

Keywords: GSK’872, human brain tissue, hyperpyrexia, methamphetamine, mixed lineage kinase domain-like protein, necrostatin-1, necroptosis, nerve regeneration, neural regeneration, rat cortical neurons, receptor-interacting protein-3, synergistic effect
Abstract
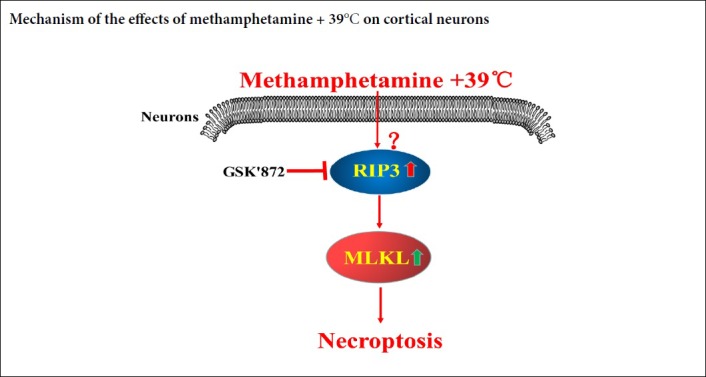
Methamphetamine is one of the most prevalent drugs abused in the world. Methamphetamine abusers usually present with hyperpyrexia (39°C), hallucination and other psychiatric symptoms. However, the detailed mechanism underlying its neurotoxic action remains elusive. This study investigated the effects of methamphetamine + 39°C on primary cortical neurons from the cortex of embryonic Sprague-Dawley rats. Primary cortex neurons were exposed to 1 mM methamphetamine + 39°C. Propidium iodide staining and lactate dehydrogenase release detection showed that methamphetamine + 39°C triggered obvious necrosis-like death in cultured primary cortical neurons, which could be partially inhibited by receptor-interacting protein-1 (RIP1) inhibitor Necrostatin-1 partially. Western blot assay results showed that there were increases in the expressions of receptor-interacting protein-3 (RIP3) and mixed lineage kinase domain-like protein (MLKL) in the primary cortical neurons treated with 1 mM methamphetamine + 39°C for 3 hours. After pre-treatment with RIP3 inhibitor GSK’872, propidium iodide staining and lactate dehydrogenase release detection showed that neuronal necrosis rate was significantly decreased; RIP3 and MLKL protein expression significantly decreased. Immunohistochemistry staining results also showed that the expressions of RIP3 and MLKL were up-regulated in brain specimens from humans who had died of methamphetamine abuse. Taken together, the above results suggest that methamphetamine + 39°C can induce RIP3/MLKL regulated necroptosis, thereby resulting in neurotoxicity. The study protocol was approved by the Medical Ethics Committee of the Third Xiangya Hospital of Central South University, China (approval numbers: 2017-S026 and 2017-S033) on March 7, 2017.
Chinese Library Classification No. R453; R364; R363
Introduction
More than 53,100 first time drug abusers were arrested in China in 2015. The percentage of abusers of amphetamines (such as methamphetamine, METH) reached 73.2% of all drug users arrested (http://news.xinhuanet.com/live/2016-02/18/c_128730815_2.htm). Such abuse leads to deleterious effects on families, major public health concerns, and the consumption of substantial resources for medical intervention (Moratalla et al., 2017; Yang et al., 2018). Most scientists focus on the addiction mechanism of METH abusers. However, apart from addiction, the damage METH causes to the central nervous system is also extremely serious. Furthermore, METH abuse is often accompanied by high body core temperature (Sanchez-Alavez et al., 2014; Harrell et al., 2015). Thus, the neurons are under the attack from both METH toxicity and hyperthermia (Shioda et al., 2010; Yang et al., 2018; Lu et al., 2019; Yang et al., 2019). Previously researchers have pointed out that METH can lead to apoptosis, autophagy and other forms of cell death (Riddle et al., 2006; Wu et al., 2007; Degterev et al., 2008; Lu et al., 2017; Xiong et al., 2017; Yang et al., 2017; Lu et al, 2019a, b). Our earlier study found that a single dose of METH can cause necroptosis of cultured rat cortical neurons in vitro (Xiong et al., 2016). However, it is unclear whether METH accompanied by hyperthermia produces necroptosis of neurons in the cortex and what molecules might be involved in the process.
Necroptosis is one type of regulated necrosis, where a type of death domain receptor is involved in cell necrosis without caspase activation (Degterev et al., 2008; Xiong et al., 2016; Cheng et al., 2018). This kind of programmed cell necrosis can be regulated by RIP1-RIP3-MLKL pathways (Ruan et al., 2015; Wang et al., 2018b, c). When the cells are subjected to various stimuli, cell death receptors activate RIP1 kinase. RIP1 interacts with RIPK3 via a shared RIP homotypic interaction motif domain to form a “necrosome” (Sun et al., 2002), which then phosphorylates RIP3. Activated RIP3 recruits and phosphorylates MLKL, resulting in cell lysis (Orzalli and Kagan, 2017). Choosing different molecular targets, which could be critical biomarkers, researchers have developed corresponding necroptotic inhibitors, such as RIP1 inhibitor necrostatin-1 (Degterev et al., 2008) and RIP3 inhibitor GSK’872 (Mandal et al., 2014; Liao et al., 2017).
Initially, necroptosis research focused mainly on fibroblasts, immune cells and tumor cells (Lau et al., 2013; Zhong et al., 2014). In recent years, increasing attention has been paid to the study of necroptosis in the nervous system and related diseases (Huang et al., 2013; Ito et al., 2016; Daniels et al., 2017). Our team is one of the first research groups to study the mechanism of neuronal necroptosis. We and other groups have found that necroptosis of neurons occurred after ischemia/reperfusion injury (Rosenbaum et al., 2010; Xu et al., 2010, 2016; Ding et al., 2015; Yin et al., 2015; Chen et al., 2016; Yang et al., 2017; Cruz et al., 2018; Wang et al., 2018a), elevated hydrostatic pressure injury (Liao et al., 2017; Shang et al., 2017), glutamate toxic injury (Wang et al., 2018d, 2019a, b) and oxidative stress damage (Jiang et al., 2014; Li et al., 2016). These studies showed that RIP3, MLKL, and calpain exerted key roles in neuronal necroptosis induced by the above injuries (Shang et al., 2014; Ding et al., 2015; Yin et al., 2015, 2018a, b, c, d, 2019b; Xu et al., 2018).
We continue to study the role of METH and hyperthermia on cortical neurons and to identify whether necroptosis plays a key role in METH and hyperthermia induced neuronal death. We have also explored the necroptotic signaling pathway of neurons involved in the toxic process. This study aims to identify cortical neuron death and the molecular mechanism underlying the action of METH and hyperthermia in vitro, verifying the molecular changes in vivo detected in post mortem brain specimens from humans who abused METH. Our investigation sheds new light on cortical neuronal injury with respect to the combined effect of METH and hyperthermia.
Materials and Methods
Primary cultured cortical neurons
Specific-pathogen-free pregnant Sprague-Dawley rats weighing 300–400 g and aged 10–12 weeks with day 18–20 (E18–20) embryos were obtained from Central South University, China. All experimental procedures were approved by the Medical Ethics Committee of the Third Xiangya Hospital of Central South University (approval No. 2017-S033) on March 7, 2017, in accordance with the experimental animal use and welfare requirements set by the Ministry of Health of China as well as the National Institutes of Health (NIH) guidelines for use and care of laboratory animals.
Animals were given free access to food and water. One E18–20 rat was used in each batch of experiments (n = 3–6). Pregnant rats were deeply anesthetized and decapitated gently and rapidly. The cortical tissue was isolated from the brain of each E18–20 fetal rat. The cortical tissue was washed three times in Hank’s balanced salt solution, digested by 2 mg/mL papain medium (Solarbio, Beijing, China) for 10 minutes at 37°C, and transferred into a fresh tube. The cortical tissue was treated with 1 mL plated medium, consisting of Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 5% horse serum (Thermo Fisher Scientific, Waltham, MA, USA), 1% penicillin-streptomycin and 1% L-cysteine (6 mg/mL), then centrifuged at 1000 r/min for 5 minutes. The supernatant was abandoned. After adding 4 mL plated medium, the tissues were resuspended and dispersed to single neurons by gently tapping 40–50 times. The cell suspension was filtered with 70 μm strainer and the liquid was transferred to a fresh tube. The filtrate was then centrifuged at 1000 r/min for 5 minutes. After removal of the supernatant, the cell sediment was resuspended with 4 mL plated medium by gently tapping several times. The resuspended sample was counted in a blood counting chamber. Finally, cells were plated onto poly-D-lysine-coated plates or dishes with plated medium pretreated with 0.1 mg/mL poly-D-lysine (Sigma St Louis, MO, USA) overnight at 37°C at 1 × 105 cells/cm2. The plated medium was replaced by neurobasal medium with 1% B27 (Thermo Fisher Scientific) after 3 hours. Subsequently, half of the medium was refreshed every other day until the 8th day when the neurons were mature. After drug treatment, samples of neurons were used for related experiments. The cells were exposed to different doses over time and divided into a normal group, METH (0, 0.25, 0.5, 1, 2, 3 mM) + 39°C (1-, 2-, 3-, 5-hour) groups, and examined by inverted light microscope. The necrosis of cells (normal group, METH + 39°C group, METH + 39°C + necrostatin-1 group) was examined using propidium iodide (PI) staining and lactate dehydrogenase (LDH) release. To further examine the effects of METH + 39°C, the RIP3 inhibitor GSK’872 was used. The cells were divided into normal group, METH group, 39°C group, METH + 39°C group, METH + 39°C + GSK’872 group.
Drug and hyperthermia treatment
The different concentrations of METH (0.25, 0.5, 1, 2, 3 mM, applied by Changsha City Public Security Bureau, China) (Huang et al., 2009; Xiong et al., 2016) were set to observe the injurious effect of METH on the cultured matured primary neurons. After giving METH, the neurons were cultured in a 5% CO2 incubator at 39°C. Primary cultured neurons were pretreated with necrostatin-1 (Sigma) 2 hours before the METH and 39°C treatment. Pretreatment with the specific RIPK3 inhibitor GSK’872 (BioVision, San Francisco, CA, USA) was applied at 0.25, 0.5 or 1 μM for 2 hours before the METH and 39°C treatment.
Propidium iodide staining
Neurons were cultured on coverslips for 8 days then used for experiments. Neuronal necrosis was analyzed by PI staining (Sigma). The slides with neurons in all groups were washed once with phosphate buffered saline (PBS) and stained with PI (2 μg/mL) at 37°C for 10 minutes, followed by three gentle washes with PBS, each for 5 minutes. The coverslips were fixed with 4% paraformaldehyde at room temperature for 15 minutes, washed three times with PBS, each for 10 minutes, and finally covered with Vector shield mounting medium H1500 (Vector Laboratories, Burlingame, CA, USA). The coverslips were observed and images were captured using a fluorescence microscope (Olympus, Tokyo, Japan) with a camera and imaging system (CellSens Standard, Olympus). PI-positive cells were counted from five fields of each coverslip, and each group contained three coverslips from three independent experiments. Cells were counted using Motic pathology image analysis software (Motic Inc., Xiamen, China).
Lactate dehydrogenase release
Cultured neurons in 96-well plates were harvested on day 8. The LDH cytotoxicity assay kit (Beyotime Biotech Inc., Shanghai, China) was used to measure LDH released from necrotic cells into the extracellular space/supernatant upon the rupture of plasma membrane after different treatments. Cell-free culture supernatants were collected from a 96-well microtiter plate and incubated with appropriate reagent mixture according to the manufacturer’s instructions, at room temperature for 30 minutes. The intensity of the red color that formed in the assay was measured at a wavelength of 490 nm using an iMark microplate reader (Bio-Rad, Berkeley, CA, USA). The intensity was proportional to both the LDH activity and percentage of necrotic cells. The necrosis rate of cortical neurons was calculated as the percentage of the color intensities of (treated cells − control cells)/(LDH releasing reagent treated cells − control cells), from four independent experiments.
Immunofluorescence staining
Cultured neurons on coverslips were harvested on day 8. Firstly, neurons were washed twice with ice-cold PBS, fixed with 4% paraformaldehyde at room temperature for 15 minutes, and washed three times with PBS, each for 10 minutes. The non-specific protein was blocked with 5% bovine serum albumin (Solarbio, Beijing, China) combined with 0.3% Triton for 1 hour at room temperature. The coverslips were incubated with the primary antibodies (rabbit monoclonal anti-RIP3, 1:200; Sigma; rabbit polyclonal anti-MLKL, 1:100; Abcam, Cambridge, UK) in immunofluorescence buffer solution (Solarbio) at 4°C overnight. In the morning, these coverslips were gently washed three times with PBS and incubated with homologous secondary 488 conjugated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch Inc., Baltimore, PA, USA) for 2 hours at room temperature. The coverslips were washed three times with PBS and covered with vector shield mounting medium. Finally, the coverslips were observed and images were captured with a fluorescence microscope (Olympus).
Western blot assay
Cultured neurons were harvested, washed twice by ice-cold PBS, and dissociated with 100 μL cell extraction buffer with 1% phenylmethanesulfonyl fluoride and 1% protease inhibitor cocktail (Thermo Fisher Scientific) for each T25 culture bottle. The neurons were scraped from the bottom with minimal force, rested on ice for 30 minutes, and centrifuged at 12,000 r/min and 4°C for 20 minutes. The supernatant was transferred into a fresh tube. The protein concentration of these samples was measured by bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA). After unifying the concentration, 5 × loading buffer was added, boiled for 5 minutes, and centrifuged at 1000 r/min for 5 minutes. The supernatant was transferred to another fresh tube. The total loading protein for each lane was 20 μg. The samples were loaded in 8–12% sodium dodecyl sulfate polyacrylamide electrophoresis gel. The protein was transferred from the gel to polyvinylidene fluoride membrane (Millipore, MA, USA) in ice cold transfer buffer. After washing once with Tris-Buffered Saline and Tween 20, the membrane was blocked with 5% skim milk at room temperature for 1–2 hours to wipe off the non-specific protein band. The membranes were incubated with primary antibodies (rabbit monoclonal anti-RIP3, 1:1000, Sigma-Aldrich; rabbit polyclonal anti-MLKL, 1:1000, Abcam; rabbit polyclonal anti-GAPDH, 1:2000, Proteintech, Wuhan, China) at 4°C overnight. The next day, the membrane was washed three times with Tris-Buffered Saline and Tween 20, incubated with homologous goat anti-rabbit IgG horseradish peroxidase-secondary antibody (1:2000, Beyotime Biotech Inc.) for 2 hours at room temperature. It was then washed three times with Tris-Buffered Saline and Tween 20, and finally developed with electrochemoluminescent (ECL) assay (CWBiotech, Beijing, China). The integrated optical density values of specific proteins were quantified using ImageJ software (National Institutes of Health, MD, USA). The relative expression levels of the proteins were normalized by calculating the ratio of the target proteins to GAPDH.
General data of human cadaver brain tissue specimens
The four human cadaver brain tissues were from autopsy cases from Xiangya Judicial Identification Center (attached to Central South University). Postmortem human brains were banked through the willed body donation programs, which exist with government (municipal police department) and university (Hunan Xiangya Forensic Center) approval to provide cadavers for teaching anatomy to medical students. The acquisition of brain tissue samples conformed to the ethical principles for medical research, with the approval from the family of the deceased, and complied with the Declaration of Helsinki and the approval of the Medical Ethics Committee of the Third Xiangya Hospital of Central South University (approval number: 2017-S026) on March 7, 2017. The bodies of four brain tissue donors were dissected by forensic experts within 48 hours after death and brain tissue samples were obtained. Forensic pathologists conducted detailed pathological examinations of the donors and concluded that no obvious neurological diseases were associated with the causes of death in these tissue donors. The death of two cases was caused by METH poisoning and one was cardiac sudden death induced by METH. The case in the normal group died of electric shock. Specific information on the samples is shown in Table 1.
Table 1.
General information of four specimen cases
| Items | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Gender | Male | Male | Female | Male |
| Age (years) | 24 | 40 | 37 | 37 |
| Duration of drug use (years) | 0 | 5 | 4 | 5 |
| Survival time from recent drug use (hours) | 0 | 24 | 39 | 3 |
| Underlying disease | N/A | N/A | N/A | Coronary heart disease |
| Cause of death | Death from electric shock (shock in the palm of the hand) | Acute METH poisoning caused acute coma, and eventually multiple organ failure | METH poisoning caused coma, hyperpyrexia, convulsion, and eventually multiple organ failure | Sudden death form METH-induced coronary heart attack |
| METH concentration in blood | 0 | 3.9 μg/mL | 6.5 μg/mL | 0.105 μg/mL |
| Pre-death temperature (°C) | N/A | 39.5 | 39.2 | N/A |
METH: Methamphetamine; N/A: not applicable.
Inclusion criteria
They were (1) intake of METH before death, (2) tissue obtained within 24 hours after death, (3) preservation of dead bodies in similar environment but without freezing, and (4) no neurological diseases diagnosed before death.
Exclusion criteria
Based on the differences of acquisition of forensic specimens and patient samples or deceased patient samples in hospitals and morphological studies, we took the characteristics of tissue autolysis, defect and decay as exclusion criteria.
Human brain tissue preparation
For anatomical examination, the frontal cortex of the relevant human brain tissue was cut into 1.5 cm × 1.5 cm × 1.0 cm squares, and fixed in 4% paraformaldehyde for 1 week. The tissue was dehydrated in conventional sugar for a further 1 week (Xu et al, 2019). The tissues were embedded in optimal-cutting-temperature medium (Sakura Finetek, Tokyo, Japan), and prepared into 20 μm thick cross-sections in a Shanton Cryostat (Thermo-Fisher Scientific Inc., San Jose, CA, USA). Sections were thaw-mounted on positively charged microslides, allowed to air-dry and then stored at –20°C before further histological processing.
Immunohistochemistry staining of human brain tissue
For immunolabeling with the avidin-biotin complex method, sections were treated in 0.3% H2O2 in 0.01 M PBS (pH 7.3) for 15 minutes to inactivate endogenous peroxidase. Non-specific antibody binding was blocked by pre-incubating the sections in 5% normal horse serum (Sigma) in PBS containing 0.3% Triton X-100 (Fluka, St.Louis, MO, USA) for 1 hour at room temperature. Sections were then incubated with rabbit monoclonal anti-RIP3 (1:1000), or rabbit polyclonal anti-MLKL (1:1000) antibody at 4°C overnight, then reacted with biotinylated horse anti-rabbit IgG (1:400; Vector Laboratories Inc, Burlingame, CA, USA) for 2 hours at room temperature. After 1-hour incubation with the avidin-biotin complex reagents (1:400; Vector Laboratories Inc.), the immunoreaction product was visualized in PBS containing 0.05% 3,3′-diaminobenzidine (Sigma) and 0.03% H2O2. Finally, the RIP3 sections were counterstained by hematoxylin, dehydrated, cleared and coverslipped. These sections were observed and images were captured using a microscope (Nikon, Tokyo, Japan).
Statistical analysis
Figure panels were assembled using Photoshop CC (Adobe Systems Incorporated, San Jose, CA, USA). The measurement data are presented as the mean ± SD. Two-way analysis of variance followed by the Bonferroni post hoc test, one-way analysis of variance followed by Tukey’s multiple comparison test for comparisons of more than two groups and independent sample t-tests were used to analyze the data, with Graph Pad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). A value of P < 0.05 was considered statistically significant.
Results
METH + 39°C can cause the necrosis-like neuronal death observed under the light microscope
To observe the general effect of cortical neuronal injury induced by METH, we set up a series of concentrations (0, 0.25, 0.5, 1, 2, 3 mM) of METH at 39°C to incubate cultured cortical neurons for 1, 2, 3, 5 hours to study the change of neuronal morphology under the light microscope. Results found that damage of the neurite was first observed after incubation with 0.25 mM of METH + 39°C for 3 hours, and the degree of damage to the neurite became more serious as the METH concentration and the duration time increased. After exposure to 1 mM of METH + 39°C for 5 hours, the neurite of the neuron was almost lost (Figure 1 big frame). The neuronal body began to swell in 0.25 mM of METH + 39°C for 5 hours and became spherical at higher concentrations (Figure 1). Neurons were severely damaged and presented an obvious necrosis-like feature after 1 mM of METH + 39°C for 5 hours. Given that molecular changes precede cell death, 1 mM of METH + 39°C for 1 and 3 hours alternatively was selected to further study the mechanism of neuronal injury, which ultimately leads to cell death. These results showed that the METH induced neuronal necrosis-like cell death at 39°C.
Figure 1.

Morphological effect of METH + 39°C on cortical neurons.
We applied 0, 0.25, 0.5, 1, 2, 3 mM of METH + 39°C for 1, 2, 3 and 5 hours in the experiment. The neuronal cells are photographed under the inverted microscope. The drug concentration and duration times in the red square boxes are our selected parameters for later experiments. The white arrows show morphological changes of cell necrosis. The larger picture in the lower right corner of panels is the enlargement of the smaller picture in the corresponding picture. Scale bar: 50 μm in all the panels. The baseline in each of the 100 μm in all enlarged pictures in the lower right corners of panels equals 100 μm. METH: Methamphetamine.
Necrostatin-1 decreases the necrosis rate of neurons induced by 1 mM METH + 39°C for 3 hours
PI staining results revealed that 1 mM METH induced PI positive cortical neurons at both 1 and 3 hours. Pretreatment with 20 mM necrostatin-1 can reduce the number of PI positive neuron after 1 mM METH at 1 and 3 hours (Figure 2A). The data of PI uptake rates showed that necrostatin-1 significantly reduced the necrosis rate of neurons by METH + 39°C at 3 hours. The difference between two groups of the blank and 1 mM METH + 39°C for 3 hours was significant (Figure 2B). Compared with the METH + 39°C group, the percentage of necrotic neurons was significantly decreased in the METH + 39°C + necrostatin-1 group (Figure 2C). Collectively, these results showed that METH + 39°C induced neuronal necrosis but that pretreatment with necrostatin-1 partially blocked the necrosis.
Figure 2.

Necrosis of rat cortical neurons induced by METH + 39°C.
(A) PI staining for cortical neurons by METH + 39°C, left. Pretreatment with 20 mM necrostatin-1, right; scale bars: 100 μm. (B) Statistics of PI uptake rate (mean ± SD, n = 5; two-way analysis of variance followed by the Bonferroni post hoc test), ***P < 0.001. (C) Necrosis rate of cortical neurons detected by lactate dehydrogenase assay after METH + 39°C for 3 hours, with and without pretreatment with necrostatin-1 (mean ± SD, n = 6, one-way analysis of variance followed by Tukey’s multiple comparison test). **P < 0.01, ***P < 0.001, ****P < 0.0001, vs. normal group; ####P < 0.0001. METH: Methamphetamine; ns: not significant; PI: propidium iodide.
Up-regulation of RIP3 and MLKL induced by 1 mM METH + 39°C
The above results revealed that METH + 39°C induced neuronal necroptosis which was blocked by necrostatin-1. We further studied whether the canonical necroptotic molecules were involved in this process. Immunofluorescence staining showed that the signals of RIP3 (green) were stronger in the METH + 39°C groups than in the normal group. Simultaneously, the dot patterns in the plasma of neurons were more obvious in the METH + 39°C groups than in the normal group (Figure 3A). The green signals of MLKL stained by immunofluorescence were also increased in the METH + 39°C groups, and the location of MLKL moved onto the cytomembrane (Figure 3B). Western blot assay results also showed that the expression of both RIP3 and MLKL increased compared to the normal group (Figure 3C). There were significant differences in RIP3 and MLKL between the METH + 39°C and normal groups (Figure 3D–E).
Figure 3.

Expression of RIP3 and MLKL in cortical neurons following METH + 39°C treatment.
(A–B) Immunofluorescence staining for RIP3 and MLKL in cortical neurons: the large frame is the 4 × magnification of related small frame, scale bar: 100 μm. (C) Western blot assay for RIP3 and MLKL in cortical neurons. (D–E) Statistics of RIP3 and MLKL expression based on western blot assay (mean ± SD, n = 3, one-way analysis of variance followed by Tukey’s multiple comparison test). *P < 0.05, **P < 0.01, vs. normal group. METH: Methamphetamine; MLKL: mixed lineage kinase domain-like protein; RIP3: receptor-interacting protein 3.
Pretreatment with GSK’872 down-regulates the neuronal necrosis rate and MLKL expression induced by 1 mM METH + 39°C treatment
To clarify the role of classical necrosis pathway, the RIP3 inhibitor, GSK’872, was used on primary cortex neurons and MLKL changes were detected downstream of RIP3 before exposure to 1 mM METH + 39°C. Different concentrations of GSK’872 are used and results are shown in Figure 4. Low concentrations have no effect, but high concentrations seem to have toxic effects on cells, which is the reason for our choosing 0.5 µM. The PI staining and its statistical results revealed that 0.5 μM GSK’872 decreased the percentage of necrotic neurons (Figure 4A and B). The LDH cytotoxicity assay results confirmed that the percentage of necrotic neurons of 0.5 μM GSK’872 group was significantly decreased compared with the METH + 39°C group (Figure 4C). However, no matter what the concentration of GSK’872 pretreatment was, the number of necrotic neurons was significantly higher in the METH groups than those of the normal group (Figure 4A–C). Western blot assay and statistical results also showed that MLKL expression was significantly reduced after pretreatment with GSK’872 compared with the METH + 39°C only group (Figure 4D, E).
Figure 4.

Pretreatment with GSK’872 down-regulates the increased MLKL expression and neuronal necrosis rate induced by 1 mM METH + 39°C treatment.
(A) Propidium iodide staining of cortical neurons pretreated with different concentrations of (0.25, 0.5 and 1 μM) of GSK’872 inhibitor before 1 mM METH + 39°C treatment for 3 hours: scale bar: 50 μm. (B) Statistics of propidium iodide uptake rate. (C) Detection of necrotic rate of cortical neurons by lactate dehydrogenase assay after pretreated different concentrations of GSK’872 inhibitor before 1 mM METH + 39°C treatment for 3 hours. (D) Western blot assay of MLKL in cortical neurons after pretreated different concentrations of GSK’872 inhibitor. (E) Statistics of expression of MLKL protein. Data are expressed as the mean ± SD (n = 3; one-way analysis of variance followed by Tukey’s multiple comparison test). ***P < 0.001, ****P < 0.0001, vs. normal group; #P < 0.05, ###P < 0.001, ####P < 0.0001. METH: Methamphetamine; MLKL: mixed lineage kinase domain-like protein.
Up-regulation of RIP3 and MLKL in the frontal cortex of specimen (METH abusers)
Because the four sources of brain tissue were taken from the corpses within 48 hours after the death of the donor, the tissue section showed a slight autolysis of the brain tissue, which is a normal phenomenon in forensic pathological anatomy (Figure 5). No obvious pathological phenomena of neurological diseases were seen in the tissue sections. RIP3 and MLKL were expressed in the neuronal cells of brain tissue sections. The expression of RIP3 and MLKL in case 1 (Control) was weakly positive. The expression of RIP3 and MLKL was more intense in case 2 and case 3 (METH-poisoning) than in case 4 (METH induced coronary heart attack) and case 1 (electric shock). In summary, the results from the brain specimens from people who had died of METH poisoning compared to those who had not showed a stronger expression of RIP3 and MLKL.
Figure 5.

Immunofluorescence staining of human cadaver frontal cortex sections.
The causes of death are listed in the Table 1. RIP3 and MLKL are stained by immunohistochemistry for each case. The small- to medium-sized frame is a local 4 × magnification of the larger one. Scale bars: 100 μm in row one and three, 50 μm in row two and four. MLKL: Mixed lineage kinase domain-like protein; RIP3: receptor-interacting protein 3.
Discussion
The concentrations of METH used in in vitro studies of toxicity are generally at millimolar levels, while the blood concentrations which are sufficient to generate neurotoxicity are identified in the range of 1 to 10 μM with an average blood concentration of 2.0 μM in METH abusers (Melega et al., 2007). The possible explanations for the orders of magnitude differences in concentrations are: (1) there is uptake of METH to the brain from the blood and this might lead to a higher concentration of METH in the central nervous system. This is supported by the fact that METH concentration was 10-fold higher in several sub-regions of rat brain in comparison with that in the plasma (Melega et al., 1995, O’Neil et al., 2006). (2) In METH-induced in vivo toxicity, systemic responses including immune response might play a vital role in the brain (Riviere et al., 2000). (3) METH abuse often results in body temperature increase and the hyperthermia may aggravate the neurotoxic effect of METH (Kiyatkin and Sharma, 2016). In consequence, the METH concentration required for producing neurotoxicity in vitro would be much higher than in in vivo studies (Nara et al., 2010). Much progress in the context of METH neurotoxicity has been achieved by using METH concentration in the millimolar range (Huang et al., 2009). It has been reported that a concentration of 3 mM is around the LC50 of METH-induced neuronal damage (e.g., 3 mM in immortalized mesencephalon neurons) (Huang et al., 2009). Our previous study showed that 4 mM METH induced necroptosis of the rat cortical neurons in vitro (Xiong et al., 2016). The present study showed that 1 mM METH combined with 39°C triggered the necrosis-like morphological change, whereas pretreatment with the necroptosis inhibitor, necrostatin-1, significantly reduced the METH + 39°C damage. These results suggested that under the condition of hyperthermia, lower concentrations of METH could lead to necroptotic neuronal death. Moreover, Stumm et al. (1999) found that 1 mM METH treatment for 96 hours induced apoptosis in primary cultured cortical neurons. As our research focuses on necroptosis, which is an early event in neurotoxicity, we investigated the effect of 1 mM METH for 1, 2, 3 and 5 hours together with 39°C to observe the damage of primary cultured cortex neurons. In our previous study, necrotic cell death was significantly increased after 12 hours of 4 mM METH exposure (Xiong et al., 2016). While evaluating the combined effect of METH and hyperthermia in our present investigation, we showed that necroptosis of cortical neurons occurred at an earlier stage, even 3 hours after METH treatment. We used METH with 39°C to treat neurons in vitro to simulate the real status of METH abusers. This is because METH abusers usually present with high fever. Taken together, the results suggest that even a relatively low concentration of METH or short exposure time might cause severe damage to cortical neurons due to the combination of drug action and high fever, which should shed new light on the therapeutic strategies concerning METH-elicited neurotoxicity.
RIP3 and MLKL are involved in the regulation of neuronal necroptosis induced by distinct models, which is suppressed by specific inhibitors (Ganjam et al., 2018; Wang et al., 2018a; Xu et al., 2018). MLKL is the final executor in necroptosis. Therefore, we used the RIP3 inhibitor GSK’872 to focus on the changes of MLKL. In our study, western blot assay and morphological results showed that RIP3 and MLKL were significantly up-regulated in the METH + 39°C-treated cortical neurons. For example, we can find the RIP3 green small dots in immunofluorescence staining and in the brown stained neurons in immunohistochemistry is the significant marker for necroptosis (Xu et al., 2016). Furthermore, the MLKL translocation to the cell membrane, shown in the immunofluorescence staining, coincides with other reports that MLKL was activated to translocate to the membrane to execute necroptosis (Cai et al., 2014; Chen et al., 2014). Our results suggested that RIP3/MLKL mediated necroptosis is caused by METH + 39°C in cultured neurons in vitro experiments and in postmortem human brain specimens. Moreover, our studies showed that GSK’872 down-regulated MLKL expression and neuronal necrosis rate. Even though we did not apply the technique of knockdown or over-expression of RIP3 and MLKL genes, the classical molecular changes on quantity and location expression of RIP3 and MLKL, combined with the RIP3 inhibition results, strongly suggested the existence of necroptosis induced by METH + 39°C. It also suggests that the RIP3/MLKL molecular pathway played an important role in METH + 39°C induced cortical neuronal necroptosis. Although 0.5 μM GSK’872 effectively inhibited neuronal necrosis, none of the concentrations (0.25–1 μM) of GSK’872 could reduce METH + 39°C induced neuronal necrosis to a normal level. This would indicate that the RIP3/MLKL pathway was not the only regulatory pathway involved in necrosis. An investigation of other molecules that might be involved in its regulated processing would be worth further study.
The two cases of METH-poisoning, accompanied by strongly positive expression of RIP3 and MLKL in neurons, had body temperatures of more than 39°C before death. This indicates that the synergistic effect of METH and hyperthermia may up-regulate the expression of RIP3 and MLKL in human cortical neurons. The expression of RIP3 and MLKL in the brain tissue sections of case 1, the control, was weakly positive, which may be due to common pathological features, including apoptosis and necroptosis that occur in the dying process from various death causes (Vandenabeele et al., 2008; Liu et al., 2015). The blood concentration of METH in case 4 (death by cardiac failure) was much lower than that of case 2 and case 3, and the expression of RIP3 and MLKL in brain tissue sections of case 4 was also lower than that of case 2 and case 3. These results suggest that METH action in the human body causes the up-regulation of RIP3 and MLKL expression and this is positively correlated with its dose. Validation will require a larger study in the future. However, it is impossible to strictly control the post-mortem interval in forensic cases and often some information is lacking (e.g., the body temperature before death in cases 1 and 4). It is possible these factors may have an impact on the results of immunohistochemistry of human brain tissue. In addition, the lack of sufficient human samples was also a limitation of this study. Our team will continue to collect more samples for data statistics and analysis in the future. Overall, the results showing that RIP3 and MLKL were highly expressed in human brain tissues of METH abusers further supports our hypothesis that METH plus hyperthermia can induce RIP3/MLKL modulated neuronal necroptosis. This may be applicable for the clinical therapeutic targets and potential diagnostic biomarkers in future.
In conclusion, METH + 39°C may lead to necroptosis of cortical neurons, and the RIP3/MLKL molecular pathway is involved in that necroptotic process.
Additional file: Open peer review reports 1 (122KB, pdf) and 2 (122.9KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This study was funded by the National Natural Science Foundation of China, No. 81971891 (to KX), No. 81571939 (to KX), No. 81772134 (to KX), 81772024 (to JY), and 81860781 (to FXL); the Key Research and Development Program of Hunan Province of China, No. 2018SK2091 (to KX); the Natural Science Foundation of Hunan Province of China, No. 2017JJ2339 (to JY); the Wu Jie-Ping Medical Foundation of the Minister of Health of China, No. 320.6750.14118 (to KX). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: The study protocol was approved by the Medical Ethics Committee of the Third Xiangya Hospital of Central South University (approval numbers: 2017-S026 and 2017-S033) on March 7, 2017.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Nathan K Evanson, Cincinnati Children’s Hospital Medical Center, USA; Rodolfo Gabriel Gatto, University of Illinois at Chicago, USA.
Funding: This study was funded by the National Natural Science Foundation of China, No. 81971891 (to KX), 81571939 (to KX), 81772134 (to KX), 81772024 (to JY), and 81860781 (to FXL); the Key Research and Development Program of Hunan Province of China, No. 2018SK2091 (to KX); the Natural Science Foundation of Hunan Province of China, No. 2017JJ2339 (to JY); the Wu Jie-Ping Medical Foundation of the Minister of Health of China, No. 320.6750.14118 (to KX).
P-Reviewers: Evanson NK, Gatto RG; C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Dawes EA, Yajima W, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen S, Yan J, Deng HX, Long LL, Hu YJ, Wang M, Shang L, Chen D, Huang JF, Xiong K. Inhibition of calpain on oxygen glucose deprivation-induced RGC-5 necroptosis. J Huazhong Univ Sci Technolog Med Sci. 2016;36:639–645. doi: 10.1007/s11596-016-1639-y. [DOI] [PubMed] [Google Scholar]
- 3.Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng SY, Wang SC, Lei M, Wang Z, Xiong K. Regulatory role of calpain in neuronal death. Neural Regen Res. 2018;13:556–562. doi: 10.4103/1673-5374.228762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cruz SA, Qin Z, Stewart AFR, Chen HH. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen Res. 2018;13:252–256. doi: 10.4103/1673-5374.226394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH, 3rd, Tait SWG, Martinez J, Gale M, Jr, Loo YM, Oberst A. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell. 2017;169:301–313. doi: 10.1016/j.cell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding W, Shang L, Huang JF, Li N, Chen D, Xue LX, Xiong K. Receptor interacting protein 3-induced RGC-5 cell necroptosis following oxygen glucose deprivation. BMC Neurosci. 2015;16:49. doi: 10.1186/s12868-015-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganjam GK, Terpolilli NA, Diemert S, Eisenbach I, Hoffmann L, Reuther C, Herden C, Roth J, Plesnila N, Culmsee C. Cylindromatosis mediates neuronal cell death in vitro and in vivo. Cell Death Differ. 2018;25:1394–1407. doi: 10.1038/s41418-017-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrell R, Speaker HA, Mitchell SL, Sabol KE. The effects of the beta1 antagonist, metoprolol, on methamphetamine-induced changes in core temperature in the rat. Neurosci Lett. 2015;609:81–86. doi: 10.1016/j.neulet.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Huang JF, Shang L, Zhang MQ, Wang H, Chen D, Tong JB, Huang H, Yan XX, Zeng LP, Xiong K. Differential neuronal expression of receptor interacting protein 3 in rat retina: involvement in ischemic stress response. BMC Neurosci. 2013;14:16. doi: 10.1186/1471-2202-14-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YN, Wu CH, Lin TC, Wang JY. Methamphetamine induces heme oxygenase-1 expression in cortical neurons and glia to prevent its toxicity. Toxicol Appl Pharmacol. 2009;240:315–326. doi: 10.1016/j.taap.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 13.Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–608. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang SH, Shang L, Xue LX, Ding W, Chen S, Ma RF, Huang JF, Xiong K. The effect and underlying mechanism of Timosaponin B-II on RGC-5 necroptosis induced by hydrogen peroxide. BMC Complement Altern Med. 2014;14:459. doi: 10.1186/1472-6882-14-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiyatkin EA, Sharma HS. Breakdown of blood-brain and blood-spinal cord barriers during acute methamphetamine intoxication: role of brain temperature. CNS Neurol Disord Drug Targets. 2016;15:1129–1138. doi: 10.2174/1871527315666160920112445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, Zhang ZX, Jevnikar AM. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13:2805–2818. doi: 10.1111/ajt.12447. [DOI] [PubMed] [Google Scholar]
- 17.Li N, Shang L, Wang SC, Liao LS, Chen D, Huang JF, Xiong K. The toxic effect of ALLN on primary rat retinal neurons. Neurotox Res. 2016;30:392–406. doi: 10.1007/s12640-016-9624-6. [DOI] [PubMed] [Google Scholar]
- 18.Liao L, Shang L, Li N, Wang S, Wang M, Huang Y, Chen D, Huang J, Xiong K. Mixed lineage kinase domain-like protein induces RGC-5 necroptosis following elevated hydrostatic pressure. Acta Biochim Biophys Sin (Shanghai) 2017;49:879–889. doi: 10.1093/abbs/gmx088. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Bao YH, Wang Y, Jiang JY. The role of necroptosis in neurosurgical diseases. Braz J Med Biol Res. 2015;48:292–298. doi: 10.1590/1414-431X20144310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu S, Yang X, Wang C, Chen S, Lu S, Yan W, Xiong K, Liu F, Yan J. Current status and potential role of circular RNAs in neurological disorders. J Neurochem. 2019a doi: 10.1111/jnc.14724. doi: 10.1111/jnc.14724. [DOI] [PubMed] [Google Scholar]
- 21.Lu S, Liao LS, Zhang B, Yan W, Chen LP, Yan H, Guo LM, Lu SS, Xiong K, Yan J. Antioxidant cascades confer neuroprotection in ethanol, morphine, and methamphetamine preconditioning. Neurochem Int. 2019b;131:104540. doi: 10.1016/j.neuint.2019.104540. [DOI] [PubMed] [Google Scholar]
- 22.Lu T, Kim P, Luo Y. Tp53 gene mediates distinct dopaminergic neuronal damage in different dopaminergic neurotoxicant models. Neural Regen Res. 2017;12:1413–1417. doi: 10.4103/1673-5374.215243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang L, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moratalla R, Khairnar A, Simola N, Granado N, Garcia-Montes JR, Porceddu PF, Tizabi Y, Costa G, Morelli M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog Neurobiol. 2017;155:149–170. doi: 10.1016/j.pneurobio.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Orzalli MH, Kagan JC. Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol. 2017;27:800–809. doi: 10.1016/j.tcb.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riddle EL, Fleckenstein AE, Hanson GR. Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J. 2006;8:E413–E418. doi: 10.1007/BF02854914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruan J, Mei L, Zhu Q, Shi G, Wang H. Mixed lineage kinase domain-like protein is a prognostic biomarker for cervical squamous cell cancer. Int J Clin Exp Pathol. 2015;8:15035–15038. [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Alavez M, Bortell N, Galmozzi A, Conti B, Marcondes MC. Reactive oxygen species scavenger N-acetyl cysteine reduces methamphetamine-induced hyperthermia without affecting motor activity in mice. Temperature (Austin) 2014;1:227–241. doi: 10.4161/23328940.2014.984556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shang L, Huang JF, Ding W, Chen S, Xue LX, Ma RF, Xiong K. Calpain: a molecule to induce AIF-mediated necroptosis in RGC-5 following elevated hydrostatic pressure. BMC Neurosci. 2014;15:63. doi: 10.1186/1471-2202-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shang L, Ding W, Li N, Liao L, Chen D, Huang J, Xiong K. The effects and regulatory mechanism of RIP3 on RGC-5 necroptosis following elevated hydrostatic pressure. Acta Biochim Biophys Sin (Shanghai) 2017;49:128–137. doi: 10.1093/abbs/gmw130. [DOI] [PubMed] [Google Scholar]
- 32.Shioda K, Nisijima K, Yoshino T, Kato S. Effect of risperidone on acute methamphetamine-induced hyperthermia in rats. Drug Alcohol Depend. 2010;111:241–249. doi: 10.1016/j.drugalcdep.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 34.Vandenabeele P, Declercq W, Vanden Berghe T. Necrotic cell death and ‘necrostatins’: now we can control cellular explosion. Trends Biochem Sci. 2008;33:352–355. doi: 10.1016/j.tibs.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Liao L, Huang Y, Wang M, Zhou H, Chen D, Liu F, Ji D, Xia X, Jiang B, Huang J, Xiong K. Pin1 is regulated by camkii activation in glutamate-induced retinal neuronal regulated necrosis. Front Cell Neurosci. 2019a doi: 10.3389/fncel.2019.00276. doi: 10.3389/fncel.2019.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Liao L, Wang M, Zhou H, Huang Y, Wang Z, Chen D, Ji D, Xia X, Wang Y, Liu F, Huang J, Xiong K. Pin1 Promotes regulated necrosis induced by glutamate in rat retinal neurons via CAST/Calpain2 pathway. Front Cell Neurosci. 2018d;11:425. doi: 10.3389/fncel.2017.00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, Huang Y, Yan Y, Zhou H, Wang M, Liao L, Wang Z, Chen D, Ji D, Xia X, Liu F, Huang J, Xiong K. Calpain2 but not calpain1 mediated by calpastatin following glutamate-induced regulated necrosis in rat retinal neurons. Ann Anat. 2019b;221:57–67. doi: 10.1016/j.aanat.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Guo LM, Wang Y, Zhou HK, Wang SC, Chen D, Huang JF, Xiong K. Inhibition of HSP90alpha protects cultured neurons from oxygen-glucose deprivation induced necroptosis by decreasing RIP3 expression. J Cell Physiol. 2018a;233:4864–4884. doi: 10.1002/jcp.26294. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z, Guo LM, Wang SC, Chen D, Yan J, Liu FX, Huang JF, Xiong K. Progress in studies of necroptosis and its relationship to disease processes. Pathol Res Pract. 2018b;214:1749–1757. doi: 10.1016/j.prp.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Guo LM, Zhou HK, Qu HK, Wang SC, Liu FX, Chen D, Huang JF, Xiong K. Using drugs to target necroptosis: dual roles in disease therapy. Histol Histopathol. 2018c;33:773–789. doi: 10.14670/HH-11-968. [DOI] [PubMed] [Google Scholar]
- 41.Wu CW, Ping YH, Yen JC, Chang CY, Wang SF, Yeh CL, Chi CW, Lee HC. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol Appl Pharmacol. 2007;220:243–251. doi: 10.1016/j.taap.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Xiong K, Liao H, Long L, Ding Y, Huang J, Yan J. Necroptosis contributes to methamphetamine-induced cytotoxicity in rat cortical neurons. Toxicol In Vitro. 2016;35:163–168. doi: 10.1016/j.tiv.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Xiong K, Long L, Zhang X, Qu H, Deng H, Ding Y, Cai J, Wang S, Wang M, Liao L, Huang J, Yi CX, Yan J. Overview of long non-coding RNA and mRNA expression in response to methamphetamine treatment in vitro. Toxicol In Vitro. 2017;44:1–10. doi: 10.1016/j.tiv.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 44.Xu J, Mo J, Liu X, Marshall B, Atherton SS, Dong Z, Smith S, Zhang M. Depletion of the receptor-interacting protein kinase 3 (RIP3) decreases photoreceptor cell death during the early stages of ocular murine cytomegalovirus infection. Invest Ophthalmol Vis Sci. 2018;59:2445–2458. doi: 10.1167/iovs.18-24086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu SY, Zhang QL, Zhang Q, Wan L, Jiang J, Tu T, Manavis J, Pan A, Cai Y, Yan XX. Regional and cellular mapping of sortilin immunoreactivity in adult human brain. Front Neuroanat. 2019;13:31. doi: 10.3389/fnana.2019.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Y, Wang J, Song X, Qu L, Wei R, He F, Wang K, Luo B. RIP3 induces ischemic neuronal DNA degradation and programmed necrosis in rat via AIF. Sci Rep. 2016;6:29362. doi: 10.1038/srep29362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang R, Hu K, Chen J, Zhu S, Li L, Lu H, Li P, Dong R. Necrostatin-1 protects hippocampal neurons against ischemia/reperfusion injury via the RIP3/DAXX signaling pathway in rats. Neurosci Lett. 2017;651:207–215. doi: 10.1016/j.neulet.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 49.Yang X, Wang C, Zhang X, Chen S, Chen L, Lu S, Lu S, Yan X, Xiong K, Liu F, Yan J. Redox regulation in hydrogen sulfide action: From neurotoxicity to neuroprotection. Neurochem Int. 2019;128:58–69. doi: 10.1016/j.neuint.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 50.Yang X, Wang Y, Li Q, Zhong Y, Chen L, Du Y, He J, Liao L, Xiong K, Yi CX, Yan J. The main molecular mechanisms underlying methamphetamine- induced neurotoxicity and implications for pharmacological treatment. Front Mol Neurosci. 2018;11:186. doi: 10.3389/fnmol.2018.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin B, Xu Y, Wei RL, He F, Luo BY, Wang JY. Inhibition of receptor-interacting protein 3 upregulation and nuclear translocation involved in Necrostatin-1 protection against hippocampal neuronal programmed necrosis induced by ischemia/reperfusion injury. Brain Res. 2015;1609:63–71. doi: 10.1016/j.brainres.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 52.Zhong CQ, Li Y, Yang D, Zhang N, Xu X, Wu Y, Chen J, Han J. Quantitative phosphoproteomic analysis of RIP3-dependent protein phosphorylation in the course of TNF-induced necroptosis. Proteomics. 2014;14:713–724. doi: 10.1002/pmic.201300326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.