

Abstract
Cellular senescence is associated with inflammation and extracellular matrix tissue remodeling through the secretion of proteins termed the senescence-associated secretory phenotype (SASP). Although osteocyte senescence in older individuals in the skeleton is well recognized, whether young alveolar osteocytes can also become senescent is unknown. This is potentially important in the context of periodontal disease, which is an inflammatory condition caused by a gradual change from symbiotic to pathogenic oral microflora that can lead to tooth loss. Our aim was to identify whether senescent osteocytes accumulate in young alveolar bone and whether bacterial-derived lipopolysaccharide (LPS) can influence cellular senescence in alveolar bone. An osteocyte-enriched cell population isolated from alveolar bone expressed increased levels of the known senescence marker p16Ink4a, as well as select SASP markers known to be implicated alveolar bone resorption (Icam1, Il6, Il17, Mmp13 and Tnfα), compared to ramus control cells. Increased senescence of alveolar bone osteocytes was also observed in vivo using the senescence-associated distension of satellites (SADS) assay and increased γH2AX, a marker of DNA damage associated with senescent cells. To approximate a bacterial infection in vitro, alveolar osteocytes were treated with LPS. We found increased expression of various senescence and SASP markers, increased γH2AX staining, increased SA-β-Gal activity and the redistribution of F-actin leading to a larger and flattened cell morphology, all hallmarks of cellular senescence. In conclusion, our data suggests a model whereby bacterial-derived LPS stimulates premature alveolar osteocyte senescence, which in combination with the resultant SASP, could potentially contribute to the onset of alveolar bone loss.
Keywords: Senescence, SASP, Inflammation, Bacteria, Periodontal Disease, Alveolar Bone, Osteocyte
1. Introduction
Periodontitis is the most common chronic bacterial infection and osteolytic disease, leading to edentulism and affecting life quality and general health [1, 2]. Transition from health to disease in the periodontium is associated with a gradual change from a symbiotic to a pathogenic host-microbial relationship [3, 4]. Inflammation contributes to this microbial homeostasis disruption, and fuels the overgrowth of dysbiotic pathogens, by providing nutrients in form of tissue breakdown products [5]. Thus, it is the host inflammatory reaction that primarily and ultimately causes alveolar bone resorption [5]. However, our understanding of the cellular mechanisms involved in the host’s response to bacterial challenge is incomplete.
In addition to replicative senescence, which is associated with aging phenotypes, stress-induced senescence is also observed following persistent DNA damage resulting from oncogenic or oxidative stress [6, 7]. A key senescent cell feature is the secretion of a complex mixture of pro-inflammatory cytokines and proteases, known as the senescence-associated secretory phenotype (SASP) [8]. One function of the SASP is to recruit phagocytic cells to initiate tissue remodeling, which includes senescent cell elimination, and prepare the tissue microenvironment for regeneration [9]. In contrast to this beneficial effect in tissue repair, senescent cells increasingly accumulate with aging and are frequently present at sites of age-related pathologies [10]. In addition, senescent cells have also been observed in young, obese mice fed a high-fat diet [11, 12], suggesting that in certain contexts premature senescence can and does occur.
Persistent exposure to gram-negative bacteria toxic products promotes cellular senescence [7, 13]. In order to invade, survive and multiply in the host’s periodontal tissues, gram-negative bacteria utilize a panel of virulence factors as inflammatory mediators [14]. Although inflammatory cytokine production is a key intermediate mechanism between bacterial challenge and host periodontal tissue destruction [15], some gram-negative products can also act as genotoxic agents inducing a DNA damage response and mitotic arrest [16]. Indeed, repeated LPS exposure, a gram-negative bacterial outer membrane component, can induce senescence in microglial [17], dental pulp [13] and pulmonary epithelial cells [18]. Therefore, sustained exposure to LPS could act as both a pro-inflammatory and genotoxic stress on periodontal and bone cells.
Osteocytes are the most abundant bone cells and crucial orchestrators of bone remodeling and mineral homeostasis [19], and because of their increased lifespan compared to other bone cells, they are more susceptible to accumulate molecular damage over time [20]. Given the anatomic proximity between alveolar bone and periodontal infection, alveolar-derived osteocytes are continuously exposed to bacteria and their toxins. Therefore, we hypothesized that senescent osteocytes accumulate prematurely at a younger age in alveolar bone as result of LPS-induced genotoxic stress. We aimed to identify and measure the prevalence of senescent osteocytes in young mouse alveolar bone. Our results indicate that senescent osteocytes prematurely accumulate in alveolar bone, and these dysfunctional cells could constitute a “non-microbial” source of inflammatory cytokines and proteolytic factors that promote alveolar bone destruction.
2. Methods
2.1. Mouse alveolar bone and ramus osteocyte-enriched cell population collection
All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) before study initiation. Whole jawbones were collected from euthanized 6 month-old C57BL/6 wild-type (WT) female mice (n=10) (Charles River Laboratories, Wilmington, MA, USA). For osteocyte-enriched cell population collection from alveolar bone and ramus, we used a process of extended collagenase digestions as previously described by the Bonewald laboratory [21]. Soft tissues and teeth were removed, and each jawbone hemi-sectioned at the symphysis level. Each hemi-jawbone was divided into alveolar and ramus sections, and the boundary between both sections determined by following previously described morphological landmarks [22]. The osteocyte-enriched population (n=6) was immediately homogenized (Tissue Tearor, Cole Parmer, IL, USA) in lysis buffer (QIAzol; Qiagen, Valencia, CA, USA) without in vitro cell culture [23] and stored at −80°C for later RNA extraction, as described below.
2.2. Senescent osteocytes analysis in vivo
Left hemi-jawbones were used to measure senescence-associated distension of satellites (SADS) in osteocytes located in cortical bone (n=10). For fluorescent in situ hybridization (FISH) staining, bone sections were processed as previously described [23]. The Cy3-labelled CENPB-specific (5’ ATTCGTTGGAAACGGGA 3’) peptide nucleic acid (PNA) FISH probe (Panagene Inc, Korea) was used, and slides were mounted with a DAPI-containing mounting media (ThermoFisher Scientific, Waltham, MA, USA). For SADS visualization, images were obtained using inverted laser scanning confocal microscope (LSM 780; Carl Zeiss Microscopy, Jena, Germany) with in-depth Z stacking. A senescent osteocyte was defined based on the number of SADS per cell, with a cut-off of ≥ 4 [23, 24]. Nuclei of at least 50 osteocytes were analyzed for each sample.
2.3. Primary jawbone osteocyte-like cell isolation and culture
For primary jawbone osteocyte-like cell isolation, soft tissues and teeth were removed and remaining bone fragments were processed using a modified protocol derived from Stern et al. and El Deeb Zakhary et al. combined methods [21, 25]. Briefly, bone fragments from alveolar bone and ramus were independently digested with collagenase (Liberase; Roche Diagnostics GmbH, Mannheim, Germany) dissolved in α-MEM and incubated at 37°C for 20 minutes (four incubations). After each sequential digestion, cells were discarded and bone particles stored. Bone fragments were washed in 1x phosphate buffered saline (PBS; pH 7), placed in 6-well plates containing alpha-minimal essential growth medium (α-MEM) supplemented with 1% penicillin and streptomycin (ThermoFisher Scientific), Glutamax, and 10% fetal bovine serum (FBS)(GE Healthcare Life Sciences HyClone Laboratories, Logan, UT, USA). Cells were incubated at 37°C and allowed to grow out from the bone fragments, and expanded for in vitro experiments. To evaluate the LPS effect, cells were cultured in supplemented α-MEM alone containing low serum (2% FBS) to reduce cell proliferation or media containing P. gingivalis LPS (10 ng/ml).
2.4. Ex vivo jawbone culture model.
We designed and performed a split mouth study using an ex vivo jawbone model (n=6). Briefly, soft tissues and teeth were removed, jawbones hemi-sectioned, and alveolar bone blocks obtained as described above. Alveolar bone blocks were rinsed with PBS and kept in supplemented α-MEM. Right alveolar bone blocks were cultured in 12-well plates and exposed to P. gingivalis LPS (10 ng/ml) as described above (6 days, media changed daily), whereas the contralateral/left blocks were cultured with supplemented media alone. Then, they were collected, rinsed in PBS, and centrifuged at 1500 rpm for 5 minutes, and homogenized in lysis buffer. Samples were storedat −80°C for later RNA extraction.
2.5. RNA Isolation and QPCR
RNA isolation and QPCR gene expression analysis was performed as previously described [26]. SYBR Green (Qiagen) was used as detection reagent. Each sample was run in triplicate and normalized to Tuba1a, which was used as the internal reference gene. Primer sequences are available upon request.
2.6. Immunofluorescence Staining (IF)
Primary osteocyte-like cells were grown with low serum as described above, and stimulated with 10 ng/mL P. gingivalis LPS for six days or medium alone as control. Dissected jawbones were fixed with 4% PFA for 48 hours, and then immersed in Decalcifier II (Leica Biosystems, Buffalo Grove, IL) for 3 days. Samples were then immersed in 15% sucrose overnight at 4° C. At that time, they were rinsed in PBS, embedded in OCT and stored at −80° C. A cryostat (CM1850 UV; Leica Biosystems) was used to cut 6-micron sections. IF was performed following previously described protocols [13, 26]. Briefly, cells were fixed in 2% PFA for 15 minutes, washed in 1X PBS twice, and permeabilized in 0.1% Triton X-100. Next, cells were blocked with 10% goat serum for 1 hour. Then, cells were incubated with γH2AX primary antibody (1:300), or p53 (phospho-S15) (1:100) for 1 hour, washed three times, and incubated with the secondary antibody (goat α-rabbit Alexa 447) for 1 hour at room temperature. To evaluate morphological changes in F-actin cytoskeleton, cells were labelled with Alexa Fluor 555 Phalloidin (25 μg/ml). DAPI Fluoromont (SouthernBiotech, Birmingham, AL) was used as mounting medium and nuclear staining. A laser confocal microscope (Carl Zeiss Microscopy) was used to detect labeled cells.
2.7. Senescence-associated β-galactosidase staining.
For senescence-associated β-galactosidase assay (SA-β-gal), cells were cultured in 6-well plates, and then the staining was performed following a previously described protocol [27]. Cells were fixed in 2% PFA for 15 minutes at room temperature, and washed twice in 1X PBS. Reagents were prepared following the manufacturer’s instructions, and 1 ml of staining solution was added to each well. The plate was covered with aluminum foil to protect of light, and incubated at 37° C for 18–24 hours. The staining solution was then removed and replaced with 1X PBS. SA-β-gal-positive cells displaying blue staining were quantified by acquiring images from six different random fields, and at least 500 cells were counted for each condition. The percentageof SA-β-gal-positive cells was determined by counting the total number of cells versus the number of SA-β-gal-positive cells. Representative images are displayed for each condition.
2.8. Statistical Analyses
Data are shown as mean ± SEM and values were considered statistically significant using Student’s t test at p < 0.05*, p < 0.01** and p < 0.001***. Analyses were performed using Microsoft Office Excel 2003 (Microsoft Corp., Redmond, WA) and GraphPad software (GraphPad Software, Inc., San Diego, CA, USA).
3. Results
3.1. Senescent osteocytes accumulate in alveolar bone from young mice
Given that p16Ink4a is one of the most robust and well-characterized senescent cell biomarkers along with p53 and p21 [28], we initially evaluated their expression in an osteocyte-enriched cell population from alveolar bone compared with similarly isolated cells from ramus bone, without in vitro culture. We found higher p16Ink4a expression, with a trend for p21 expression, in alveolar bone osteocytes compared to the ramus (Fig. 1a). Next, we evaluated the expression of twelve recognized SASP factors [23] and found that Icam1, Igfbp4, Il6, Il17, Mmp13 and Tnfα expression were significantly upregulated in alveolar bone osteocytes as compared to ramus (Fig. 1b). To validate our results, we measured SADS, a hallmark and consistent event in cellular senescence [24] in alveolar bone in vivo. We found evidence of SADS-positive, elongated/decondensed centromeres in a subset of osteocytes (Fig. 1c), where upon quantification a higher percentage of senescent osteocytes was found in alveolar bone in relation to ramus (30% versus 19%, p<0.011) (Fig. 1d). Therefore, senescent osteocytes accumulate site-specifically in young alveolar bone in vivo, where they secrete a specific subset of pro-inflammatory SASP factors.
Fig. 1.
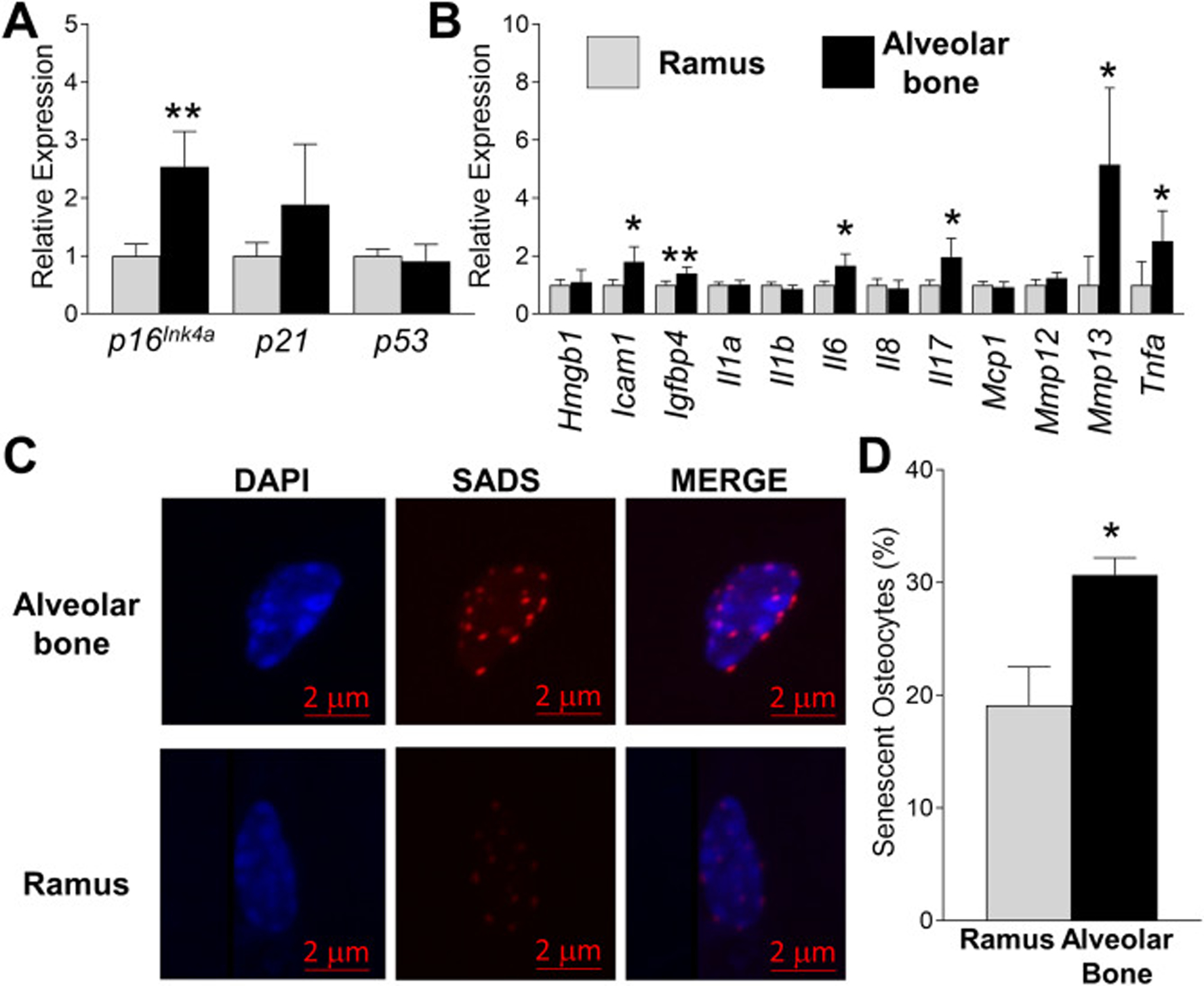
Identification of senescent cells in young alveolar bone. (a-b) Alveolar bone and ramus osteocyte-enriched cell populations were prepared from 6 month-old WT female mice and immediately assayed (no culture) for expression of selected senescent marker and SASP genes using QPCR. Data represent Mean ± SEM (n=10). *P ≤ 0.05 and **P≤ 0.01 relative to ramus control. (c) Fluorescent in situ hybridization (FISH) was used to identify SADS-positive senescent cells in ramus and alveolar bone from 6 month-old WT female mice in vivo (a representative cell is shown). (d) Quantitation of SADS-positive cells in ramus and alveolar osteocyte-enriched cell populations (50 cells counted per sample). The red bar denotes the magnification scale (2 μm). Data represent Mean ± SEM (n=10). *P ≤ 0.05 relative to ramus control.
Given that agents that cause DNA damage may lead to premature senescence [29] and repeated LPS stimulation induces premature senescence in adipocyte progenitors [30], we next examined the effects of LPS on γH2AX, a recognized marker of DNA damage [31]. IF staining showed a stronger γH2AX (Fig. 2a) signal in periodontal ligament and in the nearby osteocytes, compared to those located in ramus (Fig. 2b) or basal bone (Fig. 2c).
Fig. 2.
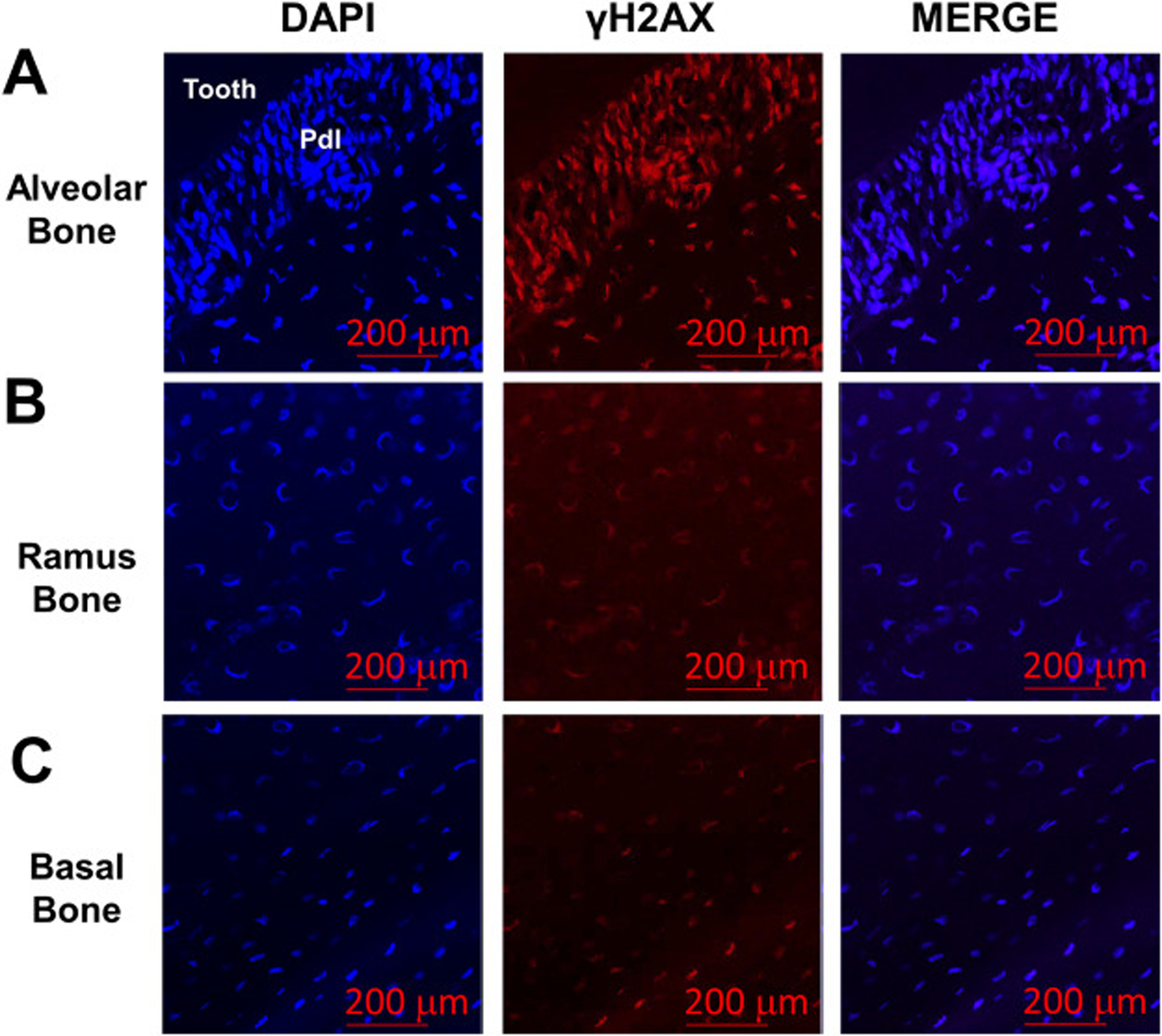
Levels of DNA damage are increased in alveolar bone osteocytes. (a) Presence of γH2AX, a marker of DNA damage, was evaluated by immunofluorescence in sections from alveolar bone from 6 month-old WT female mice in vivo. Stronger γH2AX (red signal) was observed both within the periodontal ligament (Pdl) and in neighboring alveolar osteocytes. Ramus (b) and underlying basal bone (c) displayed lower amounts of γH2AX signal. Cell nuclei were labeled with DAPI (blue signal). The red bars denote the magnification scale (200 μm).
3.2. LPS exposure increases senescence and SASP markers expression in alveolar bone cells.
Since senescent osteocytes in the murine skeleton usually do not appear until ~18 months of age [23, 32], it was curious that a significant 30% of osteocytes from young, 6 month-old alveolar bone exhibit senescent qualities, as evidenced by SADS (Fig. 1 c,d). We surmised that the close proximity of alveolar bone with periodontal pathogen infection and the toxins they produce (e.g. LPS) may explain the higher senescent cell burden observed in young alveolar bone. To confirm that LPS directly induces senescent markers and SASP factor expression, we exposed cultured primary alveolar osteocytes cells to repeated LPS stimulation. LPS produced a significant upregulation of p16Ink4, p21 and p53 expression (Fig. 3a) and a robust induction of the SASP factors Icam1, Il6, Mcp1, Mmp12 and Mmp13 (Fig. 3b). A subset of these cells were plated and stained for γH2AX [31]. Although DNA damage, as assessed by γH2AX staining, was observed in control- and LPS-treated cells (possibly due to the enzymatic nature of the cell preparation), a more localized and robust γH2AX-positive signature was observed in cells treated with LPS (Fig. 3c). These data suggest that LPS induces expression of senescent markers and SASP factors in alveolar-derived, osteocyte-like cells, possibly explaining the “premature” senescence observed in alveolar osteocytes from young animals.
Fig. 3.
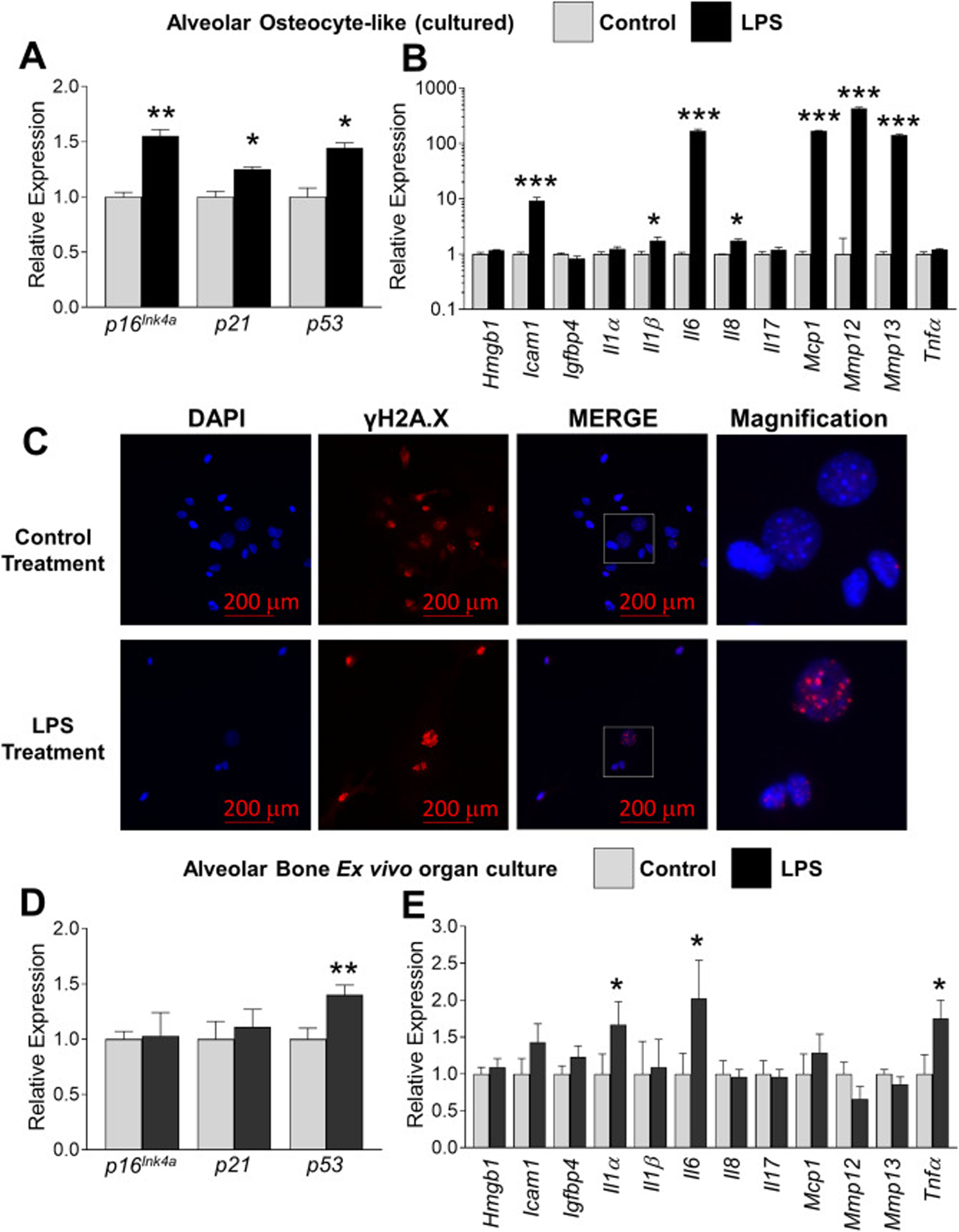
Chronic LPS exposure increases several indices of cellular senescence in alveolar bone osteocytes. (a-b) Alveolar bone osteocyte-like cells were prepared from 6 month-old WT female mice, treated with either vehicle control or P. gingivalis LPS (10 ng/ml) for six days and assayed for expression of selected senescent marker and SASP genes using QPCR. Data represent Mean ± SEM (n=10). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001 relative to vehicle control. (c) Alveolar osteocyte-like cells were treated with control or LPS as in panels a-b and stained with an antibody against γH2AX (red signal). The boxed inset in the Merge panel is shown enlarged in the Magnification panel to observe single γH2AX-stained foci. Cell nuclei were labeled with DAPI (blue signal). The red bars denote the magnification scale (200 μm). (d-e) Intact (non-digested) left and right alveolar bone blocks were treated with vehicle control or LPS, respectively, for 6 days as assayed for expression of selected senescent marker and SASP genes using QPCR. Data represent Mean ± SEM (n=6). *P ≤ 0.05, and **P ≤ 0.01 relative to vehicle control.
Ex vivo models mimic the conventional in vivo model as tissues and cells are morphologically positioned within the normal extracellular matrix [33–35]. Indeed, the ex vivo murine mandible culture model represents an “ideal system” to substitute for in vivo studies, are ethically acceptable, and have been validated for the study of inflammatory bone destruction in the presence of LPS [34, 35]. Therefore, we took advantage of this validated ex vivo model and found that alveolar blocks exposed to LPS exhibited a significantly higher p53 expression compared to the contralateral control samples (Fig. 3d). In addition, increased Il-1α, Il-6 and Tnfα expression was also observed after LPS stimulation (Fig. 3e). In contrast to what we expected, no difference was observed in p16Ink4 or p21 expression in this ex vivo model system when compared to the in vitro cellular model (Fig. 3 a–c). Nevertheless, these results indicate that p53 activation by LPS could mediate the secretion of a specific subset of SASP factors by alveolar bone osteocytes in an ex vivo or in vivo model.
Since our data suggests that in an ex vivo model p53 expression is increased in LPS-treated alveolar blocks, we examined alveolar bone sections from 6-month old mice (not LPS treated) for active p53 protein (phospho-p53; P-p53). We found significant P-p53 staining in alveolar bone (both in the periodontal ligament and in nearby osteocytes; Fig. 4a), compared with little staining in ramus bone (Fig. 4b). Interestingly, we did find P-p53 signal in osteocytes from basal bone (Fig. 4c), however this does not lead to significant a DNA damage response as basal bone osteocytes stain weakly for γH2AX (Fig. 2c). This suggests that different levels of senescence may exist in various periodontal tissues and bone compartments (alveolar vs ramus vs basal).
Fig. 4.
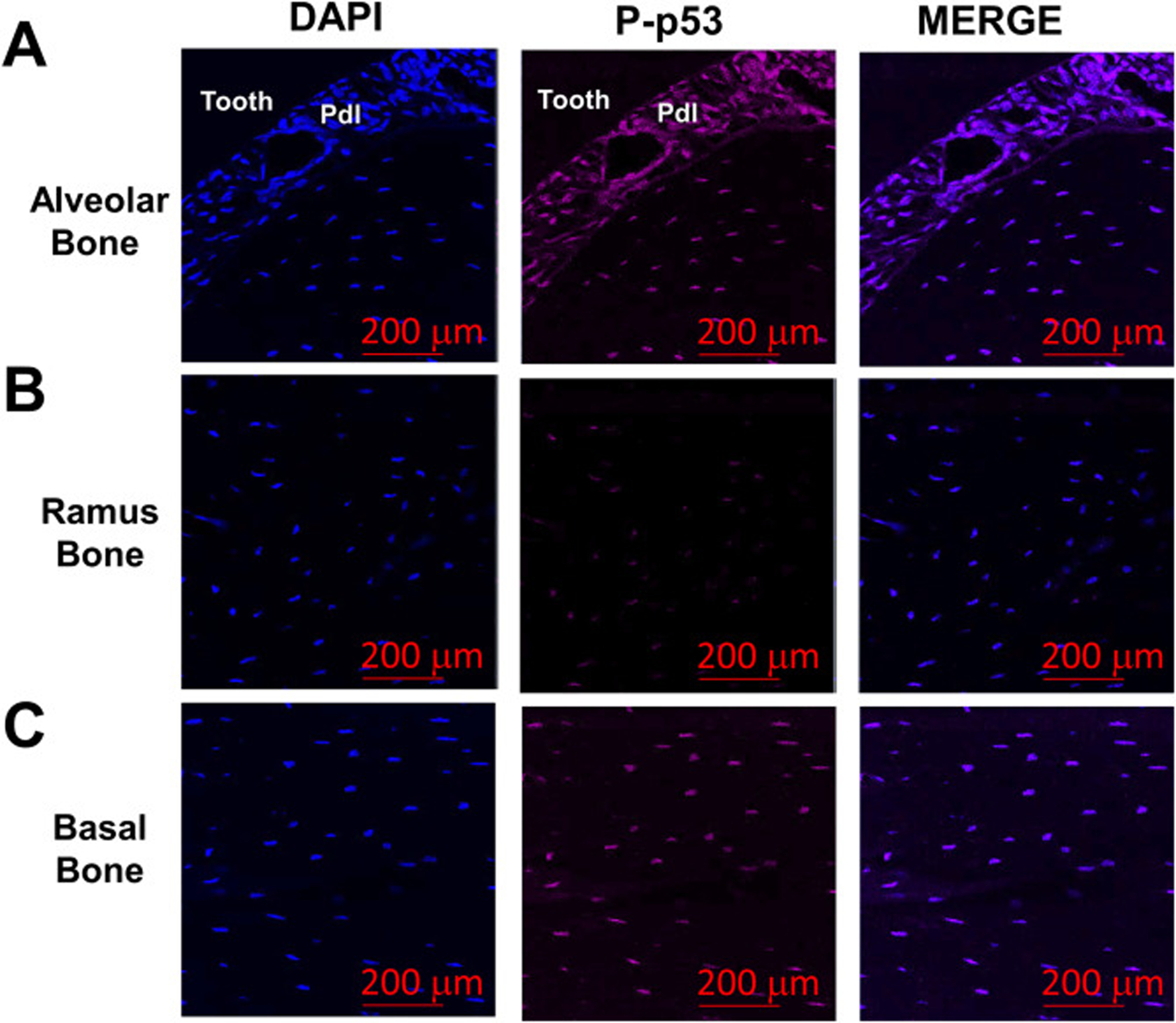
P-p53 protein levels are increased in alveolar bone osteocytes in vivo. (a-c) Bone sections from alveolar, ramus and underlying basal bone from 6 month-old WT female mice were stained for Phospho(P)-p53 levels (magenta signal). Cell nuclei were labeled with DAPI (blue signal). Cell nuclei were labeled with DAPI (blue signal). The red bars denote the magnification scale (200 μm).
To provide further evidence that LPS treatment of alveolar osteocytes in vitro induces phenotypes reminiscent of cellular senescence, we exposed cultured primary alveolar osteocytes cells to repeated LPS stimulation (as in Fig. 3) and measured F-actin redistribution and SA-β-gal activity, two additional hallmarks of the cellular senescent program [36, 37]. Indeed, enlarged and flatten cells displaying F-actin cytoskeleton alterations were observed following LPS treatment (Fig. 5a). Consistent with these findings, the percentage of SA-β-gal positive cells was also higher in LPS treated cells, even after a single exposure (Fig. 5b–c). Therefore, persistent LPS exposure may promote DNA damage that leads to p53 activation and premature osteocyte senescence.
Fig. 5.
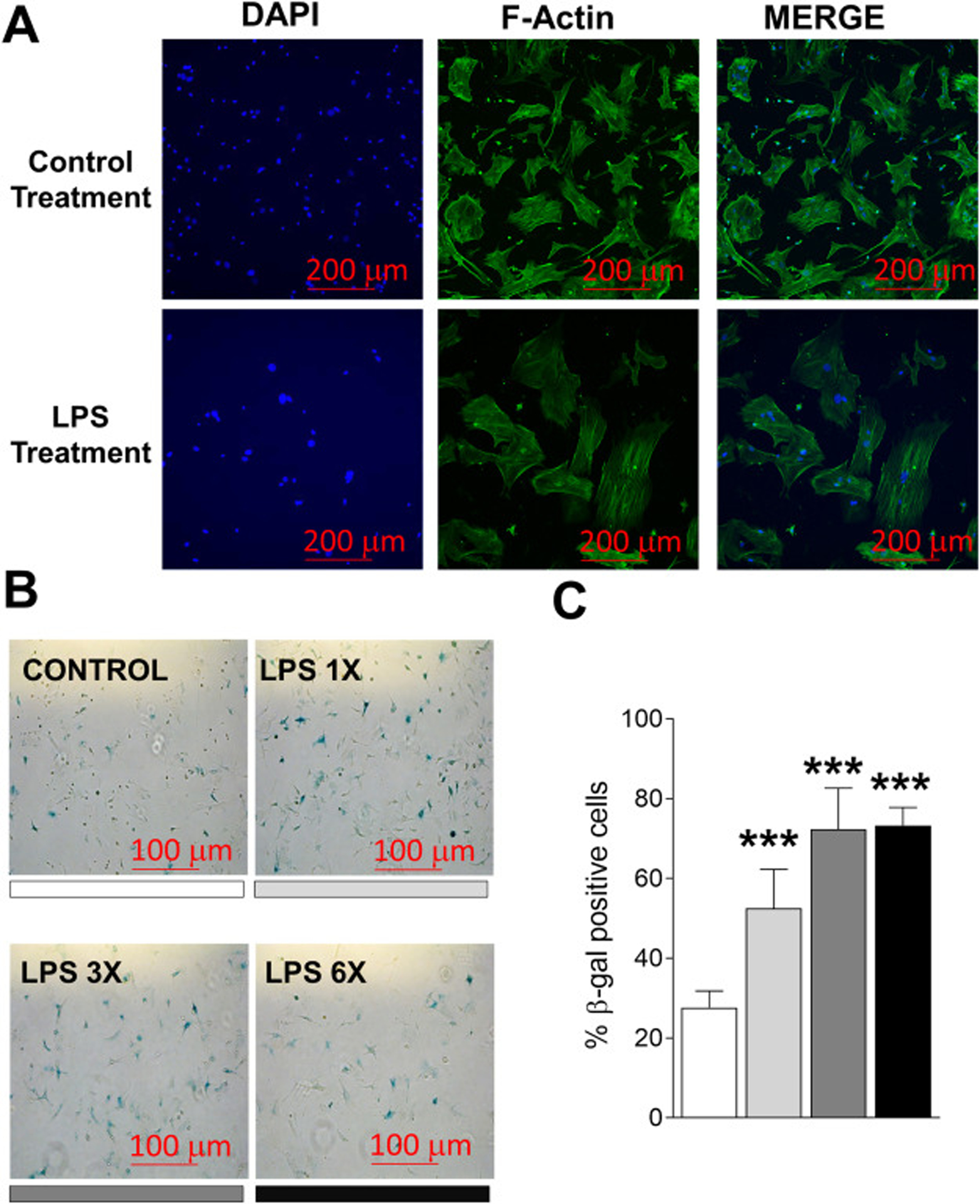
LPS treatment of alveolar bone osteocyte-like cells induces cytoskeletal alterations and the acquisition of senescence-associated SA-β-gal activity in vitro. (a) Alveolar bone osteocyte-enriched cells were prepared from 6 month-old WT female mice and treated with LPS and stained for F-Actin (green staining). (b-c) Alveolar bone osteocyte-enriched cells were cultured in supplemented α-MEM alone containing low serum (2% FBS) to reduce cell proliferation, or containing LPS (10 ng/ml). Cells were stimulated with LPS once, three times, and six times (for one day each treatment time; the shading under each image denotes the bar in panel c). The red bars denote the magnification scale (100 μm). Data represent Mean ± SEM (n=6). **P ≤ 0.001 relative to vehicle control.
4. Discussion
Inflammation promotes periodontitis-associated “inflammophilic” bacteria overgrowth by providing tissue breakdown products as nutrients, creating a positive feedback loop between pathogenic microbiota and inflammation [5]. Recently, it was reported that osteocytes play a crucial role in bacterial-induced alveolar bone destruction by expressing RANKL [38]. Whether this is caused by the acquisition of alveolar osteocyte senescence and is exacerbated by bacterial toxins such as LPS, are unknown and the subject of this report. Herein, we used multiple approaches to measure whether cellular senescence occurs alveolar ostecytes and whether LPS treatment induces indices of senesence. We provide evidence that indeed senescent alveolar osteocytes accumulate in young mice (6 months of age) and that LPS exacerbates the acquisition of various senescent markers in vitro and in an ex vivo model.
Senescent cells accumulate in aged tissues over time, and are frequently found at sites of age-related pathologies, such as in osteoporosis [23]. We previously reported that a higher number of senescent osteocytes accumulate in bone from older individuals compared with young controls [23]. In our current study, we found that a subset of senescent osteocytes prematurely accumulate in alveolar bone (30%), which is constantly exposed to bacterial infection and their genotoxins in adult young mice in vivo. Furthermore, LPS exposure upregulated p16ink4a, p21 and p53 mRNA expression, increased γH2AX levels and increased the percentage of SA-β-gal-positive osteocyte-like bone cells in vitro. The close proximity with periodontal microbial infection and produced toxins could explain the higher senescent osteocyte accumulation in alveolar bone in vivo, suggesting a bacterial-genotoxin induced premature senescence. Since premature senescence in young animals has also been observed in the context of inflammation associated with obesity [11, 12], we propose that a similar phenomenon is occurring in young alveolar bone, triggered by bacterial-derived LPS-induced genotoxicity, and constitutes a new paradigm in periodontal bone loss. This premise is reinforced when considering that pro-inflammatory cytokines and matrix-degrading enzymes secreted by senescent osteocytes not only damage their local microenvironment, but also induce “senescence-induced senescence” or bystander effects in neighboring cells [39]. Therefore, continuous exposure to SASP factors could also spread senescence toward progenitor cells, thereby promoting their premature exhaustion, limiting their proliferative potential, and leading to defective alveolar bone regeneration.
Cellular senescence is an age-associated process by which cells acquire phenotypic alterations, and contribute to alter their local microenvironment. Indeed, cellular senescence and SASP factors play a crucial role in the genesis and/or progression of age-related diseases [40]. Given that mice naturally develop periodontal disease starting around 9 months of age [41], our finding that 30% of dysfunctional senescent osteocytes are already present at 6 months appears to precede the onset of alveolar bone loss. However, further research is required to elucidate this hypothesis. On the other hand, we found that ex vivo alveolar bone culture exposed repeatedly to LPS exhibited significant higher p53, but not p16Ink4 or p21 expression. In agreement with this finding, it was recently reported that LPS induces the increased secretion of Il1α, Il6 and Tnfα through p53-dependent activation in human gingival fibroblasts [42]. Given that p16Ink4 positive cells may not always express SASP factors in vivo, but SASP is a damage response separable from growth arrest [43], we suggest that p53 activation by LPS may induce senescent osteocytes to secrete SASP factors as result of DNA damage.
The acquisition of the SASP turns senescent cells into pro-inflammatory cells that alter tissue structure and function [43]. Given that SASP factors recruit immune and inflammatory cells, senescent cell accumulation can also induce a chronic “non-microbial” inflammatory reaction. In our study, we found that Icam1, Il6, Il17 and Tnfα, key factors implicated in periodontitis, were highly expressed in osteocyte-enriched samples from alveolar bone. On the other hand, in agreement with the role of SASP factors in promoting tissue remodeling [9, 44], Mmp13 was consistently upregulated. This protease, as well as other metalloproteinases, is highly expressed in destructive periodontitis, promotes bone collagen matrix degradation, and plays a significant role in the onset of alveolar bone resorption [45, 46]. We speculate that SASP factors could contribute to change symbiotic into pathogenic bacteria by exacerbating inflammation, remodeling, and providing tissue breakdown products that fuel pathogenic bacterial overgrowth. Therefore, we suggest that the acquisition of the senescent phenotype could be both a mechanism of self-clearance (osteocytes with DNA damage) and a mechanism of protection against bacterial invasion by promoting bone resorption through the SASP. However, the resulting collagen breakdown products and other tissue debris could be used by “inflammophilic” bacteria as source of nutrients [5], resulting in a positive feedback loop between pathogenic bacteria and senescent osteocytes over time (Fig. 6). Although outside the scope of this study, this hypothesis warrants future examination.
Fig. 6.
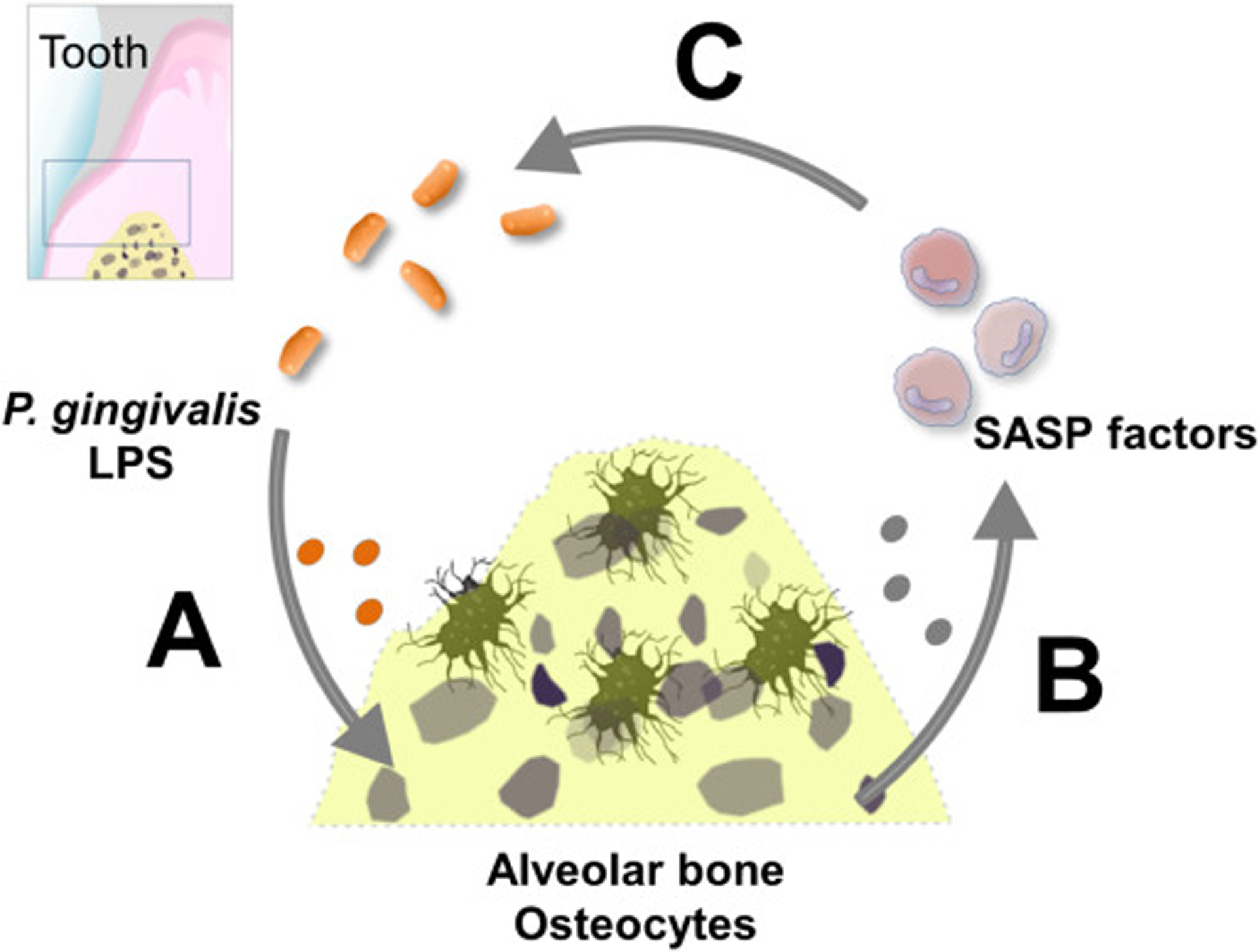
Suggested model of the potential role of LPS induced osteocyte senescence in alveolar bone. (a) Anatomical proximity between periodontal bacterial infection and alveolar bone predisposes osteocytes to undergo LPS induced genotoxic stress, promoting premature senescence over time. (b) Senescent osteocytes release SASP factors to recruit host immune and inflammatory cells to promote tissue remodeling, including its own clearance. However, senescent osteocyte accumulation might exacerbate local inflammation and increase tissue breakdown products release, which fuels dysbiotic bacteria overgrowth. (c) A positive feedback loop between pathogenic bacteria and the senescent osteocytes might be established.
The main strength of this study is that it provides the first evidence of senescent cell accumulation in alveolar bone in young adult mice (6 months of age) and suggests that bacterially-derived LPS plays a role not only in the acquisition of “premature” cellular senescence (e.g. in young adults), but also may contribute to the production of a SASP that causes localized inflammation and ultimately bone loss, a hallmark of periodontal disease. One the main limitations of this study are the somewhat discordant data with LPS treatment in either isolated alveolar osteocytes (in vitro) where increased expression of p16Ink4a, p21 and p53 was observed (Fig. 3a), whereas in alveolar bone blocks (ex vivo) only increased p53 expression was observed (Fig. 3d). This could be due to the system and length of time of LPS exposure, as both were treated for 6 days. Longer exposure of the alveolar blocks to LPS may uncover further regulation of senescence-associated gene expression. However, further ex vivo and in vivo experiments would be needed to clarify which senescence pathway is activated in alveolar bone. In any case, our data still indicates that senescence is increased, which is a novel observation.
In conclusion, our results provide evidence that senescent osteocytes accumulate in a site-specific manner in alveolar bone during young adulthood. They also indicate that proximity to periodontal bacterial infection may predispose alveolar bone osteocytes to undergo higher LPS-induced DNA damage. Premature osteocyte senescence induced by persistent LPS exposure may represent a potential novel pathogenic mechanism by which gram-negative oral bacteria promote inflammatory alveolar bone destruction. Given that osteocytes orchestrate bone remodeling, a higher number of dysfunctional senescent osteocytes in alveolar bone could jeopardize tissue homeostasis. Thus, targeting senescent cells could represent a novel approach to delay alveolar bone loss.
Highlights.
Premature osteocyte senescence is a host response to bacterial infection and a novel mechanism implicated in the pathogenesis of alveolar bone loss.
Osteocytes orchestrate bone remodeling and higher number of dysfunctional senescent osteocytes in alveolar bone could jeopardize tissue homeostasis.
Long-lived osteocytes are perhaps more susceptible to undergoing LPS-induced genotoxic stress because of their anatomical proximity to periodontal bacterial infection.
Senescent-osteocytes secreted factors represent a “non-microbial” source of pro-inflammatory factors that may exacerbate bacterial inflammation.
Premature osteocyte accumulation could be a novel mechanism implicated in the pathogenesis of juvenile and other types of early-onset periodontitis.
Acknowledgements
This work was supported by NIH Grants R01 AR068275 (DGM), P01 AG004875 (SK/DGM), P01 AG062413 (SK, JNF, DGM), R01 AG048792 (SK/DGM) and K01 AR070241 (JNF). The authors declare no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arigbede AO, Babatope BO, Bamidele MK, Periodontitis and systemic diseases: A literature review, J Indian Soc Periodontol 16 (2012) 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loesche WJ, Grossman NS, Periodontal disease as a specific, albeit chronic, infection: diagnosis and treatment, Clin Microbiol Rev 14 (2001) 727–752, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berezow AB, Darveau RP, Microbial shift and periodontitis, Periodontol 2000 55 (2011) 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajishengallis G, Periodontitis: from microbial immune subversion to systemic inflammation, Nat Rev Immunol 15 (2015) 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajishengallis G, The inflammophilic character of the periodontitis-associated microbiota, Mol Oral Microbiol 29 (2014) 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campisi J, Aging, cellular senescence, and cancer, Annu Rev Physiol 75 (2013) 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blazkova H, Krejcikova K, Moudry P, Frisan T, Hodny Z, Bartek J, Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling, J Cell Mol Med 14 (2010) 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coppe JP, Desprez PY, Krtolica A, Campisi J, The senescence-associated secretory phenotype: the dark side of tumor suppression, Annu Rev Pathol 5 (2010) 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serrano M, Senescence helps regeneration, Dev Cell 31 (2014) 671–672. [DOI] [PubMed] [Google Scholar]
- 10.McHugh D, Gil J, Senescence and aging: Causes, consequences, and therapeutic avenues, J Cell Biol 217 (2018) 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I, A crucial role for adipose tissue p53 in the regulation of insulin resistance, Nat Med 15 (2009) 1082–1087. [DOI] [PubMed] [Google Scholar]
- 12.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL, Fat tissue, aging, and cellular senescence, Aging Cell 9 (2010) 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng X, Feng G, Xing J, Shen B, Tan W, Huang D, Lu X, Tao T, Zhang J, Li L, Gu Z, Repeated lipopolysaccharide stimulation promotes cellular senescence in human dental pulp stem cells (DPSCs), Cell Tissue Res 356 (2014) 369–380. [DOI] [PubMed] [Google Scholar]
- 14.How KY, Song KP, Chan KG, Porphyromonas gingivalis: An Overview of Periodontopathic Pathogen below the Gum Line, Front Microbiol 7 (2016) 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graves D, Cytokines that promote periodontal tissue destruction, J Periodontol 79 (2008) 1585–1591. [DOI] [PubMed] [Google Scholar]
- 16.Guerra L, Guidi R, Frisan T, Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression?, FEBS J 278 (2011) 4577–4588. [DOI] [PubMed] [Google Scholar]
- 17.Yu HM, Zhao YM, Luo XG, Feng Y, Ren Y, Shang H, He ZY, Luo XM, Chen SD, Wang XY, Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells, Neuroimmunomodulation 19 (2012) 131–136. [DOI] [PubMed] [Google Scholar]
- 18.Kim CO, Huh AJ, Han SH, Kim JM, Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells, Arch Gerontol Geriatr 54 (2012) e35–41. [DOI] [PubMed] [Google Scholar]
- 19.Bonewald LF, The amazing osteocyte, J Bone Miner Res 26 (2011) 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farr JN, Khosla S, Cellular senescence in bone, Bone 121 (2019) 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stern AR, Stern MM, Van Dyke ME, Jahn K, Prideaux M, Bonewald LF, Isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice, Biotechniques 52 (2012) 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klingenberg CP, Leamy LJ, Quantitative genetics of geometric shape in the mouse mandible, Evolution 55 (2001) 2342–2352. [DOI] [PubMed] [Google Scholar]
- 23.Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, Drake MT, Tchkonia T, LeBrasseur NK, Kirkland JL, Bonewald LF, Pignolo RJ, Monroe DG, Khosla S, Identification of Senescent Cells in the Bone Microenvironment, J Bone Miner Res 31 (2016) 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swanson EC, Manning B, Zhang H, Lawrence JB, Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence, J Cell Biol 203 (2013) 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Deeb Zakhary I, Wenger K, Elsalanty M, Cray J, Sharawy M, Messer R, Characterization of primary osteocyte-like cells from rat mandibles, Oral Surg Oral Med Oral Pathol Oral Radiol 123 (2017) 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aquino-Martinez R, Farr JN, Weivoda MM, Negley BA, Onken JL, Thicke BS, Fulcer MM, Fraser DG, van Wijnen AJ, Khosla S, Monroe DG, miR-219a-5p Regulates Rorbeta During Osteoblast Differentiation and in Age-related Bone Loss, J Bone Miner Res 34 (2019) 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noren Hooten N, Evans MK, Techniques to Induce and Quantify Cellular Senescence, J Vis Exp (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waaijer MEC, Gunn DA, van Heemst D, Slagboom PE, Sedivy JM, Dirks RW, Tanke HJ, Westendorp RGJ, Maier AB, Do senescence markers correlate in vitro and in situ within individual human donors?, Aging (Albany NY) 10 (2018) 278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robles SJ, Adami GR, Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts, Oncogene 16 (1998) 1113–1123. [DOI] [PubMed] [Google Scholar]
- 30.Zhao M, Chen X, Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors, Am J Physiol Endocrinol Metab 309 (2015) E334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma A, Singh K, Almasan A, Histone H2AX phosphorylation: a marker for DNA damage, Methods Mol Biol 920 (2012) 613–626. [DOI] [PubMed] [Google Scholar]
- 32.Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S, Targeting cellular senescence prevents age-related bone loss in mice, Nat Med 23 (2017) 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abubakar AA, Noordin MM, Azmi TI, Kaka U, Loqman MY, The use of rats and mice as animal models in ex vivo bone growth and development studies, Bone Joint Res 5 (2016) 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sloan AJ, Taylor SY, Smith EL, Roberts JL, Chen L, Wei XQ, Waddington RJ, A novel ex vivo culture model for inflammatory bone destruction, J Dent Res 92 (2013) 728–734. [DOI] [PubMed] [Google Scholar]
- 35.Smith EL, Locke M, Waddington RJ, Sloan AJ, An ex vivo rodent mandible culture model for bone repair, Tissue Eng Part C Methods 16 (2010) 1287–1296. [DOI] [PubMed] [Google Scholar]
- 36.Chen QM, Tu VC, Catania J, Burton M, Toussaint O, Dilley T, Involvement of Rb family proteins, focal adhesion proteins and protein synthesis in senescent morphogenesis induced by hydrogen peroxide, J Cell Sci 113 (Pt 22) (2000) 4087–4097. [DOI] [PubMed] [Google Scholar]
- 37.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. , A biomarker that identifies senescent human cells in culture and in aging skin in vivo, Proc Natl Acad Sci U S A 92 (1995) 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graves DT, Alshabab A, Albiero ML, Mattos M, Correa JD, Chen S, Yang Y, Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL, J Clin Periodontol 45 (2018) 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T, A senescent cell bystander effect: senescence-induced senescence, Aging Cell 11 (2012) 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LeBrasseur NK, Tchkonia T, Kirkland JL, Cellular Senescence and the Biology of Aging, Disease, and Frailty, Nestle Nutr Inst Workshop Ser 83 (2015) 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oz HS, Puleo DA, Animal models for periodontal disease, J Biomed Biotechnol 2011 (2011) 754857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, Zeng J, Wang X, Zheng M, Luan Q, P53 mediates lipopolysaccharide-induced inflammation in human gingival fibroblasts, J Periodontol 89 (2018) 1142–1151. [DOI] [PubMed] [Google Scholar]
- 43.Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J, Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype, J Biol Chem 286 (2011) 36396–36403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munoz-Espin D, Serrano M, Cellular senescence: from physiology to pathology, Nat Rev Mol Cell Biol 15 (2014) 482–496. [DOI] [PubMed] [Google Scholar]
- 45.Hernandez M, Valenzuela MA, Lopez-Otin C, Alvarez J, Lopez JM, Vernal R, Gamonal J, Matrix metalloproteinase-13 is highly expressed in destructive periodontal disease activity, J Periodontol 77 (2006) 1863–1870. [DOI] [PubMed] [Google Scholar]
- 46.Franco C, Patricia HR, Timo S, Claudia B, Marcela H, Matrix Metalloproteinases as Regulators of Periodontal Inflammation, Int J Mol Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]