

Abstract
A phytopathogenic fungus, Macrophomina phaseolina, which infects a wide range of plants, is an important consideration in agronomy. A jute endophytic bacterium, Burkholderia contaminans NZ, was found to have a promising effect in controlling the fungus in in vitro culture conditions. Using the iTRAQ LC-MS/MS method for quantitative proteomics study, an analysis of the whole proteome of Macrophomina phaseolina with or without B. contaminans NZ challenge identified 2204 different proteins, of which 137 were found to have significant deviation in expression. Kyoto encyclopedia of genes and genomes pathway analysis identified most of the upregulated proteins to be functionally related to energy production (26.11%), as well as defense and stress response (23.45%), while there was significant downregulation in oxidative stress protection pathways (42.61%), growth and cell wall integrity (30.95%), and virulence (23.81%). Findings of this study suggest the development of a battle when the phytopathogen encounters the bacterium. B. contaminans NZ manages to arrest the growth of the fungus and decrease its pathogenicity, but the fungus apparently survives under “hibernating” conditions by upregulating its energy metabolism. This first ever proteomic study of M. phaseolina will go a long way in understanding and developing strategies for its effective control.
Introduction
Antagonistic fungal–bacterial interactions lie at the very heart of competitive survival for the limited resources in the bio-ecosystem. This paradigm for existence has been a long-term focus of researchers desperate for an enhanced understanding of bionetwork functions so as to develop potent biological control agents against fungal pathogens, providing alternatives to chemicals for practical agronomic purposes. Numerous examples highlight the use of bio-control agents in combating fungal phytopathogens, among them the control of Fusarium by Bacillus in cumin1 and by Pseudomonas putida in tomato2 are two from a list of many recent developments.
With respect to bio-control, some bacteria exhibit antifungal properties by producing antifungal compounds, secondary metabolites, chitinolytic enzymes, siderophores, toxins, etc.3,4 Some other bacteria like B. gladioli exhibit mycophagy against Rhizoctania solani, that is, the bacteria grow and multiply at the expense of fungal biomass.4 However, due to the pathogens’ diverse mechanisms of resistance, an efficient biological arsenal is still unavailable. Many pathogenic fungi possessing a well-equipped mechanism for thriving in a competitive environment render themselves almost invincible to control. They appear to be affected at some point by the bio-control agents but soon functionalize their genomic resources to overpower the latter.5 It is therefore necessary to understand how these pathogens respond in the presence of bio-control agents. In vitro studies on the antagonism of bacterial strains against fungal plant pathogens have revealed significant information on the genes associated and compounds involved.6 Transcriptomic analysis of the phytopathogen Rhizoctonia solani AG-3 in response to the antagonistic bacteria Serratia proteamaculans and Serratia plymuthica(7) and a dual transcriptional profiling of Collimonas fungivorans versus Aspergillus niger(8) were performed some years ago. These transcriptomic analyses revealed differential expression of genes related to xenobiotic degradation, toxin and antioxidant production, carbohydrate and lipid metabolism, and hyphal rearrangements and genes for production of putative antifungal compounds. Proteome mapping of an antagonistic F. oxysporum strain proposed candidate proteins that may play important roles in bio-control and highlight the close interrelationship between the fungus and its bacterial partners.9
Macrophomina phaseolina, a phytopathogenic fungus that affects a diverse range of host plants, poses a major worldwide threat to crop yield.10 Diseases caused by M. phaseolina include seedling blight, charcoal rot, color rot, stem rot, root rot, and damping off in more than 500 plant species, among which are economically important crops like cotton, sorghum, gerbera, soybean, potato, sunflower, chickpea, and jute, an important fiber-producing crop of Southeast Asia.11 This fungus is a major growth-limiting factor of the two most widely cultivated species of jute, Corchorus olitorius and C. capsularis.(12)
A number of reports have been made on the bio-control of M. phaseolina, some of which include control by Bacillus and Trichoderma in strawberry,13 by Trichoderma in sunflower,14 mung bean,15 and chickpea, by Trichoderma and Pseudomonas(16) in gerbera, etc. However, not much is known as to how they impede M. phaseolina from infecting plants or what the molecular mechanism of the fungal response to inhibition is.
In the present manuscript, we report the isolation of Burkholderia contaminans NZ as an endophytic bacterium from jute (Corchorus olitorius), which inhibits the growth of M. phaseolina in in vitro culture conditions. The study attempted to understand the mechanism of antifungal activity of B. contaminans NZ, and we found that the bacterium does not kill the fungus but forms and maintains a steady inhibition zone around the fungal mycelia. These mycelia are even able to germinate when transferred from the bacteria challenged plate onto fresh medium albeit with loss of pathogenicity. This bacterium–fungus interaction demonstrates the ability of M. phaseolina to withstand bacterial stress and develop strategies to remain static in the face of adversity.
Even with the availability of M. phaseolina genome sequenced in 2012,17 a proteome study is necessary to understand its response to different stimuli. We therefore employed a strategy for a relative and absolute quantification (iTRAQ)-based proteomic analysis of M. phaseolina to delineate the changes in the fungal proteome in the presence of B. contaminans NZ. The iTRAQ technique, which has a high degree of sensitivity, with amine specific isobaric reagents permitting identification and quantitation of up to eight different samples simultaneously,18 was used to obtain an extensive coverage of the M. phaseolina proteome. In this regard, we have been able to identify up to 82.4% of the total fungal proteins. A total of 2204 proteins were identified, of which 137 were found to be differentially regulated upon B. contaminans NZ challenged condition. Interestingly, most of these proteins with altered expression are related to defense, virulence, cell proliferation, and cell wall composition together with the proteins of redox and metabolic pathways.
The ability of M. phaseolina to remain alive under inhibitory conditions imposed by B. contaminans NZ points to a distinct phenomenon executed by the phytopathogen. The fungus upregulates its energy metabolic pathway at the cost of downregulating the expression of proteins involved in oxidative stress management and proteins which can lead to pathogenicity. This apparently allows M. phaseolina to lie torpid under bacterial inhibition.
Overall, the proteome data of M. phaseolina provide us with important information as to how the fungus responds to the bio-control environment.
Materials and Methods
Unless mentioned otherwise, all of the chemicals were obtained from Sigma-Aldrich, (St. Louis, MO). Culture media, Potato Dextrose Agar (PDA), and Tryptic Soya Broth (TSB) were obtained from HiMedia (HiMedia, India). Trypsin (mass spectrometry grade), RIPA (radioimmune precipitation) lysis and extraction buffer, and BCA Protein Assay Kit were purchased from Thermo Scientific (Thermo Scientific Pierce, Rockford, IL). iTRAQ 4-plex multiplex kit was purchased from AB Sciex (Framingham, MA). Protease inhibitor cocktail was purchased from Roche Diagnostics (Indianapolis, IN).
In Vitro Dual-Culture Assays
In vitro dual-culture assays were carried out on PDA plates. A 5 mm plug taken from the plate of an actively growing colony of M. phaseolina was inoculated on one side of a Petri dish. Fresh cells of B. contaminans NZ were streaked in 3 cm length parallel lines on the other side of the fungal plug. Plates containing only the fungus were also set up as controls. All plates were incubated at 28 ± 2 °C for 4–5 days.
Photographs of hyphae from both control and B. contaminans NZ challenged fungus were taken using a fluorescence microscope (EVOS FL, ThermoSci) version 3.6.0 (bright field, 20×).
Change in Pathogenicity of B. contaminans NZ Challenged M. phaseolina
To assess any changes in fungal pathogenicity, an assay was performed with jute seeds (Corchorus olitorius var. O4). The seeds were surface-sterilized by treating with 5% NaOCl for 1 min and then rinsed with autoclaved water three times for 2 min.19 The seeds were allowed to germinate on a 110 mm Whatman filter paper (moistened with sterile water) in Petri plates under two different conditions. In one plate, 1% fungal mycelial solution was inoculated, and in the other, a similar amount of mycelia from B. contaminans NZ challenged M. phaseolina was used. A seedling plate without any inoculum was used as the control.
Cell Culture and Preparation of Protein Extracts
M. phaseolina cells were incubated in 50 mL of PDB for 5 days in an incubator shaker at 28 °C at 180 rpm; 20 mL of an overnight culture of B. contaminans NZ in TSB was added to 50 mL of 3 day old M. phaseolina culture in PDB. The fungal mycelia were collected after 2 days of co-culture.
Pure and co-cultured fungal mycelia were filtered and washed three times with ice-cold 10 mM Na phosphate buffer (pH 6.0) to remove bacterial contamination and were ground in liquid nitrogen in a precooled mortar. Crushed fungal cell samples were homogenized in ice-cold RIPA buffer containing 1× concentration of a protease inhibitor cocktail. The supernatant was collected following centrifugation and subjected to three pulses of sonication on ice. Total soluble proteins were recovered by centrifugation at 14 000g for 30 min at 4 °C, precipitated overnight with 6 volumes of (vol/vol) ice-cold acetone at 4 °C, and centrifuged at 10 000g for 10 min at 4 °C. The resulting protein precipitate was washed twice with cold acetone, air-dried, and stored at −80 °C until use. Semidried pellet was dissolved using 8 M urea (Sigma-Aldrich). Total protein concentration was determined by the BCA Protein Assay kit according to the manufacturer’s instructions. The protein concentration was calculated using bovine serum albumin (BSA) as a standard. Three such experiments were carried out; a total of three biological and three technical replicates were analyzed for the elucidation of the differential proteome.
Trypsin Digestion and iTRAQ Labeling for LC-MS/MS
A total of 100 μg of proteins from each sample were reduced using 10 mM DTT (dithiothreitol) at 60 °C for 60 min and alkylated using a 20 mM IAA (iodoacetamide) at room temperature for 30 min. The proteins were then digested overnight by sequencing-grade trypsin dissolved in 1:20 w/w 50 mM TEAB (triethylammonium bicarbonate) at 37 °C according to the manufacturer’s instructions (AB Sciex, Inc.). Then, the peptides from each sample were first resuspended in 100 mM TEAB and iTRAQ reagents were dissolved in 70 μL of ethanol by vortexing for 1 min. Peptides from control and stressed samples were labeled with iTRAQ mass tag 114 and 116, respectively, at room temperature for 2 h. The reaction was stopped by adding 120 μL of H2O, followed by centrifugation at 13 800g for 1 min. The samples were then pooled together into one fresh tube, as illustrated in Figure 5, and dried in a SpeedVac concentrator. Except for the iTRAQ labeling, a similar digestion protocol was followed to prepare fungal protein samples for total protein identification.
Figure 5.

Overview of experimental design. (A) Workflow of quantitative proteomic analysis, (B) table showing the number of modulated proteins and total identified proteins, (C) histogram displaying log 2 ratios of all proteins, and (D) box plot showing the effect of bias correction (normalization) based on total reporter ion intensity.
Peptide Fractionation by High-pH RP HPLC and LC-MS/MS
Prior to mass spectrometric analysis, the pooled peptides were resuspended in 80 μL of buffer A (98% H2O, 2% acetonitrile, pH 10.0) and were fractionated using an Agilent 1200 HPLC system on high-pH reverse-phase Zorbax 300Extend-C18 column (2.1 × 100 mm, 3 μm, 150 Å, C18, Agilent Technology, Santa Clara, CA). The 60 min linear gradient was composed of 96% buffer A for 1 min; 4–19% buffer B (98% acetonitrile, 2% H2O, pH 10.0) for 30 min; then 19–95% buffer B for 23 min, followed by 95% buffer B for 5 min. The eluted fractions were collected at every 1 min interval into 48 fractions and then pooled to give a total of 12 fractions. The collected fractions were then lyophilized and stored at −20 °C until MS analysis.
For LC-MS/MS, data were acquired using 5600 TripleTOF+ (AB Sciex, Concord, Canada). The instrument was coupled with an Eksigent NanoLC-2DPlus system (Eksigent, Dublin, CA), and the samples were loaded at a flow rate of 2 μL/min for 10 min and eluted from the analytical column at a flow rate of 300 nL/min in a linear gradient of 5–35% solvent B in 60 min. Solvent A was composed of 0.1% (v/v) formic acid in water, and solvent B contained 95% (v/v) acetonitrile with 0.1% (v/v) formic acid. The TripleTOF 5600 system was run on an information-dependent acquisition (IDA) mode with a TOF/MS survey scan (350–1250 m/z) where the accumulation time was 250 ms. For fragmentation, a maximum of 10 precursor ions per cycle were selected with a total cycle time of roughly 2.3 s and each MS/MS spectrum was collected for 100 ms (100–1500 m/z). The parent ions with a charge state from +2 to +5 were included for the MS/MS fragmentation. The threshold precursor ion intensity was set at more than 120 cps (count per second) and was not present on the dynamic exclusion list. After fragmentation of an ion by MS/MS, its mass and isotopes were excluded for 10 s. The MS/MS spectra were operated in high-sensitivity mode with “adjust collision energy” when using iTRAQ reagent settings.
Protein Identification and Data Analysis
All of the wiff. files containing MS and MS/MS spectra generated from Triple TOF 5600 were submitted for database searching and quantitative analysis using the ProteinPilot v4.5 software (AB Sciex, Concord, Canada). ProteinPilot search engine was used for iTRAQ-based quantitation in a data-dependent mode. This search engine uses a sequence tag method plus protein database searching. Each MS/MS spectrum was searched against Macrophomina species from Uniprot/swissprot database (November 28, 2012; 14 056 entries) and against Burkholderia proteins (1 508 793 entries) to distinguish the bacterial proteins from those of the fungus. The search parameters were set as iTRAQ peptide label, cysteine alkylation with methyl methanethiosulfonate, trypsin digestion, and identification focus for biological modifications. The resulting dataset was auto-bias-corrected to normalize any variation arising from unequal mixing of the differently labeled samples. False discovery rate (FDR) was estimated using a target-decoy-based strategy. The proteins and peptides were filtered with 1% global protein-level FDR. For quantitation, the ratio threshold was set to >1.3 (equivalent or more than 95% confidence) and p-value < 0.05 to ensure that quantitation was based on at least two unique peptides. Proteins were considered only if they were significant in all independent biological and technical triplicate experiments. The average values of replicates were used to indicate the final protein abundance at a given time point. The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE20 partner repository with the dataset identifier PXD009121.
Statistical and Bioinformatics Analysis
A statistical analysis to compare the groups was performed using t-test (Sigma Stat, Jandel Scientific). For functional annotation and cellular location, the protein lists were analyzed according to the Blast2GO tool (https://www.blast2go.com/), Kyoto Encyclopedia of Genes and Genomes (KEGG). Predicted interacting partners were analyzed using STRING v10 database.21
Results
Effects of B. contaminans NZ on Growth and Morphology of M. phaseolina Mycelium
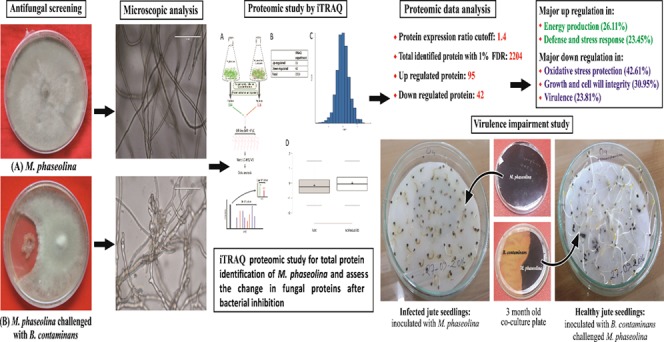
When M. phaseolina was co-cultured with B. contaminans NZ, a clear inhibition of fungal mycelial growth was evident at 4 days (Figure 1B) compared to the control without bacteria (Figure 1A). This inhibition persisted for more than 16 weeks (Figure 1C). Sclerotia collected even from a 3-month-old confrontation plates were found to retain viability and were able to germinate when transferred onto a fresh PDA plate. Microscopic observations of fungal hyphae during interactions with bacteria revealed a change in the hyphal morphology, including mycelia swelling with increased septation and branching and thickened cell walls (Figure 2B) compared to the control hyphae, which were straight having normal branching and septation (Figure 2A).
Figure 1.
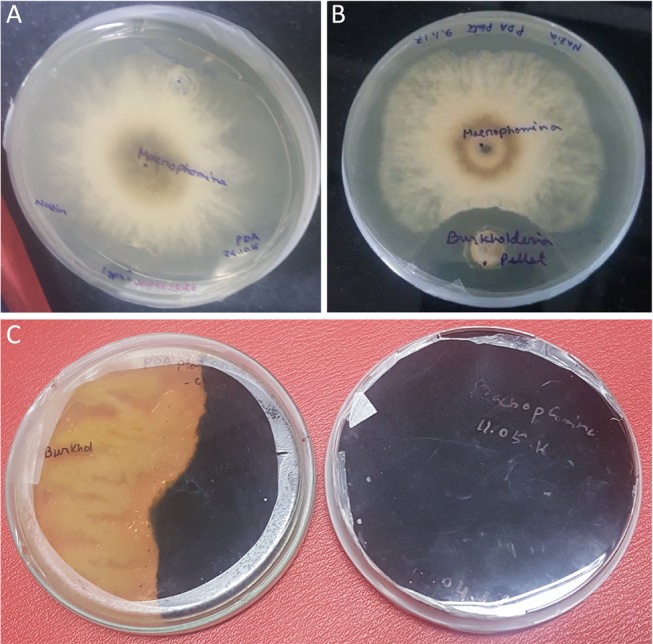
In vitro dual-culture bacterial–fungal assay. (A) Control M. phaseolina monoculture, (B) M. phaseolina challenged with B. contaminans NZ (5 days), and (C) persistent inhibition of M. phaseolina by B. contaminans NZ (16 weeks). On the right is the monoculture of M. phaseolina, and the left shows the dual culture of M. phaseolina vs B. contaminans NZ with the bacterium inhibiting the growth of M. phaseolina. This inhibition has been shown to persist for more than 16 weeks.
Figure 2.
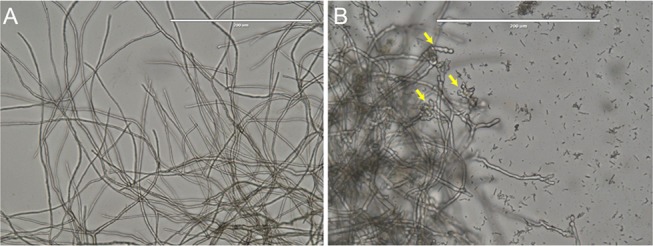
Microscopic analysis. (A) Control M. phaseolina having extended straight mycelia with normal branching and septation. (B) Microscopic examination of the mycelia of M. phaseolina challenged with B. contaminans NZ at the intersection with the zone of inhibition. The arrows showing increased frequency of septa, branching, and swollen mycelia.
Impairment of Pathogenicity in M. phaseolina
When B. contaminans NZ challenged M. phaseolina was used to inoculate jute seeds, the germinating seedlings looked healthy (Figure 3C) and were similar to the ones in which no M. phaseolina was inoculated (Figure 3A). This indicated a significant loss of virulence in the Burkholderia challenged M. phaseolina. However, M. phaseolina without the stress appeared as virulent as expected with seedlings dying from heavy infection when inoculated with the same (Figure 3B).
Figure 3.

Pathogenicity reduction test. (A) Jute (Corchorus olitorius var. O4) seedlings without any fungal inoculation. The seedlings are long and white, indicating healthy normal growth. Seedlings inoculated with (B) M. phaseolina, where the seedlings are small and brown, indicating heavy infection, and (C) B. contaminans NZ challenged M. phaseolina. Here, the seedlings are white, long, and healthy-looking, indicating a loss of pathogenicity of B. contaminans NZ challenged M. phaseolina.
Identification of M. phaseolina Proteome
A total of 2204 proteins common to the two biological replicates were identified (Figure 4A) (a list of total identified proteins is available in Supporting Information Table S1). Analysis found M. phaseolina to have a relatively high number of enzymes which belong to hydrolases, oxidoreductases, and transferases (Figure 4C). Homology-based function prediction was carried out for M. phaseolina proteome using Blast2GO. A majority of the proteins was found to belong to the transport machinery (14%) followed by cellular protein metabolic process (12%), carboxylic metabolic process (9%), and oxidation–reduction process (9%) in the category of biological process (Figure 4D). Similarly, the hydrolase activity (27%) and the oxidoreductase activity (20%) were predicted in the molecular function category (Figure 4E). Most of the proteins of the cellular compartment category were found to be localized in the membrane (Figure 4F).
Figure 4.

Analysis of identified proteins. (A) Venn diagram showing proteins identified with two biological replicates. Graph showing (B) the score distribution with different lengths of identified proteins and (C) enzyme distribution. Classification of identified proteins based on (D) biological process, (E) molecular function, and (F) cellular compartment.
Functional annotation of the total proteins identified by KEGG, classified them into relevant pathways (Supplementary Table S2).
Quantitative Proteomics of M. phaseolina under Bacterial Stress
On quantitative analysis of M. phaseolina under normal and B. contaminans NZ stressed conditions (Figure 5A), a total of 47 282 spectra were obtained from the iTRAQ LC-MS/MS experiment. After data filtering to eliminate low-scoring spectra, a total of 25 429 unique spectra with 1% false discovery rate (FDR) met the strict confidence criteria for identification. Raw data showed normal distribution of relative abundance reflecting the unchanged condition of the majority of proteins (Figure 5C); data were further normalized using the bias correction feature of the ProteinPilot v 4.5 software (Figure 5D). Spectral data were searched using Uniprot decoy database, and proteins with at least two validated peptides were considered for quantitation. iTRAQ data for all replicates were found to be similar, suggesting a change in protein abundance in M. phaseolina under B. contaminans NZ stress.
Among the proteins which showed a significant change (P < 0.05) in abundance, 137 differentially expressed proteins (DEPs) within the ratio >1.40 or <0.8 under B. contaminans NZ stress were chosen.
Profile of Differential Protein Abundance
Among the total differentially abundant proteins in all of the replicates, 95 were found to be upregulated (Figure 6A) and 42 downregulated (Figure 6B) (lists of upregulated and downregulated proteins are available in Supporting Information Tables S3 and S4, respectively). A volcano plot was used to show the log 2 ratio of the gene expression levels between the control and stressed conditions; the colored dots in green and red represent the differentially abundant proteins. P value <0.01 is represented by the blue horizontal lines, and 1.4-fold expression difference is represented by the two red vertical lines (Figure 6C). Cluster analysis based on biological process revealed that the majority of proteins belong to the metabolic processes (Figure 6D).
Figure 6.

Differentially expressed proteins detected by quantitative iTRAQ mass spectrometry. Venn diagram showing (A) up- and (B) downregulated proteins with three biological replicates. (C) Volcano plot showing the log 2 ratio of protein abundance levels between control and stressed conditions; the colored dots in green and red represent the differentially expressed proteins (P value < 0.01 represented by the black horizontal line) and 1.4-fold expression difference (represented by two red vertical lines), respectively. (D) Hierarchical clustering of differentially expressed proteins in M. phaseolina after B. contaminans NZ stress.
Differentially expressed proteins of M. phaseolina were further annotated using Blast2GO. Up (Figure 7A)- and downregulated (Figure 7B) proteins were analyzed separately into three categories of “molecular function”, “biological process”, and “cellular component”.
Figure 7.

GO-based functional annotation of (A) upregulated and (B) downregulated proteins.
Functional Classification of Differentially Expressed Proteins
Metabolic pathway enrichment analysis of responsive proteins was further carried out according to the KEGG pathway database. The differentially abundant proteins were found to be majorly classified into 12 categories according to their putative biological functions.
Upregulated DEPs: The majority of upregulated DEPs classified into three categories: energy and carbohydrate metabolism (26.11%), defense and stress response (23.45%), and amino acid metabolism (19.91%).
The other categories are: genetic information processing/transcription; translation (6.64%); cell growth and death/endocytosis/apoptosis/senescence (5.31%); signaling pathway (4.87%); folding, sorting, and degradation (3.09%); biosynthesis of other secondary metabolites (3.09%); lipid metabolism (2.66%); metabolism of cofactors and vitamins (2.21%); nucleotide metabolism (1.32%); cellular community/cell motility (0.88%); and metabolism of xenobiotics (0.44%) (Figure 8A).
Figure 8.

Functional categorization based on KEGG database of (A) upregulated and (B) downregulated proteins.
The effect of B. contaminans NZ resulted in the upregulation of several proteins involved in energy and carbohydrate metabolism, glycolysis, and citric acid cycle. Many proteins involved in defense response were also upregulated, such as the molecular chaperones, antioxidant enzymes, and heat shock proteins, indicating their crucial protective roles against biotic stress.
Downregulated DEPs: The three major categories of downregulated DEPs are defense- and stress-related (22.5%), energy and carbohydrate metabolism (16.9%), and cell growth and death/apoptosis/senescence (15.5%).
The other categories are: amino acid metabolism (9.9%), xenobiotic biodegradation and metabolism (8.4%), translation and transcription (5.6%), lipid metabolism (4.2%), biosynthesis of other secondary metabolites (4.2%), transport and cellular motility (4.2%), signal transduction (2.8%), metabolism of cofactors and vitamins (2.8%), and folding, sorting, and degradation (2.8%) (Figure 8B).
The downregulated proteins of energy and carbohydrate metabolism are mainly related to biosynthesis of disaccharides, polysaccharides, metabolism of amino sugars, fatty acid biosynthesis, and the oxidative pentose phosphate pathway.
As the enzymes essential for cell wall rigidity, cell integrity and important regulators of growth, cell division, motility, and oxidative damage protecting antioxidant systems enzymes were downregulated majorly, it is clear that B. contaminans NZ manifests profound stress on the fungal cell wall structure and organization.
Strikingly, pathogenicity regulatory genes and metabolic and signaling responsive proteins were also downregulated, indicating that M. phaseolina resorts to energy conservation as an effective strategy for countering the stress imposed by B. contaminans NZ.
Protein–protein interaction networks are important for understanding cellular processes at the systems level.22 To assess such interactive network of up- and downregulated fungal proteins, the most upregulated protein enolase was searched as the query protein in the STRING database. The predicted functional partners were found to be pyruvate kinase, phosphoglycerate kinase, and triosephosphate isomerase. In M. phaseolina, all of these proteins were found to be upregulated with enolase (Figure 9A).
Figure 9.

Interaction analysis of (A) enolase and (B) GRX1 (glutathione S-transferase) using STRING database.
As an example of a downregulated protein, glutathione S-transferase, which plays an important role in oxidative stress management, was used as another input in the STRING database. Its predicted functional partners were found to be glutaredoxin and glutathione oxidoreductase, both of which were found to be downregulated along with the input protein (Figure 9B).
Discussion
Many researchers have focused their attention on bio-control approaches for limiting the growth of Macrophomina phaseolina. In this regard, Burkholderia sp. has been known for some time to be effective in controlling the growth of this plant pathogen.23 While studying a microbiome of jute, Burkholderia contaminans NZ isolated as a jute endophyte was also found to be a potent bio-control agent effective against M. phaseolina. Since proteomics is now widely employed to recognize factors responsive to environmental or biotic stresses, a comparison of both B. contaminans NZ challenged and unchallenged proteome of M. phaseolina was expected to provide an understanding of physiological responses to this biotic stress condition and highlight the variations in the protein profile following the stress.
Besides the genome sequence of M. phaseolina that depicts its large arsenal of hydrolyzing enzymes,17 not much is known about the protein profile of the fungus. Abundance of the hydrolyzing enzymes was validated by our proteome data (Figure 4C). Upon further analyses of the differentially regulated proteins, a pattern became apparent, which allowed an understanding of how this pathogen while losing its killing prowess manages to survive under “hibernating” conditions when confronted by B. contaminans NZ.
Based on the data collected through this experiment, M. phaseolina appears to be significantly affected by the presence of B. contaminans NZ. Its proteome changes considerably with elevated expression of defense responsive genes, and it seems to apply a major emphasis on energy production through the recruitment of diverse pathways and by decreasing the anabolic system (lipid synthesis, etc). Most of the highly abundant proteins obtained under B. contaminans NZ-induced stress are found to be involved in carbohydrate and energy metabolism. The main pathways affected are the classical glycolytic pathway, TCA cycle, oxidative phosphorylation, pentose phosphate pathway, and gluconeogenesis. Further carbohydrate metabolic processes which were also affected include fructose, sucrose, starch, and other monosaccharide metabolic pathways.
Proteins Upregulated
Proteins Involved in Carbohydrate and Energy Metabolism
Enolase, an enzyme involved in glycolysis, is known to respond to stress by increasing its expression.24 This enzyme has been reported to be a multifunctional protein, upregulated in heat shock and in response to hypoxic stress and glucose deprivation.24
Thus, a 6.2-fold upregulation of enolase implies that B. contaminans NZ causes substantial stress on M. phaseolina, which alludes to an all-out effort by the latter to stay alive. It corroborates why viable fungus is obtained even from 3-month-old bacteria–fungus co-culture plates. Studies by De Backer et al. and Lo et al. showed that knockout of enolase (ENO1) gene causes a reduction in the growth rate and mycelium formation and increased drug sensitivity in C. albicans.(24)
Another upregulated enzyme, triosephosphate isomerase, crucial for glycolysis, has also been reported to be regulated in various abiotic stress responses, and the expression is highly increased under oxidative stress.25 Additionally, elevated expression of the glycolytic enzyme 2,3-bisphosphoglycerate-independent phosphoglycerate mutase observed for M. phaseolina was similar to that found for Pallisentis umbellatus, where both the enzymes and the glycolytic pathways were found to be induced under oxidative stressed condition during the initial sclerotia formation.26
Phosphoglycerate kinase, a housekeeping enzyme of the glycolytic pathway reported to protect cells against oxidants,27 was also in the list of proteins which showed an elevated expression. A proteome analysis of A. fumigatus, where 117 proteins were identified with an altered abundance in response to hypoxia, phosphoglycerate kinase was also shown to have increased activity.27
Other key glycolytic enzymes, namely, ATP-dependent 6-phosphofructokinase along with other glycolytic enzymes ketose-bisphosphate aldolase class-2, glyceraldehyde-3-phosphate dehydrogenase and pyruvate kinase, had elevated levels in M. phaseolina after B. contaminans NZ challenge.
Upregulation of the citric acid and oxidative phosphorylation pathways indicate a possible mechanism by which M. phaseolina tries to overcome B. contaminans NZ challenge. One of the most significantly upregulated proteins, citrate synthase, determines the fungal ability to survive by reducing its competition for nutrient resources.28 Upregulation of formate dehydrogenase, which has an important role in respiration and oxidative phosphorylation and is known to provide alternative metabolic pathways, is thought to help fungal survival under unfavorable conditions.29
Proteins Necessary for Defense
M. phaseolina also upregulates some of its defense-related proteins upon bacterial stress, including chaperonins and heat shock proteins, which usually function as signals to biotic stressors. Among them, Hsp 12 is solely responsible for stress tolerance, cell morphology, and adhesion, and Hsp 20 and 70 are involved in membrane and cellular protein maintenance.30 Upregulation of such proteins appears to limit the growth of M. phaseolina in the presence of B. contaminans NZ (as evident from the microscopic study of fungal filaments (Figure 2)).
Proteins Downregulated
Proteins Involved in Oxidative Stress Management
M. phaseolina fails to confront the oxidative stressed condition posed by B. contaminans NZ because of downregulation of its major oxidative stress controlling systems and the machinery that helps to maintain integrity and rigidity of its cell wall. In fungal intracellular signaling, ROS is used to decide between growth and proliferation on the one hand and growth arrest and cell differentiation on the other.31 Interestingly, most of the rate-limiting enzymes related to oxidative stress response pathway were shown to be downregulated in M. phaseolina under B. contaminans NZ stress. The rate-limiting enzyme of the pentose phosphate pathway (glucose-6-phosphate 1-dehydrogenase) responsible for NADPH production was found to be downregulated. Since a steady supply of glutathione was apparently absent due to the downregulation of glucose-6-phosphate 1-dehydrogenase, glutathione-associated enzymes like glutathione S-transferase and glutaredoxin were also found to be downregulated.
Reduced expression of proteins involved in oxidative stress management appears to be a target for B. contaminans NZ inhibition, and as a result, an array of proteins like the peroxidase, flavodoxin, glutaredoxin, etc. involved in such stress response was found to be downregulated.32
Proteins for Cellular and Structural Integrity
Proteins essential for cell wall rigidity and cell integrity, namely, tubulin, were greatly downregulated, indicating an influence on fungal cell wall structure or organization. Glycosyl transferase family 39, essential for cell wall rigidity, cell integrity and budding, growth, and adaptation to environmental stress, was also downregulated.33 Glutamine amidotransferase class-2, a downregulated enzyme, is reported to be induced by cell wall stressors.34 It catalyzes the first and rate-limiting step in the biosynthetic pathway for the synthesis of a cell wall protein, chitin. Expression of cell wall modification genes is expected to be altered when fungi are exposed to stress conditions.7 Another downregulated protein, heterokaryon incompatibility Het-C reported for regulation of cell wall assembly is essential in fungal growth and development.35
Downregulation of the main regulators of growth, cell division, motility, and development, such as RasGTPase, microtubule proteins, tubulin α chain, kinesin-like protein, etc., could also be responsible for the observed reduction in growth and marked morphological change in hyphae with swollen and balloon-shaped cells and subcellular abnormalities.36 Excessive hyphal branching close to the bacterial colony was discernible (Figure 2). Such morphological changes in the cell membrane have also been reported forF. solani and C. dematium following inhibition by Burkholderia cepacia.(37)
Proteins Involved in Producing Secondary Metabolites
Fungi, especially the filamentous ones, produce a range of secondary metabolites associated with pathogenicity through the involvement of various cytochrome P450s (CYPs).38 In F. asiaticum, it has been reported that tubulin binding cofactor A (TBCA) plays a vital role in the vegetative growth, conidiation, temperature sensitivity, and virulence.39 Downregulation of both cytochrome P450s and TBCA explain the reasons behind the observed loss of pathogenicity in the case of Burkholderia challenged M. phaseolina.
The secondary metabolite melanin is important for fungal survival and plays a crucial role in infecting hosts.40 As melanin synthesis is essential for fungal pathogenicity, enzymes involved in melanin biosynthesis like scytalone dehydratase have been considered a good target for developing control agents against fungal diseases.41 After bacterial interaction, scytalone dehydratase of M. phaseolina was found to be downregulated 7.69-fold, indicating severe impairment of melanin biosynthesis, leading to decreased stress tolerance and virulence.42
Proteins Involved in Virulence
Moreover, GTP-binding proteins like septins (downregulated in M. phaseolina after the biotic stress) are generally necessary for virulence in fungal pathogens and are directly associated in host tissue adhesion and entry.43 In C. albicans, a family of four flavodoxin-like proteins (FLPs) act as NAD(P)H quinone oxidoreductases, conferring important antioxidant effects. FLPs reduce ubiquinone (coenzyme Q), which in turn serve as a free-radical-scavenging antioxidant in the membrane and is critical for fungal virulence.32 With the downregulation of flavodoxin in M. phaseolina, B. contaminans NZ appears to exude both oxidative stress and reduced fungal pathogenicity.
In a virulence study of A. fumigatus, ribosomal biogenesis proteins, RNA-processing protein HAT helix, and signaling molecules (including G-protein, Ras protein RasGTPase, and recoverin) have been reported to increase virulence through an alteration of the metabolic response under stressed conditions.44 These same proteins found to be downregulated in M. phaseolina emphasize their effect on fungal virulence. The downregulated NmrA like protein is known to serve as a regulator of the sugar-sensor mechanism integrating carbon and nitrogen metabolism to control plant infection in the rice blast fungus Magnaporthe oryzae.(45)
Conclusions
This pioneering proteomics study has shed light on how M. phaseolina, a versatile organism in terms of sustainability, employs diverse combating mechanisms resulting in a tug of war that leads to an apparent inhibition of fungal growth. However, the phytopathogen under siege manages to stay dormant in its own territory and reverts from its arrested growth to an active life with reduced virulence once the challenge is removed. The available genome data and now this proteomic analysis have contributed to a novel understanding of how this devastating fungal pathogen responds towards bio-control and stress.
Acknowledgments
The authors thank Regional Centre for Biotechnology (RCB) for the logistic support and Ahlan Sabah Ferdous for her kind help in writing the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b01870.
List of total identified proteins in M. phaseolina (Table S1); KEGG function annotation of total identified proteins in M. phaseolina according to relevant pathways (Table S2); list of up- and downregulated proteins in B. contaminans NZ challenged M. phaseolina with three biological replicates (Table S3); and list of up- and downregulated proteins with fold changes (Table S4) (XLSX)
Author Contributions
§ N.R.Z. and B.K. contributed equally to this work.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The research work was funded by Higher Education Quality Enhancement Project (HEQEP) (grant no. CP-3250), a World Bank-financed development project.
The authors declare no competing financial interest.
Supplementary Material
References
- Gajera H.; Savaliya D. D.; Hirapara D. G.; Patel S.; Golakiya B.. Biocontrol Mechanism of Bacillus for Fusarium Wilt Management in Cumin (Cuminum cyminum L.). In Current Trends in Plant Disease Diagnostics and Management Practices; Springer, 2016; pp 29–47. [Google Scholar]
- Pastor N.; Masciarelli O.; Fischer S.; Luna V.; Rovera M. Potential of Pseudomonas putida PCI2 for the Protection of Tomato Plants Against Fungal Pathogens. Curr. Microbiol. 2016, 73, 346–353. 10.1007/s00284-016-1068-y. [DOI] [PubMed] [Google Scholar]
- Vurukonda S. S. K. P.; Giovanardi D.; Stefani E. Plant growth promoting and biocontrol activity of Streptomyces spp. as endophytes. Int. J. Mol. Sci. 2018, 19, 952. 10.3390/ijms19040952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain D. M.; Yadav S. K.; Tyagi I.; Kumar R.; Kumar R.; Ghosh S.; Das J.; Jha G. A prophage tail-like protein is deployed by Burkholderia bacteria to feed on fungi. Nat. Commun. 2017, 8, 404. 10.1038/s41467-017-00529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbore J.; Benhamou N.; Vallance J.; Le Floch G.; Grizard D.; Regnault-Roger C.; Rey P. Biological control of plant pathogens: advantages and limitations seen through the case study of Pythium oligandrum. Environ. Sci. Pollut. Res. 2014, 21, 4847–4860. 10.1007/s11356-013-1807-6. [DOI] [PubMed] [Google Scholar]
- Ballhausen M.-B.; de Boer W. The sapro-rhizosphere: Carbon flow from saprotrophic fungi into fungus-feeding bacteria. Soil Biol. Biochem. 2016, 102, 14–17. 10.1016/j.soilbio.2016.06.014. [DOI] [Google Scholar]
- Gkarmiri K.; Finlay R. D.; Alström S.; Thomas E.; Cubeta M. A.; Högberg N. Transcriptomic changes in the plant pathogenic fungus Rhizoctonia solani AG-3 in response to the antagonistic bacteria Serratia proteamaculans and Serratia plymuthica. BMC Genomics 2015, 16, 630. 10.1186/s12864-015-1758-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mela F.; Fritsche K.; De Boer W.; Van Veen J. A.; De Graaff L. H.; Van Den Berg M.; Leveau J. H. Dual transcriptional profiling of a bacterial/fungal confrontation: Collimonas fungivorans versus Aspergillus niger. ISME J. 2011, 5, 1494. 10.1038/ismej.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti M.; Grunau A.; Minerdi D.; Gehrig P.; Roschitzki B.; Eberl L.; Garibaldi A.; Gullino M. L.; Riedel K. A proteomics approach to study synergistic and antagonistic interactions of the fungal–bacterial consortium Fusarium oxysporum wild-type MSA 35. Proteomics 2010, 10, 3292–3320. 10.1002/pmic.200900716. [DOI] [PubMed] [Google Scholar]
- Kumari R.; Shekhawat K.; Gupta R.; Khokhar M. Integrated Management against Root-rot of Mungbean [Vigna radiata (L.) b Wilczek] incited by Macrophomina phaseolina. J. Plant Pathol. Microbiol. 2015, 10.4172/2157-7471.1000136. [DOI] [Google Scholar]
- Babu B. K.; Saxena A. K.; Srivastava A. K.; Arora D. K. Identification and detection of Macrophomina phaseolina by using species-specific oligonucleotide primers and probe. Mycologia 2007, 99, 797–803. 10.1080/15572536.2007.11832511. [DOI] [PubMed] [Google Scholar]
- Sharmin S.; Azam M. S.; Islam M. S.; Sajib A. A.; Mahmood N.; Hasan A. M.; Ahmed R.; Sultana K.; Khan H. Xyloglucan endotransglycosylase/hydrolase genes from a susceptible and resistant jute species show opposite expression pattern following Macrophomina phaseolina infection. Commun. Integr. Biol. 2012, 5, 598–606. 10.4161/cib.21422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana A. M.; Basallote-Ureba M. J.; Aguado A.; Akdi K.; Capote N. Biological control of strawberry soil-borne pathogens Macrophomina phaseolina and Fusarium solani, using Trichoderma asperellum and Bacillus spp. Phytopathol. Mediterr. 2016, 55, 109. [Google Scholar]
- Reetha A. K.; Pavani S. L.; Mohan S. Ecofriendly management of fungal antagonistic Trichoderma sp. against charcoal rot of sunflower caused by Macrophomina phaseolina (Tassi) Goid. J. Biopestic. 2014, 7, 73. [Google Scholar]
- Hussain S.; Ghaffar A.; Aslam M. Biological Control of Macrophomina phaseolina Charcoal Rot of Sunflower and Mung Bean. J. Phytopathol. 1990, 130, 157–160. 10.1111/j.1439-0434.1990.tb01163.x. [DOI] [Google Scholar]
- Pandey R.; Gohel N.; Jaisani P. Management of Wilt and Root Rot of Chickpea caused by Fusarium oxysporum f. sp. ciceri and Macrophomina phaseolina through Seed Biopriming and Soil Application of Bio-Agents. Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 2516–2522. 10.20546/ijcmas.2017.605.282. [DOI] [Google Scholar]
- Islam M. S.; Haque M. S.; Islam M. M.; Emdad E. M.; Halim A.; Hossen Q. M. M.; Hossain M. Z.; Ahmed B.; Rahim S.; Rahman M. S.; et al. Tools to kill: genome of one of the most destructive plant pathogenic fungi Macrophomina phaseolina. BMC Genomics 2012, 13, 493. 10.1186/1471-2164-13-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C. H.; Bern M. Recent developments in quantitative proteomics. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2011, 722, 171–182. 10.1016/j.mrgentox.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs J. T.; Franco C. M. Isolation and identification of actinobacteria from surface-sterilized wheat roots. Appl. Environ. Microbiol. 2003, 69, 5603–5608. 10.1128/AEM.69.9.5603-5608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2015, 44, D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D.; Franceschini A.; Wyder S.; Forslund K.; Heller D.; Huerta-Cepas J.; Simonovic M.; Roth A.; Santos A.; Tsafou K. P.; Kuhn M.; Bork P.; Jensen L. J.; von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–52. 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A.; Grossmann A.; Herwig R.; Stelzl U. Cluster-based assessment of protein-protein interaction confidence. BMC Bioinf. 2012, 13, 262. 10.1186/1471-2105-13-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez A.; Echavez-Badel R.; Hepperly P.; Schröder E. Inoculated common beans are protected against Macrophomina phaseolina by Burkholderia cepacia UPR 5C. Plant Soil 1994, 162, 293–297. 10.1007/BF01347716. [DOI] [Google Scholar]
- Ji H.; Wang J.; Guo J.; Li Y.; Lian S.; Guo W.; Yang H.; Kong F.; Zhen L.; Guo L.; et al. Progress in the biological function of alpha-enolase. Anim. Nutr. 2016, 2, 12–17. 10.1016/j.aninu.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Arruda Grossklaus D.; Bailão A. M.; Rezende T. C. V.; Borges C. L.; de Oliveira M. A. P.; Parente J. A.; de Almeida Soares C. M. Response to oxidative stress in Paracoccidioides yeast cells as determined by proteomic analysis. Microbes Infect. 2013, 15, 347–364. 10.1016/j.micinf.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Li B.; Tian X.; Wang C.; Zeng X.; Xing Y.; Ling H.; Yin W.; Tian L.; Meng Z.; Zhang J.; et al. SWATH label-free proteomics analyses revealed the roles of oxidative stress and antioxidant defensing system in sclerotia formation of Polyporus umbellatus. Sci. Rep. 2017, 7, 41283. 10.1038/srep41283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minic Z. Proteomic studies of the effects of different stress conditions on central carbon metabolism in microorganisms. J. Proteomics Bioinf. 2015, 8, 80. 10.4172/jpb.1000355. [DOI] [Google Scholar]
- Alekseev K.; Dubina M.; Komov V. Molecular-genetic and biochemical characteristics of citrate synthase from the citric-acid producing fungus Aspergillus niger. Appl. Biochem. Microbiol. 2016, 52, 810–817. 10.1134/S0003683816090027. [DOI] [Google Scholar]
- Ponpinit T.; Pinchai N. Phenotypic analysis of Aspergillus fumigatus strain lacking the nad-dependent formate dehydrogenase encoding gene FDH. JITMM Proc. 2014, 3, 9–15. [Google Scholar]
- Tiwari S.; Thakur R.; Shankar J. Role of heat-shock proteins in cellular function and in the biology of fungi. Biotechnol. Res. Int. 2015, 2015, 080-090. 10.1155/2015/132635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbach M.; Weber M.; Rinnerthaler M.; Karl T.; Breitenbach-Koller L. Oxidative stress in fungi: its function in signal transduction, interaction with plant hosts, and lignocellulose degradation. Biomolecules 2015, 5, 318–342. 10.3390/biom5020318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Naseem S.; Sharma S.; Konopka J. B. Flavodoxin-like proteins protect Candida albicans from oxidative stress and promote virulence. PLoS Pathog. 2015, 11, e1005147 10.1371/journal.ppat.1005147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klutts J. S.; Yoneda A.; Reilly M. C.; Bose I.; Doering T. L. Glycosyltransferases and their products: cryptococcal variations on fungal themes. FEMS Yeast Res. 2006, 6, 499–512. 10.1111/j.1567-1364.2006.00054.x. [DOI] [PubMed] [Google Scholar]
- Lee H.; Damsz B.; Woloshuk C. P.; Bressan R. A.; Narasimhan M. L. Use of the plant defense protein osmotin to identify Fusarium oxysporum genes that control cell wall properties. Eukaryotic Cell 2010, 9, 558–568. 10.1128/EC.00316-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saupe S. J.; Kuldau G. A.; Smith M. L.; Glass N. L. The product of the het-C heterokaryon incompatibility gene of Neurospora crassa has characteristics of a glycine-rich cell wall protein. Genetics 1996, 143, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fortwendel J. R. Orchestration of morphogenesis in filamentous fungi: conserved roles for Ras signaling networks. Fungal Biol. Rev. 2015, 29, 54–62. 10.1016/j.fbr.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhao Z.; Liu H.; Luo Y.; Zhou S.; An L.; Wang C.; Jin Q.; Zhou M.; Xu J.-R. Molecular evolution and functional divergence of tubulin superfamily in the fungal tree of life. Sci. Rep. 2014, 4, 6746 10.1038/srep06746. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rischitor P. E.; Konzack S.; Fischer R. The Kip3-like kinesin KipB moves along microtubules and determines spindle position during synchronized mitoses in Aspergillus nidulans hyphae. Eukaryotic Cell 2004, 3, 632–645. 10.1128/EC.3.3.632-645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanasamy P.Biological Management of Diseases of Crops: Characteristics of Biological Control Agents; Springer: The Netherlands, 2013; Vol. 1. [Google Scholar]
- Chen W.; Lee M.-K.; Jefcoate C.; Kim S.-C.; Chen F.; Yu J.-H. Fungal cytochrome p450 monooxygenases: their distribution, structure, functions, family expansion, and evolutionary origin. Genome Biol. Evol. 2014, 6, 1620–1634. 10.1093/gbe/evu132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Chen X.; Jiang J.; Yu M.; Yin Y.; Ma Z. The tubulin cofactor A is involved in hyphal growth, conidiation and cold sensitivity in Fusarium asiaticum. BMC Microbiol. 2015, 15, 35. 10.1186/s12866-015-0374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. A.; Wheeler M. H. Biosynthesis and functions of fungal melanins. Annu. Rev. Phytopathol. 1986, 24, 411–451. 10.1146/annurev.py.24.090186.002211. [DOI] [Google Scholar]
- Yamada N.; Motoyama T.; Nakasako M.; Kagabu S.; Kudo T.; Yamaguchi I. Enzymatic characterization of scytalone dehydratase Val75Met variant found in melanin biosynthesis dehydratase inhibitor (MBI-D) resistant strains of the rice blast fungus. Biosci., Biotechnol., Biochem. 2004, 68, 615–621. 10.1271/bbb.68.615. [DOI] [PubMed] [Google Scholar]
- Tseng M. N.; Chung P. C.; Tzean S. S. Enhancing the stress tolerance and virulence of an entomopathogen by metabolic engineering of dihydroxynaphthalene melanin biosynthesis genes. Appl. Environ. Microbiol. 2011, 77, 4508–4519. 10.1128/AEM.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges A. A.; Gladfelter A. S. Septin form and function at the cell cortex. J. Biol. Chem. 2015, 290, 17173–17180. 10.1074/jbc.R114.634444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazaei C. Molecular Insights into Pathogenesis and Infection with Aspergillus fumigatus. Malays. J. Med. Sci. 2017, 24, 10. 10.21315/mjms2017.24.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciandia A.; Suarez T. The NMRA/NMRAL1 homologue PadA modulates the expression of extracellular cAMP relay genes during aggregation in Dictyostelium discoideum. Dev. Biol. 2013, 381, 411–422. 10.1016/j.ydbio.2013.06.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.