

Abstract
A series of novel substituted 2-pyrimidylbenzothiazoles incorporating either sulfonamide moieties or the amino group at C2 of the pyrimidine ring were synthesized and evaluated for its antiviral potency. The novel synthesis of the ring system was carried out by reacting guanidine or N-arylsulfonated guanidine with different derivatives of ylidene benzothiazole based on Michael addition pathways. The antiviral activity of the newly synthesized compounds was examined by a plaque reduction assay against HSV-1, CBV4, HAV HM 175, HCVcc genotype 4 viruses, and HAdV7. In the case of HSV-1, it was determined that 5 out of the 21 synthesized compounds exhibited superior viral reduction in the range of 70–90% with significant IC50, CC50, and SI values as compared with acyclovir. In the case of CBV4, nine compounds have shown more than 50% reduction. Comparable results were obtained for seven of these synthesized compounds when evaluated against HAV with only a couple of them showing 50% reduction or more against HCVcc genotype 4. Remarkably, one compound, 9a, has shown broad action against all five examined viruses, rendering it as potentially an effective antiviral agent. The five potent compounds 9a, 9b, 14b, 14g, and 14h against HSV-1 have also presented inhibitory activity against the Hsp90α protein with IC50 in the range of 4.87–10.47 μg/mL. Interestingly, a combination of the potent synthesized compounds with acyclovir led to IC50 values lower than that of acyclovir alone. The potent compounds 9a, 9b, 14b, 14g, and 14h were also docked inside the active site of Hsp90α to assess the interaction pattern between the tested compounds and the active site of the protein.
Introduction
Viral infections are considered to be one of the major threats to human health. Herpes simplex virus type 1 (HSV-1), a member of the Herpesviridae family, is considered to be the cause of a range of diseases from mild uncomplicated mucocutaneous infections to more serious infections such as cold sores and encephalitis.1 Acyclovir (ACV) is normally used in treating infections caused by HSV-1.2 The most common side effects caused by ACV are nausea, diarrhea, headache, and vomiting. Coxsackievirus B4 (CBV4) virus, a member of the Picornavirus genus, has been involved in the development of insulin-dependent diabetes mellitus (IDDM) normally caused by virus-induced pancreatic cell damage.3 Thus far, there is no specific treatment or vaccine available for CBV4 infections. However, the only available treatment is directed only toward relieving the symptoms resulting from the viral infections.4 Hepatitis A (HAV) virus normally causes inflammation and may affect the liver functions5 but hardly results in serious liver damage. Hepatitis C virus (HCV), however, causes chronic viral infection and is recognized to be one of the leading causes of liver impairment such as cirrhosis and hepatocellular carcinoma.6 Remarkably, sofosbuvir (Sovaldi) is used for the treatment of HCV in combination with other medications such as ribavirin, peginterferon-alfa, simeprevir, ledipasvir, daclatasvir, or velpatasvir to reduce the amount of HCV in the effected body and thus help the liver to recover.7 However, sofosbuvir may cause some unwanted effects such as fatigue, headache, nausea, and anemia. Infections caused by human adenovirus type 7 (HAdV7) may include acute respiratory disease syndrome, pneumonia, pharyngoconjunctival fever, and diseases of the central nervous system.8 Such as the case with CBV4, there is no direct treatment against the viral infection.9 Therefore, the development of novel drugs with superior activity against these drug-resistant viruses will most certainly require extensive synthetic research and clinical assessment.
Of interest here, it has been shown on many occasions that pyrimidine, benzothiazole, and sulfonamide structural units present within various molecules have exhibited interesting antiviral activities. The pyrimidine ring, for example, is the base unit of both ACV and Sovaldi, which are used for the treatment of HSV-1 and HCV, respectively. Assessing the antiviral potency of several published compounds, shown in Figure 1, has indicated their promising activity against HSV-1.10 For example, pyrazolo[3,4-d]pyrimidine derivatives A and B as well as both dimethoxyphenylpyrimidin-5(4H)-one C and thiazolopyrimidine D all showed good antiviral activities against HSV-1 compared to ACV.11 Moreover, pyrimido[2,1-b]benzothiazole derivatives E and F showed antiviral activity against HSV-1 with 61 and 50% reduction in the viral plaques, respectively.12 The piperidinyl amidrazone derivative G was shown to have diminished the quantity of HSV-1 viral plaques by 62% as compared to the reference drug aphidicolin.13
Figure 1.

Pyrimidine, benzothiazole, and sulfonamide compounds as antiviral agents.
Inasmuch, pyrimidine nucleoside analogues, compounds H and I (Figure 1), exhibited antiviral activities against CVB4 with an EC50 value of 9.0 μg/mL in addition to compound J that showed an impressive EC50 of 1 μM and a selectivity index of 141.14,15
The pyrimidine ring bearing the benzothiazole moiety at the C5 position, compound K (Figure 1), was found to produce the optimal inhibition for HCV replication with an EC50 value of 0.03 μM in addition to a selectivity index greater than 550.16,17 Acyl sulfonamide L, thiazolone-based sulfonamides M, and 6-(indol-2-yl) pyridine-3-sulfonamide derivative N have all demonstrated remarkable inhibition against HCV.18−20 Some of the cycloalkylthiopheneimine derivatives bearing a benzothiazole moiety were also synthesized and assessed for their antiviral activities against ADV7. In this regard, compound O (Figure 1) exhibited high potent antiviral activities with an EC50 value of 10.8 μg/mL, which was better than the control compound ribavirin with an EC50 value of 27.8 μg/mL.21 This clearly demonstrated that the structural units of pyrimidine, benzothiazole, and sulfonamide present within the various molecules are a common factor among the active compounds for combating different infectious viruses.
Based on our experience in developing new synthetic approaches for the synthesis of novel benzothiazole, pyrimidine, and sulfonamide compounds in high yield,22−33 novel compounds with potent antiviral activities are planned to be further developed and assessed as potential antiviral drugs in this work. To achieve the target compounds in high yield, the pronounced reactivity of guanidine and its derivatives will also be utilized for the development of competent strategies for the synthesis of a novel benzothiazole pyrimidine sulfonamide ring system. This will be done through the reaction of ylidenes of benzothiazoles with either guanidine or sulfaguanidine derivatives. The study will be further extended to evaluate the exceptional characteristics of these compounds as antiviral agents.
Results and Discussion
Chemistry
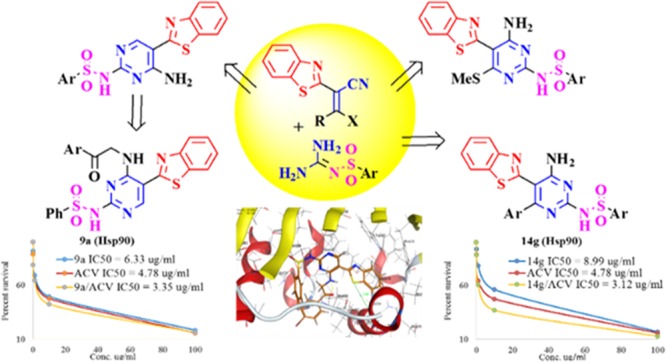
A series of 2-pyrimidylbenzothiazole derivatives were synthesized starting with the facile preparation of benzothiazol-2-yl-acetonitrile 1, which was allowed to react with N,N-dimethylformamide dimethyl acetal (DMF-DMA) 2 in ethyl alcohol at room temperature for 10 min to afford the 2-(benzo[d]thiazol-2-yl)-3-(dimethylamino)acrylonitrile 3 intermediate in high yield (Scheme 1).34 This intermediate was allowed to react further with N-arylsulfonated guanidine 4a,b under basic conditions using potassium hydroxide, forming N-(4-amino-5-(benzo[d]thiazol-2-yl)pyrimidin-2-yl)arylsulfonamide 5a,b (Scheme 1).
Scheme 1. Synthesis of N-(4-Amino-5-(benzo[d]thiazol-2-yl)pyrimidin-2-yl)arylsulfonamide 5a,b.
Structures 5a,b were elucidated on the basis of their IR, 1H NMR, and 13C NMR spectral analysis. The IR spectra of compound 5a showed characteristic absorption bands of the NH2 and NH groups in the vicinity of 3429 and 3278 cm–1, respectively. The 1H NMR spectrum revealed a multiplet signal at δ 7.30–7.99 ppm assigned to the aromatic protons, a broad band at δ 7.67 ppm assigned to the protons of the NH2 group, and a characteristic singlet signal at δ 8.32 ppm assigned to the CH proton. The 13C NMR showed 15 signals, which were attributed to the aromatic carbons of both the benzothiazole ring and phenylsulfonyl group. The reaction proceeded via Michael addition of the amino group of the N-arylsulfonated guanidine 4a,b to the double bond of the enamine with elimination of NH(CH3)2 on the first instance followed by the intramolecular cyclization through the addition of the amino group to the cyano group as to provide the pyrimidine derivatives 5a,b.
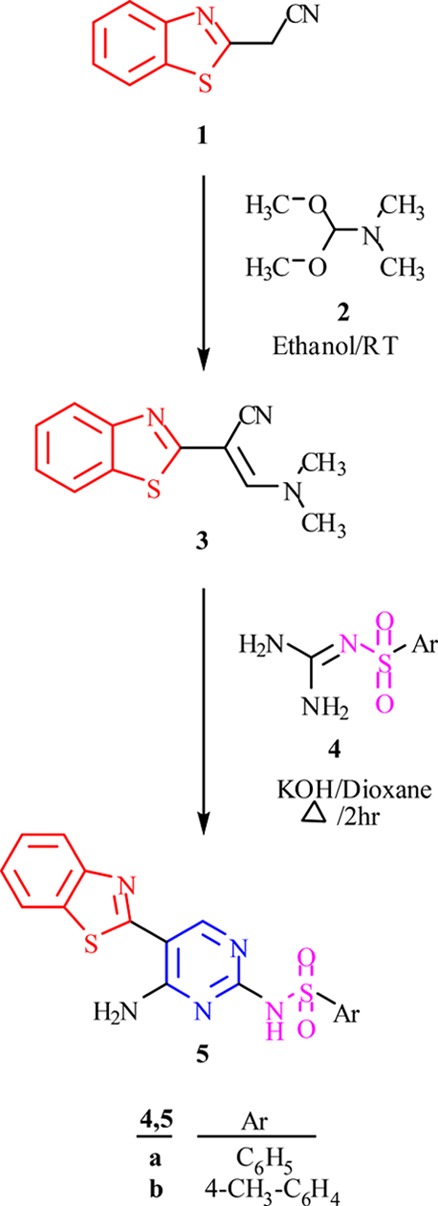
In an attempt to prepare the N-(8-(benzo[d]thiazol-2-yl)-3-methylimidazo[1,2-c]pyrimidine-5-yl)benzenesulfonamide fused-ring structure through the reaction of compound 5a with 3-chloropentane-2,4-dione 6 catalyzed by sodium hydrogen carbonate in refluxing ethanol, a different open structure compound was obtained instead. The product was analyzed spectrally and confirmed to be N-(5-(benzo[d]thiazol-2-yl)-4-(2-oxopropyl)amino)pyrimidin-2-yl)benzenesulfonamide 7 (Scheme 2). The singlet signal in the 1H NMR spectra at 4.94 ppm assigned to the two protons of the CH2 group along with the existence of the carbonyl signature at 1645 cm–1 in the IR data confirmed the structure of compound 7.
Scheme 2. Synthesis of N-(5-(Benzo[d]thiazol-2-yl)-4-(2-oxopropyl)amino) and 4-(2-Aryl-2-oxoethyl)amino) pyrimidin-2-yl)benzenesulfonamide 7 and 9a–c.
Coincidentally, N-(3-aryl-8-(benzo[d]thiazol-2-yl)imidazo[1,2-c]pyrimidine-5-yl) benzenesulfonamide was also not prepared by reacting 5a with 2-bromo-4-substituted acetophenone 8a–c using similar conditions as above. 1H NMR and IR data revealed that the products of this reaction were N-(5-(benzo[d]thiazol-2-yl)-4-(2-aryl-2-oxoethyl)amino)pyrimidin-2-yl)benzenesulfonamide 9a–c (Scheme 2). The absence of the imidazole proton signal in the 1H NMR and the presence of the singlet signal in the range of 5.61–5.63 ppm as well as a signal at 57.0 ppm in the 13C NMR assigned to the two protons of the CH2 group in addition to a band at 1650 cm–1 of the carbonyl group in the IR spectra were all consistent with the chemical structures 9a–c (Scheme 2).
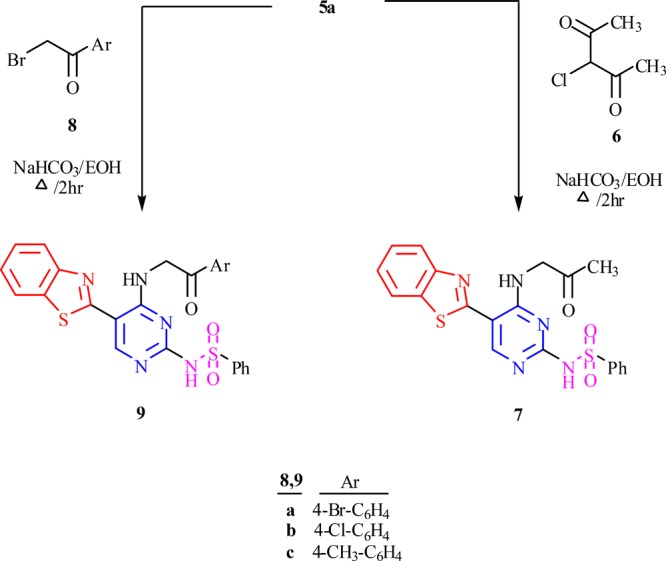
To further the study, a new series of 5-(benzo[d]thiazol-2-yl)-6-arylpyrimidine-2,4-diamine 13a–c have also been synthesized by reacting 2-(benzo[d]thiazol-2-yl)arylacrylonitrile 11c–e (prepared by reacting arylaldehyde 10a–e with benzothiazol-2-yl-acetonitrile 1) with guanidine hydrochloride 12 in the presence of potassium hydroxide in 1,4-dioxane under reflux conditions (Scheme 3). The structure of the resulting compounds was confirmed by spectral and elemental analysis. IR spectra of compound 13b, for example, showed a band for the amino group in the vicinity of 3432 cm–1. In addition, the 1H NMR spectra showed a characteristic signal for protons of the methyl group at δ 2.41 ppm and aromatic protons at δ 7.07–7.65 ppm.
Scheme 3. Synthesis of 5-(Benzo[d]thiazol-2-yl)-6-arylpyrimidine-2,4-diamine 13a–c.
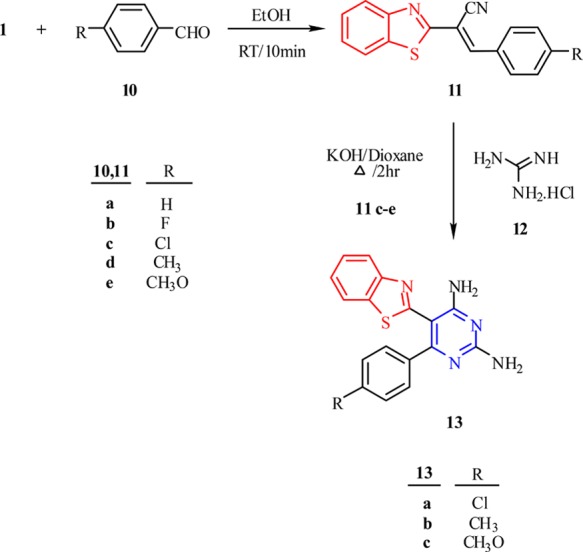
Replacement of guanidine hydrochloride in the previous reaction with N-arylsulfonated guanidine 4a,b under the same conditions afforded the N-(4-amino-5-(benzo[d]thiazol-2-yl)-6-(4-alkylbenzene)pyrimidin-2-yl)arylsulfonamide 14a–j (Scheme 4). Spectral data of compounds 14a–j were consistent with their proposed structures. The 1H NMR spectrum of 14e revealed a singlet band at δ 3.78 ppm assigned to the protons of the OCH3 group, a multiplet at δ 6.96–8.10 ppm assigned to the aromatic protons, a broad band at δ 8.59 ppm assigned to the protons of the NH2 group, and a broad band at δ 11.93 ppm characteristic of the NH proton. Additionally, 13C NMR showed a signal at δ 55.7 ppm, which was attributed to the OCH3 carbon.
Scheme 4. Synthesis of N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-substituted benzene)pyrimidin-2-yl)arylsulfonamide 14a–j.
Finally, benzothiazol-2-yl-acetonitrile 1 was reacted with carbon disulfide in the presence of sodium ethoxide for 20 min, which afforded the disodium salt that was further reacted with methyl iodide at room temperature to yield 2-(benzo[d]thiazol-2-yl)-3,3-bis(methylthio)acrylonitrile 16. The latter was reacted with the N-arylsulfonated guanidine 4a,b to afford N-(4-amino-5-(benzo[d]thiazol-2-yl)-6-(methylthio)pyrimidin-2-yl)arylsulfonamide 17a,b(35) (Scheme 5). Spectral data of compound 17b were consistent with the proposed structure. IR spectra of the latter showed characteristic absorption bands of the NH2 and NH groups at wavenumbers 3375 and 3214 cm–1, respectively. The 1H NMR spectrum revealed two singlet signals at δ 2.37 and 2.39 ppm for the protons of CH3 and SCH3 groups, respectively, a multiplet at δ 7.41–8.12 ppm for the aromatic protons, a broad band at δ 8.49 ppm for the protons of the NH2 group, and a broad band at δ 11.50 ppm characteristic of the NH proton.
Scheme 5. Synthesis of N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(methylthio)pyrimidin-2-yl)arylsulfonamide 17a,b.
Biological Evaluation
Antiviral Activity
The antiviral activities of the newly synthesized 2-pyrimidylbenzothiazoles were evaluated in vitro against a wide variety of viruses such as HSV-1, CBV4, HAV HM 175, ED-43/SG-Feo (VYG) replicon of HCV genotype 4a, and HAdV7. As it is well known that there is no specific cure available for CBV4, HCVgenotype4 and HAdV7 viruses and that available commercial remedies are only used to treat the symptoms but not the illness itself. For this particular reason, acyclovir was used as a commercial standard for HSV-1 against which our new compounds are compared. In order to study the antiviral activities, the newly synthesized compounds were first subjected to a cytotoxicity evaluation using cell lines FRHK-4, Hep2, BGM, Vero, and Huh 7.5 as described clearly in the Supporting Information. No significant difference was observed between the amounts of the nontoxic doses of the various synthesized compounds, which ranged between 60 and 120 μg/mL. The synthesized compounds showed an apparent effect on the infected viral cell lines having different types of genome. The percentage of viral replication was assessed by measuring the viral load in treated cells as compared to the untreated cell line (Table 1).
Table 1. Antiviral Mean Percent of Reduction of Nontoxic Doses of Synthesized Compounds against Herpes Simplex Virus, Coxsackievirus B4, Hepatitis A Virus HM 175, HCVcc Genotype 4, and Adenovirus Type 7a.
| mean
% of reduction |
|||||
|---|---|---|---|---|---|
| compd no. | herpes simplex virus | coxsackievirus B4 | hepatitis A virus HM 175 | HCVcc genotype 4 | adenovirus type 7 |
| 5a | 20 ± 0.9 | 60 ± 1.5 | 50 ± 1 | 30 ± 0.8 | 20 ± 0.5 |
| 5b | 40 ± 1.4 | 50 ± 1.2 | 40 ± 1.0 | 20 ± 0.8 | 20 ± 0.3 |
| 7 | 10 ± 0.5 | 13.3 ± 0.4 | 10 ± 0.2 | 10 ± 0.3 | 10 ± 0.2 |
| 9a | 90 ± 2.5 | 70 ± 1.4 | 70 ± 1.1 | 50 ± 0.9 | 50 ± 0.9 |
| 9b | 70 ± 1.5 | 70 ± 1.6 | 60 ± 1.2 | 60 ± 1.2 | 40 ± 0.8 |
| 9c | 30 ± 1.0 | 50 ± 1.1 | 40 ± 1.2 | 10 ± 0.6 | 10 ± 0.2 |
| 13b | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.2 | 10 ± 0.1 |
| 13c | 10 ± 0.3 | 10 ± 0.2 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.2 |
| 14a | 10 ± 0.2 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.2 |
| 14b | 70 ± 1.3 | 60 ± 1.1 | 50 ± 1.1 | 10 ± 0.6 | 30 ± 0.5 |
| 14c | 30 ± 0.8 | 60 ± 1.2 | 50 ± 1.0 | 30 ± 0.7 | 30 ± 0.6 |
| 14d | 10 ± 0.1 | 30 ± 0.2 | 20 ± 0.2 | 10 ± 0.1 | 10 ± 0.1 |
| 14e | 16.7 ± 0.1 | 20 ± 0.2 | 13.3 ± 0.2 | 10 ± 0.3 | 10 ± 0.1 |
| 14f | 10 ± 0.1 | 10 ± 0.2 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.2 |
| 14g | 80 ± 2.4 | 70 ± 1.5 | 60 ± 1.0 | 20 ± 0.7 | 40 ± 0.8 |
| 14h | 70 ± 2.0 | 66.7 ± 1.1 | 60 ± 0.9 | 20 ± 0.5 | 40 ± 0.7 |
| 14i | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.2 | 10 ± 0.2 | 10 ± 0.1 |
| 14j | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.3 | 10 ± 0.1 |
| 17a | 10 ± 0.3 | 10 ± 0.1 | 10 ± 0.2 | 10 ± 0.1 | 10 ± 0.2 |
| 17b | 10 ± 0.2 | 20 ± 0.2 | 10 ± 0.1 | 10 ± 0.1 | 10 ± 0.1 |
| acyclovir | 99.6 ± 2.8 | NT | NT | NT | NT |
NT = not tested.
The results indicated that nine compounds, 5a, 5b, 9a, 9b, 9c, 14b, 14c, 14g, and 14h, had remarkable antiviral effects that exceeded 50% reduction against the studied viruses (Figure 2). Error bars in the figure represent the standard deviation of the measured data. The 50% maximum cytotoxicity concentration (CC50) and the 50% maximal inhibitory concentration (IC50) as well as the selectivity index (SI), CC50/IC50 ratio, were evaluated for the nine compounds that exhibited greater than 50% viral reduction against the aforementioned tested viruses (Tables 2–4 and Figures 3–7).
Figure 2.
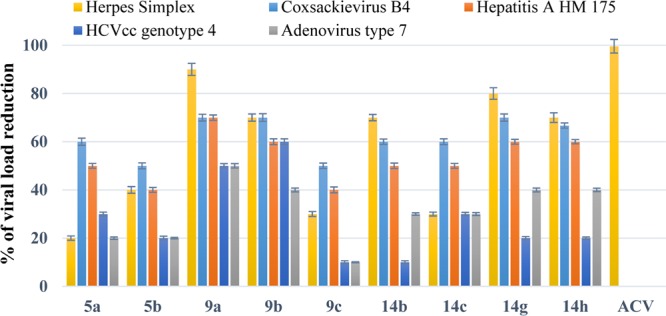
Comparison between the percent of viral load reduction of most potent compounds 5a,b, 9a–c, 14b,c, and 14g,h.
Table 2. Antiviral Activity against Herpes Simplex Virus of Compounds with Viral Reduction 50% or More in Terms of CC50, IC50 (μg/μL), and SI.
| compd No. | CC50 (μg/μL) | IC50 (μg/μL) | SI |
|---|---|---|---|
| 9a | 0.27 | 0.063 | 4.29 |
| 9b | 0.24 | 0.074 | 3.24 |
| 14b | 0.25 | 0.066 | 3.79 |
| 14g | 0.24 | 0.05 | 4.80 |
| 14h | 0.23 | 0.071 | 3.24 |
| acyclovir | 0.028 | 0.007 | 4.00 |
Table 4. Antiviral Activity against HCVcc Genotype 4 and Adenovirus Type 7 of Compounds with Viral Reduction 50% or More in Terms of CC50, IC50 (μg/μL), and SIa.
| HCVcc
genotype 4 |
adenovirus
type 7 |
|||||
|---|---|---|---|---|---|---|
| compd no. | CC50 (μg/μL) | IC50 (μg/μL) | SI | CC50 (μg/μL) | IC50 (μg/μL) | SI |
| 9a | 0.25 | 0.12 | 2.08 | 0.26 | 0.12 | 2.17 |
| 9b | 0.25 | 0.086 | 2.91 | NT | NT | |
NT = not tested.
Figure 3.
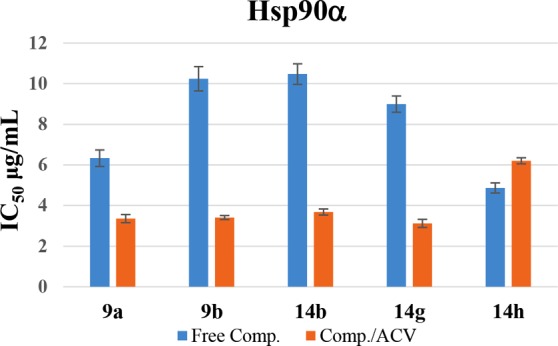
Relative IC50 of tested compounds and their combination with acyclovir on the Hsp90α protein.
Figure 7.

Best docked pose of 9b inside the binding pocket of Hsp90α (PDB ID 3b25) with a docking score of −8.3103 kcal/mol.
Five of the synthesized compounds, 9a, 9b, 14b, 14g, and 14h, showed a high level of potency against HSV-1 (Figure 2). While the viral reduction of ACV hit the 99.6% mark, compound 9a reached 90%, compound 14g reached 80%, and compounds 9b and 14h reached 70% (Figure 2). Furthermore, as shown in Table 2, the five compounds also had IC50 values ranging from 0.05 to 0.074 μg/μL, while that of ACV was 0.007 μg/μL. Although these compounds showed high performance against HSV-1when compared to the standard drug, two compounds in particular, 9a and 14g, showed much better performance than ACV in terms of its CC50. Compound 9a had a CC50 value of 0.27 μg/μL, and compound 14g had a CC50 value of 0.24 μg/μL, whereas ACV had a value of 0.028 μg/μL (Table 2). With respect to the SI factor, compound 9a gave an SI value of 4.29, and compound 14g had an SI value of 4.8, whereas ACV had an SI value of 4. These selectivity indices and cytotoxicity results as well as the viral reduction percentages indicate clearly the antiviral potency of these newly synthesized compounds, 9a and 14g, especially when compared to the standard drug, ACV.
In the case of coxsackievirus B4, nine compounds, 5a, 5b, 9a, 9b, 9c, 14b, 14c, 14g, and 14h, showed more than 50% viral reduction (Figure 2). The IC50 values of these compounds ranged from 0.074 to 0.093 μg/μL with CC50 values ranging from 0.23 to 0.17 μg/μL (Table 3). Among these nine compounds, compound 14c (CC50 = 0.23 μg/μL) showed the highest value for the cytotoxicity, whereas compound 14h showed the lowest inhibition concentration (IC50 = 0.074 μg/100 μL). Furthermore, compound 9a showed the highest SI value of 2.62.
Table 3. Antiviral Activity against Coxsackievirus B4 and Hepatitis A Virus HM 175 of Compounds with Viral Reduction 50% or More in Terms of CC50, IC50 (μg/μL), and SIa.
| coxsackievirus
B4 |
hepatitis
A virus HM 175 |
|||||
|---|---|---|---|---|---|---|
| compd no. | CC50 (μg/μL) | IC50 (μg/μL) | SI | CC50 (μg/μL) | IC50 (μg/μL) | SI |
| 5a | 0.22 | 0.085 | 2.59 | 0.22 | 0.10 | 2.20 |
| 5b | 0.20 | 0.11 | 1.82 | NT | NT | |
| 9a | 0.22 | 0.084 | 2.62 | 0.24 | 0.078 | 3.08 |
| 9b | 0.22 | 0.092 | 2.39 | 0.23 | 0.086 | 2.67 |
| 9c | 0.17 | 0.09 | 1.89 | NT | NT | |
| 14b | 0.18 | 0.093 | 1.93 | 0.22 | 0.010 | 2.20 |
| 14c | 0.23 | 0.091 | 2.53 | 0.23 | 0.010 | 2.30 |
| 14g | 0.19 | 0.082 | 2.32 | 0.23 | 0.081 | 2.84 |
| 14h | 0.19 | 0.074 | 2.57 | 0.20 | 0.074 | 2.70 |
NT = not tested.
Seven compounds, namely, 5a, 9a, 9b, 14b, 14c, 14g, and 14h, have shown more than 50% reduction against HAV as clearly indicated in the supplementary tables with compound 9a exhibiting the highest level of inhibitory activity among all others against HAV with 70% viral reduction. An IC50 value of 0.078 μg/μL, a CC50 value of 0.24 μg/μL, and an SI value of 3.08 were observed of 9a against the virus as indicated in Table 3. Although compounds 14g and 14h revealed the same average reduction around 60%, compound 14g showed higher CC50 than compound 14h.
Compound 9a showed apparent activity against both hepatitis C virus genotype 4a and adenovirus type 7, while compound 9b showed slightly higher activity against hepatitis C virus genotype 4a (Figure 7). As shown in Table 4, both compounds 9a and 9b have the same cytotoxicity concentrations (CC50 = 0.25 μg/μL) but different inhibition concentrations (IC50) with values of 0.12 and 0.086 μg/μL, respectively.
Structure–Activity Relationships
Based on the above results, the preliminary structure–activity relationships (SAR) have been established. In the case of pyrimidine substituted with phenylsulfonamide at C2, 5a, the compound exhibited higher activity when compared to pyrimidine substituted with tosylamide group, 5b (Figure 2). Additionally, the presence of a variety of amino arylethanone groups on the pyrimidine ring, 9a–c, has increased its antiviral activities when compared to that of the starting compound 5a (Figure 2). It is also clear that compound 9a with a bromide substituent at the para position of the aryl moiety showed higher activity against HSV-1, CBV4, HAV, and HAdV7 (Figure 8) than that of 9b with a chloride substituent. However, replacement of the bromide substituent on the aryl moiety with an electron-donating group such as a methyl group, 9c, has lowered its activity (Figure 2). Similar observations can be also made in the case of compounds 14a–h. The presence of electron-withdrawing groups such as a fluoride or chloride group on the para position of the aryl group (position C6 of the pyrimidine ring), 14b, 14c, 14g, and 14h, showed higher activities than those of compounds containing electron-donating groups such as a methyl or methoxy group, 14d, 14e, 14i, and 14j. Compounds with the methylthio group at C6 of the pyrimidine ring 17a,b showed very low activities against all tested viruses.
Figure 8.

Best docked pose of 14b inside the binding pocket of Hsp90α (PDB ID 3b25) with a docking score of −9.7282 kcal/mol.
Hsp90α Inhibition Assay
Heat shock protein 90 (Hsp90α) present in most cell types is important for viral protein folding, assembly, and replication.36 During infection, HSV-1, for example, uses the Hsp90α chaperone system, and the viral polymerase could be a client protein of Hsp90α.37 The possibility that inhibitors of Hsp90α would also be inhibitors of HSV-1 infection has led us to examine our newly synthesized compounds as possible novel Hsp90α inhibitors.
In order to investigate the effect of the potent HSV-1 antiviral compounds 9a, 9b, 14b, 14g, and 14h against Hsp90α, the Hsp90α (C-Terminal) inhibitor screening assay kit was used. A combination of each potent compound with ACV (reference drug), in a 1:1 ratio, was also tested. Results for all compounds were calculated as IC50 and are displayed in Figure 3. The data obtained during this study were used to graph a dose–response curve. The concentration of tested compounds that is required to inhibit 50% of the virus cell population, IC50, was evaluated (Figure 4). Error bars in the various figures represent the standard deviation of the measured data. By comparing the performance of these compounds against that of the standard drug ACV, several observations are noted. All five compounds exhibited a potent inhibitory effect toward Hsp90α and were active in the microgram per milliliter solution (Table 5).
Figure 4.

Different concentrations of 9a,b, 14b, and 14g,h and their combination with acyclovir versus the corresponding percent of cell survival using Hsp90α (C-Terminal) inhibitor screening assay. Each point is the mean (standard deviation) of three independent experiments.
Table 5. Mean ± SD of IC50 Values (the Drug Concentrations That Inhibited 50% of Cell Proliferation) and the Different Concentrations Used of the Tested Compounds and Their Combination with Acyclovir on the Hsp90α Protein.
| compd no. | 100 (μg/mL) | 10 (μg/mL) | 1 (μg/mL) | 0.1 (μg/mL) | IC50 (μg/mL) |
|---|---|---|---|---|---|
| 9a | 81.5553 | 50.3722 | 30.80014 | 8.45589 | 6.33 ± 0.4 |
| 9b | 80.37387 | 45.04568 | 18.17787 | 4.320374 | 10.24 ± 0.6 |
| 14b | 80.73341 | 43.68688 | 18.83649 | 3.165316 | 10.47 ± 0.5 |
| 14g | 82.85524 | 43.74068 | 21.44356 | 6.136187 | 8.99 ± 0.4 |
| 14h | 88.05286 | 50.34291 | 33.03409 | 9.589642 | 4.87 ± 0.25 |
| ACV | 84.06329 | 52.33982 | 35.451 | 11.2422 | 4.78 ± 0.2 |
| 9a/ACV | 83.63504 | 57.47486 | 35.49681 | 21.20627 | 3.36 ± 0.2 |
| 9b/ACV | 83.20892 | 60.08167 | 38.78328 | 14.9151 | 3.41 ± 0.1 |
| 14b/ACV | 83.78818 | 58.34788 | 37.00635 | 14.80698 | 3.68 ± 0.2 |
| 14g/ACV | 86.96226 | 62.80885 | 39.23257 | 11.46644 | 3.13 ± 0.2 |
| 14h/ACV | 75.0849 | 53.2219 | 36.75174 | 10.26611 | 6.20 ± 0.2 |
Consistent with the calculated IC50, compound 14h was the most potent Hsp90α inhibitor with an IC50 value of 4.87 μg/mL followed by 9a and 14g with IC50 values of 6.33 and 8.99 μg/mL, respectively, as compared to the ACV reference drug IC50 value of 4.78 μg/mL. It is clear that the combination of four out of the five tested compounds, namely, 9a, 9b, 14b, and 14g, with ACV has increased the potency of the compounds, which was reflected as a marked reduction in the IC50 values of the original single compounds. Notwithstanding, these combinations have also shown lower IC50 values than that of ACV itself. For example, the IC50 values of compounds 9a, 9b, 14b, and 14g have dropped from 6.33, 10.24, 10.47, and 8.99 μg/mL to 3.35, 3.41, 3.68, and 3.12 μg/mL, respectively, when combined with ACV in a 1:1 ratio. The resulting data indicated that a combination of compounds 9a, 9b, 14b, and 14g with ACV is highly recommended for use as possible potent inhibitors for Hsp90α and, consequently, inhibitors for HSV-1.
Molecular Modeling and Docking Study
To evaluate the underlying principles behind the action of these new compounds in inhibiting Hsp90α, a molecular docking study using a molecular modeling environment (MOE) was performed on the reference drug ACV and the potent compounds 9a, 9b, 14b, 14g, and 14h. The compounds were docked with the crystal structure of the Hsp90α protein (PDB ID 3b25) through the removal of the bound ligand, B2K (4-methyl-6-(toluene-4-sulfonyl)-pyrimidin-2-ylamine), to uncover the binding pattern of these compounds with the receptor. The docking study revealed that the molecules had good binding energy in the range of −5.6421 to −9.7282 kcal/mol with the receptor within the active site. The pyrimidine ring of ACV and the tested compounds 9b, 14b, 14g, and 14h showed hydrophobic interaction with the active site of Phe 138 (Figures 5–10). Only one compound, 9a, displayed one hydrogen-bonding interaction at a distance of 3.84 Å between the sulfur atom of the benzothiazole ring and amino acid residue Asp 102 (Figure 6). Moreover, compounds 9a and 9b interacted with the amino acid residues Met 98 and Tyr 139, respectively, through polar bonds (Figures 6 and 7). In addition, two aren-H interactions of the benzene ring and the amino group of compounds 14b and 14h were observed to bind to Leu 107 and Trp162, respectively (Figures 8 and 10), while compounds 9b and 14g showed one aren-H interaction with Asn 51 and Thr 184, respectively (Figures 7 and 9). The docking study revealed a docking score of −9.1024 kcal/mol for compound 14h, −8.6521 kcal/mol for compound 9a, and a docking score of −5.6421 kcal/mol for the reference drug ACV.
Figure 5.

Best docked pose of acyclovir inside the binding pocket of Hsp90α (PDB ID 3b25) with a docking score of −5.6421 kcal/mol.
Figure 10.

Best docked pose of 14h inside the binding pocket of Hsp90α (PDB ID 3b25) with a docking score of – 9.1024 kcal/mol.
Figure 6.

Best docked pose of 9a inside the binding pocket of Hsp90α (PDB ID 3b25) with a docking score of −8.6521 kcal/mol.
Figure 9.

Best docked pose of 14g inside the binding pocket of Hsp90α (PDB ID 3b25) with a docking score of −8.1512 kcal/mol.
Conclusions
In conclusion, 21 new compounds of 2-pyrimidylbenzothiazoles bearing either the amino group or sulfonamide moieties at the C2 position of the pyrimidine ring were synthesized by reacting guanidine or N-arylsulfonated guanidine with different derivatives of ylidene benzothiazole. The structures of all the compounds were confirmed spectroscopically and via elemental analyses. The newly synthesized compounds were evaluated for their antiviral activity against HSV-1, COB4, HAV HM 175, ED-43/SG-Feo (VYG) replicon of HCV genotype 4a, and HAdV7. Nine compounds exhibited remarkable activities against these viruses with high cytotoxicity concentration and more than 50% viral reduction. The most active five compounds against HSV-1 have been also evaluated against Hsp90α with their activities compared to that of the reference drug acyclovir. A combination of the tested compounds and acyclovir in a 1:1 ratio, amazingly, led to increased potency for these compounds with IC50 values lower than that of acyclovir. The work confirms that the newly synthesized novel compounds, 2-pyrimidylbenzothiazole derivatives, exhibit exceptional antiviral activities and inhibitory effect on the Hsp90α protein and thus can be useful as highly effective broad spectrum antiviral agents.
Experimental Section
Chemistry
Melting points were measured using an SMP3 apparatus. IR spectra, using KBr discs, were measured on either a Pye Unicam SP-1000 or FTIR plus 460 spectrophotometer. Both 1H and 13C spectra were recorded on a Bruker Avance (III)-400 spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR) at the Ain Shams University, Cairo, Egypt, using DMSO-d6 with Si(CH3)4 as an internal standard. Thin-layer chromatography (TLC), aluminum sheets coated with silica gel F254 (Merck), and an ultraviolet (UV) lamp were used to monitor the progress of the reactions. The elemental analyses were done at the Microanalytical Data Unit at the Cairo University and performed on Vario EI III Elemental CHNS analyzer.
General Procedure for the Synthesis of 5a,b
2-(Benzo[d]thiazol-2-yl)-3-(dimethylamino)acrylonitrile 3 (2.30 g, 0.01 mmol) was added to a stirred solution of the N-carbamimidoylarylsulfonamide 4a,b (0.01 mol) in dry dioxane (20 mL) containing potassium hydroxide (0.56 g, 0.01 mol). The reaction mixture was heated under reflux for 2 h. After completion of the reaction (TLC), the reaction mixture was cooled and poured into ice water. The resulting precipitate was filtered off, washed with water, dried, and recrystallized from DMF.
N-(4-Amino-5-(Benzo[d]thiazol-2-yl)pyrimidin-2-yl)benzenesulfonamide (5a)
Yellow crystals; (DMF); yield 74.3%; m.p. 345–346 °C; IR (KBr, cm–1): ν 3429 and 3278 (NH, NH2), 3059 (ArCH), 1599 (C=C). 1H NMR (400 MHz, DMSO-d6): δ 7.30–7.45 (m, 5H, 3Ar-H & 1benzothiazole-H), 7.67 (br s, 2H, NH2), 7.85–7.99 (m, 4H, 2Ar-H & 2benzothiazole-H), 8.32 (s, 1H, CH pyrimidine). 13C NMR (100 MHz, DMSO-d6): δ 99.9, 121.5, 122.0, 124.7, 126.7, 127.6, 127.7, 129.7, 131.7, 147.2, 153.2, 158.2, 160.1, 164.5, 166.9 (Ar-C). Anal. Calcd for C17H13N5O2S2 (383.45): C% 53.25; H% 3.42; N% 18.26. Found: C% 53.33; H% 3.40; N% 18.20.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)pyrimidin-2-yl)-4-methylbenzenesulfonamide (5b)
Yellow crystals; (DMF); yield 85.7%; m.p. 336–337 °C; IR (KBr, cm–1): ν 3397 and 3288 (NH, NH2), 3061 (ArCH), 1605 (C=C). 1H NMR (400 MHz, DMSO-d6): δ 2.31 (s, 3H, CH3), 7.21 (d, 2H, J = 6.8 Hz, Ar-H), 7.36 (t, 1H, J = 8 Hz, benzothiazole-H), 7.46 (t, 1H, J = 8 Hz, benzothiazole-H), 7.86 (d, 2H, J = 8 Hz, Ar-H), 7.92 (d, 1H, J = 8 Hz, benzothiazole-H), 8.01 (d, 1H, J = 8 Hz, benzothiazole-H), 8.19 (br s, 2H, NH2), 8.49 (s, 1H, CH pyrimidine). 13C NMR (100 MHz, DMSO-d6): δ 21.3 (CH3), 100.8, 121.9, 122.1, 125.3, 126.8, 127.9, 128.6, 132.1, 140.3, 142.7, 153.0, 156.0, 160.2, 161.0, 166.0 (Ar-C). Anal. Calcd for C18H15N5O2S2 (397.47): C% 54.39; H% 3.80; N% 17.62. Found: C% 54.30; H% 3.87; N% 17.55.
Procedure for the Synthesis of 7
To a solution of 3-chloropentane-2,4-dione 6 (1.20 mL, 0.01 mol) in 30 mL of ethanol, 5a (3.83 g, 0.01 mol) was added. After being refluxed for 1 h, sodium bicarbonate (1.30 g, 0.015 mol) was added, and the mixture was heated for additional 2 h. After completion of the reaction (TLC), the solid precipitate was filtered off, washed with water, and dried.
N-(5-(Benzo[d]thiazol-2-yl)-4-((2-oxopropyl)amino)pyrimidin-2-yl)benzenesulfonamide (7)
White crystals; (DMF); yield 80%; m.p. 291–292 °C; IR (KBr, cm–1): ν 3423 (NH), 2921 (ArCH), 1645 (C=O). 1H NMR (400 MHz, DMSO-d6): δ 2.21 (s, 3H, CH3), 4.94 (s, 2H, CH2), 7.46–7.53 (m, 5H, 3Ar-H & 2benzothiazole-H), 7.96–8.12 (m, 4H, 2Ar-H & 2benzothiazole-H), 8.64 (s, 1H, CH pyrimidine), 8.82 (br s, 1H, NH), 9.31 (br s, 1H, NH). Anal. Calcd for C20H17N5O3S2 (439.51): C% 54.65; H% 3.90; N% 15.93. Found: C% 54.60; H% 3.82; N% 15.89.
General Procedure for the Synthesis of 9a–c
To a solution of substituted phenacyl bromide 8a–c (0.01 mol) in 30 mL of ethanol, 5a (3.83 g, 0.01 mol) was added, and the mixture heated under reflux for 1 h. Sodium bicarbonate (1.30 g, 0.015 mol) was then added, and the mixture was refluxed for additional 2 h. After completion of the reaction (TLC), the solid precipitate was filtered off, washed with water, and dried.
N-(5-(Benzo[d]thiazol-2-yl)-4-((2-(4-bromophenyl)-2-oxoethyl)amino)pyrimidin-2-yl)benzenesulfonamide (9a)
White needles; (DMF); yield 80%; m.p. 294–295 °C; IR (KBr, cm–1): ν 3440 (NH), 3047 (ArCH), 1651 (C=O). 1H NMR (400 MHz, DMSO-d6): δ 5.61 (s, 2H, CH2), 7.39 (t, 2H, J = 8 Hz, Ar-H), 7.45–7.49 (m, 2H, Ar-H, benzothiazole-H), 7.55 (t, 1H, J = 8 Hz, benzothiazole-H), 7.83 (d, 2H, J = 8 Hz, Ar-H), 7.89 (d, 2H, J = 8 Hz, Ar-H), 8.00 (d, 2H, J = 8 Hz, Ar-H), 8.07 (d, 1H, J = 8 Hz, benzothiazole-H), 8.13 (d, 1H, J = 8 Hz, benzothiazole-H), 8.78 (s, 1H, CH pyrimidine), 8.88 (br s, 1H, NH), 9.37 (br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 57.0 (CH2), 101.3, 122.1, 122.3, 125.9, 126.7, 127.4, 127.7, 128.3, 130.0, 130.9, 132.0, 132.1, 133.2, 143.2, 149.7, 151.9, 152.1, 159.0, 162.9, 191.8 (Ar-C). Anal. Calcd for C25H18BrN5O3S2 (580.48): C% 51.73; H% 3.13; N% 12.06. Found: C% 51.65; H% 3.19; N% 12.14.
N-(5-(Benzo[d]thiazol-2-yl)-4-((2-(4-chlorophenyl)-2-oxoethyl)amino)pyrimidin-2-yl)benzenesulfonamide (9b)
White needles; (DMF); yield 80%; m.p. 292–293 °C; IR (KBr, cm–1): ν 3365 (NH), 3057 (ArCH), 1649 (C=O). 1H NMR (400 MHz, DMSO-d6): δ 5.63 (s, 2H, CH2), 7.41–7.53 (m, 5H, 3Ar-H, 2benzothiazole-H), 7.67 (d, 2H, J = 8 Hz, Ar-H), 7.92 (d, 2H, J = 8 Hz, Ar-H), 8.01–8.09 (m, 4H, 2Ar-H & 2benzothiazole-H), 8.75 (s, 1H, CH), 8.88 (br s, 1H, NH), 9.36 (br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 57.0 (CH2), 101.3, 122.0, 122.3, 125.8, 126.7, 127.4, 127.7, 129.1, 129.9, 130.9, 132.1, 132.9, 139.0, 143.2, 149.7, 151.9, 152.1 and 159.0 (Ar-C), 162.9 (C=O), 191.8 (Ar-C). Anal. Calcd for C25H18ClN5O3S2 (536.03): C% 56.02; H% 3.38; N% 13.07. Found: C% 56.10; H% 3.30; N% 13.10.
N-(5-(Benzo[d]thiazol-2-yl)-4-((2-(p-tolyl)-2-oxoethyl)amino)pyrimidin-2-yl)benzenesulfonamide (9c)
White needles; (DMF); yield 80%; m.p. 305–307 °C; IR (KBr, cm–1): ν 3436 (NH), 3038 (ArCH), 1648 (C=O). 1H NMR (400 MHz, DMSO-d6): δ 2.44 (s, 3H, CH3), 5.62 (s, 2H, CH2), 7.37–7.50 (m, 6H, 5Ar-H & benzothiazole-H), 7.56 (t, 1H, J = 8 Hz, benzothiazole-H), 7.88 (d, 2H, J = 8 Hz, Ar-H), 7.98 (d, 2H, J = 8 Hz, Ar-H), 8.08 (d, 1H, J = 8 Hz, benzothiazole-H), 8.14 (d, 1H, J = 8 Hz, benzothiazole-H), 8.79 (s, 1H, CH pyrimidine), 8.85 (br s, 1H, NH), 9.35 (br s, 1H, NH). Anal. Calcd for C26H21N5O3S2 (515.61): C% 60.57; H% 4.11; N% 13.58. Found: C% 60.65; H% 4.03; N% 13.63.
General Procedure for the Synthesis of 13a–c
To a stirred solution of guanidine hydrochloride (1.43 g, 0.015 mol) 12 in dry dioxane (30 mL) containing potassium hydroxide (0.84 g, 0.015 mol), 2-(benzo[d]thiazol-2-yl)-3-arylacrylonitrile 11c–e (0.01 mol) was added, and the mixture was refluxed for 2 h. After completion of the reaction (TLC), the reaction mixture was then cooled and poured into ice water. The resulting precipitate was filtered off, washed with water, dried, and recrystallized from DMF.
5-(Benzo[d]thiazol-2-yl)-6-(4-chlorophenyl)pyrimidine-2,4-diamine (13a)
Orange crystals; (DMF); yield 76%; m.p. >350 °C; IR (KBr, cm–1): ν 3431 (NH2), 2913 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 7.09 (t, 1H, J = 8 Hz, benzothiazole-H), 7.26 (t, 1H, J = 8 Hz, benzothiazole-H), 7.50–7.58 (m, 5H, 4Ar-H, benzothiazole-H), 7.69 (d, 1H, J = 8 Hz, benzothiazole-H). 13C NMR (100 MHz, DMSO-d6): δ 66.83, 81.58, 120.61, 121.30, 123.26, 125.98, 129.26, 133.87, 153.22, 167.59 (Ar-C). Anal. Calcd for C17H12ClN5S (353.83): C% 57.71; H% 3.42; N% 19.79. Found: C% 57.77; H% 3.35; N% 19.84.
5-(Benzo[d]thiazol-2-yl)-6-p-tolylpyrimidine-2,4-diamine (13b)
Orange crystals; (DMF); yield 77%; m.p. >350 °C; IR (KBr, cm–1): ν 3432 (NH2), 2919 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.41 (s, 3H, CH3), 7.07 (t, 1H, J = 8 Hz, benzothiazole-H), 7.24–7.28 (m, 3H, 2Ar-H & benzothiazole-H), 7.44 (d, 2H, J = 5.6 Hz, Ar-H), 7.57 (d, 1H, J = 8 Hz, benzothiazole-H), 7.65 (d, 1H, J = 8 Hz, benzothiazole-H). 13C NMR (100 MHz, DMSO-d6): δ 21.61 (CH3), 66.83, 81.60, 120.45, 121.18, 123.10, 125.87, 129.84, 153.18, 168.15 (Ar-C). Anal. Calcd for C18H15N5S (333.41): C% 64.84; H% 4.53; N% 21.01. Found: C% 64.90; H% 4.48; N% 21.08.
5-(Benzo[d]thiazol-2-yl)-6-(4-methoxyphenyl)pyrimidine-2,4-diamine (13c)
Orange crystals; (DMF); yield 79%; m.p. >350 °C; IR (KBr, cm–1): ν 3441 (NH2), 2915 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 3.84 (s, 3H, OCH3), 7.01–7.09 (m, 3H, 2Ar-H & benzothiazole-H), 7.25 (t, 1H, J = 8 Hz, benzothiazole-H), 7.48 (d, 1H, J = 8 Hz, benzothiazole-H), 7.58 (d, 2H, J = 8 Hz, Ar-H), 7.65 (d, 1H, J = 8 Hz, benzothiazole-H). Anal. Calcd for C18H15N5OS (349.41): C% 61.87; H% 4.33; N% 20.04. Found: C% 61.94; H% 4.26; N% 20.10.
General Procedure for the Synthesis of 14a–j
A mixture of 2-(benzo[d]thiazol-2-yl)-3-arylacrylonitrile 11a–c (0.01 mol) and N-carbamimidoylarylsulfonamide 4a,b (0.01 mol) in dry dioxane (20 mL) containing potassium hydroxide (0.56 g, 0.01 mol) was refluxed for 2 h. After completion of the reaction (TLC), the reaction mixture was then cooled and poured into ice water. The resulting precipitate was filtered off, washed with water, dried, and recrystallized from appropriate solvent.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-phenylpyrimidin-2-yl)benzenesulfonamide (14a)
Buff crystals; (Ethanol); yield 83%; m.p. 260–261 °C; IR (KBr, cm–1): ν 3419 and 3366 (NH, NH2), 3224 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 7.33–7.60 (m, 10H, 8Ar-H & 2benzothiazole-H), 7.83 (d, 1H, J = 8 Hz, benzothiazole-H), 8.02 (d, 1H, J = 8 Hz, benzothiazole-H), 8.11 (d, 2H, J = 8 Hz, Ar-H), 8.6 (br s, 2H, NH2), 11.9 (br s, 1H, NH). Anal. Calcd for C23H17N5O2S2 (459.54): C% 60.11; H% 3.73; N% 15.24. Found: C% 60.18; H% 3.68; N% 15.30.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-fluorophenyl)pyrimidin-2-yl)benzenesulfonamide (14b)
Yellow needles; (DMF); yield 76%; m.p. 281–282 °C; IR (KBr, cm–1): ν 3405 and 3368 (NH, NH2), 3191 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 7.27–7.59 (m, 9H, 7Ar-H & 2benzothiazole-H), 7.90 (d, 1H, J = 8 Hz, benzothiazole-H), 8.03 (d, 1H, J = 8 Hz, benzothiazole-H), 8.11 (d, 2H, J = 8 Hz, Ar-H), 8.62 (br s, 2H, NH2), 11.90 (br s, 1H, NH). Anal. Calcd for C23H16FN5O2S2 (477.53): C% 57.85; H% 3.38; N% 14.67. Found: C% 57.94; H% 3.32; N% 14.71.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-chlorophenyl)pyrimidin-2-yl)benzenesulfonamide (14c)
Yellow needles; (DMF); yield 81%; m.p. 298–299 °C; IR (KBr, cm–1): ν 3410 and 3362 (NH, NH2), 3181 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 7.37–7.63 (m, 9H, 7Ar-H & 2benzothiazole-H), 7.91 (d, 1H, J = 8 Hz, benzothiazole-H), 8.03 (d, 1H, J = 8 Hz, benzothiazole-H), 8.09 (d, 2H, J = 7.6 Hz, Ar-H), 8.50 (br s, 2H, NH2), 11.81 (br s, 1H, NH). Anal. Calcd for C23H16ClN5O2S2 (493.99): C% 55.92; H% 3.26; N% 14.18. Found: C% 55.98; H% 3.21; N% 14.26.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-p-tolylpyrimidin-2-yl)benzenesulfonamide (14d)
Yellow needles; (Ethanol); yield 72%; m.p. 258–259 °C; IR (KBr, cm–1): ν 3385 and 3309 (NH2, NH), 3179 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.26 (s, 3H, CH3), 7.08–7.22 (m, 5H, 4Ar-H & benzothiazole-H), 7.34–7.43 (m, 3H, Ar-H), 7.52 (t, 1H, J = 7.6 Hz, benzothiazole-H), 7.67–7.70 (m, 3H, 2Ar-H & benzothiazole-H), 7.81–7.84 (m, 3H, benzothiazole-H, NH2), 10.05 (br s, 1H, NH). Anal. Calcd for C24H19N5O2S2 (473.57): C% 60.87; H% 4.04; N% 14.79. Found: C% 61.00; H% 3.98; N% 14.84.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-methoxyphenyl)pyrimidin-2-yl)benzenesulfonamide (14e)
Buff crystals; (DMF); yield 76%; m.p. 286–287 °C; IR (KBr, cm–1): ν 3405 and 3358 (NH, NH2), 3169 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 3.78 (s, 3H, OCH3), 6.96 (d, 2H, J = 7.6 Hz, Ar-H), 7.30–7.36 (m, 3H, 2Ar-H & benzothiazole-H), 7.45–7.60 (m, 4H, 3Ar-H & benzothiazole-H), 7.87 (d, 1H, J = 7.6, benzothiazole-H), 8.02 (d, 1H, J = 6.8 Hz, benzothiazole-H), 8.10 (d, 2H, J = 7.6 Hz, Ar-H), 8.59 (br s, 2H, NH2), 11.93 (br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 55.7 (OCH3), 114.5, 122.0, 122.9, 125.8, 126.6, 128.5, 128.7, 131.8, 135.1, 151.7, 161.6, 161.9 (Ar-C). Anal. Calcd for C24H19N5O3S2 (489.57): C% 58.88; H% 3.91; N% 14.31. Found: C% 58.99; H% 3.97; N% 14.40.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-phenylpyrimidin-2-yl)-4-methylbenzenesulfonamide (14f)
Colorless needles; (Ethanol); yield 82%; m.p. 244–245 °C; IR (KBr, cm–1): ν 3440 and 3350 (NH2, NH), 3226 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.36 (s, 3H, CH3), 7.35–7.48 (m, 9H, 7Ar-H & benzothiazole-H), 7.85–7.97 (m, 4H, 2Ar-H & 2benzothiazole-H). Anal. Calcd for C24H19N5O2S2 (473.57): C% 60.87; H% 4.04; N% 14.79. Found: C% 60.97; H% 4.00; N% 14.85.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-fluorophenyl)pyrimidin-2-yl)-4-methylbenzenesulfonamide (14g)
Yellow needles; (Ethanol); yield 77.9%; m.p. 266–267 °C; IR (KBr, cm–1): ν 3400 and 3366 (NH, NH2), 3181 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.37 (s, 3H, CH3) 7.24–7.50 (m, 8H, 6Ar-H & 2benzothiazole-H), 7.89–8.04 (m, 4H, 2Ar-H & 2benzothiazole-H), 8.05 (br s, 2H, NH2), 11.75 (br s, 1H, NH). Anal. Calcd for C24H18FN5O2S2 (491.56): C% 58.64; H% 3.69; N% 14.25. Found: C% 58.73; H% 3.63; N% 14.32.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-chlorophenyl)pyrimidin-2-yl)-4-methylbenzenesulfonamide (14h)
Yellow solid; (DMF); yield 85%; m.p. 297–298 °C; IR (KBr, cm–1): ν 3404 and 3366 (NH, NH2), 3181 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.36 (s, 3H, CH3), 7.33–7.47 (m, 8H, 6Ar-H & 2benzothiazole-H), 7.89–8.03 (m, 4H, 2Ar-H & 2 benzothiazole-H). Anal. Calcd for C24H18ClN5O2S2 (508.02): C% 56.74; H% 3.57; N% 13.79. Found: C% 56.81; H% 3.53; N% 13.84.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-p-tolylpyrimidin-2-yl)-4-methylbenzenesulfonamide (14i)
White crystals; (DMF); yield 78%; m.p. 323–325 °C; IR (KBr, cm–1): ν 3453 and 3360 (NH2, NH). 1H NMR (400 MHz, DMSO-d6): δ 2.33 (s, 3H, CH3), 2.35 (s, 3H, CH3), 7.10–7.16 (m, 6H, Ar-H), 7.24 (t, 1H, J = 8 Hz, benzothiazole-H), 7.39 (t, 1H, J = 7.6 Hz, benzothiazole-H), 7.74–7.77 (m, 3H, 2Ar-H & benzothiazole-H), 7.88 (d, 1H, J = 8 Hz, benzothiazole-H). 13C NMR (100 MHz, DMSO-d6): δ 21.3, 21.4 (2CH3), 98.5, 121.5, 121.8, 124.6, 126.2, 128.0, 129.1, 130.1, 134.6, 137.2, 138.9, 139.1, 144.5, 151.9, 161.7, 167.2 (Ar-C). Anal. Calcd for C25H21N5O2S2 (487.60): C% 61.58; H% 4.34; N% 14.36. Found: C% 61.68; H% 4.29; N% 14.41.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(4-methoxyphenyl)pyrimidin-2-yl)-4-methylbenzenesulfonamide (14j)
Yellow crystals; (Ethanol); yield 79.4%; m.p. 248–249 °C; IR (KBr, cm–1) ν 3368 and 3237 (NH, NH2), 3065 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.37 (s, 3H, CH3), 3.78 (s, 3H, OCH3), 6.96 (d, 2H, J = 9.2 Hz, Ar-H), 7.32–7.37 (m, 5H, 4Ar-H & benzothiazole-H), 7.48 (t, 1H, J = 7.2 Hz, benzothiazole-H), 7.87 (d, 1H, J = 8.4 Hz, benzothiazole-H), 7.98–8.03 (m, 3H, 2Ar-H & benzothiazole-H), 8.51 (br s, 2H, NH2), 11.67 (br s, 1H, NH). Anal. Calcd for C25H21N5O3S2 (503.60): C% 59.62; H% 4.20; N% 13.91. Found: C% 59.72; H% 4.16; N% 13.97.
General Procedure for Synthesis of 17a,b
2-(Benzo[d]thiazol-2-yl)-3,3-bis(methylthio)acrylonitrile3816 (2.78 g, 0.01 mol) was added to a stirred solution of the N-carbamimidoylarylsulfonamide 4a,b (0.01 mol) in dry dioxane (20 mL) containing potassium hydroxide (0.56 g, 0.01 mol), and the reaction mixture was refluxed for 2 h. After completion of the reaction (TLC), the solid precipitate was filtered off and then recrystallized using an appropriate solvent.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(methylthio)pyrimidin-2-yl)benzenesulfonamide (17a)
Colorless crystal; (DMF); yield 75%; m.p. 244–245 °C; IR (KBr, cm–1): ν 3431 and 3874 (NH, NH2). 1H NMR (400 MHz, DMSO-d6): δ 2.19 (s, 3H, CH3), 7.32–7.39 (m, 4H, 3Ar-H & benzothiazole-H), 7.46 (t, 1H, J = 8 Hz, benzothiazole-H), 7.83–7.85 (m, 2H, Ar-H), 7.92 (d, 1H, J = 8 Hz, benzothiazole-H), 8.01 (d, 1H, J = 8 Hz, benzothiazole-H). Anal. Calcd for C18H15N5O2S3 (429.54): C% 50.33; H% 3.52; N% 16.30. Found: C% 50.43; H% 3.48; N% 16.35.
N-(4-Amino-5-(benzo[d]thiazol-2-yl)-6-(methylthio)pyrimidin-2-yl)-4-methylbenzenesulfonamide (17b)
Yellow crystals; (Ethanol); yield 75%; m.p. 238–239 °C; IR (KBr, cm–1): ν 3375 and 3321 (NH, NH2), 3161 (ArCH). 1H NMR (400 MHz, DMSO-d6): δ 2.37 (s, 3H, CH3), 2.39 (s, 3H, CH3), 7.41–7.53 (m, 4H, 2Ar-H & 2benzothiazole-H), 7.94–8.12 (m, 4H, 2Ar-H & 2benzothiazole-H), 8.49 (br s, 2H, NH2), 11.50 (br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 14.2 (SCH3), 21.4 (CH3), 122.1, 122.7, 125.7, 126.8, 127.9, 128.4, 129.2, 129.7, 134.5, 138.3, 143.7, 151.7, 154.6, 162.3, 168.5 (Ar-C). Anal. Calcd for C19H17N5O2S3 (443.57): C% 51.45; H% 3.86; N% 15.79. Found: C% 51.52; H% 3.82; N% 15.86.
Biological Evaluation
Cytotoxicity Test
Cytotoxicity and antiviral tests were carried out at The National Research Center, Cairo, Egypt. Cytotoxicity was done according to the literature.39−41 To prepare for the tests, 50 mg of each sample was allowed to dissolve in 1 mL of DMSO. To avoid possible contamination of the samples, 24 μL of 100× of an antibiotic–antimycotic mixture was added to 1 mL of each sample. Next, 100 μL of each sample was subjected to bi-fold dilutions followed by inoculating 100 μL of each dilution in Hep-2, Vero, BGM, FRHK4, and Huh 7.5 cell lines previously cultured in 96-multiwell plates to determine the nontoxic dose of the examined samples. The well plates were provided from Greiner Bio-One, Germany, while the cell lines were obtained from the Holding Company for Biological Products & Vaccines VACSERA, Egypt. To complete the cytotoxicity assay, cell morphology evaluation using an inverted light microscope and the cell viability test through the application of trypan blue dye exclusion method was utilized.
Cell Morphology Evaluation by Inverted Light Microscopy
As mentioned earlier, cultures of the cell lines (2 × 105 cells/mL) were prepared. The cultures were then incubated at a temperature of 37 °C for 24 h in a humidified CO2 atmosphere with a 5% ratio (v/v). This is to allow for the cell monolayers to be confluent. From each well separately, the medium was then removed and subsequently replenished with 100 μL of bi-fold dilutions, prepared in DMEM (GIBCO BRL), of the different tested samples. One hundred microliters of DMEM was used as the cell control without sample addition. All cell cultures were incubated in a humidified 5% (v/v) CO2 atmosphere at a temperature of 37 °C for 72 h. On a daily basis, the cell morphology was assessed for any possible morphological alterations on a microscopic scale such as cell rounding and shrinking, loss of confluence, and cytoplasm granulation and vacuolization. The morphological changes were thus scored.40
Cell Viability Assay
Trypan blue dye exclusion method was used in this assay.42 The aforementioned cell cultures (2 × 105 cells/mL) were grown in 12-well tissue culture plates. One hundred microliters of bi-fold dilutions of the tested samples was applied after 24 h incubation for each well as described previously. The medium was then removed after 72 h, which is followed by trypsinizing the cells with an equal volume of 0.4% (w/v) of the trypan blue dye aqueous solution. Viable cells were assessed using a phase contrast microscope.
Determination of Coxsackievirus B4, HAV HM175, Adenovirus 7, and HSV-1 Titers Using Plaque Assay
One hundred microliters of nontoxic dilutions was mixed with 100 μL of the different doses of HSV-1, HAdV7, HAV HM175, and CBV4 (1 × 105, 1 × 106, and 1 × 107). Each mixture was incubated for half an hour at a temperature of 37 °C. One hundred microliters of 10-fold dilutions of the untreated and treated adenovirus 7, HAV HM175, CBV4, and HSV-1 was inoculated separately into Hep-2, FRHK4, BGM, and Vero cell lines in 12-multiwell plates, respectively. The samples were incubated without constant rocking as to allow for the adsorption for 1 h in a 5% CO2-water vapor atmosphere at a temperature of 37 °C as to mimic the human body temperature. To keep the cells from drying, the plates were occasionally rocked. One milliliter of 2X DMEM media (Dulbecco’s modified Eagle’s medium supplied from Gibco-BRL) in addition to another 1 mL of 1% agarose was added to each well after the adsorption is complete. The plates were then incubated in a 5% CO2-water vapor atmosphere at a temperature of 37 °C. Following the incubation, the sample cells were stained with 0.4% crystal violet after fixation with formalin, the number of plaques was counted, and the titers were also calculated. The latter was expressed in terms of plaque-forming units per milliliter (pfu/mL).43,44 CC50 and IC50 were evaluated for the promising materials (viral reduction 50% or more). CC50 referring to the 50% cytotoxic concentration of the test extract is defined as the concentration that reduces the OD492 of the treated uninfected cells to half the OD492 of the untreated uninfected cells. IC50 refers to the concentration at which the compound plaque reduction rate reaches halfway between the baseline and the maximum. All data were taken as the average of three measurements (triplicates).
Antiviral Bioassay of Tested Compounds against ED-43/SG-Feo (VYG) Replicon of HCV Genotype 4a
To carry out the antiviral assay, a nontoxic dose of the tested compounds was used against ED-43/SG-Feo (VYG) replicon of HCV genotype 4a. To quantify HCV RNA, qRT-PCR supplied from Taqman probe kit (Qiagen) was used. This was done in algal extracts treated with Huh 7.5-infected cells. According to the literature and following the manufacturer’s instructions, a dose-dependent decrease in subgenomic RNA copies was shown.45
Hsp90α (C-Terminal) Inhibitor Screening Assay
The Hsp90α (C-Terminal) inhibitor screening assay was used to assess the inhibition of Hsp90α binding to its target protein cyclophilin D (PPID). A solution of 3X Hsp90α assay buffer 2 was first diluted with water to 1x Hsp90α assay buffer 2. Four microliters of the diluted Hspα protein (1.5 ng/μL) was added to each well designated “Blank” and “Substrate Control”. Two microliters of the same solution without an inhibitor (inhibitor buffer) was added to wells assigned to “Positive Control”, “Substrate Control”, and “Blank”. To initiate the enzymatic reaction, 4 μL of diluted PPID was added to each well designated “Substrate control”, “Positive Control”, and “Test Inhibitor” and then incubated at room temperature for 30 min. The total volume for each well was 10 μL. An amount of 10 μL of diluted glutathione (250-fold with 1x detection buffer) was added to each well. Ten microliters of diluted streptavidin-conjugated donor beads was also added to wells in a 96-well plate. Alpha-counts were then read. The percentage inhibition was calculated for the different concentrations tested against the control, and the IC50 values against the HSPα protein were calculated from the concentration–inhibition response curve.
Molecular Modeling
Docking simulations were performed using the crystal structure of Hsp90α (PDB ID 3b25) in complex to 4-methyl-6-(toluene-4-sulfonyl)-pyrimidin-2-ylamine (B2K).46 The PDB file was retrieved from the Protein Data Bank. The structure of chain A was processed using the Structure Preparation application in an MOE (molecular operating environment, 2014), and the ligand molecule was removed from the protein active site. Subsequently, the Protonate 3D application of the MOE was used to add the missing hydrogens and properly assign the ionization states. The default procedure in the MOE Dock application was used to find the favorable binding configurations of the studied ligands. Initial placement poses generated by the Alpha Triangle matcher were rescored and filtered using the London dG Scoring method to pick those exhibiting maximal hydrophobic, ionic, and hydrogen-bond contacts to the protein. This was followed by a refinement stage. The generated poses were energy minimized using the MMFF94x force field. Finally, the optimized poses were ranked using the GBVI/WSA DG free-energy estimates. Docking poses were visually inspected, and interactions with binding pocket residues were analyzed.47
Acknowledgments
The authors thank the Faculty of Science of Helwan University for the support provided to complete this study.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03706.
IR, 1H NMR, and 13C NMR spectra for the synthesized compounds, table of the nontoxic doses of tested compounds on FRHK-4, Hep2, BGM, Vero, and Huh 7.5 cell lines, and 3D interaction of acyclovir, 9a, 9b, 14b, 14g, and 14h inside the binding pocket of Hsp90α (PDB ID 3b25) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kukhanova M. K.; Korovina A. N.; Kochetkov S. N. Human herpes simplex virus: life cycle and development of inhibitors. Biochemistry (Moscow) 2014, 79, 1635–1652. 10.1134/S0006297914130124. [DOI] [PubMed] [Google Scholar]
- Birkmann A.; Zimmermann H. HSV antivirals–current and future treatment options. Curr. Opin. Virol. 2016, 18, 9–13. 10.1016/j.coviro.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Triantafilou K.; Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J. Virol. 2004, 78, 11313–11320. 10.1128/JVI.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg A. K.; Olsson A.; Korsgren O.; Frisk G. Antiviral treatment of Coxsackie B virus infection in human pancreatic islets. Antiviral Res. 2007, 74, 65–71. 10.1016/j.antiviral.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Thursz M. R.; Richardson P.; Allison M.; Austin A.; Bowers M.; Day C. P.; Downs N.; Gleeson D.; MacGilchrist A.; Grant A.; Hood S.; Masson S.; McCune A.; Mellor J.; O’Grady J.; Patch D.; Ratcliffe I.; Roderick P.; Stanton L.; Vergis N.; Wright M.; Ryder S.; Forrest E. H.; Prednisolone or pentoxifylline for alcoholic hepatitis. N. Engl. J. Med. 2015, 372, 1619–1628. 10.1056/NEJMoa1412278. [DOI] [PubMed] [Google Scholar]
- Berenguer M.; Wright T. L. Hepatitis B and C viruses: molecular identification and targeted antiviral therapies. Proc. Assoc. Am. Physicians. 1998, 110, 98–112. [PubMed] [Google Scholar]
- Sheridan C. FDA approvals usher in the post-interferon era in HCV. Nat. Biotechnol. 2014, 32, 3–5. 10.1038/nbt0114-3. [DOI] [PubMed] [Google Scholar]
- Gerber S. I.; Erdman D. D.; Pur S. L.; Diaz P. S.; Segreti J.; Kajon A. E.; Belkengren R. P.; Jones R. C. Outbreak of adenovirus genome type 7d2 infection in a pediatric chronic-care facility and tertiary-care hospital. Clin. Infect. Dis. 2001, 32, 694–700. 10.1086/319210. [DOI] [PubMed] [Google Scholar]
- Chen S.; Tian X. Vaccine development for human mastadenovirus. J. Thorac. Dis. 2018, 10, S2280. 10.21037/jtd.2018.03.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashad A. E.; Hegab M. I.; Abdel-Megeid R. E.; Micky J. A.; Abdel-Megeid F. M. E. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg. Med. Chem. 2008, 16, 7102–7106. 10.1016/j.bmc.2008.06.054. [DOI] [PubMed] [Google Scholar]
- Mohamed S. F.; Flefel E. M.; Amr A. E.-G. E.; Abd El-Shafy D. N. Anti-HSV-1 activity and mechanism of action of some new synthesized substituted pyrimidine, thiopyrimidine and thiazolopyrimidine derivatives. Eur. J. Med. Chem. 2010, 45, 1494–1501. 10.1016/j.ejmech.2009.12.057. [DOI] [PubMed] [Google Scholar]
- El-Sherbeny M. A. Synthesis of certain pyrimido[2,1-b]benzo-thiazole and benzothiazolo [2,3-b]quinazoline derivatives for in vitro antitumor and antiviral activities. Arzneimittelforschung 2000, 50, 848–853. 10.1055/s-0031-1300300. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziza H. A.; Abdel-Wahab B. F.; Badria F. A. Stereoselective synthesis and antiviral activity of (1E,2Z,3E)-1-(piperidin-1-yl)-1-(arylhydrazono)-2-[(benzoyl/benzothiazol-2-oyl)hydrazono]-4-(aryl1)but-3-enes. Arch. Pharm. Chem. Life Sci. 2010, 343, 152–159. 10.1002/ardp.200900195. [DOI] [PubMed] [Google Scholar]
- Dejmek M.; Hřebabecký H.; Šála M.; Dračínský M.; Procházková E.; Leyssen P.; Neyts J.; Balzarini J.; Nencka R. From norbornane-based nucleotide analogs locked in South conformation to novel inhibitors of feline herpes virus. Bioorg. Med. Chem. 2014, 22, 2974–2983. 10.1016/j.bmc.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Tǎnase C. I.; Drǎghici C.; Cojocaru A.; Galochkina A. V.; Orshanskaya J. R.; Zarubaev V. V.; Shova S.; Enache C.; Maganu M. New carbocyclic N6-substituted adenine and pyrimidine nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; synthesis, antiviral, anticancer activity and X-ray crystallography. Bioorg. Med. Chem. 2015, 23, 6346–6354. 10.1016/j.bmc.2015.08.033. [DOI] [PubMed] [Google Scholar]
- Arasappan A.; Bennett F.; Girijavallabhan V.; Huang Y.; Huelgas R.; Alvarez C.; Chen L.; Gavalas S.; Kim S.-H.; Kosinski A.; Pinto P.; Rizvi R.; Rossman R.; Shankar B.; Tong L.; Velazquez F.; Venkatraman S.; Verma V. A.; Kozlowski J.; Shih N.-Y.; Piwinski J. J.; MacCoss M.; Kwong C. D.; Clark A. T.; Fowler A. T.; Geng F.; Kezar H. S. III; Roychowdhury A.; Reynolds R. C.; Maddry J. A.; Ananthan S.; Secrist J. A. III; Chase C. L.; Curry S.; Huang H.-C.; Tong X.; Njoroge F. G. 5-Benzothiazole substituted pyrimidine derivatives as HCV replication (replicase) inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3229–3234. 10.1016/j.bmcl.2012.03.036. [DOI] [PubMed] [Google Scholar]
- Li K.; Frankowski K. J.; Belon C. A.; Neuenswander B.; Ndjomou J.; Hanson A. M.; Shanahan M. A.; Schoenen F. J.; Blagg B. S. J.; Aubé J.; Frick D. N. Optimization of potent hepatitis C virus NS3 helicase inhibitors isolated from the yellow dyes thioflavine S and primuline. J. Med. Chem. 2012, 55, 3319–3330. 10.1021/jm300021v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannopoulos C. G.; Xu P.; Ni F.; Chan L.; Pereira O. Z.; Reddy T. J.; Das S. K.; Poisson C.; Nguyen-Ba N.; Turcotte N.; Proulx M.; Halab L.; Wang W.; Bédard J.; Morin N.; Hamel M.; Nicolas O.; Bilimoria D.; L’Heureux L.; Bethell R.; Dionne G. HCV NS5B polymerase-bound conformation of a soluble sulfonamide inhibitor by 2D transferred NOESY. Bioorg. Med. Chem. Lett. 2004, 14, 5333–5337. 10.1016/j.bmcl.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Smith K. L.; Varaprasad C. V. N. S.; Chang E.; Alexander J.; Yao N. Synthesis of thiazolone-based sulfonamides as inhibitors of HCV NS5B polymerase. Bioorg. Med. Chem. Lett. 2007, 17, 841–845. 10.1016/j.bmcl.2006.08.104. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Zhang X.; Zhu J.; Turpoff A.; Chen G.; Morrill C.; Huang S.; Lennox W.; Kakarla R.; Liu R.; Li C.; Ren H.; Almstead N.; Venkatraman S.; Njoroge F. G.; Gu Z.; Clausen V.; Graci J.; Jung S. P.; Zheng Y.; Colacino J. M.; Lahser F.; Sheedy J.; Mollin A.; Weetall M.; Nomeir A.; Karp G. M. Structure–activity relationship (SAR) optimization of 6-(indol-2-yl) pyridine-3-sulfonamides: identification of potent, selective, and orally bioavailable small molecules targeting hepatitis C (HCV) NS4B. J. Med. Chem. 2014, 57, 2121–2135. 10.1021/jm401621g. [DOI] [PubMed] [Google Scholar]
- Ke S.; Wei Y.; Yang Z.; Wang K.; Liang Y.; Shi L. Novel cycloalkylthiophene–imine derivatives bearing benzothiazole scaffold: Synthesis, characterization and antiviral activity evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 5131–5134. 10.1016/j.bmcl.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Azzam R. A. Tailored-design synthesis of sulfapyrimidine derivatives. J. Heterocycl. Chem. 2019, 56, 619–627. 10.1002/jhet.3439. [DOI] [Google Scholar]
- Azzam R. A.; Elgemeie G. H.; Elsayed R. E.; Jones P. G. Crystal structure of N-[6-amino-5-(benzo[d]thiazol-2-yl)-3-cyano-4-methylsulfanyl-2-oxo-1,2-dihydropyridin-1-yl]-4-methylbenzenesulfonamide dimethylformamide monosolvate. Acta. Crystallogr., Sect. E: Crystallogr. Commun. 2017, 73, 1820–1822. 10.1107/S2056989017015778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam R. A.; Elgemeie G. H.; Elsayed R. E.; Jones P. G. Crystal structure of N’-[2-(benzo[d]thiazol-2-yl)acetyl]-4-methylbenzenesulfonohydrazide. Acta Cryst. E. 2017, 73, 1041–1043. 10.1107/S2056989017008738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgemeie G. H.; Azzam R. A.; Elsayed R. E. Sulfa drug analogs: new classes of N-sulfonyl aminated azines and their biological and preclinical importance in medicinal chemistry (2000–2018). Med. Chem. Res. 2019, 28, 1099–1131. 10.1007/s00044-019-02378-6. [DOI] [Google Scholar]
- Elgemeie G.; Abu-Zaied M.; Azzam R. Antimetabolites: A first synthesis of a new class of cytosine thioglycoside analogs. Nucleosides Nucleotides Nucleic Acids 2016, 35, 211–222. 10.1080/15257770.2015.1127961. [DOI] [PubMed] [Google Scholar]
- Elgemeie G. H.; Shams H. Z.; Elkholy Y. M.; Abbaas N. Novel synthesis of N-amino-2-pyridones and cycloalkane ring-fused Pyridines containing benzothiazole moiety. Het. Commun 2000, 6, 363–268. [Google Scholar]
- Azzam R. A.; Elgemeie G. H. Synthesis and antimicrobial evaluation of novel N-substituted 4-ethylsulfanyl-2-pyridones and triazolopyridines. Med. Chem. Res. 2019, 28, 62–70. 10.1007/s00044-018-2264-z. [DOI] [Google Scholar]
- Elgemeie G. H.; Abu-Zaied M. A.; Loutfy S. A. 4-Aminoantipyrine in carbohydrate research: design, synthesis and anticancer activity of a novel class of derivatives of 4-aminoantipyrine thioglycosides and their corresponding pyrazolopyrimidine and pyrazolopyridine thioglycosides. Tetrahedron 2017, 73, 5853–5861. 10.1016/j.tet.2017.08.024. [DOI] [Google Scholar]
- Elgemeie G.; Altalbawy F.; Alfaidi M.; Azab R.; Hassan A. Synthesis, characterization and antimicrobial evaluation of novel 5-benzoyl-N-substituted amino- and 5-benzoyl-N-sulfonylamino-4-alkylsulfanyl-2-pyridones. Drug Des., Dev. Ther. 2017, 11, 3389–3399. 10.2147/DDDT.S149615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdallah A. E. M.; Elgemeie G. H. Design, synthesis, docking, and antimicrobial evaluation of some novel pyrazolo[1,5-a]pyrimidines and their corresponding cycloalkane ring-fused derivatives as purine analogs. Drug Des. Devel. Ther. 2018, 12, 1785–1798. 10.2147/DDDT.S159310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Zaied M. A.; Elgemeie G. H. A facile synthesis of novel pyrazolopyrimidine thioglycosides as purine thioglycoside analogues. Nucleosides, Nucleotides Nucleic Acids 2018, 37, 67–77. 10.1080/15257770.2017.1419254. [DOI] [PubMed] [Google Scholar]
- Elgemeie G. H.; Salah A. M.; Abbas N. S.; Hussein H. A.; Mohamed R. A. Pyrimidine non-nucleoside analogs: A direct synthesis of a novel class of N-substituted amino and N-sulfonamide derivatives of pyrimidines. Nucleosides, Nucleotides Nucleic Acids 2017, 36, 213–223. 10.1080/15257770.2016.1257808. [DOI] [PubMed] [Google Scholar]
- Dawood K. M.; Kandeel Z. E.; Farag A. M. Heterocyclic synthesis via enaminonitriles: a convenient route to some new pyrazole, isoxazole, pyrimidine, pyrazolo[1,5-a]pyrimidine, pyrimido[1,2-a]benzimidazole and pyrido[1,2-a]-benzimidazole derivatives. J. Chem. Res. 1998, 208–209. 10.1039/a707362c. [DOI] [Google Scholar]
- Azzam R. A.; Elgemeie G. H.; Osman R. R.; Jones P. G. Crystal structure of potassium [4-amino-5-(benzo-[d]thiazol-2-yl)-6-(methylsulfanyl)pyrimidin-2-yl]-(phenylsulfonyl)-azanide dimethylformamide monosolvate hemihydrate. Acta. Crystallogr., Sect. E: Crystallogr. Commun. 2019, 75, 367–371. 10.1107/S2056989019002275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney A.; Workman P. Hsp90 as a new therapeutic target for cancer therapy: the story unfolds. Expert. Opin. Biol. Ther. 2002, 2, 3–24. 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- Geller R.; Taguwa S.; Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta, Mol. Cell Res. 2012, 1823, 698–706. 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda A. A.; Amer F. A.; Zaki M. E. A.; Samir K. H. Revised synthesis of some new derivatives of biological interest 2-heterocyclic benzothiazolyl derivatives of biological interest. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 155, 59–66. 10.1080/10426509908044970. [DOI] [Google Scholar]
- Shaker Y. M.; Omar M. A.; Mahmoud K.; Elhallouty S. M.; El-Senousy W. M.; Ali M. M.; Mahmoud A. E.; Abdel-Halim A. H.; Soliman S. M.; El Diwani H. I. Synthesis, in vitro and in vivo antitumor and antiviral activity of novel 1-substituted benzimidazole derivatives. J. Enzyme Inhib. Med. Chem. 2015, 30, 826–845. 10.3109/14756366.2014.979344. [DOI] [PubMed] [Google Scholar]
- Simões C. M. O.; Amoros M.; Girre L. Mechanism of antiviral activity of triterpenoid saponins. Phytother. Res. 1999, 13, 323–328. . [DOI] [PubMed] [Google Scholar]
- Walum E.; Stenberg K.; Jenssen D.. Understanding cell toxicology: principles and practice; Ellis Horwood: New York,1990,101–117. [Google Scholar]
- Schmidtke M.; Knorre C.; Blei L.; Stelzner A.; Birch-Hirschfeld E. Penetration and antiviral activity of coxsackie virus B3-specific phosphorothioate oligodeoxynucleotides (PS-ODN). Nucleosides Nucleotsides 1998, 17, 1557–1566. 10.1080/07328319808004686. [DOI] [Google Scholar]
- Azzam R. A.; Elgemeie G. H.; Osman R. R. Synthesis of novel pyrido [2,1-b] benzothiazole and N-substituted 2-pyridylbenzothiazole derivatives showing remarkable fluorescence and biological activities. J. Mol. Struct. 2020, 1201, 127194. 10.1016/j.molstruc.2019.127194. [DOI] [Google Scholar]
- Saeed M.; Scheel T. K. H.; Gottwein J. M.; Marukian S.; Dustin L. B.; Bukh J.; Rice C. M. Efficient replication of genotype 3a and 4a HCV replicons in human hepatoma cells. Antimicrob. Agents Chemother. 2012, 56, 5365–5373. 10.1128/AAC.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T.; Fukami T. A.; Hasegawa K.; Ono N.; Suda A.; Shindo H.; Yoon D. O.; Kim S. J.; Na Y. J.; Aoki Y.; Shimma N.; Tsukuda T.; Shiratori Y. Lead generation of heat shock protein 90 inhibitors by a combination of fragment-based approach, virtual screening, and structure-based drug design. Bioorg. Med. Chem. Lett. 2011, 21, 5778–5783. 10.1016/j.bmcl.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Ju H.-Q.; Xiang Y.-F.; Xin B.-J.; Pei Y.; Lu J.-X.; Wang Q.-L.; Xia M.; Qian C.-W.; Ren Z.; Wang S.-Y.; Wang Y.-F.; Xing G.-W. Synthesis and in vitro anti-HSV-1 activity of a novel Hsp90 inhibitor BJ-B11. Bioorg. Med. Chem. Lett. 2011, 21, 1675–1677. 10.1016/j.bmcl.2011.01.098. [DOI] [PubMed] [Google Scholar]
- Elgemeie G. H.; Fathy N. M.; Farag A. B.; Alkhursani S. A. Design, synthesis, molecular docking and anti-hepatocellular carcinoma evaluation of novel acyclic pyridine thioglycosides. Nucleosides Nucleotides Nucleic Acids 2018, 37, 186–198. 10.1080/15257770.2018.1450508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.