

Abstract
Background
To examine whether MLKL participated in the invasion of radiosensitive nasopharyngeal carcinoma (NPC) cell (CNE-2) and radioresistant NPC cell (CR) through regulating epithelial-mesenchymal transition (EMT).
Methods
siRNA and CRISPR/Cas9 technique were used to decrease MLKL expression in NPC cell (CNE-2 and CR). Trans-well assay was conducted to evaluate invasion. Gene expression profiling was performed using Human U133 2.0 plus arrays (Affymetrix). Kyoto Encyclopedia of Genes and Genomes (KEGG) was adopted to analyze gene expression profiling. Hub genes at a functional level were accessed by protein-to-protein network (PPI). Quantitative real-time PCR and Western blot were used to access EMT markers.
Results
Invasion of CR was about 3~fold change higher than that of CNE-2. Silencing MLKL by siRNA inhibited invasion of CR, not CNE-2. Further, depleting MLKL by CRISPR-Cas9 in CR (CR-MLKL KO) also inhibited its invasion. KEGG pathway analysis showed invasion-related pathways were altered, such as adherent junction, TGF-β signaling pathway. PPI demonstrated that compared with CNE-2, CR showed 9 elevated hub genes including EGFR, JUN, CD44, SPP1, VIM, IL-8, BCL2, WDFY2, PIK3CD and 1 downregulated hub gene CDH1. After MLKL depletion, 8 hub genes were downregulated (EGFR, JUN, CD44, SPP1, VIM, FGF13, PLAU, MMP1) and 2 hub genes were upregulated (MMP9, CDH1). Quantitative real-time PCR results showed that compared with CNE-2, CR displayed decreased epithelial markers significantly (E-Cadherin) and increased mesenchymal markers significantly (Vimentin, N-Cadherin, Zeb1), indicating irradiation-induced EMT. After depletion of MLKL in CR, the expression of E-Cadherin, Vimentin, N-Cadherin, Zeb1 was reversed to the level of CNE-2. Western blot confirmed the results from qRT-PCR.
Conclusions
Depletion of MLKL efficiently inhibits invasion of radioresistant NPC by suppressing EMT. MLKL may be an important target to suppress distant metastasis of NPC patients who relapsed after radiotherapy.
Keywords: Nasopharyngeal carcinoma (NPC), necroptosis, epithelial-mesenchymal transition (EMT)
Introduction
Nasopharyngeal carcinoma (NPC) is one of the most commonly diagnosed head and neck malignancies in Southern China and Southeast Asia (1). The incidence of NPC is as high as 18.7/100,000 in endemic regions. The prevailing use of intensity-modulated radiotherapy (IMRT) and combined chemoradiation therapy have substantially improved the outcomes of NPC in terms of overall survival and disease control (2). Despite of the aggressive chemotherapy strategies used, distant metastases remain the most important reason of treatment failure (3-6).
Cancer metastasis is comprised of numerous steps. It is usually initiated by the gaining of invasive phenotype of the cancer cells (7). Epithelial-mesenchymal transition (EMT) plays a significant role in tumor invasion, which is characterized by decreased expression of epithelial markers and increased expression of mesenchymal markers (8,9). Tumor cells that undergo EMT acquire mesenchymal phenotype and become more invasive. The association between EMT and metastasis in NPC as well as its mechanisms have been previously reported (10,11). Recently, an increasing number of studies have demonstrated treatment-induced invasion, including those induced by ionizing radiotherapy, involves EMT through the TGF-β/SMAD pathway (12-15). Results from in vitro studies have indicated that the suppression of EMT could potentially lead to the inhibition of tumor metastasis in a number of malignancies including breast and prostate cancer (16,17). Therefore, suppressing EMT is considered as a promising strategy to inhibit metastasis of cancers including NPC.
Tumor necrosis, traditionally regarded as an un-programmed process which can be induced by ionizing radiation, is known to play an important role in tumorigenesis (18). Recently, “necroptosis”, i.e., a regulated form of necrosis which involves receptor interacting protein kinases 1 and 3 (RIPK1 and RIPK 3) and mixed lineage kinase like (MLKL) has been reported (19). Upon necroptotic stimuli, RIP1 recruits RIP3, forming necrosome leading to RIP3 phosphorylation, which then activates MLKL by phosphorylation (20). Activated MLKL translocates to the plasma membranes and makes pore structures in the membrane, which eventually leads to the disruption of membrane permeability. MLKL has been demonstrated to activate cell-surface proteases of ADAM family promoting cell invasion in colon cancer cell (HT-29) (21), while MLKL-depletion in breast cancer cells (MVT-1) has been found to reduce the metastatic foci in lung (22). Taken together, MLKL appears to play an important role in cancer invasion and metastasis.
Results of studies for non-cancer human diseases have suggested an association between MLKL and the regulators of epithelial/mesenchymal cell status (23,24). For example, a negative relationship between p-MLKL and E-cadherin was observed in intestinal mucosal samples of pediatric patients with inflammatory bowel disease, while activated MLKL was found to alter E-cadherin and reduce cell-cell adhesion in vitro (23). On the other hand, necrosulfonamide, a pharmacological inhibitor of MLKL has been found to decrease α-SMA, coll1α, and vimentin expressions (involved in EMT) in LX-2 cell line and impair wound healing (24). However, it is unclear whether MLKL regulates invasion through EMT in any cancer cells. Moreover, the role of MLKL in the invasion and metastasis of NPC has never been investigated. Therefore, the present study was conducted to investigate whether MLKL can regulate invasion of NPC through EMT.
Methods
Cell culture
CNE-2 cells (radiosensitive NPC cells) were kindly provided by Xiangya Hospital (Changsha, Hunan, China). CR cells (radioresistant NPC cells) were established by repeated X-ray irradiation as previously reported (25). All cells were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 1% penicillin/streptomycin, at 37 °C in a humidified atmosphere containing 5% CO2.
Invasion assay
Cell invasion was measured in Trans-well chambers with 8.0 µm pore size (Falcon). Fifty thousand (50,000) cells were seeded on filters coated with 50 µg/cm2 of reconstituted Matrigel basement membrane (BD Biosciences) with 10% FBS-containing 1640 medium in the lower chamber and serum-free medium in the upper chamber. After 24 hours of incubation, cells were fixed using methanol, stained with crystal violet 0.5% and counted in five random fields under a light microscope (Nikon ECLIPSE Ni).
Lung metastasis model
Animal experiments were approved by animal ethics committee of Shanghai Proton and Heavy Ion Center (SPHIC). Female BALB/c nude mice (6 weeks) were purchased from The Lingchang Bio-Technology Company (Shanghai, China). Fifty thousand (500,000) cells suspended in 200 µL PBS were intravenously injected into nude mice through the tail vein. The mice were sacrificed 10 weeks later and their lungs were fixed, paraffin-embedded, sectioned, and stained with H&E.
Silencing of MLKL
SiRNA was used for silencing of MLKL. CNE-2 and CR cells seeded onto 6-well plates were transfected with MLKL siRNA (5'-CCUGCGUAUAUUUGGGAUUTT-3', 5'-AAUCCCAAAUAUACGCAGGTT-3') using Lipofectamine-2000 (Invitrogen). After 6 hours of incubation with serum-free medium and another 48 h in 10% serum-containing medium, protein was isolated and Western blot was performed to access transfection efficiency.
Gene editing
The MLKL knocked-out CR cells (CR-MLKL KO) were constructed by Shanghai Sunbio Medical Biotechnology. The following sgRNA sequences were used: sgRNA1: GGCAGCTGGAGCCACGTCGG; sgRNA2: GAGAAGACCTAGAACTGAGG. Annealed sgRNA oligonucleotides targeting MLKL were cloned into pSB1198 plasmid (Sunbio Medical Biotechnology, Shanghai, China). Transfection was carried out using Lipofectamine 2000 (Life technologies) at approximately 60% confluence, according to the manufacturer’s instructions. Cells were incubated and subsequently single-cell-diluted in 96-well plates. Clones without MLKL were selected on the basis of Western blot analysis.
Global gene expression by DNA microarray analysis
Cells from CNE-2, CR, CR-MLKL KO were scraped and centrifuged. The cell pellets were flash frozen by immediately transferring to liquid nitrogen and held for at least 15 minutes. All samples were hybridized to Human U133 2.0 plus arrays (Affymetrix). We defined differential expression gene (DEG) as genes with a fold change of over 3 compared with the respective control group. KEGG pathway enrichment was performed. Enrich factor used to evaluate the extent of enrichment was defined as (the number of DEG in specific term/total DEG)/(the number of total genes in specific term of the database/the number of total genes in the database). Protein-to-protein interactions network was generated by uploading DEG in STRING database (http://string-db.org/cgi/input.pl) and drawn by Cytoscape.
qRT-PCR
Total RNA was extracted from cells using TIANGEN DNA & RNA Isolation Kit. RNA quantity and quality were assessed using the Nanodrop-Lite (Thermo Scientific). cDNA was synthesized using a Primescript RT reagent kit with gDNA eraser (TAKARA). RT-qPCR analyses were performed using TB Green Premix EX Taq (TAKARA) to determine the gene expression of E-Cadherin, Keratin 18, N-Cadherin, Vimentin, TWIST1, SNAIL1, ZEB1, ZEB2, β-catenin and results were normalized to β-Actin housekeeping gene transcripts. Primers were listed in Table 1.
Table 1. Primers.
| Gene | Sequence (5'-3') |
|---|---|
| E-Cadherin-F | CGAGAGCTACACGTTCACGG |
| E-Cadherin-R | GGGTGTCGAGGGAAAAATAGG |
| KRT18-F | TCGCAAATACTGTGGACAATGC |
| KRT18-R | GCAGTCGTGTGATATTGGTGT |
| N-Cadherin-F | TGCGGTACAGTGTAACTGGG |
| N-Cadherin-R | GAAACCGGGCTATCTGCTCG |
| Vimentin-F | AGTCCACTGAGTACCGGAGAC |
| Vimentin-R | CATTTCACGCATCTGGCGTTC |
| TWIST1-F | GTCCGCAGTCTTACGAGGAG |
| TWIST1-R | GCTTGAGGGTCTGAATCTTGCT |
| SNAI1-F | TCGGAAGCCTAACTACAGCGA |
| SNAI1-R | AGATGAGCATTGGCAGCGAG |
| ZEB1-F | TTACACCTTTGCATACAGAACCC |
| ZEB1-R | TTTACGATTACACCCAGACTGC |
| ZEB2-F | GCGATGGTCATGCAGTCAG |
| ZEB2-R | CAGGTGGCAGGTCATTTTCTT |
| CTNNB1-F | CATCTACACAGTTTGATGCTGCT |
| CTNNB1-R | GCAGTTTTGTCAGTTCAGGGA |
| β-Actin-F | CATGTACGTTGCTATCCAGGC |
| β-Actin-R | CTCCTTAATGTCACGCACGAT |
Protein analysis by Western blotting
Proteins from 20 µg of whole cell lysate were separated by 7.5% SDS-PAGE (Bio-Rad) and transferred to PVDF membranes (Bio-Rad). Membranes were blocked for 1 hour, followed by incubation with rabbit polyclonal antibodies against MLKL, E-Cadherin, N-Cadherin and β-Actin (Cell Signaling Technology) overnight at 4 °C. The membranes were then washed three times and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technology) for 1 hour. After three final washes, the blots were visualized using an ECL detection system (Bio-Rad).
Statistical analysis
We evaluated the statistical significance using Student’s t‐test by the GraphPad. Prism (GraphPad Software, San Diego, California, USA). P<0.05 was considered as statistically significant. Each experiment was repeated at least three times unless otherwise specified. Microarray analysis was conducted once.
Results
Silencing MLKL inhibited invasion of CR
Activation of MLKL by phosphorylation is known to result in necroptosis, which has been linked to MLKL mediated cell migration (21,22). Therefore, we analyzed the p-MLKL status in these cells and found no detectable p-MLKL in CNE-2 and CR cells (Figure 1A). Further, the endogenous level of MLKL was marginally (~20%) higher in CR as compared to CNE-2 (Figure 1B). To further investigate whether MLKL is associated with invasion of NPC cells, we transiently knockdown the expression of MLKL by using siRNA in both CNE-2 and CR and achieved a silencing efficiency of 70% (Figure 1B). Interestingly, trans-well assay carried out to evaluate tumor cell invasion showed a higher degree of invasion (3-fold) of CR cells as compared to CNE-2 (Figure 1C,D). Silencing MLKL profoundly inhibited invasion in CR while a significant change was not observed in CNE-2 (Figure 1C,D), indicating MLKL may play more important role in the invasion of radioresistant NPC cells. These observations suggested that repeated irradiation of NPC cells leads to enhanced invasive capacity, besides the acquisition of radio-resistance reported earlier (25).
Figure 1.

Silencing MLKL inhibited invasion of CR, not CNE-2. (A) Phosphorylated MLKL was not detected in CNE-2 and CR; (B) Western blot analysis of MLKL in CNE-2, MLKL-silenced CNE-2, CR, and MLKL-silenced CR. The level of MLKL in CNE-2 was set as 1; (C) Trans-well assay was used to compare the invasive capabilities of cells; (D) quantification of Trans-well assay. *, significant difference (P<0.05). MLKL, mixed lineage kinase like.
Depletion of MLKL impaired invasion of CR
To investigate the mechanisms underlying MLKL mediated cell invasion in the radioresistant NPC cells (i.e., CR), we used CRISPR/Cas9 to knockout MLKL in CR. The depletion of MLKL in CR (referred to as CR-MLKL KO) was confirmed by Western blot (Figure 2A). MLKL knockout also led to changes in morphological features of CR-MLKL KO with less pseudopodia and more round-like shape compared with CR (Figure 2B). The spindle-like cell shape and pseudopodia are well-known morphological features of mesenchyme cells (12). These observations suggested that MLKL depletion may have an influence on epithelial to mesenchyme transformation (EMT).
Figure 2.
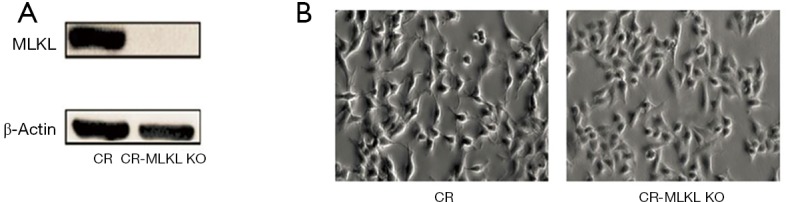
Depletion of MLKL (by CRISPER-Cas9) was associated with morphological changes. (A) Depletion of MLKL confirmed by Western blot; (B) morphological features showing less pseudopodia and more round-like shape in CR-MLKL KO by light microscopy. Original magnification: 200×. MLKL, mixed lineage kinase like.
In vitro assay demonstrated that the invasion of CR-MLKL KO was substantially reduced as compared with CR (Figure 3A), consistent with our observations with siRNA. We also studied the impact of MLKL on lung metastasis in vivo by injecting CNE-2, CR and CR-MLKL KO through the tail vein and enumerating metastasis foci in the lung. No significant lung metastasis foci could be observed in CR-MLKL KO group at 10 weeks following injection, while foci were evident in CNE-2 and CR groups (Figure 3B). However, no significant difference between CR and CNE-2 groups were observed. Thus, both in vitro and in vivo results revealed that MLKL depletion reduced the invasive behavior of CR.
Figure 3.

Deletion of MLKL impaired invasion of CR both in vitro and in vivo. (A) Stained images from Trans-well assay showing changes in the degree of invasive capabilities of CNE-2, CR, CR-MLKL KO; (B) representative image of H&E stained lung metastasis. Arrow indicates lung metastasis foci. *, significant difference (P<0.05). Magnification: 200×. MLKL, mixed lineage kinase like.
Differential gene expression between CNE-2, CR and CR-MLKL KO
To explore potential mechanisms underlying the regulation of invasion by MLKL in NPC cells, we carried out global gene expression using microarray and conducted KEGG pathway enrichment to gain insight into alterations in the pathways indicated by changes in the gene expression after MLKL depletion. Some of the invasion-associated pathways such as adherent junction, TGF-β signaling pathway, ECM-receptor interaction were among the top 10 enrichment pathways affected following repeated irradiation and MLKL depletion (Table 2). These pathways are reported to be closely related to irradiation-induced EMT (26,27).
Table 2. KEGG pathway enrichment analysis.
| Pathway ID | Description | Up-regulated genes | Down-regulated genes | Enrichment factor | P |
|---|---|---|---|---|---|
| CNE-2 vs. CR | |||||
| hsa00100 | Steroid biosynthesis | SQLE, LSS, MSMO1, TM7SF2, CYP2R1, CEL, FDFT1 | 2.7 | 0.0108 | |
| hsa04520 | Adherens junction | CDC42, FYN, LEF1, FGFR1, PARD3, PVRL1, SMAD4, PTPRJ, SNAI1, IGF1R, LMO7, CTNNB1, TCF7L2, INSR, ACTN1, SMAD3, TGFBR1, PTPRM, CSNK2A2, EGFR | ERBB2, CDH1, SORBS1, PTPRB | 2.5 | 5.78E-05 |
| hsa05219 | Bladder cancer | CXCL8, RPS6KA5, MDM2, MMP1, TYMP, HBEGF, RASSF1, EGFR | THBS1, ERBB2, MMP9 CDH1, ARAF | 2.44 | 0.002628 |
| hsa05213 | Endometrial cancer | LEF1, PIK3CD, CTNNB1, PIK3R1, TCF7L2, AKT3, AXIN2, EGFR | AKT2, APC2, PTEN, ERBB2, CDH1, ARAF, ELK1, PIK3R5 | 2.37 | 0.001392 |
| hsa05210 | Colorectal cancer | LEF1, TGFB3, SMAD4, PIK3CD, CTNNB1, PIK3R1, TCF7L2, BCL2, AKT3, JUN, SMAD3, TGFBR1, AXIN2, TGFB1 | AKT2, APC2, ARAF, CYCS, PIK3R5 | 2.36 | 0.000598 |
| hsa00051 | Fructose and mannose metabolism | HKDC1, ALDOC, DAK | ALDOB, PMM2, C12ORF5, FPGT, AKR1B10, TPI1 | 2.1 | 0.02581 |
| hsa05211 | Renal cell carcinoma | CDC42, ARNT2, TGFB3, GAB1, ETS1, PIK3CD, PIK3R1, AKT3, JUN, PAK1, TGFB1 | AKT2, TCEB2, CUL2, RAP1A, ARAF, PAK6, PIK3R5 | 2.07 | 0.003591 |
| hsa04512 | ECM-receptor interaction | COL4A6, HSPG2, CD44, COL4A5, ITGA1, THBS3, ITGB4, LAMA2, CD47, SPP1, LAMB2, ITGA5, TNC, COL6A2, ITGA3, COL4A1, COL6A1, COL4A2 | THBS1, GP6, ITGA9, ITGB1 | 2.07 | 0.001506 |
| hsa05100 | Bacterial invasion of epithelial cells | CDC42, SEPT6, CLTA, GAB1, PIK3CD, CTNNB1, PIK3R1, SEPT8, ITGA5, DNM3, CTTN, SHC4 | SEPT3, ARPC3, CAV2, CDH1, ITGB1, ARPC5L, ARHGEF26, PIK3R5 | 1.98 | 0.003974 |
| hsa03320 | PPAR signaling pathway | PLIN2, EHHADH, SLC27A1, CPT1C, MMP1, ACOX3, ACSL4, PPARD, PPARA, PCK1, PCK2, ADIPOQ, ACSL5 | OLR1, FABP5, RXRA, SORBS1, SLC27A2 | 1.93 | 0.007707 |
| CR vs. CR-MLKL KO | |||||
| hsa00740 | Riboflavin metabolism | RFK, TYR | 3.66 | 0.03656 | |
| hsa00730 | Thiamine metabolism | ALPI, ALPP, AK1, ALPPL2, NTPCR, THTPA | 2.74 | 0.01473 | |
| hsa04350 | TGF-beta signaling pathway | ID2, ID3, BAMBI, SMAD2, BMP2, SMAD6, SMAD9, INHBB, CREBBP | BMP5, TGFB3, SMURF1, TGFB2, THBS1, ID4, DCN, SMAD4, BMPR1B, RBL1, FST, ACVR1B, ZFYVE16, TGFB1, SMURF2, TGFBR1, SMAD3, INHBA | 2.35 | 6.27E-05 |
| hsa05219 | Bladder cancer | MMP9, CDH1 | RPS6KA5, THBS1, CXCL8, ERBB2, FGFR3, MDM2, VEGFA, HBEGF, MMP1, TYMP, EGFR | 2.32 | 0.004185 |
| hsa00531 | Glycosaminoglycan degradation | HYAL3, ARSB | HEXA, IDUA, IDS, SGSH | 2.31 | 0.03468 |
| hsa04520 | Adherens junction | FYN, SMAD2, CREBBP, CDH1, PTPRB, SORBS1 | MET, CDC42, LEF1, ERBB2, RAC2, CTNNB1, FGFR1, PVRL1, BAIAP2, SMAD4, SSX2IP, PTPRF, TCF7L2, EGFR, TGFBR1, SMAD3, PTPRM | 2.27 | 0.00032 |
| hsa05210 | Colorectal cancer | APC2, SMAD2, GSK3B, BAX | TGFB3, TGFB2, LEF1, RAC2, CTNNB1, SMAD4, JUN, PIK3R1, FOS, RALGDS, TCF7L2, TGFB1, APC, TGFBR1, SMAD3 | 2.24 | 0.001124 |
| hsa04512 | ECM-receptor interaction | ITGB1 | CD44, COL4A5, THBS3, THBS1, HSPG2, CD47, ITGA6, LAMB1, LAMC3, LAMA3, ITGB4, ITGA5, LAMC2, TNC, ITGA3, COL6A2, SPP1, LAMA4, LAMB3, ITGA2, COL6A1, COL4A2, COL4A1 | 2.14 | 0.000589 |
| hsa05216 | Thyroid cancer | CDH1, RXRA | RXRB, LEF1, CTNNB1, TFG, TPM3, TCF7L2 | 2.02 | 0.04142 |
| hsa05211 | Renal cell carcinoma | CUL2, TCEB2, RAP1A, CREBBP | PDGFB, ARNT2, TGFB3, MET, TGFB2, CDC42, VHL, GAB1, VEGFA, JUN, PIK3R1, TGFB1, SLC2A1, PAK1 | 1.97 | 0.006236 |
DEGs, differentially expressed genes; KEGG, Kyoto encyclopedia of genes and genomes.
To further examine the most relevant genes that are the affected by repeated irradiation (CNE-2 vs. CR) or MLKL depletion (CR vs. CR-MLKL KO), we analyzed protein to protein interactions using String database and Cytoscape software (Figure 4). Among the top 10 hub genes, CR showed elevation in 9 hub genes including EGFR, JUN, CD44, SPP1, VIM, IL-8, BCL2, WDFY2, PIK3CD and 1 downregulated hub gene CDH1 (E-cadherin) as compared to CNE-2. On the other hand, MLKL depletion resulted in the down-regulation of 8 hub genes (EGFR, JUN, CD44, SPP1, VIM, FGF13, PLAU, MMP1), while 2 hub genes were upregulated (MMP9, CDH1). As CDH1 (E-cadherin) and VIM (Vimentin) are important markers of EMT (28), these observations suggest that EMT is significantly influenced by repeated irradiation and MLKL depletion.
Figure 4.

Protein-to-protein interaction analysis showing EMT is influenced by repeated irradiation and MLKL depletion. (A) Top 10 hub genes between CR and CNE-2; (B) top 10 hub genes between CR and CNE-2. Red indicts upregulation. Green indicates downregulation. EMT, epithelial-mesenchymal transition; MLKL, mixed lineage kinase like.
EMT is induced in radio-resistant cells and suppressed by MLKL depletion
To validate the findings of microarray suggesting MLKL-mediated suppression of EMT and to study the effects of irradiation, we carried out qRT-PCR for the expression of selected genes and their protein levels by Western blot analysis. Based on the literature, we selected nine EMT markers to evaluate their expression that included two epithelial markers (E-Cadherin, and keratin 18) and seven mesenchymal markers (Vimentin, N-Cadherin, β-catenin, Snail1, Zeb1, Zeb2, and Twist) (29). Results demonstrated that the epithelial marker E-Cadherin was significantly decreased, while mesenchymal markers (Vimentin, N-Cadherin, Zeb1) were increased in CR cells created by repeated irradiation (25) (Figure 5). These observations were consistent with the differences in the migration (Figure 1) and earlier reports on radiation-induced EMT (30). In addition, lack of significant changes in some of the markers such as keratin 18, β-catenin, Snail1, and Twist suggested that these cytoskeletal markers or transcription factors did not participate in radiation-induced EMT in NPC. In contrast, the expressions of EMT markers in CR-MLKL KO were similar to CNE-2, thereby strengthening the suggestion that MLKL depletion inhibited EMT in CR cells.
Figure 5.
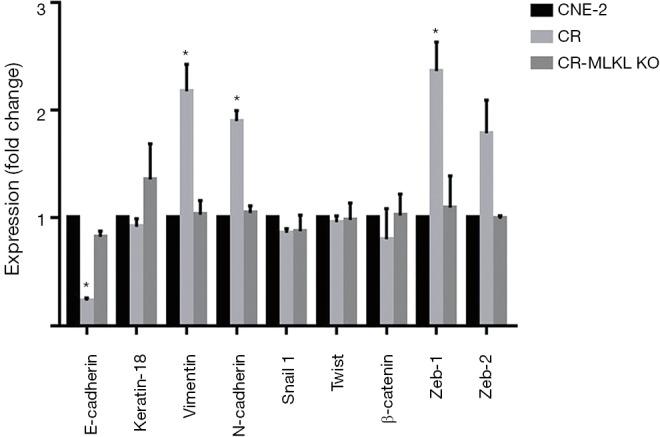
EMT is induced by irradiation and suppressed by MLKL deletion. Expression levels of EMT related genes analyzed by qRT-PCR in CNE-2, CR, CR-MLKL KO. Results are expressed as means ± SD. Statistical analysis was carried out with Student’s t‐test. *, significant difference compared with CNE-2 (P<0.05). EMT, epithelial-mesenchymal transition; MLKL, mixed lineage kinase like.
Analysis of protein by Western blot showed that the levels of E-Cadherin, Vimentin and Zeb1 which were elevated in the radioresistant CR cells as compared to CNE-2, were restored to levels comparable with CNE-2 in CR-MLKL KO (Figure 6). Significant changes were not observed in N-cadherin due to large variation in three independent experiments. Taken together, both mRNA and protein level data clearly showed knockout of MLKL suppressed EMT in radioresistant NPC cells.
Figure 6.
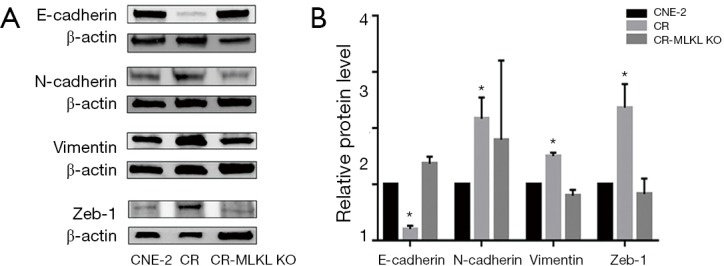
Western blot of EMT markers in CNE-2, CR and CR-MLKL KO. (A) Representative band of Western blot; (B) quantification of Western blot. *, significant difference (P<0.05). β-actin was used as a loading control. EMT, epithelial-mesenchymal transition; MLKL, mixed lineage kinase like.
Discussion
Acquisition of metastatic phenotype during advanced stages of malignancy as well as induction of invasion following photon-based radiotherapy contributes to poor prognosis (12,31). The role of MLKL in tumor invasion has been poorly investigated with limited studies suggesting a potential link between necroptosis mediated by activated MLKL and tumor invasion as well as metastasis (21,22). In the present study, a robust invasion of untreated CR cells as compared to radiosensitive CNE-2 cells was not associated with phosphorylated MLKL, which was negligible in both cells. However, the level of total MLKL in highly invasive CR cells was moderately higher (~20%) than CNE-2 cells. Most importantly, depletion of MLKL either by siRNA or by CRISPER-Cas9 severely compromised invasion of CR accompanied by reverse of EMT, suggesting that total MLKL played significant role in invasion of radioresistant NPC cell. To the best of our knowledge, this is the first experimental study showing the role of total MLKL in EMT, cell invasion and metastasis, thereby suggesting that MLKL as a potential target for reducing treatment-induced invasion and metastasis of radioresistant NPC.
EMT is one of the most significant mechanisms involved in cancer invasion, with epithelial cells transitioning to mesenchymal cells losing polarity to become more motile (28). The process of EMT is regulated by several proteins located on cell-surface (N-Cadherin, E-Cadherin etc.), cytoskeleton (Vimentin, Keratin 18, α-SMA etc.), transcription factors (Zeb1, Snail etc.) and extra-cellular matrix (Fibronectin etc.) (29). Photon beam irradiation is well known to induce EMT in various types of tumors, including glioma (32), lung cancer (33), esophageal cancer (13), gastric cancer (34), liver cancer (35), colorectal cancer (12), cervical cancer (14), endometrial cancer (15), which is partly responsible for poor prognosis and treatment-induced metastasis. In line with these observations, the radioresistant NPC cells (CR) generated by continuous irradiation of CNE-2 cells showed EMT and a higher degree of invasion as well as metastasis in the present study. While the role of MLKL in EMT has not been investigated in tumors so far, its activated (phosphorylated) form has been implicated in EMT during hepatic fibrosis as blocking necrosome formation with necrosulfonamide that prevents activation and translocation MLKL to the cell membrane suppressed mesenchymal phenotype of hepatic stellate cell (LX-2) (24). In the present study, EMT in CR was inhibited by MLKL depletion with either siRNA or CRISPER-Cas9 (CR-MLKL KO) indicating a possible involvement of MLKL in EMT and invasion. Moreover, alterations in invasion-associated pathways such as adherent junction, TGF-β signaling pathway observed here following MLKL depletion (Table 2) that are reported to be linked to irradiation-induced EMT (12) also lend support to the involvement of MLKL in EMT and invasion. Recently, an increasing number of therapeutic agents that inhibit EMT has been under investigation. For instance, withaferin A, purified from the plant withania somnifera, impaired EMT by targeting vimentin in breast cancer (16). Monoclonal antibody targeting N-cadherin inhibited invasion of prostate cancer cell (17). Additionally, an endogenous estradiol metabolite-2-Methoxyestradiol was demonstrated to reverse EMT in NPC cells (36) and is now under clinical trials (NCT00030095). Further, besides EMT, the protein-protein interaction network analysis also revealed a differential pattern between CNE-2 vs. CR and CR vs. CR-MLK KO in certain markers of stem-like cells (CD44), radioresistance (EGFR), angiogenesis (PLAU), suggesting pleiotropic functions of MLKL. Therefore, development of potential treatment strategies by targeting MLKL has a great promise in NPC therapeutics. Mechanisms underlying MLKL-mediated invasion and metastasis independent of its activation (phosphorylation) and correlations with EMT are presently not clear. Whether it is linked to the endosomal trafficking and extracellular vesicle generation (37), which have been recently shown to be mediated by MLKL independent of its activation needs to be investigated in future.
Earlier studies have shown that MLKL may have either anti-tumor or pro-tumor effects depending on the type of cancer or clinical scenario. For instance, lower MLKL has been found to be associated with reduced overall and relapse-free survival in patients with resected pancreatic adenocarcinoma, especially following adjuvant chemotherapy (38), while a better prognosis has been observed in patients with higher MLKL in ovarian (39), cervical (40) and gastric cancers (41). However, a negative correlation between MLKL and prognosis has also been reported in esophageal and colon cancers (42). Similarly, our recent study has also shown that higher MLKL expression is related to worse overall survival and disease-free survival in low grade glioma and glioblastoma (43). Relationship between MLKL level and invasion as well as metastasis observed among CNE-2, CR and CR-MLKL KO cells here suggests that MLKL may be a negative prognostic indicator in NPC purely from the view point of disease dissemination.
Conclusions
In conclusion, MLKL depletion inhibits EMT in NPC cells and abolishes invasion of radioresistant NPC cells, thereby suggesting MLKL as a potentially promising molecular target for developing therapeutic strategies in the future.
Acknowledgments
Funding: This work was supported by National Key Research and Development Program of China (Project No. 2017YFC0108603), Science and Technology Commission of Shanghai Municipality (Project No. 19JC1414800). Science and Technology Development Fund of Shanghai Pudong New Area (Project No. PKJ2016-Y41). Shanghai Municipal Health Commission (Project No. 201640024).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Animal experiments were approved by animal ethics committee of Shanghai Proton and Heavy Ion Center (SPHIC).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Tang LL, Chen WQ, Xue WQ, et al. Global trends in incidence and mortality of nasopharyngeal carcinoma. Cancer Lett 2016;374:22-30. 10.1016/j.canlet.2016.01.040 [DOI] [PubMed] [Google Scholar]
- 2.Chua MLK, Wee JTS, Hui EP, et al. Nasopharyngeal carcinoma. Lancet 2016;387:1012-24. 10.1016/S0140-6736(15)00055-0 [DOI] [PubMed] [Google Scholar]
- 3.Lee N, Xia P, Quivey JM, et al. Intensity-modulated radiotherapy in the treatment of nasopharyngeal carcinoma: an update of the UCSF experience. Int J Radiat Oncol Biol Phys 2002;53:12-22. 10.1016/S0360-3016(02)02724-4 [DOI] [PubMed] [Google Scholar]
- 4.Ng WT, Lee MC, Hung WM, et al. Clinical outcomes and patterns of failure after intensity-modulated radiotherapy for nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys 2011;79:420-8. 10.1016/j.ijrobp.2009.11.024 [DOI] [PubMed] [Google Scholar]
- 5.Lai SZ, Li WF, Chen L, et al. How does intensity-modulated radiotherapy versus conventional two-dimensional radiotherapy influence the treatment results in nasopharyngeal carcinoma patients? Int J Radiat Oncol Biol Phys 2011;80:661-8. 10.1016/j.ijrobp.2010.03.024 [DOI] [PubMed] [Google Scholar]
- 6.Tan WL, Tan EH, Lim DT, et al. Advances in systemic treatment for nasopharyngeal carcinoma. Chin Clin Oncol 2016;5:21. 10.21037/cco.2016.03.03 [DOI] [PubMed] [Google Scholar]
- 7.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011;331:1559-64. 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420. 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang Z, Liang W, Luo L. HSP27 promotes epithelial-mesenchymal transition through activation of the β-catenin/MMP3 pathway in pancreatic ductal adenocarcinoma cells. Transl Cancer Res 2019;8:1268-78. 10.21037/tcr.2019.07.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie LQ, Bian LJ, Zhi L, et al. Altered expression of e-cadherin by hepatocyte growth factor and effect on the prognosis of nasopharyngeal carcinoma. Ann Surg Oncol 2010;17:1927-36. 10.1245/s10434-010-0922-6 [DOI] [PubMed] [Google Scholar]
- 11.Horikawa T, Yang J, Kondo S, et al. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res 2007;67:1970-8. 10.1158/0008-5472.CAN-06-3933 [DOI] [PubMed] [Google Scholar]
- 12.Zhou YC, Liu JY, Li J, et al. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys 2011;81:1530-7. 10.1016/j.ijrobp.2011.06.1956 [DOI] [PubMed] [Google Scholar]
- 13.He E, Pan F, Li G, et al. Fractionated ionizing radiation promotes epithelial-mesenchymal transition in human esophageal cancer cells through PTEN deficiency-mediated AKT activation. PLoS One 2015;10:e0126149. 10.1371/journal.pone.0126149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan S, Wang Y, Yang Q, et al. Low-dose radiation-induced epithelial-mesenchymal transition through NF-kB in cervical cancer cells. Int J Oncol 2013;42:1801-6. 10.3892/ijo.2013.1852 [DOI] [PubMed] [Google Scholar]
- 15.Tsukamoto H, Shibata K, Kajiyama H, et al. Irradiation-induced epithelial-mesenchymal transition (EMT) related to invasive potential in endometrial carcinoma cells. Gynecol Oncol 2007;107:500-4. 10.1016/j.ygyno.2007.08.058 [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Hahm ER, Marcus AI, et al. Withaferin A inhibits experimental epithelial-mesenchymal transition in MCF-10A cells and suppresses vimentin protein level in vivo in breast tumors. Mol Carcinog 2015;54:417-29. 10.1002/mc.22110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka H, Kono E, Tran CP, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 2010;16:1414-20. 10.1038/nm.2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards CH, Mohammed Z, Qayyum T, et al. The prognostic value of histological tumor necrosis in solid organ malignant disease: a systematic review. Future Oncol 2011;7:1223-35. 10.2217/fon.11.99 [DOI] [PubMed] [Google Scholar]
- 19.Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol 2015;33:79-106. 10.1146/annurev-immunol-032414-112248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012;148:213-27. 10.1016/j.cell.2011.11.031 [DOI] [PubMed] [Google Scholar]
- 21.Cai Z, Zhang A, Choksi S, et al. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res 2016;26:886-900. 10.1038/cr.2016.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao D, Cai Z. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res 2018;28:868-70. 10.1038/s41422-018-0058-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negroni A, Colantoni E, Pierdomenico M, et al. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig Liver Dis 2017;49:1201-10. 10.1016/j.dld.2017.08.017 [DOI] [PubMed] [Google Scholar]
- 24.Saeed W, Jun D, Kim E, et al. Necroptosis signalling pathway in hepatic fibrosis; role of receptor-interacting serine-threonine kinase 3 and mixed lineage kinase domain-like in cirrhosis. J Hepatol 2018;68:S462 10.1016/S0168-8278(18)31170-X [DOI] [Google Scholar]
- 25.Bao C, Sun Y, Dong Y, et al. The relative biological effectiveness of proton and carbon ion beams in photon-sensitive and resistant nasopharyngeal cancer cells. Transl Cancer Res 2018;7:170-9. 10.21037/tcr.2018.01.25 [DOI] [Google Scholar]
- 26.Rajput S, Kumar BN, Banik P, et al. Thymoquinone restores radiation-induced TGF-β expression and abrogates EMT in chemoradiotherapy of breast cancer cells. J Cell Physiol 2015;230:620-9. 10.1002/jcp.24780 [DOI] [PubMed] [Google Scholar]
- 27.Zhu H, Chen H, Wang J, et al. Collagen stiffness promoted non-muscle-invasive bladder cancer progression to muscle-invasive bladder cancer. Onco Targets Ther 2019;12:3441-57. 10.2147/OTT.S194568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brabletz T, Kalluri R, Nieto MA, et al. EMT in cancer. Nat Rev Cancer 2018;18:128-34. 10.1038/nrc.2017.118 [DOI] [PubMed] [Google Scholar]
- 29.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 2009;119:1429-37. 10.1172/JCI36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su Z, Li G, Liu C, et al. Ionizing radiation promotes advanced malignant traits in nasopharyngeal carcinoma via activation of epithelial-mesenchymal transition and the cancer stem cell phenotype. Oncol Rep 2016;36:72-8. 10.3892/or.2016.4768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnomet A, Syne L, Brysse A, et al. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene 2012;31:3741-53. 10.1038/onc.2011.540 [DOI] [PubMed] [Google Scholar]
- 32.Xiong Y, Ji W, Fei Y, et al. Cathepsin L is involved in X-ray-induced invasion and migration of human glioma U251 cells. Cell Signal 2017;29:181-91. 10.1016/j.cellsig.2016.10.012 [DOI] [PubMed] [Google Scholar]
- 33.Liu W, Huang YJ, Liu C, et al. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3β and repression of ZEB1. Lab Invest 2014;94:362-70. 10.1038/labinvest.2013.153 [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Zheng L, Sun Y, et al. Tangeretin enhances radiosensitivity and inhibits the radiation-induced epithelial-mesenchymal transition of gastric cancer cells. Oncol Rep 2015;34:302-10. 10.3892/or.2015.3982 [DOI] [PubMed] [Google Scholar]
- 35.Pan Y, Zhou C, Yuan D, et al. Radiation Exposure Promotes Hepatocarcinoma Cell Invasion through Epithelial Mesenchymal Transition Mediated by H2S/CSE Pathway. Radiat Res 2016;185:96-105. 10.1667/RR14177.1 [DOI] [PubMed] [Google Scholar]
- 36.Wu SL, Li YJ, Liao K, et al. 2-Methoxyestradiol inhibits the proliferation and migration and reduces the radioresistance of nasopharyngeal carcinoma CNE-2 stem cells via NF-kappaB/HIF-1 signaling pathway inactivation and EMT reversal. Oncol Rep 2017;37:793-802. 10.3892/or.2016.5319 [DOI] [PubMed] [Google Scholar]
- 37.Yoon S, Kovalenko A, Bogdanov K, et al. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 2017;47:51-65.e7. 10.1016/j.immuni.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 38.Colbert LE, Fisher SB, Hardy CW, et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 2013;119:3148-55. 10.1002/cncr.28144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He L, Peng K, Liu Y, et al. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther 2013;6:1539-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruan J, Mei L, Zhu Q, et al. Mixed lineage kinase domain-like protein is a prognostic biomarker for cervical squamous cell cancer. Int J Clin Exp Pathol 2015;8:15035-8. [PMC free article] [PubMed] [Google Scholar]
- 41.Ertao Z, Jianhui C, Kang W, et al. Prognostic value of mixed lineage kinase domain-like protein expression in the survival of patients with gastric cancer. Tumour Biol 2016;37:13679-85. 10.1007/s13277-016-5229-1 [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Zhou M, Mei L, et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget 2016;7:22219-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong Y, Sun Y, Huang Y, et al. Upregulated necroptosis-pathway-associated genes are unfavorable prognostic markers in low-grade glioma and glioblastoma multiforme. Transl Cancer Res 2019;8:821-7. 10.21037/tcr.2019.05.01 [DOI] [PMC free article] [PubMed] [Google Scholar]