

SUMMARY
Regulatory T cells (Tregs) can impair anti-tumor immune responses and are associated with poor prognosis in multiple cancer types. Tregs in human tumors span diverse transcriptional states distinct from those of peripheral Tregs, but their contribution to tumor development remains unknown. Here, we use single-cell RNA sequencing (RNA-seq) to longitudinally profile dynamic shifts in the distribution of Tregs in a genetically engineered mouse model of lung adenocarcinoma. In this model, interferon-responsive Tregs are more prevalent early in tumor development, whereas a specialized effector phenotype characterized by enhanced expression of the interleukin-33 receptor ST2 is predominant in advanced disease. Treg-specific deletion of ST2 alters the evolution of effector Treg diversity, increases infiltration of CD8+ T cells into tumors, and decreases tumor burden. Our study shows that ST2 plays a critical role in Treg-mediated immunosuppression in cancer, highlighting potential paths for therapeutic intervention.
Graphical Abstract
In Brief
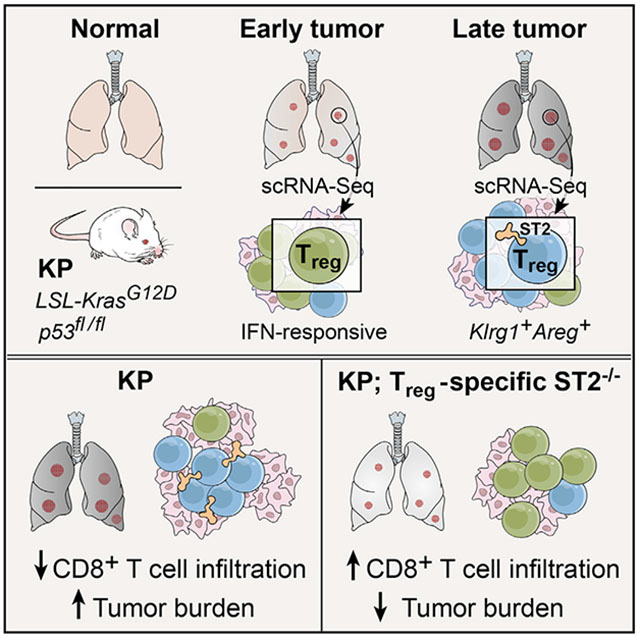
Li et al. show in a genetic mouse model of lung adenocarcinoma that during tumor development regulatory T cell (Treg) diversity shifts from an interferon-responsive to a ST2-positive, Klrg1+Areg+ effector-like phenotype. Treg-specific deletion of ST2 alters Treg heterogeneity, increases tumor infiltration by CD8+ T cells, and decreases tumor burden.
INTRODUCTION
The clinical success of immune checkpoint inhibitors in the treatment of non-small cell lung cancer (NSCLC) highlights how targeting immunosuppression in the tumor microenvironment can be an effective therapeutic strategy (Makkouk and Weiner, 2015; Soria et al., 2015). However, only some patients respond to immune therapies, suggesting that an improved understanding of other immunosuppressive mechanisms is needed for effective treatment.
One major mechanism of immunosuppression is posed by CD4+ regulatory T cells (Tregs), which can impair anti-tumor immune responses (Tanaka and Sakaguchi, 2017). Tregs are critical for maintaining immune tolerance and preventing autoimmunity (Josefowicz et al., 2012). Tregs are associated with poor prognosis in several cancers, including lung adenocarcinoma (Shang et al., 2015; Suzuki et al., 2013). In mouse models, Treg depletion can enhance anti-tumor immunity (Bos et al., 2013; Joshi et al., 2015; Marabelle et al., 2013), and antibodies directed against CTLA-4 act in part by depleting Tregs (Simpson et al., 2013).
Due to their phenotypic diversity, Tregs differentially impact tumor immune responses, such that effector Tregs promote tumor growth (Green et al., 2017), whereas poorly immunosuppressive Tregs contribute to anti-tumor immunity (Overacre-Delgoffe et al., 2017; Saito et al., 2016). This functional diversity may be reflected in their transcriptional programs. Tregs in distinct tissues and inflammatory contexts have transcriptional profiles related to their tissue-resident functions (Panduro et al., 2016). In human tumors, Tregs have a program that may be shared across cancer types and is associated with clinical outcome (De Simone et al., 2016; Magnuson et al., 2018; Plitas et al., 2016). Characterization of pro-tumorigenic Treg subsets may guide efforts to target these populations.
Inducible, autochthonous models of cancer are ideal for studying mechanisms of tumor tolerance because they recapitulate the longitudinal development and features of the endogenous tumor microenvironment better than transplanted, more “foreign” tumors (Dranoff, 2011). Our group has previously developed a model of lung adenocarcinoma in which activation of oncogenic K-rasG12D and loss of Trp53 are driven by intratracheal delivery of a lentivirus expressing Cre recombinase (KP: LSL-KrasG12D, p53fl/fl) (DuPage et al., 2009; Jackson et al., 2005). Using a lentivirus that also expresses known T cell antigens (LucOS: luciferase fused to chicken ovalbumin [Ova] and the peptide SIYRYYGL), we can monitor tumor-specific T cell responses (DuPage et al., 2011). T cell infiltration of these tumors delays tumor growth, but the number and activity of anti-tumor CD8+ T cells decline over time, and the development of immune tolerance is partly due to the expansion of lung Tregs (Joshi et al., 2015). Treg depletion results in T cell infiltration of tumors, suggesting that Tregs actively suppress anti-tumor immune responses. Because Treg-depleted animals succumb to systemic autoimmunity, a strategy targeting features of lung tumor-specific Tregs is required to minimize self-directed cytotoxicity.
Here, we map the diversity of conventional CD4+ T cells (Tconvs) and Tregs throughout tumor development in the KP model using single-cell RNA sequencing (scRNA-seq). Whereas Tconv subsets were stable overtime, Treg diversity changed with tumor progression. At early time points, Tregs expressed genes associated with interferon (IFN) signaling, whereas mice with advanced disease had more killer cell lectin-like receptor 1 (Klrg1)+ and amphiregulin (Areg)+ Tregs. Analyzing these data, we identified ST2 as a potential mediator of effector Treg phenotypes during tumor development. Treg-specific ablation of ST2 altered longitudinal patterns of Treg diversity, increased CD8+ T cell infiltration of tumors, and reduced tumor size. Our high-resolution characterization of Treg diversity in the tumor microenvironment thus allows us to refine ways to target Treg function in cancer.
RESULTS
scRNA-Seq Reveals Lung-Specific Transcriptional Programs for Tumor-Associated CD4+ Tconvs and Tregs
Consistent with prior reports that lung Tregs expand during KP tumor development (Joshi et al., 2015), the fraction of Ki-67+ Tregs by flow cytometry was elevated in lungs with early tumors (Figure 1A), whereas the fraction of Ki-67+ Tconvs was modestly increased at 5 and 8 weeks but returned to baseline by 12 weeks (Figure S1A).
Figure 1. scRNA-Seq Reveals Distinctive Lung CD4+ T Cell Signatures and Overlapping Tconv and Treg Diversity.

(A) Treg proliferation peaks early in tumor development. Percent Ki-67+ Tregs throughout KP tumor development from two to three experiments (dot: one mouse). Error bars: SEM. ***p < 0.001, Tukey’s multiple comparisons test. NS, non-significant.
(B) Experiment overview. KP, Foxp3GFP mice were harvested at the indicated weeks after tumor induction with Lenti-LucOS. 1,254 Tconvs and 1,679 Tregs from lung and msLNs were profiled by plate-based scRNA-seq.
(C) Lung-specific gene expression programs include genes shared by, and unique to, Tconvs and Tregs. Genes (rows, row-normalized) differentially expressed (STAR Methods) between cells from lung versus msLNs for Tregs and Tconvs (columns). Left black bars indicate significantly differentially expressed Treg and Tconv genes. Bottom: cell expression scores for corresponding lung and LN signatures. Color indicates cell type and tissue of origin.
(D) Lung cells show particular diversity. Diffusion component (DC) embedding of all cells (dots), colored by cell type and tissue of origin (top left), or Z score of the lung (bottom left) or msLN (bottom right) programs. Top right: distribution of DC scores.
(E) Lung Tregs and Tconvs have highly correlated programs. Spearman’s correlation coefficient (color bar) of Tconv expression Z scores for Tconv programs (columns) and Treg programs (rows) (STAR Methods).
We hypothesized that this early proliferation of Tregs may be associated with changes in Treg diversity. We used scRNA-seq to characterize heterogeneity in tumor-associated CD4+ T cells over time and the relationship between Treg and Tconv diversity. We profiled by full-length scRNA-seq 1,254 Tconvs and 1,679 Tregs from the lungs and mediastinal lymph nodes (msLNs) of non-tumor-bearing and tumor-bearing KP, Foxp3GFP mice along a time course after tumor induction (Figure 1B).
Tissue-specific programs included both genes shared by lung Tconvs and Tregs and genes uniquely upregulated in each (Figure 1C; Table S1). Lung Tregs expressed high levels of Il1rl1, Cxcr4, Areg, and Klrg1 compared with msLN Tregs, whereas Tconvs expressed Cd44, Ccr4, and Itgb1 (Figure 1C). Gene programs associated with a recently described transcriptional trajectory of tissue-resident Tregs (Miragaia et al., 2019) were consistent with those highlighted by our scRNA-seq profiles of lung cells (Figure S1B). msLN Tregs and Tconvs expressed genes associated with a naive or central memory phenotype, including Lef1, Sell, and Ccr7 (Figures 1C and S1C), whereas lung cells were more activated (Figure 1C). Subsets of lung Tconvs and Tregs that scored high for the msLN signature also expressed genes associated with T cell receptor (TCR) signaling, including Nr4a1 and Junb, suggesting that they may be recently activated (Figure S1C).
Both lung and msLN cells spanned a spectrum of cell states, with lung cells showing higher diversity. This was apparent when lung and msLN signature genes were used to create a diffusion map (Figures 1D and S1D; STAR Methods; Haghverdi et al., 2015).
Lung Tconv and Treg Subsets Share a Limited Number of Expression Programs, Including a Th17-like Phenotype
To assess the different transcriptional programs of Tconv and Treg subsets in the lung, we performed PAthway and Gene set Over-Dispersion Analysis (PAGODA) (Fan et al., 2016) to identify groups of genes with co-varying expression (STAR Methods; Figures S1E and S1F; Table S2). The relative proportions of cells expressing markers of different Tconv programs remained stable during tumor development (Figure S1G). Tconv and Treg subsets expressed several overlapping programs, including programs associated with naive/resting T cells and IFN signaling (Figure 1E).
Of the Tconv programs associated with effector CD4+ T cell subsets, only the Th17 program was correlated with a Treg program (program 13; Figure 1E). Program 13 marks Tregs that express Rorc and Il17a (Figure S1H), reminiscent of Th17-like effector Tregs (Tr17), which are thought to inhibit Th17 responses (Kim et al., 2017). By flow cytometry, RORγt+ Tregs comprise ~10% of lung Tregs throughout tumor development (Figure S1l). Expression of program 13 and lung Treg signature genes was inversely correlated (Figures S1J and S1K), suggesting that Tr17-like cells represent a distinct state.
Remarkably, TCR clonotypes shared between Tregs and Tconvs were predominantly Tr17-like and Th17-like cells, respectively. Twelve TCR clonotypes were shared across Tregs and Tconvs (Table S3; STAR Methods). Of the 19 Tregs and 20 Tconvs belonging to these shared TCR clonotypes, 13 Tregs were Tr17-like (Figures S1L and S1M). Due to the small number of identified clonotypic families, no temporal trend could be reliably detected. Overall, this suggests that Tr17 differentiation may reflect a shared clonal origin with Th17 cells.
A Klrg1+Areg* Effector-like Treg Program Becomes Predominant during Tumor Development
In contrast with Tr17-like cells, which represented a fixed proportion of lung Tregs during tumor development, other Treg programs changed in prominence over time (Figure 2A). After 8 weeks, there was decreased expression of programs 1, 3, 8, and 9, which marked cycling cells (Figure 2A), corresponding to the decline in Ki67+ Tregs (Figure 1A). Two other programs also changed over time, reflecting an IFN response (programs 6 and 23; Figures 2A–2C; Figure S2A) and a Klrg1+Areg+ (KA) effector-like program (programs 12 and 21; Figures 2A–2C; Figures S2A and S2B).
Figure 2. A Klrg1+Areg+ Treg Phenotype Becomes Dominant during Tumor Development.

(A) Changes in prominence of cycling, IFN-stimulated, and Treg effector-like programs with tumor development. Linear regression analysis of program expression Z scores as a function of time since tumor initiation. Dot plot shows for each program (row) and time point (column) the coefficient of the time point covariate (color scale) with non-tumor-bearing lung as reference and the percentage of cells with Z score > 1.5 (dot size).
(B and C) An IFN and a Klrg1+Areg+ effector-like program peak early and late in tumor development, respectively. Two-dimensional force-directed layout embedding of lung Tregs colored by normalized program Z score for the KA_TR program (B, top, programs 12 and 21), IFNstim_TR program (B, bottom, programs 6 and 23), and time point (C).
(D) Percentage of Tregs expressing the indicated protein (y axis) throughout KP tumor development (x axis) from two to three experiments (dot: one mouse). Error bars: SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001, Tukey’s multiple comparisons test.
The IFN-responsive Treg program (“IFNstim_TR”) included many IFN-stimulated genes (ISGs) downstream of either type I or II IFN signaling. Twenty-eight genes from the IFNstim_TR program were significantly downregulated in Tregs during tumor progression (Figure S2C; STAR Methods). IFNγ promotes a T-bet+CXCR3+ Treg population that can suppress Th1 responses (Hall et al., 2012; Koch et al., 2009, 2012). Neither Cxcr3 nor Tbx21 are IFNstim_TR genes, but IFNstim_TR expression was correlated with Tbx21 expression (Figure S2D). Moreover, cells scoring highly for the IFNstim_TR program also scored highly for a lymphoid tissue Treg program (Figure S2E), and msLN Tregs had higher expression of IFNstim_TR genes compared with lung Tregs at 12 and 20 weeks post induction (p.i.) (Figure S2F). Taken together, Tregs expressing the IFN-responsive program (“IR Tregs”) were most prevalent early in tumor development and in msLNs, and may thus have recently arrived to the lung.
Meanwhile, the Klrg1+Areg+ effector-like Treg program (“KA_TR”) included genes upregulated in Tregs from mouse non-lymphoid tissues and human cancers (Figure S2E; STAR Methods). Tregs expressing the KA_TR program (“KA Tregs”) expressed Ccr6, but not Cxcr3, representing a population distinct from IR Tregs (Figure S2G). Klrg1 and Areg expression have been associated with Treg differentiation and tissue repair, respectively (Arpaia et al., 2015; Burzyn et al., 2013; Cheng et al., 2012). 40% of lung Tregs from KP mice with advanced disease have been shown to be CD103+KLRG1+ (double-positive [DP]) (Joshi et al., 2015). The KA_TR program was enriched for genes upregulated in DP Tregs (Figures S2H and S2I; STAR Methods), including genes associated with T cell activation and putative Treg effector functions (e.g., Nr4a1, Cd69, Il1rl1, Areg, Srgn, and Fgl2). KA and DP Tregs are highly similar and are likely representative of a KLRG1+ effector Treg population.
The IR and KA Treg programs represented distinct Treg phenotypes within each time point and followed opposite temporal patterns: expression of IFNstim_TR genes was highest in cells from week 5 and declined thereafter, whereas expression of KA_TR genes increased and remained elevated (Figures 2A–2C). This trend was reflected by individual genes: Cxcr3 expression decreased, whereas Pdcd1 and Lilrb4 expression increased during tumor development (Figure S2J). More generally, KA_TR genes were upregulated in DP Tregs, whereas Cxcr3 and IFNstim_TR genes were significantly downregulated (Figures S2H and S2K). Indeed, CXCR3 protein levels decreased, and proteins encoded by KA_TR genes, including CD85k, CD69, CXCR6, PD-1, and ST2, increased during tumor progression (Figure 2D). Taken together, our data suggest that tumor progression may be associated with a shift from the IR to KA Treg programs. We hypothesize that the immunosuppression associated with late-stage tumors may be because of the prevalence of KA Tregs.
ST2 May Promote the KA Treg Phenotype in Mice Bearing Advanced Lung Tumors
Il1rl1, a KA_TR gene that encodes the interleukin-33 (IL-33) receptor ST2, marked a heterogeneous Treg population that had higher expression of KA_TR genes. ST2 was most highly expressed in DP lung Tregs (Figure 3A), consistent with prior data that ST2 marks a tissue Treg program that expresses KLRG1 and GATA3 (Delacher et al., 2017). Il1rl1+ and Il1rl1− Tregs both spanned a full spectrum of cell states (Figure S3A) and had similar transcriptional diversity (Figure S3B; STAR Methods). Nevertheless, Il1rl1+ Tregs had higher expression of KA_TR and DP genes and lower expression of Th17-like and resting Treg genes (Figures 3B, S3C, and S3D). Il1rl1+ Tregs also had lower expression of IFNstim_TR genes compared with Il1rl1− Tregs in non-tumor-bearing lungs (Figure S3C). Il1rl1+ and Il1rl1− Tregs had similar expression of cell-cycle genes and Ki-67 (Figures S3C and S3E), suggesting that proliferation does not account for the observed differences in phenotype. Genes differentially expressed between Il1rl1+ and Il1rl1− Tregs from human colon cancer were also enriched for KA_TR genes (Figure 3C; STAR Methods). ST2 signaling may thus be a conserved pathway in human and mouse Tregs that promotes the KA/DP Treg phenotype and/or the proliferation of KA/DP Tregs. Consistent with the presence of ST2 signaling throughout tumor development, IL-33, the only known ligand of ST2, was highly expressed in normal lung and in early and late KP tumors (Figure 3D). IL-33 was predominantly expressed on surfactant protein C (SPC+) type II epithelial (AT2) cells in normal lung (Figure S3F), and AT2 and mesenchymal cells in tumor-bearing lungs (Figure S3G), consistent with prior reports (Treutlein et al., 2014).
Figure 3. ST2 Marks a Diverse Population of KA/DP Tregs in Lung Tumor-Bearing Mice.

(A) ST2 is most highly expressed in DP lung Tregs. Representative distributions of ST2 expression on CD103−KLRG1− (DN, gray), CD103+KLRG1− (SP, blue), and CD103+KLRG1+ (DP, red) Tregs isolated from tumor-bearing lungs.
(B) KA_TR genes are upregulated in Il1rl1+ Tregs throughout tumor development. Empirical cumulative distribution functions (ECDFs) of the scores of programs 12 (top) and 21 (bottom) of Il1rl1+ (blue) versus Il1rl1− Tregs (gray) by time point after tumor induction.
(C) Il1rl1+ Tregs in human colon cancer have higher expression of KA_TR genes. Overlap of genes upregulated in I11rl1+ Tregs in human colon cancer (blue) and programs 12 (top, p = 1.5 × 10−5) and 21 (bottom, p = 5.3 × 10−6) genes (STAR Methods). p values: hypergeometric test.
(D) IL-33 is highly expressed in lung adenocarcinoma. Immunohistochemistry (IHC) staining of tumor-bearing lungs from KP mice at weeks 13 and 22 p.i. with Lenti-LucOS. Two representative images are shown per time point. Scale bar: 20μm.
(E) Lung Tregs are enriched for ST2+ cells in late-stage tumors. Percent ST2+ among lung and msLN Tregs and Tconvs from tumor-bearing KP mice at week 20 p.i. as measured by flow cytometry. Error bars: SEM. ****p < 0.0001, *p < 0.05, Tukey’s multiple comparisons test.
ST2 protein was preferentially expressed by lung Tregs late in tumor development. ST2 levels on lung Tregs increased with time (Figure 2D), and ST2 was expressed primarily by Tregs, with lower expression in CD8+ T cells and Tconvs (Figures 3E and S3H). We hypothesized that the expansion of ST2+ Tregs may drive the increase in KA/DP Tregs during lung tumor development.
Treg-Specific ST2 Is Required for the Increase in DP Tregs during Tumor Progression
To test whether ST2 signaling was necessary to develop the KA/DP Treg response, we studied the effects of Treg-specific Il1rl1 deletion. We used a modified KP model, where FlpO recombinase induces expression of oncogenic K-ras and loss of p53 (KPfrt: FSF-KrasG12D, p53frt/frt) (Lee et al., 2012), allowing us to use the Cre-lox system to delete Il1rl1 in Tregs. We crossed KPfrt and Foxp3YFP-Cre, Il1rl1fl/fl mice to model lung adenocarcinoma in the setting of Treg-specific ST2 deficiency (Figure 4A). We infected the mice with a lentivirus expressing FlpO recombinase and GFP fused to Ova and SIYRGYYL (FlpO-GFP-OS) in order to induce tumors that would express the same T cell antigens as those in the LucOS model. Confirming Treg-specific recombination of the Il1rl1 locus, ST2 expression was unchanged in CD8+ T cells and Tconvs (Figure S4A).
Figure 4. Treg-Specific ST2 Ablation Alters Treg Diversity and Enhances CD8+ T Cell Infiltration of Tumors.

(A) Experiment overview. KPF and KPF-ST2FL mice were infected with FlpO-GFP-OS.
(B, C, and E) Flow cytometric analyses of KPF-ST2FL and KPF mice at 24–25 weeks p.i. All data are from two to three experiments; n = 3–5 mice per group. Error bars: SEM.
(B) Percent Tregs (left) and Tconvs (right) of CD4+ lung cells. *p < 0.05, two-tailed Student’s t test.
(C) Percent CD103−KLRG1+ (gray), DN (black), SP (blue), and DP (red) of Tregs. ****p < 0.0001, *p < 0.05, Sidak’s multiple comparisons test.
(D) Bulk RNA-seq identifies expression signature distinguishing KPF from KPF-ST2FL Tregs from tumor-bearing mice. Row-normalized expression (Z score) of select signature genes (rows, STAR Methods) across Treg populations (columns, colored as in C).
(E) Percent CXCR3+CCR6− (left) and CXCR3−CCR6+ (right) of Tregs. **p < 0.01, two-tailed Student’s t test.
(F) Increased CD8+T cell infiltration in KPF-ST2FL mice. CD8+ cells per tumor area (left) and CD8/Treg ratio (right) in pooled tumors from KPF-ST2FL and KPF mice as measured by immunohistochemistry (IHC) staining of cross sections of tumor-bearing lungs. Error bars: SEM. **p < 0.01, ****p < 0.0001, Mann-Whitney test.
(G) Reduced tumor burden in KPF-ST2FL mice. Percent of total lung occupied by tumor (left, p = 0.0315) and average tumor size (right, p = 0.0106) in KPF-ST2FL and KPF mice. Error bars: SEM. Mann-Whitney test was used.
NS, non-significant.
Early-stage KPfrt, Foxp3YFP-Cre, Il1rl1fl/fl (KPF-ST2FL) and KPfrt, Foxp3YFP-Cre (KPF) mice had similar fractions of Tconvs and Tregs (Figure S4B), but late in tumor progression KPF-ST2FL mice had a lower proportion of Tregs, of which fewer were DP Tregs (Figures 4B and 4C). Notably, DP Tregs from KPF and KPF-ST2FL mice had similar Ki-67 expression, suggesting that the decreased fraction of DP Tregs in KPF-ST2FL mice was not due to impaired proliferation (Figure S4C). msLNs and splenic Tregs did not have fewer DP Tregs (Figure S4D). Proportions of Th1, Th17, CD8+ T cells, tumor antigen-specific CD8+ T cells, and alveolar macrophages were also comparable among KPF-ST2FL mice and controls (Figures S4E-S4H).
Bulk RNA-seq of DP, single-positive (SP), and double-negative (DN) Tregs from KPF-ST2FL and KPF mice identified an expression signature lower in KPF-ST2FL versus KPF Tregs and highest among KPF DP Tregs (Figures 4D and S4I). The signature was enriched for DP signature genes, including Dgat2, Furin, and Nfkbia, genes preferentially expressed in Il1rl1+ Tregs (p = 1.2 × 10−13, hypergeometric test) and genes upregulated by Tregs in human NSCLC (Figures S4J and S4K). KPF-ST2FL Tregs also showed higher expression of some genes, including Itgb1, Il10, Klf6, and Fos (Figure 4D), suggesting that they may adopt alternative phenotypes.
We hypothesized that KPF-ST2FL mice may have altered proportions of Tr17-like and CXCR3+ Tregs. Indeed, CXCR3+CCR6− Tregs were increased, whereas CXCR3−CCR6+ Tregs were decreased, in KPF-ST2FL mice compared with KPF mice (Figures 4E and S4L). However, RORγt expression was unchanged (Figure S4M), suggesting that a CCR6+ Treg population exclusive of Tr17-like cells decreases in KPF-ST2FL mice. Earlier in tumor development, CXCR3+ Tregs from KPF-ST2FL mice also had increased fluorescence intensity of CXCR3 (Figure S4N). Taken together, our data support the hypothesis that ST2 regulates Treg diversity over time by promoting the KA/DP Tregs over alternate phenotypes.
Treg-Specific ST2 Ablation Leads to Increased CD8+ T Cell Infiltration and a Reduction in Tumor Burden
Tumors from KPF-ST2FL mice had >50% higher CD8+ T cell infiltration than tumors from KPF mice, resulting in higher CD8/Treg ratios (Figure 4F). KPF-ST2FL mice also had a lower tumor burden and smaller tumors compared with controls (Figures 4G and S4O), suggesting that greater CD8+ T cell infiltration of tumors may result in better inhibition of tumor growth. Moreover, tumor infiltration by Foxp3+ T cells was also greater in KPF-ST2FL mice (Figure S4P), supporting the hypothesis that loss of ST2 signaling encourages a pro-inflammatory Treg phenotype rather than reducing Treg numbers. Overall, our study suggests that Treg-specific inhibition of ST2 signaling may result in a less immunosuppressive tumor microenvironment characterized by increased anti-tumor CD8+ T cell activity and lower tumor burden.
DISCUSSION
Mice with Treg-specific ST2 deficiency have impaired growth of transplanted and chronic inflammation-associated tumors (Ameri et al., 2019; Magnuson et al., 2018; Pastille et al., 2019). We show in an autochthonous mouse model of oncogene-driven lung adenocarcinoma that Treg-specific ST2 loss altered Treg diversity and increased CD8+ T cell infiltration, suggesting that KA Tregs curb anti-tumor CD8+ T cell activity. Lung CD8+ T cells express low levels of ST2, suggesting that the observed phenotype is not due to increased ST2 signaling in CD8+ T cells. Indeed, the proportion and phenotype of CD8+ T cells in KPF-ST2FL mice are similar to that of control mice (data not shown). Our data point to the potential value of disrupting ST2 signaling in cancer, especially in concert with other immunotherapies that improve CD8+ T cell function.
We observed a slight reduction in lung Tregs in KPF-ST2FL mice, consistent with reports that IL-33 can stimulate TCR-independent expansion of Tregs (Arpaia et al., 2015; Kolodin et al., 2015). However, we did not find differences in proliferation between Tregs from KPF-ST2FL mice and controls. Instead, ST2-deficient Tregs may adopt multiple alternate states because of loss of IL-33 signaling. In contrast with a recent report that colon ST2+ Tregs have lower expression of Th17-associated genes, and that recombinant IL-33 inhibits Tr17 differentiation (Pastille et al., 2019), KPF-ST2FL mice did not have a greater proportion of RORγt+ or IL-17+ Tregs. Rather, loss of IL-33 signaling favored a CXCR3+ phenotype, and DP Tregs from KPF-ST2FL mice had lower expression of DP genes, suggesting that ST2 may help regulate the KA/DP Treg phenotype. IL-33 has been shown to increase expression of Foxp3 and GATA-3 (Kolodin et al., 2015; Vasanthakumar et al., 2015), transcription factors critical for Treg differentiation. KA/DP Tregs have similar features to previously described “tissue-protective” Tregs in muscle, lung, and tumors (Arpaia et al., 2015; Burzyn et al., 2013; Green et al., 2017), providing a basis for how ST2-mediated promotion of these Tregs may aid tumor growth. Indeed, KA/DP Tregs express a program similar to that of Tregs in human cancers (De Simone et al., 2016; Guo et al., 2018), including TNFRSF9+ Tregs in human NSCLC (Zheng et al., 2017).
CXCR3 directs Tregs to sites of Th1 inflammation (Koch et al., 2009), which may explain the prominence of the IFNstim_TR program during early tumorigenesis at the peak of CD8+ T cell tumor infiltration and IFN signaling (DuPage et al., 2011). CXCR3 may mark recently arrived Tregs that have distinct functions from KA Tregs, and temporal shifts in IFNstim_TR and KA_TR gene expression may reflect Treg adaptation to the tumor microenvironment over time. Alternatively, the decline in CXCR3+ Tregs during tumor development may reflect cellular turnover and/or outgrowth of KA Tregs because of reduced IFN even as IL-33 remains abundant. Increased expression of CXCR3 in Tregs in KPF-ST2FL mice compared with controls suggests that loss of IL-33 signaling results in greater IFN signaling, which is likely associated with enhanced infiltration of tumors by CD8+ T cells, a major source of IFNg (data not shown). Differential expression of CXCR3 and CCR6 also suggests that Treg localization may be altered in KPF-ST2FL mice, consistent with greater Foxp3+ cell infiltration observed in their tumors. Several reports have described an IFN signature or a distinct population of CXCR3+ Tregs in human tumors, although their significance is not well defined (Halim et al., 2017; Johdi et al., 2017; Redjimi et al., 2012).
Longitudinal scRNA-seq in the KP model provides a window into the natural history of Tconv and Treg diversity in cancer that is challenging to achieve using bulk populations or patient samples. Tr17-like, IFN-responsive, and KA/DP effector Treg populations have been described previously in human tumors, and we show that these states exist simultaneously and their relative proportions vary with tumor development and ST2 activity. Moreover, loss of ST2 signaling in Tregs can alter Treg composition and ultimately impact tumor growth. Although Treg transcriptional heterogeneity may pose a challenge for targeting tumor Treg activity, our study provides proof of concept that pathways that control Treg diversity, maturation, and function may be useful targets for future therapies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Tyler Jacks (tjacks@mit.edu). Plasmids generated in this study are being submitted to Addgene. All unique/stable reagents generated in this study are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
KP, KPfrt, Foxp3GFP, Foxp3RFP, Foxp3YFP/Cre, and Il1rl1fl/fl mice, all on a C57BL/6 background, have been previously described (Bettelli et al., 2006; Chen et al., 2015; DuPage et al., 2011; Rubtsov et al., 2008; Wan and Flavell, 2005; Young et al., 2011). Both male and female mice were used for all experiments, and mice were gender and age-matched within experiments. Experimental and control mice were co-housed whenever appropriate. All studies were performed under an animal protocol approved by the Massachusetts Institute of Technology (MIT) Committee on Animal Care. Mice were assessed for morbidity according to MIT Division of Comparative Medicine guidelines and humanely sacrificed prior to natural expiration.
METHOD DETAILS
Mouse studies
For in vivo labeling of circulating immune cells, anti-CD4-PE (eBioscience, RM4-4, 1:400) and anti-CD8β-PE (eBioscience, 1:400) were diluted in PBS and administered by IV injection 5 minutes before harvest (Anderson et al., 2012). Alternatively, anti-CD45-PE-CF594 (30-F11, BD Biosciences, 1:200) was also used for intravascular labeling and was administered 2 minutes before sacrifice.
Lentiviral production and tumor induction
The lentiviral backbone Lenti-LucOS has been described previously (DuPage et al., 2011). Lentiviral plasmids and packaging vectors were prepared using endo-free maxiprep kits (QIAGEN). The pGK::GFP-LucOS::EFS::FlpO lentiviral plasmid was cloned using Gibson assembly (Akama-Garren et al., 2016; Gibson et al., 2009). Briefly, GFP-OS was created as a protein fusion of GFP and ovalbumin257-383, which includes the SIINFEKL and AAHAEINEA epitopes, and SIYRYYGL antigen. Lentiviral plasmids and packaging vectors were prepared using endo-free maxiprep kits (QIAGEN). Lentiviruses were produced by co-transfection of 293FS* cells with Lenti-LucOS or FlpO-GFP-OS, psPAX2 (gag/pol), and VSV-G vectors at a 4:3:1 ratio, respectively, with Mirus TransIT LT1 (Mirus Bio, LLC). Virus-containing supernatant was collected 48 and 72h after transfection and filtered through 0.45mm filters before concentration by ultracentrifugation (25,000 RPM for 2 hours with low decel). Virus was then resuspended in 1:1 Opti-MEM (GIBCO) - HBSS. Aliquots of virus were stored at −80°C and titered using the GreenGo 3TZ cell line (Sánchez-Rivera et al., 2014).
For tumor induction, mice between 8-15 weeks of age received 2.5 × 104 PFU of Lenti-LucOS or 4.5 × 104 PFU of FlpO-GFP-OS intratracheally as described previously (DuPage et al., 2009).
Tissue isolation and preparation of single cell suspensions
After sacrifice, lungs were placed in 2.5mL collagenase/DNase buffer (Joshi et al., 2015) in gentleMACS C tubes (Miltenyi) and processed using program m_impTumor_01.01. Lungs were then incubated at 37°C for 30 minutes with gentle agitation. The tissue suspension was filtered through a 100 μm cell strainer and centrifuged at 1700 RPM for 10 minutes. Red blood cell lysis was performed by incubation with ACK Lysis Buffer (Life Technologies) for 3 minutes. Samples were filtered and centrifuged again, followed by resuspension in RPMI 1640 (VWR) supplemented with 1% heat-inactivated FBS and 1X penicillin-streptomycin (GIBCO), and 1X L-glutamine (GIBCO).
Spleens and lymph nodes were dissociated using the frosted ends of microscope slides into RPMI 1640 supplemented with 1% heat-inactivated FBS and 1X penicillin-streptomycin (GIBCO), and 1X L-glutamine (GIBCO). Spleen cell suspensions were spun down at 1500 RPM for 5 minutes, and red blood cell lysis with ACK Lysis Buffer was performed for 5 minutes. Cells were filtered through 40 μm nylon mesh and, after centrifugation, resuspended in supplemented RPMI 1640. Lymph node suspensions were filtered through a 40 μm nylon mesh, spun down at 1500 RPM for 5 minutes, and resuspended in supplemented RPMI 1640.
For ex vivo T cell stimulation experiments to detect intracellular cytokines, 0.5 × 105 cells were plated in a 96-well U-bottom plate (BD Biosciences) in RPMI 1640 (VWR) supplemented with 10% heat-inactivated FBS, 1X penicillin-streptomycin (GIBCO), 1X L-glutamine (GIBCO), 1X HEPES (GIBCO), 1X GlutaMAX (GIBCO), 1mM sodium pyruvate (Thermo Fisher), 1X MEM non-essential amino acids (Sigma), 50μM β-mercaptoethanol (GIBCO), 1X Cell Stimulation Cocktail (eBioscience), 1X monensin (BioLegend), and 1X brefeldin A (BioLegend). Cells were incubated in a tissue culture incubator at 37°C with 5% CO2 for 4 hours.
Staining for flow cytometric analysis
Approximately 0.5-1 × 106 cells were stained for 15-30 minutes at 4°C in 96-well U-bottom plates (BD Biosciences) with directly conjugated antibodies (Table S7). SIINFEKL-Kb tetramer was prepared using streptavidin-APC (Prozyme) and SIINFEKL-Kb monomer from the NIH Tetramer Core.
After staining, cells were fixed with Cytofix/ Cytoperm Buffer (BD). Samples that were destined for Foxp3 or other transcription factor staining were fixed with the Foxp3 Transcription Factor Staining Buffer Kit (eBioscience). Intracellular cytokine and transcription factor staining were performed right before analysis using either the BD Perm/Wash Buffer (BD) or the Foxp3 Transcription Factor Staining Buffer Kit (eBioscience); staining was performed for 45 minutes at 4°C. Analysis of Tregs (i.v.negCD4+Foxp3+) and Tconv (i.v.negCD4+Foxp3−) was performed on an LSR II (BD) with 405, 488, 561, and 635 lasers. Data analysis was performed using FlowJo software.
Isolation of Treg populations for bulk RNA-Seq
For sequencing of CD103−KLRG1− (DN), CD103+KLRG1− (SP), and CD103+KLRG1+ (DP)Tregs: 100-200 DP, SP, and DNTreg cells from LucOS-infected, KP, Foxp3RFP mice were sorted using a MoFlo Astrios cell sorter. cDNA was prepared by the SMART-Seq2 protocol (Picelli et al., 2013) with the following modifications: RNA was purified using 2.2X RNAclean SPRI beads (Beckman Coulter) without final elution, after which beads were air-dried and immediately resuspended with water and oligoDT for annealing, and 18 cycles of preamplification were used for cDNA. cDNA was then mechanically sheared and prepared into sequencing libraries using the Thru-Plex-FD Kit (Rubicon Genomics). Sequencing was performed on an Illumina HiSeq 2000 instrument to obtain 50 nt paired-end reads.
For comparison of KPF and KPF-ST2FLTregs: 100-200 DP, SP, and DN Tregs were sorted into Buffer TCL (QIAGEN) plus 1% β-mercaptoethanol and cDNA was prepared with 14 cycles of preamplification. Nextera library preparation was performed as previously described (Picelli et al., 2013) and sequencing was performed with 50 × 25 paired end reads using two kits on the NextSeq500 5 instrument.
Single-cell sorting of Tconv and Treg populations for RNA sequencing
Tconv (DAPIneg, i.v.neg, Thy1.2+CD4+Foxp3−GFPneg) and Treg (DAPIneg, i.v.neg, Thy1.2+CD4+Foxp3−GFPpos) cells were isolated from ~4 mice per time point and single-cell sorted into Buffer TCL (QIAGEN) plus 1% β-mercaptoethanol in 96-well plates using a MoFlo Astrios cell sorter. Each plate had a 30-100 cell population well and an empty well as controls. Following sorting, plates were spun down for 1” at 2000 RPM and frozen immediately at −80C.
Droplet-based scRNA-seq of CD45+ and CD45− populations from tumor-bearing lungs
Tumors were microdissected under dissection microscope and dissociated into single cell suspensions as previously described. Samples were pelleted at 1700 RPM for 5 minutes and resuspended in 500ul of MACS buffer containing PBS, 0.5% bovine serum albumin (BSA), and 2mM EDTA. CD45+ and CD45− cells were then magnetically separated using MACS CD45 MicroBeads (Miltenyi Biotec) as per manufacturer’s instructions. Briefly, cells were stained with CD45 MicroBeads for 15 minutes at 4°C. Samples were washed with MACS buffer and pelleted at 1700rpm for 5 minutes. Samples were resuspended in 1ml of MACS buffer and added to LS MACS column on LS Separator magnet (Miltenyi Biotec). Flow through was collected as CD45− population. Columns were washed 3× with MACS buffer and flow-through was added to CD45− population. 5ml of MACS buffer was then then added to column, the column was removed from the magnet, and cells were expelled from column into conical using plunger; this was the CD45+ sample. CD45+ and CD45− samples were pelleted at 1700RPM for 5 minutes and resuspended in PBS with 0.01% BSA before proceeding to droplet based scRNaseq.
Single cells were processed through the 10X Genomics Single Cell 3′ platform using the Chromium Single Cell 3′ Library & Gel Bead Kit V2 kit (10X Genomics), per manufacturer’s protocol. Briefly, 6,000 cells were loaded onto each channel and partitioned into Gel Beads in Emulsion in the Chromium instrument. Cell lysis and barcoding occur, followed by amplification, fragmentation, adaptor ligation and index library PCR. Libraries were sequenced on an Illumina HiSeqX at a read length of 98 base pairs.
Preparation of scRNaseq libraries
Plates were thawed and RNA was purified using 2.2X RNAclean SPRI beads (Beckman Coulter) without final elution (Shalek et al., 2013). SMART-seq2 and Nextera library preparation was performed as previously described (Picelli et al., 2013), with some modifications as described in a previous study (Singer et al., 2017). Plates were pooled into 384 single-cell libraries, and sequenced 50 × 25 paired end reads using a single kit on the NextSeq500 5 instrument.
Quantitative PCR for validation of RNA-Seq experiments
Quantitative PCR was performed using various primer sets (Table S7). 1ng of cDNA generated using SMART-Seq2 was included in a reaction with 1μL of each primer (2μM stock) and 5μL of KAPA SYBR Fast LightCycler 480 (KAPA Biosystems). Cp values were measured using a LightCycler 480 Real-Time PCR System (Roche). Relative fold-change in expression values were calculated using the following formula: 2(ΔCp(Sample) - ΔCp(Spleen)), where ΔCp(Sample) = Sample CpGene of interest - Sample CpGAPDH, and ΔCp(Spleen) = Spleen CpGene of Interest - Spleen CpGAPDH.
Immunohistochemistry (IHC) and immunofluorescence staining
Lung lobes and spleens allocated for IHC and IF were perfused with 4% paraformaldehyde in PBS and fixed overnight at 4°C. Lung lobes and/ or spleen were transferred to histology cassettes and stored in 70% ethanol until paraffin embedding and sectioning (KI Histology Facility). H&E stains were performed by the core facility using standard methods.
For IHC, 5 μm unstained slides were dewaxed, boiled in citrate buffer (1 g NaOH, 2.1 g citric acid in 1L H2O, pH 6), for 5 minutes at 125°C in a decloaking chamber (Biocare Medical), washed with 3X with 0.1% Tween-20 (Sigma) in TBS, and blocked and stained in Sequenza slide racks (Thermo Fisher). Slides were blocked with Dual Endogenous Peroxidase and Alkaline Phosphatase Block (Dako) and then with 2.5% Horse Serum (Vector Labs). Slides were incubated in primary antibody overnight, following by washing and incubation in HRP-polymer-conjugated secondary antibodies (ImmPRESS HRP mouse-adsorbed anti-rat and anti-goat, Vector Laboratories). Slides were developed with ImmPACT DAB (Vector Laboratories). Primary antibodies used were goat anti-IL-33 (R&D, AF3626), rat anti-CD8a (Thermo Fisher, 4SM16), and rat anti-Foxp3 (Thermo Fisher, FJK-16 s). Stains were counterstained with hematoxylin using standard methods before dehydrating and mounting.
After fixation, lung lobes and spleen allocated for IF were perfused with 30% sucrose in PBS for cryoprotection for 6-8h at 4°C. Tissues were then perfused with 30% optimum cutting temperature (O.C.T.) compound (Tissue-Tek) in PBS and frozen in 100% O.C.T in cryomolds on dry ice. 6μm sections were cut using a CryoStar NX70 cryostat (Thermo), and air-dried for 60-90 minutes at room temperature. Sections were incubated in ice-cold acetone (Sigma) for 10 minutes at −20°C and then washed 3 × 5 minutes with PBS. Samples were permeabilized with 0.1% Triton X-100 (Sigma) in PBS followed by blocking with 0.5% PNB in PBS (Perkin Elmer). Primary antibodies were incubated overnight. Primary antibodies used were rabbit anti-prosurfactant protein C (SPC) (Millipore, AB3786, 1:500) and goat anti-IL-33 (R&D, AF3626, 1:200). After washing 3 × 5 minutes, samples were incubated in species-specific secondary antibodies conjugated to Alexa Fluor 568 and Alexa Fluor 488, respectively, at 1:500. Sections were then fixed in 1% PFA and mounted using Vectashield mounting media with DAPI (Vector Laboratories).
Immunohistochemistry and immunofluorescence tissue section images were acquired using a Nikon 80 Eclipse 80i fluorescence microscope using 10x and 20x objectives and an attached Andor camera. Stained IHC slides were scanned using the Aperio ScanScope AT2 at 20X magnification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bulk RNA-seq data pre-processing
Bulk RNA-Seq reads that passed quality metrics were mapped to the annotated UCSC mm9 mouse genome build (http://genome.ucsc.edu/) using RSEM (v1.2.12) (http://deweylab.github.io/RSEM/) Li and Dewey, 2011) using RSEM’s default Bowtie (v1.0.1) alignment program (Langmead et al., 2009). Expected read counts estimated from RSEM were upper-quartile normalized to a count of 1000 per sample (Bullard et al., 2010). Genes with normalized counts less than an upper-quartile threshold of 20 across all samples were considered lowly expressed and excluded from further analyses to increase the robustness of signature scoring, as previously described (Rau et al., 2013; Sha et al., 2015). As outlined below, signature analyses were conducted either on a log2 transformed version of the filtered gene expression matrix to overcome data skewness, or on the non-transformed version for increased sensitivity by avoiding compression of weaker signals (Ashour et al., 2015; Singh and Shree, 2016).
Signature analysis in bulk RNA-Seq
Signature analyses between bulk Treg cell populations were performed using a blind source separation methodology based on Independent Component Analysis (ICA) (Hyvärinen and Oja, 2000), using the R implementation of the core JADE algorithm (Joint Approximate Diagonalization of Eigenmatrices) (Biton et al., 2014; Miettinen et al., 2017; Rutledge and Jouan-Rimbaud Bouveresse, 2013) along with custom R utilities. Multi-sample signatures were visualized using relative signature profile boxplots (Li et al., 2018). Heat-maps were generated with the Heatplus package in R using agglomerative hierarchical clustering with default euclidean distance measure, Ward’s minimum variance method for row-clustering, and complete linkage for column clustering (Figures 4D and S2H).
DP Treg signature
We identified a signature distinguishing CD103+KLRG1+ lung Tregs from other populations. The non-transformed expression matrix was decomposed using ICA with the JADE algorithm (described above) as: E = AS where E is the expression matrix (input), A is the mixing matrix (mixing weights, basis vectors), and S is the signature matrix (independent components or latent variables yielding standardized gene-scores per signature). Biologically relevant signatures were identified through two approaches: (1) Quantitative assessment of significance using a 2-sample Mann-Whitney-Wilcoxon non-parametric test between mixing weights (from A) grouped by biological condition per signature; and (b) visual inspection of a Hinton plot derived from the mixing matrix A. Corresponding signatures from S were selected for downstream analyses. Up and down genes per signature were selected using a |gene-score| > = 3 threshold (standardized score, #s.d. above/below mean). Genes with |z-score| > 3 were selected for downstream analysis (75 upregulated and 31 downregulated genes). An additional expression level filter was implemented to narrow the list of genes of interest. For upregulated genes, expression in all CD103+KLRG1+ lung Treg samples had to be greater than all but a maximum of 3 other samples (3 out of a total 8 other samples). A similar filtering scheme was employed in the other direction for downregulated genes. This yielded a total of 43 upregulated and 2 downregulated genes in CD103+KLRG1+ lung Tregs. This set of genes was used to illustrate gene expression level changes across samples (Figure S2H).
ST2-deficient Tregs Signature
A signature distinguishing ST2-deficient Tregs from wild-type Tregs (Table S5) was identified using ICA on the non-transformed expression matrix. To identify particular genes of interest, signature genes (|z-score| > 3) were filtered to include only genes that had an absolute fold change exceeding 1.5x within any of the CD103+KLRG1+ (DP), CD103+KLRG1− (SP), CD103−KLRG1− (DN) sample types between wild-type and ST2-deficient Tregs. These gene lists were further filtered to retain only those genes that appeared in at least two of the three sample types (i.e., up/downregulated in wild-type or ST2-deficient in at least two of DP/DN/SP comparisons). Genes with opposite directionality across the three sample types (n = 5 genes) were dropped. Expression levels of the resulting curated set of 14 genes were visualized using a row-normalized heatmap (Figure 4D). Signature correlation scores (z-scores) for each gene are included in Table S5.
Gene Set Enrichment Analysis (GSEA)
Selected signatures (from S) were run through the Gene Set Enrichment Analysis (GSEA) using the rank-based input format. All genes per signature were used, ranked by gene-scores from S. We used gene-sets from MsigDB v5.1 (Subramanian et al., 2005). Custom gene set additions were made to version 4.0 of the MSigDB immunologic signatures library (c7) (Table S6). Normalized Enrichment Score (NES), p values and FDR for the custom gene-sets were calculated in the context of the combined c7 v4.0 MSigDB collection.
Network representations of GSEA results were generated using EnrichmentMap (http://www.baderlab.org/Software/EnrichmentMap) for Cytoscape v3.3.0 (https://www.cytoscape.org/).
Pre-processing of SMART-Seq2 scRNA-seq data
BAM files were converted to de-multiplexed FASTQs using the Illumina-provided Bcl2Fastq software package v2.17.1.14. Paired-end reads were mapped to the UCSC mm10 mouse transcriptome using Bowtie with parameters ‘-n 0 -m 10’, which allows alignment of sequences with zero mismatches and allows for multi-mapping of a maximum of ten times.
Expression levels of genes were quantified using TPM values calculated by RSEM v1.2.8 in paired-end mode. For each cell, the number of detected genes (TPM > 0) was calculated and cells with less than 600 or more than 4,000 genes detected were excluded as well as cells that had a mapping rate to the transcriptome below 15%. To further remove potential doublets (mostly of B cells and epithelial cells), we calculated the sum log2(TPM+1) over Cd79a, Cd19, Lyz1, Lyz2 and Sftpc, and excluded any cell that scored higher than 3. We retained only genes expressed above log2TPM of 3 in at least five cells in the whole dataset.
Since we could not sort for Treg for two of the mice (#336 and #338), we had to infer which cells are Tregs from these data. To this end, we trained a random forest classifier for mice for which we have sorted both Tconv and Tregs, using the train function from the caret package in R, based on the expression of the following genes: Foxp3, Ikzf2, Areg, Il1rl1, Folr4, Wls, Tnfrsf9, Klrg1, Il2ra, Dusp4, Ctla4, Neb, Itgb1, and Cd40lg. The labeled data was partitioned into training and test sets. The model has a sensitivity and specificity above 90% in cross validation. We then applied the classifier on the unlabeled data and cells with a probability above 0.6 to be either Tconv or Treg were given the corresponding label. The remaining 4% of cells were discarded as unambiguous.
Identifying tissue-specific gene programs for Treg and Tconv
To identify genes that are differentially expressed between lung and msLN in Treg and/or Tconv, we performed a regression analysis. We focused on the proportion of cells expressing a gene, and hence on logistic regression. We performed logistic regression using the bayesglm function from the arm package in R, including only those mice (# 338, #3642, #3839, #3889) for which we had matched cells from both lung and msLN, as well as for Treg and Tconv, and excluding all genes expressed in > 95% or < 5% of cells in lung and msLN. We ran the logistic regression with expression data binarized at a log2(TPM+1) of 2 and using the following full model: gene expression ~genes detected + batch effect + tissue versus a reduced model: gene expression ~genes detected + batch effect. We corrected for multiple hypothesis by computing an FDR of the likelihood ratio test p value, and retained genes as differentially expressed between lung and msLN with p < 10−5 and an |coefficient| > 2.
Comparing the extent of cell heterogeneity between lung and msLN
Diffusion components were calculated on a gene expression matrix limited to genes that were differentially expressed between lung and msLN using the DiffusionMap function from the destiny package in R (Angerer et al., 2016) with a k of 30 and a local sigma. In order to be able to compare the variance in distributions in diffusion component 1 and 2 between lung and msLN Treg/Tconv, we downsampled the cells from the lung to the (lower) numbers of cells from the msLN. To test for significant differences in variance in the distributions of lung and msLN Treg/Tconv, we used Levene’s test for the equality of variances on the distributions of the coefficients of the downsampled cells in each of diffusion components 1 and 2.
Identifying gene programs and their time dependence
Gene programs were identified using PAGODA using the scde R package version 2.6.0. (Fan et al., 2016) on the counts table from RSEM after cleaning the data using the clean.counts function (min.lib.size = 600,min.detected = 5). The knn.error.model function was run using a k of 30, which is much lower than default, but yields statistically indistinguishable results from the default k (# cells / 4). We then ran the pagoda.varnorm to normalize gene expression variance, and the pagoda.subtract.aspect function to control for sequencing depth which then allowed us to run pagoda.gene.clusters which identifies de-novo correlated genes in the dataset. We forced PAGODA to return 100 programs. We identified programs with a significance z.score above 1.96. We removed several highly significant newly identified gene programs consisting of paralog groups with high expression correlation, likely because of multimapping of reads.
Mean program expression was calculated by averaging over the genes in each program of the centered and scaled gene expression table and transforming to a z-score over 1,000 randomly selected gene sets with matched mean-variance patterns. First, genes were grouped into 10 bins based on their mean expression, and into 10 (separate) bins based on their variance of expression across all cells. Given a list of genes (e.g., genes in a program), a cell-specific signature score was computed for each cell as follows: First, 1,000 random gene lists were generated, where each instance of a random gene-list was generated by sampling (with replacement) for each gene in the gene-list a gene from the equivalent mean and variance bin it was placed in. Then, the sum of centered and scaled gene expression in the given cell was computed for all 1000 random gene-lists generated and the z-score of the original gene-list for the generated 1,000 sample distribution is returned, as in (Singer et al., 2017).
Another program of highly correlated genes identified by PAGODA showed no biological relevance based on gene annotation, but was associated with cells processed on specific dates, suggested they reflect a contamination or batch effect. We scored each cell for this program with the above described method for scoring cells for gene signatures. When testing for differential gene expression over tumor development (described below), we included this batch effect score as a covariate in the regression analysis to control for genes that are correlated with it.
To test if a program’s expression changes over the course of tumor development, we estimated a linear model for each program and compared with a likelihood ratio test a full model: program.activity ~detected genes + time point to a reduced model: program.activity ~detected genes. For the time point covariate, healthy lung was taken as reference. We corrected the likelihood ratio test p values for multiple hypotheses for the number of programs using the p.adjust function computing the false discovery rate in the stats package.
Dimensionality reduction using diffusion component analysis
Diffusion components were calculated on a gene expression matrix limited to genes from programs of interest: programs 1,4,5,14,15 and 21 for Tconv, and programs 1,3,6,8,9,12,13,18,21,23 and 26 for Treg. Gene expression was scaled for Tregs only across all cells. Diffusion components were calculated using the DiffusionMap function from the destiny package in R (Angerer et al., 2016) with a k of 30 and a local sigma. Significant diffusion components identified by the elbow in the eigenvalues were further used for dimensionality reduction to two dimensions. The eigenvectors of the significant diffusion components were imported into gephi 0.9.2 and a force directed layout using forceatlas 2 was run until it converged to get a two dimensional embedding.
Testing for differential gene expression during tumor development
To test whether individual genes change in gene expression over the course of tumor growth, we performed a two-step regression analysis. We focused on the proportion of cells expressing a gene, and hence on logistic regression. We performed logistic regression using the bayesglm function from the arm package in R. Because gender is often confounded with a particular time point in our experiment, we did not include it as a covariate in the model, but did remove all Y chromosome genes from analysis. We also excluded all genes expressed in > 95% or < 5% of cells in each mouse. We ran the logistic regression with expression data binarized at a log2(TPM+1) of 2 and using the following full model: gene expression ~genes detected + batch effect + week p.i. (healthy lung as reference) versus a reduced model: gene expression ~genes detected + batch effect. We identified a threshold for significance by the elbow method, identifying the peak of the second derivative of the ordered fdr distribution of the likelihood ratio test for each time point. To remove significant genes whose signal was driven by only one mouse, we performed another logistic regression using a mixed effect model, accounting for mouse variability: To this end, we added to the significant genes 1,000 randomly selected genes that were non-significant by the initial test to serve as background genes, and performed a mixed effect logistic regression using the glmer function of the lme4 package in R, with the model gene expression ~tmp + (1|mouse), allowing the intercept to vary by mouse. We combined the elbow method above and the background genes to select an FDR cutoff for significance of 0.01. A gene was classified as significantly varying during tumor development if it passed this FDR cutoff in at least one time point.
T cell receptor (TCR) reconstruction and clonotype calling
TCR were reconstructed using Tracer (Stubbington et al., 2016), run in short read mode with the following settings ‘-inchworm_only = T-trinity_kmer_length = 17’. To call shared clonotypes between Treg and Tconv cells, we required all cells of a clone to have identical productive TCRA and TCRB.
Comparison of bulk and scRNA-seq signatures to published signatures
Lists of differentially expressed genes in human cancer Tregs, mouse tissue Tregs, Tr17 cells from mice, and mouse activated Tregs (Table S4) were collected either from the supplementary tables of the relevant publications, or generously provided by the authors upon request (De Simone et al., 2016; Guo et al., 2018; Kim et al., 2017; Magnuson et al., 2018; Miragaia et al., 2019; Plitas et al., 2016; Tan et al., 2016; Zheng et al., 2017).
ST2 transcriptional programs in human colorectal cancer Tregs
To examine the generalizability of our findings and their relevance to human cancer, we identified gene programs that co-vary with ST2 expression in human colorectal cancer Tregs (Zhang et al., 2018). We compared cells in which ST2 was detected (ST2+) and cells in which ST2 was not detected (ST2−) to identify an ST2+ program. Differential expression analysis was performed using t test on the log-transformed TPM values. We confirmed that the program was not confounded by cell quality and ensured that it captured differences between ST2+ and ST2− cells within each tumor (data not shown). To this end, we first computed the overall expression (OE) of the program across the relevant T cells, in a way that eliminates technical noise, as previously described (Jerby-Arnon et al., 2018). We then tested whether the OE of the program was higher in ST2+ cells compared to ST2- by using a mix-effected multilevel (random intercepts) regression model, where the program OE is the dependent variable and ST2 detection is provided as a binary covariate. The model included patient-specific intercepts to control for the dependency between the scRNA-seq profiles of cells from the same tumor, and controlled for cell complexity with a covariate that denotes the number of genes detected in each cell. The model was implemented using the lme4 and lmerTest R packages (https://cran.r-project.org/web/packages/lme4/index.html ).
Processing and analysis of droplet-based scRNA-seq
De-multiplexing, alignment to the mm10 transcriptome and unique molecular identifier (UMI)-collapsing were performed using the Cellranger toolkit from 10X Genomics version 1.1.0. For each cell, we quantified the number of genes for which at least one read was mapped, and then excluded all cells with fewer than 500 detected genes. Genes that were detected in less than 3 cells were excluded. Expression values Ei,j for gene i in cell j were calculated by dividing UMI counts for gene i by the sum of the UMI counts in cell j, to normalize for differences in coverage, and then multiplying by 10,000 to create TPM-like values (TP10K), and finally computing log2(TP10K + 1).
Selection of variable genes was performed by fitting a logistic regression to the cellular detection fraction (often referred to as a), using the total number of UMIs per gene as a predictor (Montoro et al., 2018). Outliers from this curve are genes that are expressed in a lower fraction of cells than would be expected given the total number of UMIs mapping to that gene, that is, likely cell-type or state-specific genes. We used a threshold of deviance of < −0.15 and a minimum of 100 total UMIs. We restricted the expression matrix to this subset of variable genes and values were centered and scaled and capped at a z-score of 10.
We restricted the expression matrix to the subsets of variable genes and high-quality cells noted above, and then centered and scaled values before inputting them into principal component analysis (PCA), implemented using ‘RunPCA’ in Seurat which runs the irlba function. After PCA, significant principal components were identified using the elbow-method when looking at the distribution of singular values. Scores from only those significant principal components were used as the input to further analysis. For visualization purposes, the dimensionality of the datasets was further reduced to 2D embeddings using the RunUMAP() function on the first 24 PCs and clusters were identified using the FindNeighbors() and FindClusters() functions of the Seurat package in R. Clusters were post hoc merged to six major cell populations using canonical markers for all cell types detected.
Analysis of IHC Images
QuPath software was used to annotate tumor and lobe areas (Bankhead et al., 2017). CD8-stained and Foxp3-stained images were standardized to a common set of stain vector parameters. CD8+ cell detection was performed using the PositiveCellDetection plugin with the following parameters:
runPlugin(‘qupath.imagej.detect.nuclei.PositiveCellDetection’, ‘{“detectionImageBrightfield”: “Optical density sum,” “requestedPixelSizeMicrons”: 0.5, “backgroundRadiusMicrons”: 8.0, “medianRadiusMicrons”: 0.0, “sigmaMicrons”: 1.5, “minAreaMicrons”: 7.0, “maxAreaMicrons”: 125.0, “threshold”: 0.3, “maxBackground”: 2.0, “watershedPostProcess”: true, “excludeDAB”: false, “cellExpansionMicrons”: 2.0, “includeNuclei”: false, “smoothBoundaries”: false, “makeMeasurements”: true, “thresholdCompartment”: “Cytoplasm: DAB OD max,” “thresholdPositive1”: 0.7, “thresholdPositive2”: 0.4, “thresholdPositive3”: 0.6, “singleThreshold”: true}’);
Foxp3+ cell detection was performed using the PositiveCellDetection plugin with the following parameters:
runPlugin(‘qupath.imagej.detect.nuclei.PositiveCellDetection’, ‘{“detectionImageBrightfield”: “Optical density sum,” “requestedPixelSizeMicrons”: 0.5, “backgroundRadiusMicrons”: 8.0, “medianRadiusMicrons”: 0.0, “sigmaMicrons”: 1.5, “minAreaMicrons”: 7.0, “maxAreaMicrons”: 125.0, “threshold”: 0.3, “maxBackground”: 2.0, “watershedPostProcess”: true, “excludeDAB”: false, “cellExpansionMicrons”: 2.0, “includeNuclei”: false, “smoothBoundaries”: false, “makeMeasurements”: true, “thresholdCompartment”: “Cell: DAB OD mean,” “thresholdPositive1”: 0.3, “thresholdPositive2”: 0.4, “thresholdPositive3”: 0.6, “singleThreshold”: true}’);
Scored cells were normalized to tumor area.
Additional statistical analyses
Unpaired, two-tailed Student’s t tests, Mann-Whitney tests, Tukey’s multiple comparisons tests, and Sidak’s multiple comparisons tests were used for all statistical comparisons using GraphPad Prism software.
DATA AND CODE AVAILABILITY
The accession number for the bulk and scRNA-Seq data reported in this paper is GEO: GSE139232. All code is available upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| KLRG1 2F1 PE-Cy7 | Thermo Fisher | RRID: AB_1518768 |
| CD103 2E7 APC | BioLegend | RRID: AB_1227502 |
| CD4 RM4-5 APC-eFluor780 | Thermo Fisher | RRID: AB_1272183 |
| Foxp3 FJK-16 s FITC | Thermo Fisher | RRID: AB_465243 |
| IL-17 eBio17B7 PerCP-Cy5.5 | Thermo Fisher | RRID: AB_925753 |
| CD44 IM7 Alexa Fluor 700 | Thermo Fisher | RRID: AB_494011 |
| CD62L MEL-14 eFluor450 | Thermo Fisher | RRID: AB_1963590 |
| CCR6 29-2L17 PE/Dazzle 594 | BioLegend | RRID: AB_2687019 |
| RORgt Q31-378 Alexa Fluor 647 | BD Biosciences | RRID: AB_2738916 |
| T-bet O4-46 PE | BD Biosciences | RRID: AB_10564071 |
| PD-1 J43 PE-Cy7 | BioLegend | RRID: AB_572017 |
| CD69 H1.2F3 BV785 | BioLegend | RRID: AB_2629640 |
| CXCR3 CXCR3-173 BV421 | BD Biosciences | RRID: AB_10900974 |
| ST2 U29-93 Brilliant Blue 700 | BD Biosciences | RRID: AB_2743483 |
| CD85k H1.1 PE | Biolegend | RRID: AB_2561653 |
| Ki-67 B56 BV786 | BD Biosciences | RRID: AB_2732007 |
| CD45.2 104 V500 | BD Biosciences | RRID: AB_10897142 |
| Thy1.2 30-H12 APC-eFluor780 | Thermo Fisher | RRID: AB_1272187 |
| CD103 2E7 BV510 | BioLegend | RRID: AB_2562713 |
| CD4 RM4-5 BUV737 | BD Biosciences | RRID: AB_2738734 |
| CD8a 53-6.7 BUV395 | BD Biosciences | RRID: AB_2739421 |
| CD45 30-F11 PE-CF594 | BD Biosciences | RRID: AB_11154401 |
| CD45 30-F11 APC-Ef780 | Thermo Fisher | RRID: AB_1548781 |
| CXCR6 SA051D1 PE/Dazzle 594 | Biolegend | RRID: AB_2721700 |
| KLRG1 2F1 BV711 | BioLegend | RRID: AB_2629721 |
| CD11c HL3 PE-Cy7 | BD Biosciences | RRID: AB_469590 |
| Siglec F E50-2440 PE | BD Biosciences | RRID: AB_394341 |
| CD4 RM4-4 PE | Biolegend | RRID: AB_313691 |
| CD8b eBioH35-17.2 PE | Thermo Fisher | RRID: AB_657768 |
| IHC and IF: Goat anti-mouse IL-33 | R&D Biosystems | RRID: AB_884269 |
| IHC: Rat anti-mouse CD8alpha | Thermo Fisher | RRID: AB_2637159 |
| IF: Rabbit anti-mouse proSP-C | Millipore | RRID: AB_91588 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant mouse IL-33 | BioLegend | Biolegend 580506 |
| Cell Stimulation Cocktail | Thermo Fisher | Cat: 00-4970-03 |
| SIINFEKL-Kb monomer | NIH Tetramer Core | |
| Critical Commercial Assays | ||
| Thru-Plex-FD Library Prep Kit | Rubicon Genomics | |
| Nextera XT Library Prep Kit | Illumina | FC-131-1096 |
| Deposited Data | ||
| Bulk and single cell RNA sequencing | GEO | GEO: GSE139232 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J | RRID: IMSR_JAX:008179 | |
| Mouse: B6.129P2-Trp53tm1Brn/J | Jackson Laboratory | RRID: IMSR_JAX:008462 |
| Mouse: C57BL/6-Foxp3tm1Flv/J | RRID: IMSR_JAX:008374 | |
| Mouse: Foxp3tm1Kuch | Bettelli et al., 2006 | MGI:3718527 |
| Mouse: B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J | Jackson Laboratory | RRID: IMSR_JAX:016959 |
| Mouse: Il1rl1tm1.1Rlee | Chen et al., 2015 | MGI:5818148 |
| Oligonucleotides | ||
| See Table S7. | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: Lenti-LucOS | Addgene | Addgene_22777 |
| Plasmid: pGK::GFP-LucOS::EFS::FlpO (FlpO-GFP-OS) | This paper | Available upon request |
| Software and Algorithms | ||
| Code generated for this study | This paper | Available upon request |
| SCDE | Fan et al., 2016 | http://hms-dbmi.github.io/scde/ |
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| RSEM v1.2.8 | Li and Dewey, 2011 | https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-323 |
| JADE v1.1.0 | Miettinen et al. 2017 | https://cran.r-project.org/web/packages/JADE/index.html |
| Tracer | Stubbington et al., 2016 | https://github.com/Teichlab/tracer |
| Gephi | Bastian et al., 2009 | https://gephi.org |
| EnrichmentMap (Cytoscape) | Merico et al., 2010 | https://www.cytoscape.org |
| QuPath | Bankhead et al., 2017 | https://qupath.github.io/ |
| Prism | GraphPad |
Highlights.
Single-cell profiling of CD4 T cells along tumor development in a mouse model
Treg diversity shifts to a Klrg1+Areg+ (KA) effector phenotype in advanced tumors
Il1rl1 (encoding ST2)+ Tregs have higher expression of KA effector Treg genes
Treg-specific ST2 loss enhances CD8+ T cell infiltration and decreases tumor burden
ACKNOWLEDGMENTS
We thank N. Joshi, N. Marjanovic, R. Satija, D. Gennert, C. Jin, and S. Riesenfeld for thoughtful discussion and technical advice; J. Park, J. Wilson, and N. Cheng for technical assistance; S. Levine at the Massachusetts Institute of Technology (MIT) BioMicro Center for sequencing support; C. Otis and S. Saldi in the Broad Flow Cytometry Core and G. Paradis in the Koch Institute Flow Cytometry Facility for flow cytometry assistance; K. Cormier and C. Condon from the Hope Babette Tang (1983) Histology Facility for histology assistance; L. Gaffney and A. Hupalowska for artwork and advice on figures; A. Sharpe for Foxp3GFP mice; D. Mathis for critical reading of the manuscript and Il1rl1fl/fl mice; and K. Anderson, J. Teixeira, M. Magendantz, and K. Yee for administrative and logistical support. This work was supported by the Howard Hughes Medical Institute (T.J. and A.R.), a Margaret A. Cunningham Immune Mechanisms in Cancer Research Fellowship Award (A.L.), the David H. Koch Graduate Fellowship Fund (A.L.), National Cancer Institute (NCI) Cancer Center Support Grant P30-CA1405, an Advanced Medical Research Foundation grant (D.C.), a Cancer Research Institute (CRI) Irvington Postdoctoral Fellowship (L.C.), a National Science Foundation Graduate Fellowship (D.C.), grants T32GM007753 (A.L.) and T32GM007287 (A.L. and D.C.) from the National Institute of General Medical Sciences (NIGMS), and the Klarman Cell Observatory at the Broad Institute (A.R.). A.R. and T.J. are Howard Hughes Medical Institute Investigators. T.J. is the David H. Koch Professor of Biology and a Daniel K. Ludwig Scholar.
DECLARATION OF INTERESTS
T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific. He is co-founder of Dragonfly Therapeutics and T2 Biosystems, and SAB member for Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. T.J.’s laboratory also receives funding from the Johnson and Johnson Lung Cancer Initiative and Calico that did not support the research described in this manuscript. A.R. is a co-founder and equity holder in Celsius Therapeutics and an SAB member for Thermo Fisher, Neogene Therapeutics, and Syros Pharmaceuticals. A.L., R.H.H., A.R., and T.J. are co-inventors on a US provisional patent application (62/788,952) directed to overcoming immunosuppression.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.120.
REFERENCES
- Akama-Garren EH, Joshi NS, Tammela T, Chang GP, Wagner BL, Lee D-Y, Rideout WM 3rd, Papagiannakopoulos T, Xue W, and Jacks T (2016). A Modular Assembly Platform for Rapid Generation of DNA Constructs. Sci. Rep 6, 16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri AH, Moradi Tuchayi S, Zaalberg A, Park JH, Ngo KH, Li T, Lopez E, Colonna M, Lee RT, Mino-Kenudson M, and Demehri S (2019). IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc. Natl. Acad. Sci. USA 116, 2646–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, and Masopust D (2012). Cutting edge: intravascular staining redefines lung CD8 T cell responses. J. Immunol 189, 2702–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32,1241–1243. [DOI] [PubMed] [Google Scholar]
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY (2015). A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour AS, Samanta S, Dey N, Kausar N, Abdessalemkaraa WB, and Hassanien AE (2015). Computed tomography image enhancement using cuckoo search: a log transform based approach. J. Signal Inf. Process 6, 244. [Google Scholar]
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian M, Heymann S, and Jacomy M (2009). In Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media. [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441,235–238. [DOI] [PubMed] [Google Scholar]
- Biton A, Bernard-Pierrot I, Lou Y, Krucker C, Chapeaublanc E, Rubio-Pérez C, López-Bigas N, Kamoun A, Neuzillet Y, Gestraud P, et al. (2014). Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 9, 1235–1245. [DOI] [PubMed] [Google Scholar]
- Bos PD, Plitas G, Rudra D, Lee SY, and Rudensky AY (2013). Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J. Exp. Med 210, 2435–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, and Dudoit S (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, and Mathis D (2013). A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W-Y, Hong J, Gannon J, Kakkar R, and Lee RT (2015). Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc. Natl. Acad. Sci. USA 112, 7249–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Yuan X, Tsai MS, Podack ER, Yu A, and Malek TR (2012). IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J. Immunol 189, 1780–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, et al. (2016). Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 45, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Träger U, Hofer A-C, Kägebein D, Wang Q, Frauhammer F, et al. (2017). Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol 18, 1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G (2011). Experimental mouse tumour models: what can be learnt about human cancer immunology? Nat. Rev. Immunol 12, 61–66. [DOI] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan J-B, Zhang K, Chun J, and Kharchenko PV (2016). Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Green JA, Arpaia N, Schizas M, Dobrin A, and Rudensky AY (2017). A nonimmune function of T cells in promoting lung tumor progression. J. Exp. Med 214, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985. [DOI] [PubMed] [Google Scholar]
- Haghverdi L, Buettner F, and Theis FJ (2015). Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998. [DOI] [PubMed] [Google Scholar]
- Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, Hoong S-J, Yuksel M, Jassem W, Hannen RF, et al. (2017). An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell Rep. 20, 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, et al. (2012). The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvärinen A, and Oja E (2000). Independent component analysis: algorithms and applications. Neural Netw. 13, 411–430. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, and Jacks T (2005). The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 65, 10280–10288. [DOI] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, et al. (2018).A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johdi NA, Ait-Tahar K, Sagap I, and Jamal R (2017). Molecular Signatures of Human Regulatory T Cells in Colorectal Cancer and Polyps. Front. Immunol 8, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu L-F, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43, 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B-S, Lu H, Ichiyama K, Chen X, Zhang Y-B, Mistry NA, Tanaka K, Lee Y-H, Nurieva R, Zhang L, et al. (2017). Generation of RORgt+ Antigen-Specific T Regulatory 17 Cells from Foxp3+ Precursors in Autoimmunity. Cell Rep. 21, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, and Campbell DJ (2012). T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor β2. Immunity 37, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, and Mathis D (2015). Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab. 21, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-L, Moding EJ, Huang X, Li Y, Woodlief LZ, Rodrigues RC, Ma Y, and Kirsch DG (2012). Generation of primary tumors with Flp recombinase in FRT-flanked p53 mice. Dis. Model. Mech 5, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zeng Q, Bhutkar A, Galván JA, Karamitopoulou E, Noordermeer D, Peng M-W, Piersigilli A, Perren A, Zlobec I, et al. (2018). GKAP Acts as a Genetic Modulator of NMDAR Signaling to Govern Invasive Tumor Growth. Cancer Cell 33, 736–751. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz-Lopez A, Kilcoyne A, Paoluzzi-Tomada E, Weissleder R, Mathis D, et al. (2018). Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc. Natl. Acad. Sci. USA 115, E10672–E10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkouk A, and Weiner GJ (2015). Cancer immunotherapy and breaking immune tolerance: new approaches to an old challenge. Cancer Res. 75,5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, et al. (2013). Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J. Clin. Invest 123, 2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 11, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen J, Nordhausen K, and Taskinen S (2017). Blind Source Separation Based on Joint Diagonalization in R: The Packages JADE and BSSasymp. Journal of Statistical Software 76. [Google Scholar]
- Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, Whibley N, Tucci A, Chen X, Lindeman I, et al. (2019). Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity 50, 493–504. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. (2017). Interferon-γ Drives Treg Fragility to Promote Anti-tumor Immunity. Cell 169, 1130–1141. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M, Benoist C, and Mathis D (2016). Tissue Tregs. Annu. Rev. Immunol 34, 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastille E, Wasmer M-H, Adamczyk A, Vu VP, Mager LF, Phuong NNT, Palmieri V, Simillion C, Hansen W, Kasper S, et al. (2019). The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 12, 990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, and Rudensky AY (2016). Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 45, 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau A, Gallopin M, Celeux G, and Jaffrézic F (2013). Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 29, 2146–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K, Valmori D, and Ayyoub M (2012). CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res. 72, 4351–4360. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. [DOI] [PubMed] [Google Scholar]
- Rutledge DN, and Jouan-Rimbaud Bouveresse D (2013). Independent Components Analysis with the JADE algorithm. Trends Analyt. Chem 50, 22–32. [Google Scholar]
- Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, et al. (2016). Two FOXP3(+) CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med 22, 679–684. [DOI] [PubMed] [Google Scholar]
- Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, and Jacks T (2014). Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Y, Phan JH, and Wang MD (2015). Effect of low-expression gene filtering on detection of differentially expressed genes in RNA-seq data. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2015, 6461–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. (2013). Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang B, Liu Y, Jiang S-J, and Liu Y (2015). Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci. Rep 5, 15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med 210,1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2017). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 171, 1221–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, and Shree R (2016). Analysis and effects of speckle noise in SAR images. In Proceedings of the 2016 2nd International Conference on Advances in Computing, Communication, Automation (ICACCA), pp. 1–5. [Google Scholar]
- Soria J-C, Marabelle A, Brahmer JR, and Gettinger S (2015). Immune checkpoint modulation for non-small cell lung cancer. Clin. Cancer Res 21, 2256–2262. [DOI] [PubMed] [Google Scholar]
- Stubbington MJT, Lönnberg T, Proserpio V, Clare S, Speak AO, Dougan G, and Teichmann SA (2016). T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods 13, 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kadota K, Sima CS, Nitadori J, Rusch VW, Travis WD, Sadelain M, and Adusumilli PS (2013). Clinical impact of immune microenvironment in stage I lung adenocarcinoma: tumor interleukin-12 receptor β2 (IL-12Rβ2), IL-7R, and stromal FoxP3/CD3 ratio are independent predictors of recurrence. J. Clin. Oncol 31, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TG, Mathis D, and Benoist C (2016). Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc. Natl. Acad. Sci. USA 113, 14103–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, and Sakaguchi S (2017). Regulatory T cells in cancer immunotherapy. Cell Res. 27, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, and Quake SR (2014). Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, et al. (2015). The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol 16, 276–285. [DOI] [PubMed] [Google Scholar]
- Wan YY, and Flavell RA (2005). Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. USA 102, 5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young NP, Crowley D, and Jacks T (2011). Uncoupling cancer mutations reveals critical timing of p53 loss in sarcomagenesis. Cancer Res. 71, 4040–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, et al. (2018). Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272. [DOI] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356. e16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the bulk and scRNA-Seq data reported in this paper is GEO: GSE139232. All code is available upon request.