , Weihe, E. , & Krieglstein, J. (1999). Clenbuterol induces growth factor mRNA, activates astrocytes, and protects rat brain tissue against ischemic damage. European Journal of Pharmacology, 379(1), 33–45. 10.1016/S0014-2999(99)00452-5 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi, M. , Hallett, P. J. , Koprich, J. B. , Chung, C. Y. , & Isacson, O. (2010). The Toll‐like receptor‐3 agonist polyinosinic: Polycytidylic acid triggers nigrostriatal dopaminergic degeneration. Journal of Neuroscience, 30(48), 16091–16101. 10.1523/JNEUROSCI.2400-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson, D. W. (2012). Parkinson's disease and parkinsonism: Neuropathology. Cold Spring Harbor Perspectives in Medicine, 2(8), a009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, P. , & Pugin, J. (2000). β‐Adrenergic agonists exert their “anti‐inflammatory” effects in monocytic cells through the IκB/NF‐κB pathway. American Journal of Physiology‐Lung Cellular and Molecular Physiology, 279(4), L675–L682. [DOI] [PubMed] [Google Scholar]
- Feinstein, D. L. , Heneka, M. T. , Gavrilyuk, V. , Russo, C. D. , Weinberg, G. , & Galea, E. (2002). Noradrenergic regulation of inflammatory gene expression in brain. Neurochemistry International, 41(5), 357–365. 10.1016/S0197-0186(02)00049-9 [DOI] [PubMed] [Google Scholar]
- Gleeson, L. C. , Ryan, K. J. , Griffin, É. W. , Connor, T. J. , & Harkin, A. (2010). The β2‐adrenoceptor agonist clenbuterol elicits neuroprotective, anti‐inflammatory and neurotrophic actions in the kainic acid model of excitotoxicity. Brain, Behavior, and Immunity, 24(8), 1354–1361. 10.1016/j.bbi.2010.06.015 [DOI] [PubMed] [Google Scholar]
- Guimarães, S. & Moura, D. (2001). Vascularadrenoceptors: anupdate. Pharmacological Reviews, 53(2), 319-356. [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, D. B. , Connaughton, E. , Connaughton, C. , Hogan, G. , Thornton, C. , Mulcahy, P. , … Dowd, E. (2013). Further characterisation of the LPS model of Parkinson's disease: A comparison of intra‐nigral and intra‐striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain, Behavior, and Immunity, 27, 91–100. 10.1016/j.bbi.2012.10.001 [DOI] [PubMed] [Google Scholar]
- Jiang, L. , Chen, S. H. , Chu, C. H. , Wang, S. J. , Oyarzabal, E. , Wilson, B. , … Hong, J. S. (2015). A novel role of microglial NADPH oxidase in mediating extra‐synaptic function of norepinephrine in regulating brain immune homeostasis. Glia, 63(6), 1057–1072. 10.1002/glia.22801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker, V. , Becker, A. , Hühne, R. , Zembatov, M. , Ravati, A. , Culmsee, C. , & Krieglstein, J. (2002). Stimulation of β‐adrenoceptors activates astrocytes and provides neuroprotection. European Journal of Pharmacology, 446(1‐3), 25–36. 10.1016/S0014-2999(02)01814-9 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin, N. W. , & Sanders, V. M. (2006). It takes nerve to tell T and B cells what to do. Journal of Leukocyte Biology, 79(6), 1093–1104. [DOI] [PubMed] [Google Scholar]
- Kirik, D. , Rosenblad, C. , Burger, C. , Lundberg, C. , Johansen, T. E. , Muzyczka, N. , … Björklund, A. (2002). Parkinson‐like neurodegeneration induced by targeted overexpression of α‐synuclein in the nigrostriatal system. Journal of Neuroscience, 22(7), 2780–2791. 10.1523/JNEUROSCI.22-07-02780.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Jiao, Q. , Du, X. , Bi, M. , Han, S. , Jiao, L. , & Jiang, H. (2018). Investigation of behavioral dysfunctions induced by monoamine depletions in a mouse model of Parkinson's disease. Frontiers in Cellular Neuroscience, 12, 241 10.3389/fncel.2018.00241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer, P. L. , & McGeer, E. G. (2004). Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism & Related Disorders, 10, S3–S7. 10.1016/j.parkreldis.2004.01.005 [DOI] [PubMed] [Google Scholar]
- McNamee, E. N. , Griffin, É. W. , Ryan, K. M. , Ryan, K. J. , Heffernan, S. , Harkin, A. , & Connor, T. J. (2010). Noradrenaline acting at β‐adrenoceptors induces expression of IL‐1β and its negative regulators IL‐1ra and IL‐1RII, and drives an overall anti‐inflammatory phenotype in rat cortex. Neuropharmacology, 59(1–2), 37–48. 10.1016/j.neuropharm.2010.03.014 [DOI] [PubMed] [Google Scholar]
- McNamee, E. N. , Ryan, K. M. , Griffin, É. W. , González‐Reyes, R. E. , Ryan, K. J. , Harkin, A. , & Connor, T. J. (2010). Noradrenaline acting at central β‐adrenoceptors induces interleukin‐10 and suppressor of cytokine signaling‐3 expression in rat brain: Implications for neurodegeneration. Brain, Behavior, and Immunity, 24(4), 660–671. 10.1016/j.bbi.2010.02.005 [DOI] [PubMed] [Google Scholar]
- McNamee, E. N. , Ryan, K. M. , Kilroy, D. , & Connor, T. J. (2010). Noradrenaline induces IL‐1ra and IL‐1 type II receptor expression in primary glial cells and protects against IL‐1β‐induced neurotoxicity. European Journal of Pharmacology, 626(2–3), 219–228. 10.1016/j.ejphar.2009.09.054 [DOI] [PubMed] [Google Scholar]
- Michelson, D. , Adler, L. , Spencer, T. , Reimherr, F. W. , West, S. A. , Allen, A. J. , … Milton, D. (2003). Atomoxetine in adults with ADHD: Two randomized, placebo‐controlled studies. Biological Psychiatry, 53(2), 112–120. 10.1016/S0006-3223(02)01671-2 [DOI] [PubMed] [Google Scholar]
- Mittal, S. , Bjørnevik, K. , Im, D. S. , Flierl, A. , Dong, X. , Locascio, J. J. , … Scherzer, C. R. (2017). β2‐Adrenoreceptor is a regulator of the α‐synuclein gene driving risk of Parkinson's disease. Science, 357(6354), 891–898. 10.1126/science.aaf3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya, C. P. , Campbell‐Hope, L. J. , Pemberton, K. D. , & Dunnett, S. B. (1991). The “staircase test”: A measure of independent forelimb reaching and grasping abilities in rats. Journal of Neuroscience Methods, 36(2–3), 219–228. 10.1016/0165-0270(91)90048-5 [DOI] [PubMed] [Google Scholar]
- Nelson, J. C. , Mazure, C. M. , Jatlow, P. I. , Bowers, M. B. Jr. , & Price, L. H. (2004). Combining norepinephrine and serotonin reuptake inhibition mechanisms for treatment of depression: A double‐blind, randomized study. Biological Psychiatry, 55(3), 296–300. 10.1016/j.biopsych.2003.08.007 [DOI] [PubMed] [Google Scholar]
- Olsson, M. , Nikkhah, G. , Bentlage, C. , & Bjorklund, A. (1995). Forelimb akinesia in the rat Parkinson model: Differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. Journal of Neuroscience, 15(5), 3863–3875. 10.1523/JNEUROSCI.15-05-03863.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill, E. , & Harkin, A. (2018). Targeting the noradrenergic system for anti‐inflammatory and neuroprotective effects: Implications for Parkinson's disease. Neural Regeneration Research, 13(8), 1332–1337. 10.4103/1673-5374.235219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan, J. B. , Ryan, K. M. , Curtin, N. M. , Harkin, A. , & Connor, T. J. (2009). Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: Implications for depression and neurodegeneration. International Journal of Neuropsychopharmacology, 12(5), 687–699. 10.1017/S146114570800967X [DOI] [PubMed] [Google Scholar]
- Palkovits, M. , & Brownstein, M. J. (1988). Maps and guide to microdissection of the rat brain. New York: Elsevier. [Google Scholar]
- Paxinos, G. , & Watson, C. (1986). The rat brain in stereotaxic coordinates (2nd ed.). Sydney: Academic Press. [DOI] [PubMed] [Google Scholar]
- Perry, V. H. , & Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nature Reviews Neurology, 10(4), 217–224. 10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- van der Poll, T. , Jansen, J. , Endert, E. , Sauerwein, H. P. , & van Deventer, S. J. (1994). Noradrenaline inhibits lipopolysaccharide‐induced tumor necrosis factor and interleukin 6 production in human whole blood. Infection and Immunity, 62(5), 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott Godoy, M. C. , Tarelli, R. , Ferrari, C. C. , Sarchi, M. I. , & Pitossi, F. J. (2008). Central and systemic IL‐1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain, 131(7), 1880–1894. 10.1093/brain/awn101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, L. , Wu, H. M. , Chen, S. H. , Zhang, D. , Ali, S. F. , Peterson, L. , … Flood, P. M. (2011). β2‐adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. Journal of Immunology, 186(7), 4443–4454. 10.4049/jimmunol.1002449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhall, L. , Elvstrand, A. , Tilling, B. , Vinge, I. , Jemsby, P. , Stahl, E. , … Bergqvist, P. B. (2003). One‐year safety and efficacy of budesonide/formoterol in a single inhaler (Symbicort® Turbuhaler®) for the treatment of asthma. Respiratory Medicine, 97(6), 702–708. 10.1053/rmed.2003.1504 [DOI] [PubMed] [Google Scholar]
- Rosenhall, L. , Heinig, J. H. , Lindqvist, A. , Leegaard, J. , Ståhl, E. , & Bergqvist, P. B. (2002). Budesonide/formoterol (Symbicort) is well tolerated and effective in patients with moderate persistent asthma. International Journal of Clinical Practice, 56(6), 427–433. [PubMed] [Google Scholar]
- Ryan, K. J. , Griffin, É. , Yssel, J. D. , Ryan, K. M. , McNamee, E. N. , Harkin, A. , & Connor, T. J. (2013). Stimulation of central β2‐adrenoceptors suppresses NFκB activity in rat brain: A role for IκB. Neurochemistry International, 63(5), 368–378. 10.1016/j.neuint.2013.07.006 [DOI] [PubMed] [Google Scholar]
- Schallert, T. , Fleming, S. M. , Leasure, J. L. , Tillerson, J. L. , & Bland, S. T. (2000). CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology, 39(5), 777–787. 10.1016/S0028-3908(00)00005-8 [DOI] [PubMed] [Google Scholar]
- Semkova, I. , Schilling, M. , Henrich‐Noack, P. , Rami, A. , & Krieglstein, J. (1996). Clenbuterol protects mouse cerebral cortex and rat hippocampus from ischemic damage and attenuates glutamate neurotoxicity in cultured hippocampal neurons by induction of NGF. Brain Research, 717(1–2), 44–54. 10.1016/0006-8993(95)01567-1 [DOI] [PubMed] [Google Scholar]
- Song, S. , Jiang, L. , Oyarzabal, E. A. , Wilson, B. , Li, Z. , Shih, Y. I. , … Hong, J. S. (2019). Loss of brain norepinephrine elicits neuroinflammation‐mediated oxidative injury and selective caudo‐rostral neurodegeneration. Molecular Neurobiology, 56(4), 2653–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K. F. , Kashima, H. , Suzuki, H. , Ono, K. , & Sawada, M. (2002). Existence of functional β1‐and β2‐adrenergic receptors on microglia. Journal of Neuroscience Research, 70(2), 232–237. 10.1002/jnr.10399 [DOI] [PubMed] [Google Scholar]
- Tansey, M. G. , & Goldberg, M. S. (2010). Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease, 37(3), 510–518. 10.1016/j.nbd.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, Y. D. , Choi, H. , Huang, W. , Onario, R. C. , Frontera, W. R. , Snyder, E. Y. , & Sabharwal, S. (2006). Therapeutic effects of clenbuterol in a murine model of amyotrophic lateral sclerosis. Neuroscience Letters, 397(1–2), 155–158. 10.1016/j.neulet.2005.12.007 [DOI] [PubMed] [Google Scholar]
- Theodore, S. , Cao, S. , McLean, P. J. , & Standaert, D. G. (2008). Targeted overexpression of human α‐synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. Journal of Neuropathology & Experimental Neurology, 67(12), 1149–1158. 10.1097/NEN.0b013e31818e5e99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Kao, S. Y. , Lee, F. J. , Song, W. , Jin, L. W. , & Yankner, B. A. (2002). Dopamine‐dependent neurotoxicity of α‐synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nature Medicine, 8(6), 600–606. 10.1038/nm0602-600 [DOI] [PubMed] [Google Scholar]
- Yssel, J. D. , O'Neill, E. , Nolan, Y. M. , Connor, T. J. , & Harkin, A. (2018). Treatment with the noradrenaline re‐uptake inhibitor atomoxetine alone and in combination with the α2‐adrenoceptor antagonist idazoxan attenuates loss of dopamine and associated motor deficits in the LPS inflammatory rat model of Parkinson's disease. Brain, Behavior, and Immunity, 69, 456–469. 10.1016/j.bbi.2018.01.004 [DOI] [PubMed] [Google Scholar]
- Zarow, C. , Lyness, S. A. , Mortimer, J. A. , & Chui, H. C. (2003). Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Archives of Neurology, 60(3), 337–341. 10.1001/archneur.60.3.337 [DOI] [PubMed] [Google Scholar]
- Zhu, Y. , Culmsee, C. , Semkova, I. , & Krieglstein, J. (1998). Stimulation of β2‐adrenoceptors inhibits apoptosis in rat brain after transient forebrain ischemia. Journal of Cerebral Blood Flow & Metabolism, 18(9), 1032–1039. 10.1097/00004647-199809000-00013 [DOI] [PubMed] [Google Scholar]