

Abstract
Background
Triple-negative breast cancers (TNBCs) are initially responsive to chemotherapy, but most recurrent TNBCs develop resistance. Autophagy is believed to play dual roles in cancer and might contribute to chemoresistance. In this study, we aimed to investigate the role of autophagy and its regulator, eukaryotic elongation factor 2 kinase (eEF2K), in determining the biological nature of TNBC.
Methods
We used in vitro models of TNBC, namely, paclitaxel-resistant cell lines derived from sensitive cell lines. Various approaches to measuring autophagy flux were applied. We assessed the effects of inhibiting autophagy and silencing eEF2K on cell viability, tumor formation and invasion. We also collected residual tumor samples from 222 breast cancer patients who underwent neoadjuvant chemotherapy and measured eEF2K and LC3 expression levels by immunohistochemistry (IHC). Multivariate survival analysis was used to determine prognostic variables.
Results
Compared to the parental lines, the chemoresistant lines exhibited enhanced starvation-stimulated autophagy and showed significant decreases in cell viability, growth and invasion upon treatment with autophagy inhibitors. eEF2K silencing also resulted in the suppression of autophagic activity and in aggressive biological behavior. In the survival analysis, residual tumor LC3 (P=0.001) and eEF2K (P=0.027) expression levels were independent prognostic factors for patients who underwent neoadjuvant chemotherapy, especially in those with TNBC.
Conclusions
Our study indicated that eEF2K and autophagy play key roles in the maintenance of aggressive tumor behavior and chemoresistance in resistant TNBC. eEF2K silencing may be a novel strategy for the treatment of TNBC.
Keywords: Autophagy, triple-negative breast cancer (TNBC), eukaryotic elongation factor 2 kinase (eEF2K), chemo-resistant, neoadjuvant chemotherapy (NAC)
Introduction
Triple-negative breast cancer (TNBC), which lacks expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor-2 (HER2), accounts for 15–20% of all breast cancers and is distinctly aggressive (1). For TNBC, chemotherapy is currently the only treatment option. Notably, TNBC has a higher response rate to chemotherapy than other breast cancer subtypes; however, for those TNBCs that are not sensitive to chemotherapy, the overall survival (OS) is usually poor (2). Thus, TNBC is a heterogeneous disease comprising multiple subtypes with different clinical outcomes. Investigations using gene expression microarrays have resulted in profound changes in the understanding of TNBC (3), specifically revealing that TNBC is not responsive to single-targeted therapy according to its genetic heterogeneity. Therapeutic strategies to prevent angiogenesis (4), block androgen receptor signaling (5), inhibit poly-ADP-ribose polymerase (6), and inhibit EGFR (7) have been reported; however, few approaches have been designed to target chemoresistant TNBC clones.
Macroautophagy, hereafter termed autophagy, is an evolutionarily conserved lysosomal degradation pathway regulated by autophagy-related (ATG) proteins (8). Autophagy is stimulated under conditions of metabolic stress, such as hypoxia, nutrient deprivation or oxidative stress, and autophagic cargos are degraded to redistribute nutrients and eliminate toxic cytoplasmic constituents (9). It is believed that autophagy can suppress tumorigenesis at the early stage of tumor (10,11), autophagy enables tumor cell survival under stressful conditions, especially those caused by antineoplastic therapies. In contrast, the prolonged activation of autophagy may also lead to cell death by cellular self-degradation (12). Thus, although autophagy may play an important role in treatment resistance (13,14), whether the inhibition of autophagy sensitizes resistant tumor cells to cytotoxic therapy remains unknown.
The consumption of metabolic energy, most of which is used for peptide chain elongation modulated by eukaryotic elongation factor 2 (EEF2) and its kinase (eEF2K), is critical for cell survival. eEF2K functions to control the rate of protein chain elongation by phosphorylating/inactivating EEF2 at Thr56 (15). eEF2K may be a potential molecular driver of several human cancers (16-18) and may also induce autophagy to promote cell survival under conditions of starvation, hypoxia or metabolic stress (19). Therefore, eEF2K silencing might suppress autophagy and potentiate the efficacy of anticancer agents. However, conflicting results have been observed (20,21). Thus, the exact role of eEF2K in cancer cell survival and the mechanisms regulating autophagy require further study.
Recent years, one of the main issues in TNBC treatment is the resistance of chemotherapy. Although some studies have suggested that eEF2K might function in tumor progression, invasion and drug resistance in TNBC (22,23), the relationship between eEF2K and autophagy and whether this relationship has therapeutic implications remain controversial. In this study, we aimed to investigate the roles of eEF2K and autophagy in determining the biological nature of TNBC and to examine the potential therapeutic utility of eEF2K in paclitaxel-resistant TNBC. We demonstrated the prognostic value of ATG biomarkers in TNBC patients with residual disease after neoadjuvant chemotherapy (NAC). Our findings provide new evidence of the sustained autophagic potential of chemoresistant TNBCs and show the potential of targeting eEF2K to reverse the aggressive nature of TNBC.
Methods
Antibodies and reagents
Rabbit polyclonal anti-LC3B, anti-ATG7, anti-ATG5, anti-eEF2, anti-phospho-eEF2, and anti-eEF2K antibodies were supplied by Abcam (Cambridge, MA, USA). Chloroquine and Earle’s balanced salt solution (EBSS) were purchased from Sigma (St. Louis, USA).
Cell culture
MDA-MB-231 and MDA-MB-468 cells were obtained from the Shanghai Cell Bank Type Culture Collection Committee (Shanghai, China), and cultured in complete growth medium according to recommendation of the distributor. The MDA-231 and MDA-468 Taxol-resistant sublines (231/Tax and 468/Tax, respectively) were established according to a previous study (24) by stepwise selection over 12 cycles with increasing concentrations of paclitaxel (Bristol-Myers Squibb) ranging from 1 to 12 nM in medium. During each cycle, we cultured 231 and 468 cells with different concentrations of paclitaxel for 2 weeks until new chemo-resistant clones were present, and the concentration of paclitaxel was increased after 2–3 stable generations.
eEF2K shRNA
Lentivirus-based shRNA targeting eEF2K and the controls were obtained from Genechem Co., Ltd. (Shanghai, China). For sheEF2K-1, sheEF2K-2, sheEF2K-3, and sheEF2K-4, the sequences were 5'-ACTCATACAGTAATCGGAA-3', 5'-ATGAGGAAGGTTACTTCAT-3', 5'-ATCCAGAGACCATGATCAT-3', and 5'-GGAAGATATTGCCACCGAA-3', respectively.
Autophagy detection using the monomer red fluorescent protein (mRFP)-green fluorescent protein (GFP) adenoviral vector
Adenoviral infection using mRFP-GFP-LC3B adenoviral vectors was performed under instructions of the manufacturer (HanBio Technology Co. Ltd., Shanghai, China) (25). A total of 1×106 infected MDA-MB-231 or MDA-MB-468 cells were seeded in six-well plates and cultured for 24 hours. Autophagic flux was determined through counting of puncta number of GFP and mRFP in the red and green channels of fluorescence microscope produced by Olympus (BX 51, Tokyo, Japan). The number of puncta in the red channel minus the number in the green channel (net red puncta) was determined manually for 50–100 cells.
3D culture
3D cell culture was carried out according to the previously described “on top” assay protocol. Briefly, 8-well culture plates were coated with 0.5 mL of chilled Matrigel® (BD Biosciences, Oxford, UK) and then incubated at 37 °C for 30 minutes for polymerization. The cells were trypsinized, counted and seeded into the coated plates at 1×105 cells per well, and the remaining chilled medium with 10% Matrigel® was added. After incubation for 3 days, cell clusters were visualized, and photos were taken every 24 hours using an inverted microscope.
Cytotoxicity and invasion assays
Cells in the logarithmic growth phase were used for these assays. To determine the half maximal inhibitory concentration (IC50), cells were seeded into a 96-well plate. The complete medium (CM) was replaced with medium containing serial dilutions of a chemotherapeutic reagent (paclitaxel). The Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Kumamoto, Japan) was used to construct the dose-response curve for each compound. By nonlinear regression analysis with Prism 5.0 (GraphPad Software Inc.), the IC50 was measured. For the invasion assays, Transwell assays (BD Biosciences, Oxford, UK) were performed under standard procedure. A total of 106 cells/mL were seeded in the top chamber and incubated for 8 hours. The average number of infiltrating cells was determined in five randomly selected fields per filter. Each experiment was repeated three times.
Patient cohort and samples
In all, 222 patients with residual disease after NAC were included in this study. All patients were treated from 2003 to 2012 at Fudan University Cancer Center. All patients underwent radical surgery after completion of NAC. After operation, additional cycles of chemotherapy were performed for patients to complete a total of 6 cycles. The NAC regimen comprised paclitaxel and carboplatin (paclitaxel 80 mg/m2 and carboplatin area under the curve (AUC) =2 on days 1, 8, and 15 of a 28-day cycle). And the median cycles for NAC was 4 cycles with a range of 3–6 cycles. All surgical samples were confirmed as residual tumor [non-pathological complete response (non-pCR)]. Data on ER, PR, and HER2 expression were from the pathology at our center. 1% of tumor cells with positive nuclear staining in immunohistochemistry (IHC) was defined as the cut-off value of positivity for both ER and PR. The overexpression of HER2 was defined as 3+ in IHC staining or amplification confirmed by fluorescent in situ hybridization. IHC analyses of tumor samples were also performed to determine LC3 and eEF2K expression.
Western blotting and IHC
Western blotting was performed using a general method. Densitometric analysis was performed using Image-Pro Plus software (v6.0, MD, USA). IHC was performed during standard protocol (2-step, GT Visiontm) on formalin-fixed, paraffin-embedded tissue. LC3B and eEF2K antibodies for IHC were supplied by Abcam (Cambridge, MA, USA). Positive and negative controls were performed according to the instructions of the manufacturer. The H score was used to determine the intensity of staining by the percentage of the positive cells. The H score ranged from 0 to 300, and the samples were characterized according to the H score: 0–100, negative (−); 100–200, moderate positive (+); and 200–300, strong positive (++). Representative immunohistochemical pictures of LC3B and eEF2K staining are shown in Figure S1.
Figure S1.
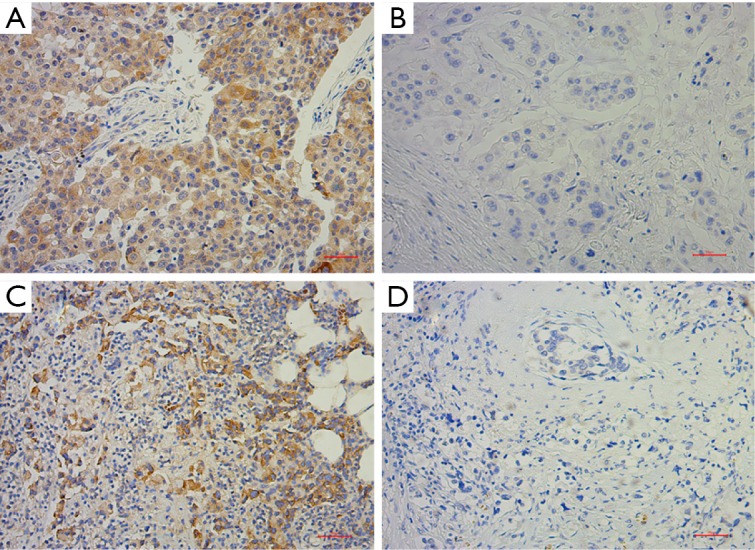
Expression of eEF2K and LC3B in post-NAC breast cancer samples. (A) Representative immunohistochemical pictures of positive eEF2K staining; (B) representative immunohistochemical pictures of negative eEF2K staining; (C) representative immunohistochemical pictures of positive LC3B staining; (D) representative immunohistochemical pictures of positive LC3B staining. Scale bar, 50 µm. eEF2K, eukaryotic elongation factor 2 kinase; NAC, neoadjuvant chemotherapy.
Statistics
The two-tailed Student’s t-test was used for comparisons of two groups. Disease-free survival (DFS) was defined as time from the operation to the first disease relapse or death from any cause. Candidates without any events were censored at the last follow-up. Survival analyses were performed by the Cox regression model, whereas Kaplan-Meier method was used to compare the differences between groups according to patient’s outcome. Two-sided P values less than 0.05 were considered significant. All statistical tests were done by SPSS (version 19.0).
Results
Autophagy affects the characteristics of chemoresistant cell lines
Using the mRFP-GFP adenoviral vector, we measured the basic autophagic level of parental MDA-MB-231 cells and MDA-MB-468 cells, and found a similar low level of LC3 dots. To evaluate the role of autophagy in paclitaxel resistance, we derived polyclonal paclitaxel-resistant TNBC cell lines. The IC50 of paclitaxel in MDA-MB-231-Taxol-resistant (231/Tax) cells was 120-fold higher than that in parental MDA-MB-231 cells [231/Tax: IC50 144.90 nM, 95% confidence interval (CI): 115.80–181.40 nM vs. 231: IC50 1.209 nM, 95% CI: 0.975–1.500 nM, P<0.001, Figure 1A,B]. Similarly, the paclitaxel IC50 in MDA-MB-468-Taxol-resistant (468/Tax) cells was 43-fold higher than that in parental MDA-MB-468 cells (468/Tax: IC50 40.51 nM, 95% CI: 32.37–50.70 nM vs. 468: IC50 0.936 nM, 95% CI: 0.738–1.186 nM, P<0.001, Figure 1C,D). To determine whether autophagy is responsible for the viability and aggressive behavior of TNBC cells, we inhibited autophagy with chloroquine, a commonly used autophagy inhibitor. The IC50 of paclitaxel in 231/Tax cells cotreated with chloroquine was markedly reduced to 11.48 nM, which was 12-fold lower than the IC50 in cells treated with paclitaxel alone. Similarly, the IC50 of paclitaxel in 468/Tax cells cotreated with chloroquine dropped to 10.35 nM, which was 4-fold lower than the IC50 in cells treated with paclitaxel alone. However, chloroquine slightly reduced the IC50 of paclitaxel in the parental cell lines (Figure 1). Furthermore, larger cell spheroids were observed for the chemoresistant lines compared to the parental lines after 3 days in the 3D cell culture system (described in the Methods). Spheroid formation was suppressed significantly by chloroquine in both 231/Tax (P<0.01) and 468/Tax cells (P<0.05), whereas chloroquine slightly weakened the colony formation ability of all parental cell lines (Figure S2A,B). Chloroquine also significantly suppressed the invasive potential of chemoresistant TNBC cells, as measured in Matrigel® Transwell assays (Figure S2C,D).
Figure 1.

The IC50 of paclitaxel (95% CI) in the breast cancer cell lines. (A) MDA-MB-231 parental cells: 1.209 nM (0.975–1.500 nM); (B) MDA-MB-231 resistant cells: 144.90 nM (115.80–181.40 nM); (C) MDA-MB-468 parental cells: 0.936 nM (0.738–1.186 nM); (D) MDA-MB-468 resistant cells: 40.51 nM (32.37–50.70 nM). In the presence of chloroquine, the IC50 value was markedly decreased to 11.48 nM [231] and 10.35 nM [468] in the resistant cell lines and was slightly reduced in the parental cell lines. CI, confidence interval.
eEF2K silencing and autophagic activity
Previous studies have shown that eEF2K can effectively induce autophagy in breast cancer cells. Therefore, we investigated whether eEF2K is a potential therapeutic target in chemoresistant TNBC cells. Silencing eEF2K by shRNA efficiently blocked phosphorylation of its downstream target, eEF2, in 231/Tax and 468/Tax cells, as confirmed by Western blot analysis (Figure S3). However, ATG genes, namely, ATG5, ATG7 and Beclin1, were not affected. As shown in Figure 2, silencing eEF2K markedly suppressed autophagy flux, as shown by the decreases in LC3 dots and LC3-II protein accumulation in eEF2K-depleted cells at both baseline and after EBSS treatment (P<0.001 and P<0.01, respectively). We also examined cell viability and invasion after silencing eEF2K. The IC50 of paclitaxel was 39.5% lower in eEF2K-depleted 231/Tax cells than in control cells (68.24 vs. 112.8 nM, P<0.001) and was 68.5% lower in eEF2K-depleted 468/Tax cells than in control cells (11.86 vs. 37.62 nM, P<0.001, Figure 3A). Additionally, the colony formation of eEF2K-depleted chemoresistant cells was also suppressed after 3 and 6 days in culture (Figure 3B). Transwell assays showed the weakened invasive potential of eEF2K-depleted chemoresistant cells (Figure 3C). The knockdown of eEF2K in TNBC cell lines had decreased cell viability and aggressive biological behavior.
Figure S3.

Western blot analysis of autophagy-related proteins in eEF2K-silenced or control 231 and 468 cells. eEF2K, eukaryotic elongation factor 2 kinase; ATG, autophagy-related.
Figure 2.

Silencing eEF2K reduces autophagic flux in paclitaxel-resistant TNBC cell lines. MDA-MB-231 and MDA-MB-468 paclitaxel-resistant cells were transfected with nontargeting control shRNA (shCTL) and shRNA targeting eEF2K (sheEF2K). Silencing eEF2K markedly suppressed autophagic flux, as shown by the decreased number of LC3 dots and accumulation of LC3-II protein in eEF2K-depleted cells at baseline and after EBSS treatment. (A) Western blot analysis of LC3 I, LC3-II, and eEF2K levels in 231 resistant (231/Tax, left) and 469 resistant (468/Tax, right) cells. The results are representative of three independent experiments (Student’s t-test; **, P<0.01); (B) representative images showing the distribution of LC3 dots (autophagy marker) in eEF2K knockdown cells expressing mRFP-GFP-LC3. Cells were incubated in the presence of Baf A1 (final concentration: 100 nM). The number of GFP+/mRFP+ (yellow) dots was determined in 50 to 100 cells. The data are presented as the mean ± SD of three independent experiments (Student’s t-test; ***, P<0.001). eEF2K, eukaryotic elongation factor 2 kinase; CM, complete medium; EBSS, Earle’s balanced salt solution; TNBC, triple-negative breast cancer; mRFP, monomer red fluorescent protein; GFP, green fluorescent protein; SD, standard deviation.
Figure 3.

Silencing eEF2K reduces cell viability and invasion. (A) The IC50 of paclitaxel in paclitaxel-resistant TNBC cells with or without eEF2K silencing. The IC50 was 39.5% lower in eEF2K-depleted 231/Tax cells than in control cells (68.24 vs. 112.8 nM, P<0.001) and was 68.5% lower in eEF2K-depleted 468/Tax cells than in control cells (11.86 vs. 37.62 nM, P<0.001); (B) spheroid formation of TNBC cells in the 3D culture system. The cells were incubated for 6 days and photographed on days 3 and 6 (scale bar: 100 µm). Colony size was measured as the average area of a single spheroid. The data are presented as the mean ± SD of three independent experiments, and the results were analyzed using Student’s t-test (***, P<0.001). The formation of tumor clones of eEF2K-depleted chemoresistant cells was suppressed at 3 and 6 days; (C) Transwell assays of paclitaxel-resistant TNBC cells (scale bar: 200 µm). These results suggest that silencing eEF2K significantly suppresses the invasive potential of chemoresistant TNBC cells. Data are presented as the mean number of cells/field from three independent experiments, and the results were analyzed using Student’s t-test (**, P<0.01). eEF2K, eukaryotic elongation factor 2 kinase; PF, power field; TNBC, triple-negative breast cancer; SD, standard deviation.
eEF2K and LC3 as prognostic markers in non-pCR responders to NAC
We collected data and surgical specimens from 222 HER2-negative breast cancer patients undergone NAC at our center. All enrolled patients underwent NAC regimens with paclitaxel and carboplatin and were considered non-pCR responders. The median age of patients was 48. Based on the hormone receptor (HR) status, patients were classified into two subtypes: luminal-like (64.9%) and TNBC (35.1%). eEF2K positivity was observed in 126 patients (53 + and 73 ++), and LC3 positivity was observed in 135 patients (70 + and 55 ++). Table 1 shows the relationships between eEF2K expression and patient characteristics. We fail to detect the relationship between eEF2K expression and most of the clinical and pathological features. However, the positivity of eEF2K was more commonly observed in HR− patients (TNBC) than in HR+ patients (luminal-like).
Table 1. Patients characteristics and eEF2K expression.
| Variables | All | eEF2K expression | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Negative (n=96) | Positive (n=126) | P value | |||||||
| No. of patients | % | No. of patients | % | No. of patients | % | ||||
| Age, years | 0.481 | ||||||||
| <40 | 46 | 20.7 | 22 | 22.9 | 24 | 19.0 | |||
| ≥40 | 176 | 79.3 | 74 | 77.1 | 102 | 81.0 | |||
| Menopausal status | 0.164 | ||||||||
| Pre | 127 | 57.2 | 60 | 62.5 | 67 | 53.2 | |||
| Post | 95 | 42.8 | 36 | 37.5 | 59 | 46.8 | |||
| Initial tumor status | 0.137 | ||||||||
| T2 | 72 | 32.4 | 38 | 39.6 | 34 | 27.0 | |||
| T3 | 123 | 55.4 | 48 | 50.0 | 75 | 59.5 | |||
| T4 | 27 | 12.2 | 10 | 10.4 | 17 | 13.5 | |||
| Residual tumor size, cm | 0.111 | ||||||||
| ≤2 | 81 | 36.5 | 42 | 43.8 | 39 | 31.0 | |||
| 2–5 | 113 | 50.9 | 45 | 46.9 | 68 | 54.0 | |||
| >5 | 28 | 12.6 | 9 | 9.4 | 19 | 15.1 | |||
| Residual involved nodes | 0.140 | ||||||||
| 0 | 49 | 22.1 | 20 | 20.8 | 29 | 23.0 | |||
| 1–3 | 57 | 25.7 | 31 | 32.3 | 26 | 20.6 | |||
| ≥4 | 116 | 52.3 | 45 | 46.9 | 71 | 56.3 | |||
| HR* status | 0.006 | ||||||||
| Negative | 78 | 35.1 | 24 | 25.0 | 54 | 42.9 | |||
| Positive | 144 | 64.9 | 72 | 75.0 | 72 | 57.1 | |||
| Vascular invasion | 0.441 | ||||||||
| Negative | 175 | 78.8 | 78 | 81.3 | 97 | 77.0 | |||
| Positive | 47 | 21.2 | 18 | 18.8 | 29 | 23.0 | |||
| Grade | 0.781 | ||||||||
| I–II | 189 | 85.1 | 81 | 84.4 | 108 | 85.7 | |||
| III | 33 | 14.9 | 15 | 15.6 | 18 | 14.3 | |||
| Ki-67 | 0.281 | ||||||||
| Negative (<15%) | 125 | 56.3 | 58 | 60.4 | 67 | 53.2 | |||
| Positive (≥15%) | 97 | 43.7 | 38 | 39.6 | 59 | 46.8 | |||
| LC3 | 0.136 | ||||||||
| Negative | 87 | 39.2 | 43 | 44.8 | 44 | 34.9 | |||
| Positive | 135 | 60.8 | 53 | 55.2 | 82 | 65.1 | |||
*, HR-positive was defined as ER- and/or PR-positive, HR-negative was defined as ER-negative and PR-negative. eEF2K, eukaryotic elongation factor 2 kinase; HR, hormone receptor; ER, estrogen receptor; PR, progesterone receptor.
We then performed survival analyses to determine DFS rates. Multivariate Cox model with backward selection was used to investigate the prognostic value of variables. Table 2 shows the P values, hazard ratios and 95% CIs for all variables. LC3 (P=0.001), eEF2K (P=0.027), Ki-67 (P=0.005) and residual node status (P<0.001) were independent predictors of DFS. Survival distributions according to LC3 and eEF2K status for the different breast cancer subtypes are shown in Figure 4. The patients with LC3+ cancer had a relatively worse DFS than those with LC3− cancer; this trend was significant for the TNBCs (P=0.005) but not for the luminal-like tumors (P=0.162) (Figure 4A,B). Similarly, eEF2K positivity was correlated with poor survival, and this correlation was more significant in the TNBC group (P=0.009) than in the luminal-like tumor group (P=0.036) (Figure 4C,D). Based on the combination of LC3 and eEF2K expression, all TNBC patients were classified into the following four subgroups: eEF2K−/LC3− (n=8); eEF2K−/LC3+ (n=16); eEF2K+/LC3− (n=15); and eEF2K+/LC3+ (n=39). The survival curves of the four risk groups are shown in Figure 4E.
Table 2. Univariate and multivariate survival analysis of factors associated with survival.
| Factors | DFS | ||
|---|---|---|---|
| Univariate (P value) | Multivariate | ||
| P value | Hazard ratio (95% CI) | ||
| Age (<40 vs. ≥40 years) | 0.564 | 0.757 | – |
| Menopausal status (pre vs. post) | 0.803 | 0.305 | – |
| Initial tumor status (T2 vs. T3 vs. T4) | 0.028 | 0.067 | – |
| Residual tumor size (≤2 vs. 2–5 vs. >5 cm) | 0.286 | 0.303 | – |
| Residual involved nodes (0 vs. 1–3 vs. ≥4) | <0.001 | <0.001 | 1.000 |
| 2.344 (0.898–6.118) | |||
| 4.427 (1.890–10.370) | |||
| HR* status (negative vs. positive) | 0.412 | 0.912 | – |
| Vascular invasion (negative vs. positive) | 0.186 | 0.532 | – |
| Grade (I–II vs. III) | 0.885 | 0.580 | – |
| Ki-67 (low vs. high) | 0.011 | 0.005 | 1.968 (1.231–3.144) |
| eEF2K (− vs. + vs. ++) | 0.002 | 0.027 | 1.000 |
| 1.668 (0.860–3.234) | |||
| 2.156 (1.209–3.846) | |||
| LC3 (− vs. + vs. ++) | 0.002 | 0.001 | 1.000 |
| 2.156 (1.128–4.119) | |||
| 3.114 (1.675–5.788) | |||
*, HR-positive was defined as ER- and/or PR-positive, HR-negative was defined as ER-negative and PR-negative. DFS, disease-free survival; CI, confidence interval; HR, hormone receptor; eEF2K, eukaryotic elongation factor 2 kinase; ER, estrogen receptor; PR, progesterone receptor.
Figure 4.

DFS according to LC3 and eEF2K status for different breast cancer subtypes. (A) DFS according to LC3 in luminal-like tumors (P=0.162); (B) DFS according to LC3 in TNBC tumors; LC3 positivity was correlated with poor survival (P=0.005); (C) DFS according to eEF2K in luminal-like tumors; eEF2K positivity was correlated with poor survival (P=0.036); (D) DFS according to eEF2K in TNBC tumors; eEF2K positivity was correlated with poor survival (P=0.009); (E) DFS according to the risk groups classified by LC3 and eEF2K. All TNBC patients were classified into the following four subgroups: eEF2K−/LC3− (n=8); eEF2K−/LC3+ (n=16); eEF2K+/LC3− (n=15); and eEF2K+/LC3+ (n=39) (P=0.004). DFS, disease-free survival; eEF2K, eukaryotic elongation factor 2 kinase; TNBC, triple-negative breast cancer.
Discussion
TNBC is a heterogeneous disease that comprising multiple intrinsic subtypes with different clinical outcomes (3). Owing to its genetic heterogeneity and acquired resistance, TNBC has a relatively higher relapse rate and shorter OS compared to other types of breast cancer. Since there is a lack of definitive targeted therapies for TNBC, the treatment options for TNBC are limited. Optimal chemotherapy agents for TNBC remain unknown, and thus, TNBC is managed with standard chemotherapy comprising paclitaxel in combination with anthracyclines or platinum drugs. At present time, our study is the first to demonstrate the effectiveness of inhibiting autophagy in paclitaxel-resistant TNBC in vitro and in patients who underwent NAC.
Numerous reviews have highlighted a correlation between the autophagy and apoptosis signaling pathways in tumor therapy (26,27). Although autophagy induction is generally observed as a response to diverse anticarcinogenic treatments, studies have reported conflicting results on the effects of autophagy on cell death and survival (28-30). Autophagy may hinder tumorigenesis initially but subsequently enable cancer cell survival in established tumors; moreover, long-term autophagy induction may result in apoptosis. Therefore, the question remains whether autophagy contributes to cell death after cytotoxic therapy or whether autophagy represents a resistance mechanism. The controversy might be due to different models used under different circumstances.
Previous studies have indicated that autophagy is often elevated in chemoresistant tumors since autophagy induction by cytotoxic tumor therapies is evident (31). Chittaranjan et al. reported that epirubicin-resistant TNBC lines exhibited at least 1.5-fold increased basal autophagy levels and showed a significant loss in viability when treated with autophagy inhibitors, indicating the dependence of resistant cells on autophagy for survival (32). In this study, we also focused on autophagy levels in highly resistant TNBC cells derived from sensitive TNBC lines. Autophagic flux was most markedly elevated in starved chemoresistant TNBC cells; however, basal autophagy was only mildly elevated (Figure 1). Therefore, the sustained potential for inducing autophagy under stressful circumstances might be an important feature of chemoresistant TNBC cells and may contribute to aggressive behavior. In concordance with this hypothesis, we observed significant reductions in tumor growth and invasion after inhibiting autophagy with chloroquine in 3D tumor formation and Transwell assays. Our findings also suggest that the induction of autophagy in response to paclitaxel may contribute to the maintenance of chemoresistance. The IC50 values of paclitaxel in the 231/Tax and 468/Tax cell lines cotreated with chloroquine were markedly decreased; however, chloroquine only slightly reduced the IC50 in the parental cell lines, suggesting the therapeutic utility of chloroquine for autophagy inhibition in addition to cytotoxicity.
eEF2K is a well-known negative regulator of protein translation and functions by inactivating eEF2 via phosphorylation at Thr56. eEF2K activity is increased in many malignancies, as eEF2K plays a key cytoprotective role in cancer cells under conditions of nutrient starvation (33). However, the precise functions of eEF2K in carcinogenesis and cancer growth remain unknown. Some studies have suggested that eEF2K may promote autophagy (34,35), which enables the survival of nutrient-starved cells. However, some studies have reported opposite conclusions (21,36). For example, Xie et al. reported that silencing eEF2K enhances ATG gene expression and promotes cell survival via the AMPK-ULK1-dependent pathway (21). To date, no mechanism has been identified to explain how eEF2K regulates autophagy. In this study, we demonstrated that knocking down eEF2K markedly suppressed autophagic flux, as the number of LC3 dots and the accumulation of LC3-II protein were decreased in eEF2K-depleted cells at baseline and after EBSS treatment (Figure 2). Our findings suggest that eEF2K plays a positive regulatory role in inducing autophagy in highly chemoresistant breast cancer.
The clinical utility of eEF2K as a therapeutic target remains unknown. Zhu et al. reported that eEF2K overexpression results in radioresistance, radiation-induced autophagy, and reduced radiation-induced apoptosis compared with the control but that eEF2K silencing promotes radiosensitivity and apoptosis in esophageal squamous cell carcinoma (34). To date, our study is the first to demonstrate the role of eEF2K in modulating the behavior of chemoresistant cells. By silencing eEF2K, we remarkably suppressed the tumor formation, invasion and chemoresistance of the resistant cell lines (Figure 3). Our findings suggest that eEF2K may contribute to the maintenance of the aggressive nature of chemoresistant TNBC, perhaps due to the interaction between eEF2K and ATG pathways since inhibiting autophagy had similar effects on chemoresistant cells. Although the exact signaling mechanism through which eEF2K controls autophagy remains unknown, eEF2K might play a key role in the crosstalk between chemoresistance and autophagy in cancer, which makes eEF2K a novel therapeutic target for TNBC.
Additionally, we investigated eEF2K expression by IHC in samples from 222 patients with HER2− residual tumors after chemotherapy. NAC is widely accepted as the standard treatment for locally advanced breast cancer. Remarkably, despite a relatively higher pCR rate, the OS of TNBC is still unfavorable compared to other breast cancer subtypes. It might have something to do with the acquired treatment resistance to chemotherapy of the non-pCR responders (37). The nature of chemoresistant residual tumors is critical for identifying unique aggressive clones of TNBC. In this study, eEF2K positivity was more commonly observed in the HR− group (TNBC) than in the HR+ group (luminal-like). We also performed a multivariate survival analysis and demonstrated that LC3 (P=0.001), eEF2K (P=0.027), Ki-67 (P=0.005) and residual node status (P<0.001) were independent predictors of DFS. The prognostic values of Ki-67 and node status have already been reported by various studies (38-40), and in our previous study, we showed that LC3 expression was correlated with the survival of non-pCR responders (41). eEF2K positivity was correlated with poor survival, and this correlation was stronger in patients with TNBC (P=0.009) than in those with luminal-like tumors (P=0.036). We also demonstrated that all TNBC patients could be classified into subgroups with different risk levels for disease relapse based on the combination of LC3 and eEF2K expression. Our findings further provide evidence that eEF2K mediated autophagy in residual TNBC tumor cells might result in the resistance to further systemic treatment, and the detection of its expression might help to the predictions of subsequent disease progression. We also suggest that IHC analysis of eEF2K and LC3 can be used to identify particular TNBC clones and show that specific resistant clones have more aggressive biologic behavior than other resistant cancer cell clones.
In conclusion, the current study has shown that eEF2K and autophagy play key roles in the maintenance of aggressive tumor behavior and in the chemoresistance of highly resistant TNBC. Measurement of eEF2K and the autophagy biomarker LC3 can further classify chemoresistant TNBC patients into different risk groups. Our findings further the understanding of the role of autophagy in breast cancer and of the therapeutic vulnerability of TNBC. Given that autophagy is regulated by multiple pathways, we suggest that silencing eEF2K is a potential novel strategy for the treatment of TNBC. Thus, the concept of approaches employing drugs targeting eEF2K as follow-up or rescue treatment for refractory breast cancer is possible.
Figure S2.

Inhibiting autophagy decreases tumor formation and invasiveness in TNBC cells. (A,B) Spheroid formation of MDA-MB-231 (A) and MDA-MB-468 (B) cells in the 3D culture system. Cells were incubated with or without chloroquine (Chlo, 6 µmol/L) for 6 days and photographed on day 4 (scale bar: 100 µm). Colony size was measured as the average area of a single spheroid. The data are presented as the mean ± SD from three independent experiments, and the results were analyzed using Student’s t-test (*, P<0.05; **, P<0.01). Transwell invasion assays of MDA-MB-231 (C) and MDA-MB-468 (D) cells showed that Chlo significantly suppressed the invasive potential of chemoresistant TNBC cells (scale bar: 200 µm). Data are presented as the mean number of cells/field from three independent experiments, and the results were analyzed using Student’s t-test (***, P<0.001). NS, not significant; PF, power field; TNBC, triple-negative breast cancer.
Acknowledgments
Funding: This research was supported by the National Natural Science Foundation of China (No. 81872134), Natural Science Foundation of Shanghai (No. 17ZR1405900), Research Foundation of Shanghai Municipal Commission of Health and Family Planning (No. 20164Y0200), Project of Excellent Youth of Shanghai Municipal Commission of Health and Family Planning (No. 2018YQ42), and Shanghai Sailing Program (No. 18YF1405000). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Our study was approved by the independent ethical committee/institutional review board of Fudan University Shanghai Cancer Center (Shanghai Cancer Center Ethical Committee) (No. 050402-4-1805C). All patients gave their written informed consent before inclusion in this study.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Bauer KR, Brown M, Cress RD, et al. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer 2007;109:1721-8. 10.1002/cncr.22618 [DOI] [PubMed] [Google Scholar]
- 2.Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007;13:2329-34. 10.1158/1078-0432.CCR-06-1109 [DOI] [PubMed] [Google Scholar]
- 3.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121:2750-67. 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackey JR, Ramos-Vazquez M, Lipatov O, et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J Clin Oncol 2015;33:141-8. 10.1200/JCO.2014.57.1513 [DOI] [PubMed] [Google Scholar]
- 5.Gucalp A, Tolaney S, Isakoff SJ, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res 2013;19:5505-12. 10.1158/1078-0432.CCR-12-3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Shaughnessy J, Schwartzberg L, Danso MA, et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol 2014;32:3840-7. 10.1200/JCO.2014.55.2984 [DOI] [PubMed] [Google Scholar]
- 7.Baselga J, Gomez P, Greil R, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol 2013;31:2586-92. 10.1200/JCO.2012.46.2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993;333:169-74. 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- 9.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728-41. 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 10.Liang XH, Yu J, Brown K, et al. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res 2001;61:3443-9. [PubMed] [Google Scholar]
- 11.Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003;1603:113-28. [DOI] [PubMed] [Google Scholar]
- 12.Apel A, Zentgraf H, Buchler MW, et al. Autophagy-A double-edged sword in oncology. Int J Cancer 2009;125:991-5. 10.1002/ijc.24500 [DOI] [PubMed] [Google Scholar]
- 13.John S, Nayvelt I, Hsu HC, et al. Regulation of estrogenic effects by beclin 1 in breast cancer cells. Cancer Res 2008;68:7855-63. 10.1158/0008-5472.CAN-07-5875 [DOI] [PubMed] [Google Scholar]
- 14.Al-Ejeh F, Kumar R, Wiegmans A, et al. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 2010;29:6085-98. 10.1038/onc.2010.407 [DOI] [PubMed] [Google Scholar]
- 15.Kenney JW, Moore CE, Wang X, et al. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv Biol Regul 2014;55:15-27. 10.1016/j.jbior.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 16.Ashour AA, Abdel-Aziz AA, Mansour AM, et al. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis 2014;19:241-58. 10.1007/s10495-013-0927-2 [DOI] [PubMed] [Google Scholar]
- 17.Hamurcu Z, Ashour A, Kahraman N, et al. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016;7:16619-35. 10.18632/oncotarget.7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu XY, Zhang L, Wu J, et al. Inhibition of elongation factor-2 kinase augments the antitumor activity of Temozolomide against glioma. PLoS One 2013;8:e81345. 10.1371/journal.pone.0081345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hait WN, Wu H, Jin S, et al. Elongation factor-2 kinase: its role in protein synthesis and autophagy. Autophagy 2006;2:294-6. 10.4161/auto.2857 [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Gopalakrishnan SM, Bui MH, et al. 1-Benzyl-3-cetyl-2-methylimidazolium iodide (NH125) induces phosphorylation of eukaryotic elongation factor-2 (eEF2): a cautionary note on the anticancer mechanism of an eEF2 kinase inhibitor. J Biol Chem 2011;286:43951-8. 10.1074/jbc.M111.301291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie CM, Liu XY, Sham KW, et al. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy 2014;10:1495-508. 10.4161/auto.29164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu JC, Voisin V, Wang S, et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol Med 2014;6:1542-60. 10.15252/emmm.201404402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tekedereli I, Alpay SN, Tavares CD, et al. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS One 2012;7:e41171. 10.1371/journal.pone.0041171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsaur I, Makarevic J, Juengel E, et al. Resistance to the mTOR-inhibitor RAD001 elevates integrin alpha2- and beta1-triggered motility, migration and invasion of prostate cancer cells. Br J Cancer 2012;107:847-55. 10.1038/bjc.2012.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu X, Fu Y, Tong J, et al. MicroRNA-216b/Beclin 1 axis regulates autophagy and apoptosis in human Tenon's capsule fibroblasts upon hydroxycamptothecin exposure. Exp Eye Res 2014;123:43-55. 10.1016/j.exer.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 26.Kim R, Emi M, Tanabe K, et al. The role of apoptotic or nonapoptotic cell death in determining cellular response to anticancer treatment. Eur J Surg Oncol 2006;32:269-77. 10.1016/j.ejso.2005.12.006 [DOI] [PubMed] [Google Scholar]
- 27.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005;12 Suppl 2:1509-18. 10.1038/sj.cdd.4401751 [DOI] [PubMed] [Google Scholar]
- 28.Bursch W, Ellinger A, Kienzl H, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 1996;17:1595-607. 10.1093/carcin/17.8.1595 [DOI] [PubMed] [Google Scholar]
- 29.Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004;6:1221-8. 10.1038/ncb1192 [DOI] [PubMed] [Google Scholar]
- 30.Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007;117:326-36. 10.1172/JCI28833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondo Y, Kanzawa T, Sawaya R, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005;5:726-34. 10.1038/nrc1692 [DOI] [PubMed] [Google Scholar]
- 32.Chittaranjan S, Bortnik S, Dragowska WH, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res 2014;20:3159-73. 10.1158/1078-0432.CCR-13-2060 [DOI] [PubMed] [Google Scholar]
- 33.Leprivier G, Remke M, Rotblat B, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013;153:1064-79. 10.1016/j.cell.2013.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu H, Song H, Chen G, et al. eEF2K promotes progression and radioresistance of esophageal squamous cell carcinoma. Radiother Oncol 2017;124:439-47. 10.1016/j.radonc.2017.04.001 [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y, Ren X, Zhang Y, et al. Integrated regulation of autophagy and apoptosis by EEF2K controls cellular fate and modulates the efficacy of curcumin and velcade against tumor cells. Autophagy 2013;9:208-19. 10.4161/auto.22801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore CE, Wang X, Xie J, et al. Elongation factor 2 kinase promotes cell survival by inhibiting protein synthesis without inducing autophagy. Cell Signal 2016;28:284-93. 10.1016/j.cellsig.2016.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nahleh Z. Neoadjuvant chemotherapy for “triple negative” breast cancer: a review of current practice and future outlook. Med Oncol 2010;27:531-9. 10.1007/s12032-009-9244-6 [DOI] [PubMed] [Google Scholar]
- 38.Miglietta L, Morabito F, Provinciali N, et al. A prognostic model based on combining estrogen receptor expression and Ki-67 value after neoadjuvant chemotherapy predicts clinical outcome in locally advanced breast cancer: extension and analysis of a previously reported cohort of patients. Eur J Surg Oncol 2013;39:1046-52. 10.1016/j.ejso.2013.06.024 [DOI] [PubMed] [Google Scholar]
- 39.Miglietta L, Vanella P, Canobbio L, et al. Prognostic value of estrogen receptor and Ki-67 index after neoadjuvant chemotherapy in locally advanced breast cancer expressing high levels of proliferation at diagnosis. Oncology 2010;79:255-61. 10.1159/000322189 [DOI] [PubMed] [Google Scholar]
- 40.Wang RX, Chen S, Jin X, et al. Value of Ki-67 expression in triple-negative breast cancer before and after neoadjuvant chemotherapy with weekly paclitaxel plus carboplatin. Sci Rep 2016;6:30091. 10.1038/srep30091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen S, Jiang YZ, Huang L, et al. The residual tumor autophagy marker LC3B serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Clin Cancer Res 2013;19:6853-62. 10.1158/1078-0432.CCR-13-1617 [DOI] [PubMed] [Google Scholar]