

Abstract
Peters plus syndrome (MIM #261540 PTRPLS), characterized by defects in eye development, prominent forehead, hypertelorism, short stature and brachydactyly, is caused by mutations in the β3-glucosyltransferase (B3GLCT) gene. Protein O-fucosyltransferase 2 (POFUT2) and B3GLCT work sequentially to add an O-linked glucose β1-3fucose disaccharide to properly folded thrombospondin type 1 repeats (TSRs). Forty-nine proteins are predicted to be modified by POFUT2, and nearly half are members of the ADAMTS superfamily. Previous studies suggested that O-linked fucose is essential for folding and secretion of POFUT2-modified proteins and that B3GLCT-mediated extension to the disaccharide is essential for only a subset of targets. To test this hypothesis and gain insight into the origin of PTRPLS developmental defects, we developed and characterized two mouse B3glct knockout alleles. Using these models, we tested the role of B3GLCT in enabling function of ADAMTS9 and ADAMTS20, two highly conserved targets whose functions are well characterized in mouse development. The mouse B3glct mutants developed craniofacial and skeletal abnormalities comparable to PTRPLS. In addition, we observed highly penetrant hydrocephalus, white spotting and soft tissue syndactyly. We provide strong genetic and biochemical evidence that hydrocephalus and white spotting in B3glct mutants resulted from loss of ADAMTS20, eye abnormalities from partial reduction of ADAMTS9 and cleft palate from loss of ADAMTS20 and partially reduced ADAMTS9 function. Combined, these results provide compelling evidence that ADAMTS9 and ADAMTS20 were differentially sensitive to B3GLCT inactivation and suggest that the developmental defects in PTRPLS result from disruption of a subset of highly sensitive POFUT2/B3GLCT targets such as ADAMTS20.
Introduction
Protein glycosylation encompasses a diverse set of post-translational modifications with an extraordinary breadth of impact on the modified proteins, including their biophysical properties, intermolecular interactions and folding (1). Key roles for specific glycosyltransferases have been revealed by a range of congenital disorders of glycosylation (2). Among them, Peters plus syndrome (PTRPLS, MIM#261540) is a rare genetic disorder characterized by the presence of anterior eye segment abnormalities (typically Peters anomaly), dysmorphic facial features, short stature, brachydactyly and developmental delay (3). Peters eye anomaly is a fusion of the cornea and lens due to persistence of the embryonic lens stalk, and it results in corneal opacity, with severe visual impairment and other eye complications. Children affected by PTRPLS also frequently have additional congenital defects including cleft palate and cardiovascular and urogenital malformations (4–9). Increased rates of miscarriage in the second and third trimesters in PTRPLS families suggest that PTRPLS can also lead to intrauterine fetal death (10). Consistent with this prediction, postmortem analysis of PTRPLS fetuses identified additional abnormalities including growth retardation, hydrocephalus, agenesis of the corpus callosum, Dandy-Walker cyst and gut anomalies (11).
PTRPLS is caused by loss-of-function mutations in the β3-glucosyltransferase gene (B3GLCT, previously named B3GALTL) (5, 6, 8, 9). B3GLCT works together with protein O-fucosyltransferase-2 (POFUT2) to add an O-linked glucose-β1-3fucose disaccharide to thrombospondin type I repeats (TSRs) (12). In the endoplasmic reticulum (ER), POFUT2 recognizes its consensus sequence in the context of a properly folded TSR (C1-X-X-S/T-C2 for group 1 TSRs or C2-X-X-S/T-C3 for group 2 TSRs, where Cs are conserved cysteines) and then utilizes guanosine diphosphate-L-fucose as a sugar donor to add O-linked fucose to the serine or threonine (S/T) in the consensus sequence (13,14). B3GLCT then transfers glucose to the O-fucosylated TSRs to form the disaccharide (15,16).
The POFUT2 O-fucosylation consensus site is present in just 49 TSR-containing human proteins. All these predicted targets are secreted or membrane-anchored proteins that modulate properties of the extracellular matrix (ECM) or cell/matrix interactions (17). Members of the ADAMTS (A Disintegrin-like And Metalloproteinase domain with ThromboSpondin type 1 motifs) superfamily comprise nearly 50% of POFUT2 targets. The various secreted ADAMTS proteases and non-catalytic members of the family (ADAMTS-like proteins) are implicated in controlling the structural properties of the ECM, organogenesis, tissue organization and cell signaling (reviewed in 18–21) as well as in the genesis of several major adult-onset disorders (21). Cell-based secretion assays and high-performance liquid chromatography (HPLC)-based in vitro folding and unfolding assays provided evidence that the O-linked fucose disaccharide stabilizes the TSR fold and accelerates the overall rate of TSR folding in the ER, thus promoting secretion of the POFUT2/B3GLCT target proteins (12). This model is supported by the observation that the disaccharide functions as a surrogate amino acid that makes contacts with neighboring amino acids in a TSR (22), suggesting a structural role for the disaccharide (reviewed in 23). All targets tested in vitro to date require O-fucose for secretion, whereas only a subset of targets is sensitive to the failure to extend O-fucose with glucose (12, 24–29). This observation combined with the marked difference between the Pofut2 mouse knockout (early embryo lethal due to loss of ADAMTS9 function (24, 30)) and PTRPLS phenotypes suggests that the congenital abnormalities present in PTRPLS patients result from reduced function of a subset of POFUT2/B3GLCT targets that are highly sensitive to absence of the glucose caused by loss of B3GLCT (12). We predicted that POFUT2/B3GLCT targets implicated in single gene defects, which have eye, palate, cardiovascular, and/or skeletal abnormalities, will be sensitive to B3GLCT loss (Table 1). To determine whether loss of B3GLCT differentially affects the function of POFUT2/B3GLCT targets and gain insight into the origin of the developmental defects present in PTRPLS, we developed and characterized two mouse B3glct knockout alleles. Combined, results from phenotypic and genetic analyses of B3glct mutants, mass-spectral analysis and cell-based secretion assays provided compelling evidence that ADAMTS9 and ADAMTS20, two closely related POFUT2/B3GLCT-modified proteins, were differentially sensitive to B3GLCT inactivation. We suggest that the developmental defects in PTRPLS result from disruption of a subset of highly sensitive POFUT2/B3GLCT targets such as ADAMTS20.
Table 1.
Single-gene mutations in POFUT2/B3GLCT targets causing a subset of defects in PTRPLS
| System affected in PTRPLS | Human | Mouse and other species |
|---|---|---|
| Ocular | ADAMTS9 (59), ADAMTS10 (60), ADAMTS17 (61), ADAMTS18 (62, 63), ADAMTSL1 (64), ADAMTSL4 (65, 66) | Adamts9 (25), Adamts10 (67, 68), Adamts17 (69,70), Adamts18 (71), Adamtsl4 (72) |
| Craniofacial | ADAMTS3 (51), ADAMTSL1 (64), ADAMTSL2 (73) | Adamts9 (34), Adamts20 (34), Ccn2 (74, 75) |
| Skeletal | ADAMTS10 (60), ADAMTS17 (61), ADAMTSL1 (64), ADAMTSL2 (73) | Adamts10 (67), Adamts17 (69, 70), Adamtsl2 (76, 77), Ccn1 (78, 79), Ccn2 (74), Tsp1 (80) |
| Genitourinary | ADAMTSL1 (64), ADAMTS9 (59) | Adamts1 (81), Adamts4 (82), Adamts18 (71) |
| Cardiac | ADAMTS10 (60), ADAMTS17 (61), ADAMTSL2 (73), THSD1 (83) | Adamts1 (84), Adamts5 (85), Adamts6 (86), Adamts9 (87), Ccn1 (78, 79). |
Results
B3glct mutations caused craniofacial changes and shortening of long bones and digits in mice
To begin to understand the mechanism by which loss of B3GLCT leads to PTRPLS symptoms, we generated two mouse lines predicted to disrupt B3GLCT function (Supplementary Material, Figs S1 and S2 and Supplementary Material, Tables S1 and S2). Using these independent knockout lines allowed us to establish that differences between the mouse and human phenotypes were specifically related to disruption of B3glct in mouse and not attributed to potential off-target effects of the specific mutation. The B3glct-LacZ-Δ4 allele was generated by deleting exon 4 and insertion of a LacZ reporter. This allele is comparable to two human PTRPLS mutations that introduce premature stop codons in exon 4 (9). The B3glct-Δ11-12 allele resulted from removal of exons 11 and 12 that encode the DDD amino acids essential for sugar transfer catalysis by B3GLCT (15, 16). Homozygotes of both alleles were viable, although slightly underrepresented (Supplementary Material, Table S3). Homozygotes were consistently smaller than littermates at weaning, with a greater effect seen in females (Fig. 1A and B and Supplementary Material, Fig. S3).
Figure 1.
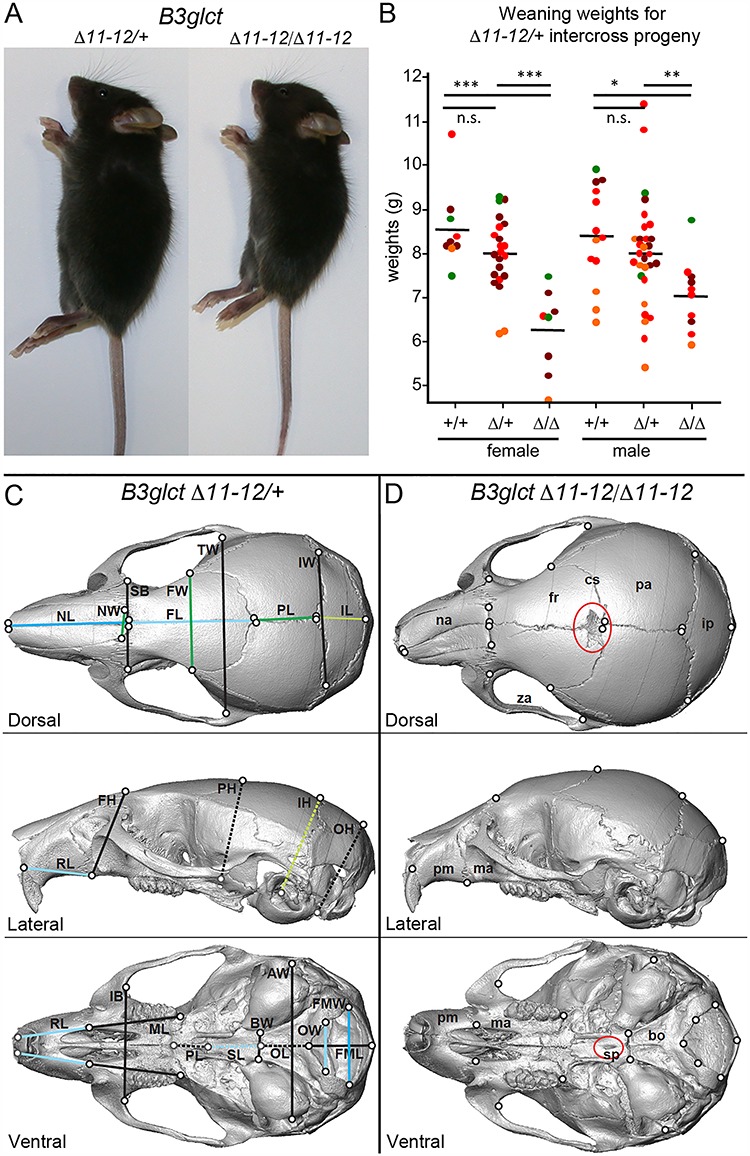
B3glct-Δ11–12 mutants were runted and have craniofacial changes relevant to PTPLS. (A) Comparison of 6-week B3glct-Δ11-12 heterozygous control (left) and B3glct-Δ11-12 mutants (right) and (B) weaning weights from intercross progeny. The mean weaning weight is indicated by horizontal bar. Significance of weaning weight differences was evaluated using unpaired, one-tailed t-test; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, n.s. not significant. Postnatal dates at which weights were measured are indicated by color; green, P23; brown, P22; red, P21 and orange, P20. Refer to Supplementary Material, Figure S3 for B3glct-LacZ-Δ4. (C, D) Micro-CT-based comparison of 6-week-old skull surfaces of B3glct-Δ11-12 heterozygotes (C) and homozygotes (D) from dorsal, lateral and ventral views. Black outlined circles indicate the position of identified landmarks on the surface of the skull, while gray outlined circles indicate landmarks that are deep to the surface being displayed (Supplementary Material, Fig. S4 and Supplementary Material, Table S4). Lines indicate a subset of linear distances compared between genotypes using Wilcoxon tests, with dashed lines connecting landmarks that are located within the displayed surface. Blue lines indicate distances reduced in homozygotes compared to heterozygotes, and green lines indicate that distances expanded in homozygotes relative to heterozygotes. Dark blue and dark green linear distances are significantly different between genotypes after Bonferroni correction for multiple testing, while light blue and green are significantly different before Bonferroni correction. Red ovals highlight abnormal features mentioned in the text. Uppercase abbreviations label linear distances (Supplementary Material, Table S5). Lowercase abbreviations: na, nasal; fr, frontal; cs, coronal suture; pa, parietal; ip, interparietal; za, zygomatic arch; pm, premaxilla; ma, maxilla; sp, sphenoid; bo, basioccipital.
We used landmark-based morphometric analyses of skull microcomputed tomography (μCT) images to determine whether loss of B3GLCT caused craniofacial changes similar to those observed in PTRPLS patients (Fig. 1C and D and Supplementary Material, Figs S4–S6 and Supplementary Material, Table S4). Comparison of overall skull shape indicated that skulls in 3- and 6-week-old B3glct-Δ11-12 homozygotes were relatively shorter, taller and wider compared to littermate controls (Supplementary Material, Figs S5 and S6 and Supplementary Material, Table S6). By comparing absolute linear distances between selected landmarks (Supplementary Material, Tables S4 and S5), we determined that 3-week-old mutants had shorter faces, cranial vaults and cranial bases along the anterior–posterior axis when compared to controls (Supplementary Material, Figs S5 and S6 and Supplementary Material, Table S5). Between 3 and 6 weeks of age, the faces of control mice took on the adult morphology of longer faces relative to skull height (Fig. 1C). In comparison, the 6-week mutant skulls retained a more juvenile appearance of a relatively short face and round skull (Fig. 1D). Similar shortened and round skulls with shortened nasal and frontal bones were observed in Spry2 loss-of-function mutations (31) and chondrocyte-specific activation of FGFR2 (Col2a1- Fgfr2P253R) (32).
Mutations in several ADAMTS proteins are known to influence the assembly and abundance of fibrillin microfibrils and could affect cell signaling pathways mediated by members of the TGF-β superfamily, which are known to bind these microfibrils (21,33). This is one potential mechanism whereby changes in the matrix properties could influence signaling during skull development. When linear distances were compared between landmarks, B3glct mutants had absolutely shorter anterior facial, frontal and sphenoid bone lengths combined with an expansion and rounding of the cranial vault with increases in the vault length and height (Fig. 1C and D and Supplementary Material, Table S5). Furthermore, greater width of the nasal (NW) and frontal bones (FW) and wider (although not significantly) superior bicanthic breadth (SB) in 6-week mutants are analogous to hypertelorism noted in PTRPLS patients.
Other shared features of the two lines of B3glct mutant mice not observed in control animals included squared nasal bones (18/18), a lack of frontal bone ossification near the midline coronal suture (18/18), a hole in the center of sphenoid bone (14/18) and nasal asymmetry (5/18) (Fig. 1D and Supplementary Material, Fig. S6). Nasal asymmetry was also associated with asymmetry in other parts of the face, including the upper jaw and zygomatic arch. A similar hole in the sphenoid bone was described in Adamts20-bt/bt;Adamts9-del/+ neonates (34). Bent nasal bone morphology was noted in several mouse models. These include mutations in Papps2, which causes a major reduction in the growth of the chondrocranium (35), a dominant Fgfr2P244R mutation, which activates FGF signaling and caused premature fusion of the facial sutures, and bridged or closed spheno-occipital synchondrosis (36) and mutations that alter euchromatin histone methylation (37).
Comparison of skeletal preps from B3glct mutants and littermates illustrated the negative effects of the B3glct-Δ11-12 mutation on bone growth (n = 6 pairs) (Fig. 2A and B and Supplementary Material, Fig. S7). At 6 weeks of age, the skeletons of B3glct mutants were normal with respect to patterning, but visibly smaller than wild-type littermates (Fig. 2A). The mutant skulls frequently maintained the more juvenile upturned snout (3/4) (Fig. 2A, right). The long bones and digits of mutants were shorter than littermate controls (Fig. 2B and Supplementary Material, Fig. S7). The reduced size also extended to other bones (i.e. scapula, Fig. 2B, top). Histological comparison of proximal tibial growth plates from 21-day-old females revealed well organized but slightly thinner growth plates in B3glct-Δ11-12 homozygotes compared to littermates (Fig. 2C–H′).
Figure 2.
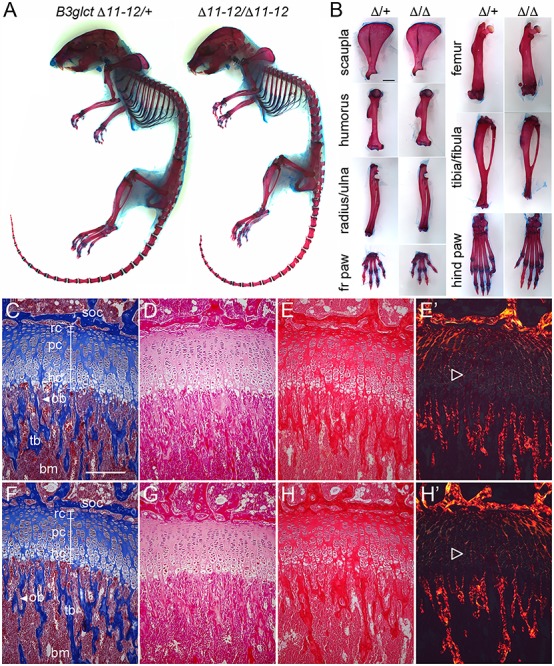
B3glct mutants had shorter bones with well-organized growth plates. (A) Comparison of skeletal preparations and (B) isolated individual bones from 48-day-old female littermates, B3glct-Δ11-12/+ (left) and B3glct-Δ11-12/Δ11–12 (right). Comparison of average bone measurements from four pairs of females is shown in Supplementary Material, Figure S7. (C–H′) Comparison of B3glct-Δ11-12/+ (C–E′) and B3glct-Δ11–12/Δ11–12 (F–H′) tibia growth plates from 21-day-old females stained with Masson trichrome (C and F), hematoxylin and eosin (D and G), picrosirius red (E and H) or picrosirius red visualized under polarized light (E′ and H′). Growth plates are indicated by white brackets and consist of resting chondrocytes (rc), proliferating chondrocytes (pc) and hypertrophic chondrocytes (hc). Secondary ossification (SOC), trabecular bone (tb) and bone marrow (bm) are indicated. Open triangles indicate collagen networks in growth plate, visualized under polarized light. On average, growth plates were 15% narrower in female homozygotes compared to controls (unpaired, one-tailed t-test P = 1.4 E−10), and 17% narrower in male homozygotes compared to controls (unpaired, one-tailed t-test P = 3.3 E−12). Scale bar in (B) is 0.2 mm and (C) is 0.1 mm.
Loss of B3glct combined with reduced Adamts9 caused eye malformations
In contrast to PTRPLS patients (9) or Adamts9 del/+ heterozygous mice (25), we did not observe corneal opacity indicative of defects in the anterior chamber in B3glct mutants. Neither B3glct-Δ11-12 nor LacZ-Δ4 homozygotes developed corneal opacity (at 3–6 weeks of age), and optical computed tomography (OCT) analysis of two mutants showed normal anterior chambers (Fig. 3A–D). This contrasts with the observation that animals heterozygous for an Adamts9 deletion (Adamts9 del/+) have congenital corneal opacity resulting from anterior segment defects, including Peters anomaly (25). Eye defects were also not reported in zebrafish B3glct mutants (38), suggesting that vertebrate species have different sensitivities to loss of B3GLCT. Since ADAMTS9 is modified by POFUT2 and B3GLCT, and siRNA reduction of B3GLCT partially reduces the amount of secreted ADAMTS9 in cell-based secretion assays (25), the lack of overt eye phenotypes in B3glct mutants suggested preservation of greater than 50% ADAMTS9 function (i.e. greater than heterozygous levels).
Figure 3.
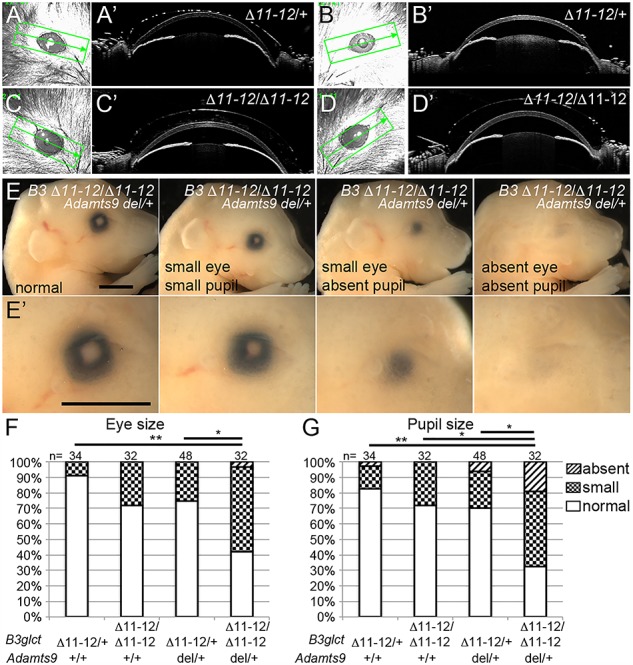
Genetic reduction of Adamts9 in B3glct mutants impaired eye development.
(A–D′) Comparison of eyes from B3glct-Δ11-12 heterozygotes (Δ11–12/+) (A-B′) and homozygotes (Δ11–12/Δ11-12) (C–D′) using OCT at 21 days of age. Scanned region is indicated by rectangle in (A–D). A′–D′ OCT sections at level indicated by the line bisecting rectangle panels in A–D. (E, E′) Range of eye phenotypes observed in E18.5 B3glct-Δ11–12 homozygotes missing one copy of Adamts9 (B3glct-Δ11-12/Δ11-12;Adamts9 del/+). Eye size was determined by measuring maximum pigmented diameter and pupil size by measuring maximum pupil diameter. (F, G) Comparison of the effects of reducing Adamts9 in B3glct-Δ11-12 heterozygotes and homozygotes on eye (F) and pupil (G) size. Eye size was categorized as small if measurements were less than 1 standard deviation from the mean of B3glct-Δ11-12 heterozygous controls. *P ≤ 0.05, **P ≤ 0.01 using Chi-square test.
We hypothesized that genetic reduction of Adamts9 in B3glct mutants would further reduce ADAMTS9 levels and could exacerbate the B3glct mutant phenotype. Consistent with this prediction, reducing one copy of Adamts9 in B3glct mutants (B3glct-Δ11-12/Δ11-12;Adamts9-del/+) caused neonatal lethality with 100% penetrance of cleft palate (Supplementary Material, Fig. S8 and Supplementary Material, Table S7). Moreover, late gestation embryos had a significant increase in eye abnormalities, ranging from small eye and/or pupil to absent pupil and/or eye, compared to littermates (Fig. 3E–G). In comparison to the previous observed eye anomalies in Adamts9 hemizygous mutants (25), concomitant B3glct deletion led to marked worsening of the eye phenotype, which could reflect a further reduction of ADAMTS9 function in the eye or an impact on another POFUT2/B3GLCT targets such as ADAMTS10, ADAMTS17, ADAMTS18, ADAMTSL1 and/or ADAMTSL4 (Table 1). Notably, cleft palate was previously observed in 100% of Adamts20-bt/bt mice heterozygous for mutations in Adamts9 (34,39). For this reason, cleft palate in B3glct/Adamts9 mutants suggests that loss of B3glct markedly reduces ADAMTS20 function.
Belting, hydrocephalus and soft tissue syndactyly provided evidence for impaired ADAMTS20 function in B3glct mutants.
Loss-of-function mutations in Adamts20 (Adamts20 bt/bt) cause the mouse belted phenotype (40,41) (Fig. 4A and A′), a white spotting defect that is considerably enhanced by concomitant deletion of one Adamts9 allele (39,41). Consistent with the prediction that loss of B3glct impairs ADAMTS20 function, we observed highly penetrant white spotting in the lumbar region of B3glct mutants (Fig. 4B and B′). Ventral white spotting was present in all B3glct mutants, and the majority also showed some degree of dorsal white spotting (Fig. 4B and Supplementary Material, Fig. S9A). Since white spotting was not reported in Adamts20-bt/+ heterozygotes (40,41), and spotting was exacerbated by reduction of ADAMTS9 or in Adamts20-bt/bt (41), the spotting defects in B3glct mutants likely resulted from marked reduction of ADAMTS20 and/or ADAMTS9 function. This prediction was consistent with the observation that white spotting in B3glct mutants did not increase in severity when one copy of Adamt20 was removed. Notably, Adamts20 (4/9) and B3glct (11/20) mutants each developed hydrocephalus (determined by isolation and bisection of brain) between 3 and 6 weeks of age (Fig. 4C and D′), suggesting that loss of ADAMTS20 function significantly contributes to development of hydrocephalus in B3glct mutants.
Figure 4.

B3glct mutants displayed white spotting, hydrocephalus and soft tissue syndactyly similar to Adamts20 bt/bt mutants. (A, A′) Typical pigmentation deficit seen in Adamts20 bt/bt animals (no spotting was observed in heterozygotes, not shown). (B, B′) Range of ventral and dorsal lumbar white spotting observed in wild-type controls (+/+) compared to B3glct-LacZ-Δ4 mutants. Similar white spotting is observed in B3glct-Δ11-12 homozygotes (not shown). Frequency and distribution of white spotting are shown in Supplementary Material, Figure S9A. (C, D′) hydrocephalus was observed in 4 of 9 Adamts20 bt/bt (C, C′), 5 of 8 B3glct-Δ11-12 mutants (D, D′) and 5 of 11 LacZ-Δ4/LacZ-Δ4 (not shown) dissected and bisected brains. Representative whole brains (C, D) were bisected and anterior halves (C′, D′) are shown. Scale bars are 2 mm. (E) Range of soft tissue syndactyly observed in B3glct-Δ11-12 homozygotes in the forelimb (above) and hindlimb (below). Scoring system used to quantitate soft tissue syndactyly (no syndactyly, 0; or severity of syndactyly, a–c) was previously described (42). Frequency and severity of syndactyly across all interdigital spaces are shown in Supplementary Material, Figure S9B and C. Scale bars are 5 mm.
In addition, we observed soft tissue syndactyly in front and rear paws of B3glct mutants (Fig. 4E and Supplementary Material, Fig. S9B and C). In contrast to B3glct mutants, Adamts20 mutants were reported to have exclusively front paw syndactyly (42). The observation that most B3glct mutants had some degree of soft tissue syndactyly was consistent with functional reduction of ADAMTS20, ADAMTS9 and/or ADAMTS5, as similar penetrance of soft tissue syndactyly was reported for Adamts20-bt/bt mutants lacking one copy of Adamts9 or null for Adamts5 (42).
In B3glct/Adamts20 intercrosses, the B3glct-Δ11-12/Δ11-12;Adamts20-bt/+ animals were underrepresented and B3glct-Δ11-12/Δ11-12;Adamts20-bt/bt animals were absent (Supplementary Material, Table S8). To Investigate why B3glct/Adamts20 double homozygotes were missing, we isolated litters at E18.5 from B3glct-Δ11-12/+;Adamts20-bt/bt X B3glct-Δ11-12/+;Adamts20-bt/+ crosses and E19.5 B3glct-Δ11-12/+;Adamts20-bt/bt intercrosses. In these litters, we observed cleft palate in B3glct-Δ11-12/Δ11-12;Adamts20-bt/bt (9/11) and B3glct-Δ11-12/Δ11-12;Adamts20-bt/+ (1/4) embryos (Supplementary Material, Fig. S8B and Supplementary Material, Tables S9 and S10). In contrast, cleft palate was not present in any of the 13 embryos homozygous for the Adamts20-bt mutation (it has been previously seen in only 3% of bt/bt mice (34)). This observation demonstrated that loss of B3glct simultaneously impairs function of multiple ADAMTS members, and in the case of the palate, likely ADAMTS9 and ADAMTS20, as cleft palate was previously noted in Adamts20 mutants having one copy of inactivated Adamts9 (34) or the hypomorphic Adamts9-Gt/+ mutation (39). However, since not all B3glct/Adamts20 double homozygotes developed cleft palate, defects in other organ systems such as the heart and lungs could likely contribute to lethality of B3glct/Adamts20 double homozygotes.
Loss of B3glct differentially affected ADAMTS9 and ADAMTS20 secretion
The abnormalities present in B3glct mutants and compound B3glct/Adamts9 or B3glct/Adamts20 mutants provided evidence that loss of B3GLCT significantly impacted ADAMTS20 function but had only limited effects on ADAMTS9. ADAMTS9 and ADAMTS20 are highly conserved, sharing similar domain organization, high sequence similarity and ability to cleave versican (43) (compare Fig. 5A and B), prompting further investigation into a possible differential sensitivity to loss of B3GLCT. Potential differences in sensitivity could result from differences in TSR modification by POFUT2/B3GLCT; for example, ADAMTS9-TSR10 and ADAMTS20-TSR14 lack a POFUT2 consensus sequence (Fig. 5A and B). Alternatively, different sensitivities to loss of B3GLCT could be due to inherent differences in TSR stability that make them more or less dependent upon the disaccharide for secretion (12).
Figure 5.

Loss of B3GLCT differentially affected secretion of ADAMTS9 and ADAMTS20. (A, B) Cartoons illustrate the similar domain structure of ADAMTS9 (A) and ADAMTS20 (B). Each protein contains 15 TSRs that are depicted by ovals. Red ovals indicate TSRs that have consensus sequences for POFUT2 mediated O-fucosylation (C1-X-X-S/T-C2 for the Group 1 TSR in ADAMTS9 and 20). Gray ovals are TSRs without a consensus sequence for O-fucosylation. The presence of glucoseβ1-3fucose disaccharide on TSRs of ADAMTS9 was previously confirmed by mass spectral analysis (25), and these modifications are indicated above ADAMTS9 TSRs by a solid red triangle for fucose and solid blue circle for glucose. Mass spectral analysis of ADAMTS20 TSRs 1 and 2–8 is described in Supplementary Material, Figure S10. TSRs 1, 4, 5, 7 and 8 were stoichiometrically modified with fucose and glucose and the O-linked disaccharide. The half-filled blue circle above TSR6 indicates that only 50% of O-linked fucose (solid red circle) on this TSR was extended with glucose. Quantification of the efficiency of fucose and glucose addition is shown in Fig. S10G. (C–F) Cell-based secretion assays to measure the effects of POFUT2 and B3GLCT mutations on secretion of ADAMTS9 TSR 2-8-Myc-His6 (C, E) or ADAMTS20 TSR 2-8-Myc-His6 (D,F) in wild-type HEK293T, CRISPR-Cas9 POFUT2 or CRISPR-Cas9 B3GLCT mutagenized HEK293T cells or mutagenized cells rescued by co-transfection with plasmids encoding wild-type POFUT2 or B3GLCT. (C, D) Representative western blots of the medium (above) and cell lysate (below); the red channel is anti-Myc and the green channel is anti-Ig used as a transfection control. (E, F) Quantification of western blots of medium (above) and cell lysate (below). Data were evaluated for statistical significance using unpaired, one-tailed t-test. *P ≤ 0.05, **P ≤ 0.01, n.s. not significant.
To evaluate the effects of glucosylation on ADAMTS9 and ADAMTS20 function, we first examined whether the TSRs are modified with O-fucose glycans. All ADAMTS9 TSRs with a POFUT2 consensus sequence were previously shown to be modified with the glucose-β1-3fucose disaccharide (25). We used a similar approach to determine whether ADAMTS20 TSR 2-8 and TSR1 were similarly modified with fucose and glucose (Supplementary Material, Fig. S10). These fragments were examined because insufficient full-length ADAMTS20 was secreted for mass spectral analysis. A summary of modifications detected is shown in Fig. 5B. With the exception of ADAMTS20 TSR6 where the O-fucose was only partially (50%) extended to the disaccharide, the remaining TSRs containing POFUT2 consensus sequences were fully O-fucosylated and elongated to the disaccharide by B3GLCT (Fig. 5B and Supplementary Material, Fig. S10A–G).
To test if loss of the disaccharide directly affected secretion of ADAMTS9 and 20, we evaluated the secretion of TSRs 2 through 8 (TSR 2–8) from these targets in HEK293T cells in which POFUT2 or B3GLCT was inactivated by CRISPR-Cas9 (24,26). Both ADAMTS9 and ADAMTS20 TSRs 2–8 were efficiently secreted from HEK293T wild-type cells (Fig. 5C–F). In contrast, knockout of POFUT2 completely blocked secretion of both ADAMTS9 and 20 TSRs 2–8. Secretion was restored by co-transfection with POFUT2 (Fig. 5C–F). Therefore, both ADAMTS9 and ADAMTS20 required the O-fucose for secretion. These results are consistent with results from evaluation of ADAMTS9 secretion in POFUT2 siRNA knockdown experiments (25). In contrast, the loss of B3GLCT had contrasting effects on ADAMTS9 and ADAMTS20 secretion. In B3GLCT mutant cells, ADAMTS9 secretion was reduced to 80% of wild-type (Fig. 5C and E). In contrast, loss of B3GLCT blocked ADAMTS20 secretion, and secretion was restored by co-transfection with B3GLCT (Fig. 5D and F). Since ADAMTS9 and 20 TSRs 2–8 have similar patterns of O-linked disaccharide modifications, these observations strongly suggested that inherent sequence differences between these proteins made a major contribution to differences in sensitivities to loss of B3GLCT.
Discussion
PTRPLS is an autosomal recessive disorder caused by mutations in B3GLCT (6,8,9,44). Classic PTRPLS patients have anterior eye chamber, craniofacial and skeletal abnormalities (3). In this study, we demonstrated that the mouse B3glct knockout models display craniofacial and skeletal abnormalities analogous to PTRPLS patients (analysis of B3glct alleles summarized in Supplementary Material, Table S11). In addition, these mutants exhibit a high penetrance of hydrocephalus, white spotting in the lumbar region and soft tissue syndactyly in B3glct mutants. While hydrocephalus is not a defining characteristic of PTRPLS patients, it was recently described in fetuses with PTRPLS (11), and there is a high rate of miscarriage/stillbirth reported in some PTRPLS families (GeneReviews, http://www.genereviews.org/10, 45)).
The presence of hydrocephalus in Adamts20 mutants (Fig. 4) suggested that a reduction of ADAMTS20 contributes significantly to development of hydrocephalus in mouse B3glct mutants and PTRPLS patients. Using genetic interaction studies (removing either Adamts9 or Adamts20 in B3glct mutants), we demonstrated that loss of B3GLCT sensitized Adamts9-del/+ and Adamts20-bt/bt embryos to development of cleft palate (Supplementary Material, Tables S7, S9 and S10). Overall, the lack of eye defects and presence of white spotting and soft tissue syndactyly in B3glct mutants were consistent with a partial loss of ADAMTS9 function and a more significant reduction in functional ADAMTS20. Results from cell-based secretion assays suggested that this difference was due to distinct effects of the B3GLCT mutation on secretion of ADAMTS9 and ADAMTS20 (Fig. 5). This observation was striking considering the similar domain organization and O-fucosylation patterns of ADAMTS9 and ADAMTS20 (Fig. 5A and B).
POFUT2-mediated O-linked fucosylation is unusual compared to other forms of glycosylation in that it requires a folded TSR as the substrate. The observation that the O-linked disaccharide interacts directly with the C2-C6 disulfide bond of the ADAMTS13 TSR (22) and raises the possibility that the disaccharide functions as a surrogate amino acid to stabilize the newly folded TSR in the ER. Stabilization would prevent re-entry of the protein into the folding cycle and promote its secretion (23). The phenotypic similarities between B3glct and ADAMTS mutants combined with the results from secretion assays suggested that fucosylated ADAMTS20 folds less efficiently compared to ADAMTS9. We hypothesize that one or more ADAMTS20 TSRs are inherently less stable than ADAMTS9 TSRs and require(s) the disaccharide for efficient folding. This prediction is consistent with the observation that O-fucosylated TSRs unfold significantly slower than unmodified modules and that the addition of glucose has a further stabilizing effect on the TSR (12).
The distinct sensitivities of ADAMTS9 and ADAMTS20 to loss of B3GLCT raise the possibility that the developmental defects in PTRPLS patients and B3glct mutants could result from reduced function of sensitive target proteins or alternatively could result from unresolved response to the accumulation of misfolded POFUT2/B3GLCT targets. However, the normal arrangement of cells and collagen matrix organization in the growth plates of B3glct mutants (Fig. 2) contrasted markedly with the disorganized growth plates observed in Col2a1 or Col10a1 (13del) mutants, in which protein misfolding contributes to skeletal growth abnormalities (46–48). This difference in growth plate organization suggested that selective loss or reduction of target protein function rather than effects of protein misfolding was responsible for growth defects in B3glct mutants.
Theoretically, the PTRPLS/B3glct mutant symptoms could result from the combined effects of loss or reduced function of up to 49 target proteins that contain consensus sites for O-fucose modification by POFUT2. In reality, the developmental defects likely result from dysfunction of a limited number of targets that are sensitive to the inability to extend the O-linked fucose to the disaccharide. This hypothesis was supported by the observation that ADAMTS9 and ADAMTS20 secretion were differentially affected by loss of B3GLCT (Fig. 5). The argument for a small number of affected targets is also supported by the absence of phenotypes associated with single mutations in several POFUT2/B3GLCT target proteins in PTRPLS patients or B3glct mutants. For example, loss of B3GLCT does not result in thrombotic thrombocytopenic purpura caused by ADAMTS13 deficiency (49, 50), lymphangiectasia-lymphedema caused by ADAMTS3 deficiency (51) or dermatosparaxis caused by ADAMTS2 deficiency in humans (52) and other species such as sheep (53). We predict that the POFUT2/B3GLCT targets most likely to be affected by loss of B3GLCT in PTRPLS patients include those listed in Table 1, where single gene mutations cause abnormalities related to PTRPLS. The inherent differential sensitivity of ADAMTS9 and ADAMTS20 to loss of B3GLCT provides clues for studying the impairment of other target proteins in future.
Materials and Methods
Ethics statement
All animal work described herein was conducted according to relevant national and international guidelines and under approved protocols. Stony Brook University animal facilities were approved by the NIH Office of Laboratory Animal Welfare (OLAW), assurance number D-16-00006. The animal studies were approved by the Stony Brook University Institutional Animal Care and Use and Committee (IACUC), which follow all the guidance set forth in: Public Health Service Policy on Humane Care and Use of Laboratory Animals distributed by Office of Laboratory Animal Welfare, NIH; Animal Welfare Act and Animal Welfare Regulations distributed by United States Department of Agriculture and Guide for the Care and Use of Laboratory Animals distributed by the National Research Council. Stony Brook University animal facilities are accredited with AAALAC International (Association for the Assessment and Accreditation of Laboratory Animal Care International). B3glcttm1Nari alleles were generated at Laboratory Animal Resource Center of the University of Tsukuba. All experiments in the University of Tsukuba were approved by the Institutional Animal Care and Use Committees of the University of Tsukuba and were conducted according to related guidelines and applicable laws in Japan.
Mice and genotyping
Maps of B3glct alleles with locations of primers used for genotyping are depicted in Supplementary Material, Figs. S1 and S2. Details of B3glct targeting are expanded in Supplementary Material. For simplicity in the text, the null LacZ-reporter allele (B3glctTm1b(KOMP)Wtsi (MGI:6274693)) is referred to as B3glct-LacZ-Δ4 (Supplementary Material, Fig. S1). The null allele (B3glcttm1.3Nari (MGI:6277079)) is referred to as B3glct-Δ11-12 (Supplementary Material, Fig. S2). Primers and conditions for genotyping B3glct alleles are listed in Supplementary Material, Tables S1 and S2. All B3glct alleles were maintained at Stony Brook University by backcrossing to C57BL/6J. At weaning, diet was supplemented with DietGel® 76A (purified water, molasses, protein (animal or wheat), corn syrup, AIN mineral mix, food acids, hydrocolloids, vegetable oil, AIN vitamin mix, corn fiber), from ClearH20; Cat# 72-07-5022 to ensure that animals affected by craniofacial or skeletal abnormalities received adequate hydration and nutrition. Adamts9tm1.2Apte (Adamts9-del) (54) and Adamts20bt-Bei1 (Adamts20-bt) (40) null alleles were previously described and were maintained at Stony Brook University by backcross to C57BL/6J.
Histology and skeletal preparations
Bones isolated from 3- to 6-week-old mice (males and females) were fixed in 4% paraformaldehyde, decalcified using EDTA, dehydrated, embedded in paraffin, sectioned and stained using hematoxylin and eosin, Masson’s trichrome or picrosirius red. Sections were photographed using a Nikon Optiphot microscope (with or without polarizing filter), AxioCam MRc camera and AxioVisionLE program (Zeiss). ImageJ (http://imagej.net/) was used to measure the tibial proximal growth plates from 3-week-old males and females in Masson’s trichrome-stained tibial sections. Three measurements were taken on each of five 4 μm sections that were separated by 40 μm. Data were evaluated for significance using the Student t-test.
Skeletal preparations from 3- to 6-week-old mice were prepared as previously described (55). Skeletons and disarticulated bones were photographed in glycerol using a Zeiss Discovery V8 microscope, AxioCam MRc camera and AxioVisionLE program (Zeiss). Whole skeleton images were reconstructed from two or three images using Adobe Photoshop photomerge feature. For bone measurement, skeletons were disarticulated and individual limb bones were photographed and measured using ImageJ (http://imagej.net/). Data were evaluated for significance using the Student t-test.
Microcomputed tomography
Skulls of 3- and 6-week-old B3glct-Δ11-12 heterozygous (n = 8 at 3-week and n = 7 at 6-week) and homozygous mice (n = 8 at 3-week and n = 10 at 6-week) were fixed in 70% ethanol after removal of surrounding soft tissue. Micro CT images of ethanol-fixed skulls were collected using a Scanco μ40 with 55 kV/145 μA at 20mm3 voxel size. Craniofacial landmarks were collected as the basis for linear distance-based and Procrustes superimposition-based morphometric comparisons. Because of the small sample size, non-parametric Wilcoxon tests were completed to identify significant differences (α = 0.05) in skull linear distances between genotypes at both ages, with Bonferroni correction for multiple testing of linear distances. Additional morphometric method details and a description of Procrustes superimposition-based methods (31) are found in Supplementary Methods.
Optical computed tomography
Non-mydriatic anterior segment OCT images were taken using the Spectralis OCT2 SD-OCT (Heidelberg Engineering, Heidelberg, Germany) with the anterior segment lens. Immediately after sedation with xylazine/ketamine, and until imaging was completed, artificial tear drops (normal saline) were applied every 3–5 minutes to prevent corneal dehydration and cataract. Sedated animals were placed in a customized fixation tube on an adjustable height stage that kept the animal secure, comfortable and allowed for optimal tilt and angulation of the eye for imaging. OCT acquisition software settings were Application—Cornea, Preset—Dense and ART—30. Cross-sectional scans were obtained across the entire corneal surface along the horizontal meridian by aligning scan angle to coincide with the nasal and temporal canthus. After acquisition, each scan line of every image was reviewed for the presence of iris, lens, or corneal abnormalities.
Cell-based secretion assays and western blot analyses
Wild-type HEK293T and CRISPR-Cas9-generated POFUT2 and B3GLCT knockout HEK293T cells were validated, cultured, transfected and analyzed as previously described (24, 26), with additional details provided in the Supplementary Methods. Briefly, cells were transfected with a total of 1.54 μg of DNA including: 1.2 μg of pSecTag2 encoding human ADAMTS9-TSR2-8 or mouse Adamts20-TSR2-8 or 1.2 μg of empty vector, plus 0.24 μg of full-length pcDNA4 encoding mouse POFUT2 (30), 0.24 μg pcDNA3.1 encoding human full-length B3GLCT (16) or 0.24 μg of empty vector, plus 0.1 μg of plasmid encoding hIgG (56, 57). ADAMTS9 and ADAMTS20 expression plasmids used for transfection are described in Supplementary Methods. Three biological replicates were performed for each condition. For western analysis, ADAMTS proteins were detected using a 1:2000 dilution of anti-Myc antibody (9E10, Stony Brook University Cell Culture/Hybridoma facility) and 1:10000 dilution of the goat anti-mouse Alexafluor680 (Life Technologies) secondary antibody. IgG loading control was detected using IRDye800 goat anti-human IgG (Rockland Immunochemicals). The membrane was imaged and quantified using the Odyssey CLx system and analyzed with Image Studio Lite software (LI-COR Biosciences).
Analysis of ADAMTS20 O-fucosylation
Constructs encoding mouse Myc-His tagged ADAMTS20 TSR1 or ADAMTS20 TSR2-8 were transfected into HEK293T cells separately and the proteins purified from conditioned medium using Ni-NTA chelating chromatography resin (G-Biosciences, St. Louis, MO) as described (58). Purified proteins were subjected to in-gel or in-solution digestion as described (58), and the resulting peptides were injected into an EASY-nLC 1000 HPLC system (Thermo Fisher Scientific) for peptide separation using a C18 reverse-phase column attached to a Q EXACTIVE PLUS Orbitrap mass spectrometer (Thermo Fisher Scientific) for peptide detection (details in Supplementary Material). Extracted ion chromatograms of all identified (glyco) peptides were generated using Xcalibur v.4.0.27.19 (Thermo Fisher Scientific). Three biological replicates were performed.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We wish to thank the Holdener and Haltiwanger laboratory members for their suggestions for experiments and manuscript revisions, and Joel Israel from McClain Laboratories for technical assistance with bone histology.
Funding
National Institutes of Health (R01HD090156 and R01HD096030 to R.S.H. and B.C.H.); Georgia Research Alliance (to R.S.H.); National Institutes of Health (EY024943 to S.S.A.).
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1. Varki A. (2017) Biological roles of glycans. Glycobiology, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Freeze H.H., Chong J.X., Bamshad M.J. and Ng B.G. (2014) Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am. J. Hum. Genet., 94, 161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Schooneveld M.J., Delleman J.W., Beemer F.A. and Bleeker-Wagemakers E.M. (1984) Peters'-plus: a new syndrome. Ophthalmic Paed. Gen., 4, 141–145. [DOI] [PubMed] [Google Scholar]
- 4. Maillette de Buy Wenniger-Prick L.J. and Hennekam R.C. (2002) The Peters' plus syndrome: a review. Ann. Genet., 45, 97–103. [DOI] [PubMed] [Google Scholar]
- 5. Dassie-Ajdid J., Causse A., Poidvin A., Granier M., Kaplan J., Burglen L., Doummar D., Teisseire P., Vigouroux A., Malecaze F. et al. (2009) Novel B3GALTL mutation in Peters-plus syndrome. Clin. Genet., 76, 490–492. [DOI] [PubMed] [Google Scholar]
- 6. Hess D., Keusch J.J., Oberstein S.A., Hennekam R.C. and Hofsteenge J. (2008) Peters plus syndrome is a new congenital disorder of glycosylation and involves defective O-glycosylation of thrombospondin type 1 repeats. J. Biol. Chem., 283, 7354–7360. [DOI] [PubMed] [Google Scholar]
- 7. Heinonen T.Y. and Maki M. (2009) Peters'-plus syndrome is a congenital disorder of glycosylation caused by a defect in the beta1,3-glucosyltransferase that modifies thrombospondin type 1 repeats. Ann. Med., 41, 2–10. [DOI] [PubMed] [Google Scholar]
- 8. Lesnik Oberstein S.A., Kriek M., White S.J., Kalf M.E., Szuhai K., den Dunnen J.T., Breuning M.H. and Hennekam R.C. (2006) Peters plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet., 79, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weh E., Reis L.M., Tyler R.C., Bick D., Rhead W.J., Wallace S., McGregor T.L., Dills S.K., Chao M.C., Murray J.C. et al. (2014) Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin. Genet., 86, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hennekam R.C., Van Schooneveld M.J., Ardinger H.H., Van Den M.J., Friedburg D., Rudnik-Schoneborn S., Seguin J.H., Weatherstone K.B., Wittebol-Post D. and Meinecke P. (1993) The Peters'-plus syndrome: description of 16 patients and review of the literature. Clin. Dysmorphol., 2, 283–300. [PubMed] [Google Scholar]
- 11. Schoner K., Kohlhase J., Muller A.M., Schramm T., Plassmann M., Schmitz R., Neesen J., Wieacker P. and Rehder H. (2013) Hydrocephalus, agenesis of the corpus callosum, and cleft lip/palate represent frequent associations in fetuses with Peters' plus syndrome and B3GALTL mutations. Fetal PPS phenotypes, expanded by Dandy Walker cyst and encephalocele. Prenat. Diagn., 33, 75–80. [DOI] [PubMed] [Google Scholar]
- 12. Vasudevan D., Takeuchi H., Johar S.S., Majerus E. and Haltiwanger R.S. (2015) Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol., 25, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luo Y., Nita-Lazar A. and Haltiwanger R.S. (2006) Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J. Biol. Chem., 281, 9385–9392. [DOI] [PubMed] [Google Scholar]
- 14. Vasudevan D. and Haltiwanger R.S. (2014) Novel roles for O-linked glycans in protein folding. Glycoconj. J., 31, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kozma K., Keusch J.J., Hegemann B., Luther K.B., Klein D., Hess D., Haltiwanger R.S. and Hofsteenge J. (2006) Identification and characterization of a beta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. J. Biol. Chem., 281, 36742–36751. [DOI] [PubMed] [Google Scholar]
- 16. Sato T., Sato M., Kiyohara K., Sogabe M., Shikanai T., Kikuchi N., Togayachi A., Ishida H., Ito H., Kameyama A. et al. (2006) Molecular cloning and characterization of a novel human beta1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology, 16, 1194–1206. [DOI] [PubMed] [Google Scholar]
- 17. Schneider M., Al-Shareffi E. and Haltiwanger R.S. (2017) Biological functions of fucose in mammals. Glycobiology, 27, 601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dancevic C.M., McCulloch D.R. and Ward A.C. (2016) The ADAMTS hyalectanase family: biological insights from diverse species. Biochem. J., 473, 2011–2022. [DOI] [PubMed] [Google Scholar]
- 19. Dubail J. and Apte S.S. (2015) Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix biol., 44-46, 24–37. [DOI] [PubMed] [Google Scholar]
- 20. Kelwick R., Desanlis I., Wheeler G.N. and Edwards D.R. (2015) The ADAMTS (a Disintegrin and metalloproteinase with Thrombospondin motifs) family. Genome Biol., 16, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mead T.J. and Apte S.S. (2018) ADAMTS proteins in human disorders. Matrix biol., 71-72, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akiyama M., Takeda S., Kokame K., Takagi J. and Miyata T. (2009) Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc. Natl. Acad. Sci. U. S. A., 106, 19274–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holdener B.C. and Haltiwanger R.S. (2019) Protein O-fucosylation: structure and function. Curr. Opin. Struct. Biol., 56, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benz B.A., Nandadasa S., Takeuchi M., Grady R.C., Takeuchi H., LoPilato R.K., Kakuda S., Somerville R.P., Apte S.S., Haltiwanger R.S. et al. (2016) Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev. Biol., 416, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dubail J., Vasudevan D., Wang L.W., Earp S.E., Jenkins M.W., Haltiwanger R.S. and Apte S.S. (2016) Impaired ADAMTS9 secretion: a potential mechanism for eye defects in Peters plus syndrome. Sci. Rep., 6, 33974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hubmacher D., Schneider M., Berardinelli S.J., Takeuchi H., Willard B., Reinhardt D.P., Haltiwanger R.S. and Apte S.S. (2017) Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci. Rep., 7, 41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niwa Y., Suzuki T., Dohmae N. and Simizu S. (2015) O-fucosylation of CCN1 is required for its secretion. FEBS Lett., 589, 3287–3293. [DOI] [PubMed] [Google Scholar]
- 28. Ricketts L.M., Dlugosz M., Luther K.B., Haltiwanger R.S. and Majerus E.M. (2007) O-fucosylation is required for ADAMTS13 secretion. J. Biol. Chem., 282, 17014–17023. [DOI] [PubMed] [Google Scholar]
- 29. Wang L.W., Dlugosz M., Somerville R.P., Raed M., Haltiwanger R.S. and Apte S.S. (2007) O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J. Biol. Chem., 282, 17024–17031. [DOI] [PubMed] [Google Scholar]
- 30. Du J., Takeuchi H., Leonhard-Melief C., Shroyer K.R., Dlugosz M., Haltiwanger R.S. and Holdener B.C. (2010) O-Fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev. Biol., 346, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Percival C.J., Marangoni P., Tapaltsyan V., Klein O. and Hallgrimsson B. (2017) The interaction of genetic background and mutational effects in regulation of mouse craniofacial shape. G3, 7, 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luo F., Xie Y., Xu W., Huang J., Zhou S., Wang Z., Luo X., Liu M., Chen L. and Du X. (2017) Deformed skull morphology is caused by the combined effects of the Maldevelopment of calvarias, Cranial Base and brain in FGFR2-P253R mice mimicking human Apert syndrome. Int. J. Biol. Sci., 13, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hubmacher D. and Apte S.S. (2015) ADAMTS proteins as modulators of microfibril formation and function. Matrix biol., 47, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Enomoto H., Nelson C.M., Somerville R.P., Mielke K., Dixon L.J., Powell K. and Apte S.S. (2010) Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation. Development, 137, 4029–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hallgrimsson B., Brown J.J., Ford-Hutchinson A.F., Sheets H.D., Zelditch M.L. and Jirik F.R. (2006) The brachymorph mouse and the developmental-genetic basis for canalization and morphological integration. Evol. Dev., 8, 61–73. [DOI] [PubMed] [Google Scholar]
- 36. Nah H.D., Koyama E., Agochukwu N.B., Bartlett S.P. and Muenke M. (2012) Phenotype profile of a genetic mouse model for Muenke syndrome. Childs. Nerv. Syst., 28, 1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balemans M.C., Ansar M., Oudakker A.R., van Caam A.P., Bakker B., Vitters E.L., van der Kraan P.M., de Bruijn D.R., Janssen S.M., Kuipers A.J. et al. (2014) Reduced Euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev. Biol., 386, 395–407. [DOI] [PubMed] [Google Scholar]
- 38. Weh E., Takeuchi H., Muheisen S., Haltiwanger R.S. and Semina E.V. (2017) Functional characterization of zebrafish orthologs of the human Beta 3-Glucosyltransferase B3GLCT gene mutated in Peters plus syndrome. PLoS One, 12, e0184903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nandadasa S., Nelson C.M. and Apte S.S. (2015) ADAMTS9-mediated extracellular matrix dynamics regulates umbilical cord vascular smooth muscle differentiation and rotation. Cell Rep., 11, 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rao C., Foernzler D., Loftus S.K., Liu S., McPherson J.D., Jungers K.A., Apte S.S., Pavan W.J. and Beier D.R. (2003) A defect in a novel ADAMTS family member is the cause of the belted white-spotting mutation. Development, 130, 4665–4672. [DOI] [PubMed] [Google Scholar]
- 41. Silver D.L., Hou L., Somerville R., Young M.E., Apte S.S. and Pavan W.J. (2008) The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet., 4, e1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCulloch D.R., Nelson C.M., Dixon L.J., Silver D.L., Wylie J.D., Lindner V., Sasaki T., Cooley M.A., Argraves W.S. and Apte S.S. (2009) ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev. Cell, 17, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Somerville R.P., Longpre J.M., Jungers K.A., Engle J.M., Ross M., Evanko S., Wight T.N., Leduc R. and Apte S.S. (2003) Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J. Biol. Chem., 278, 9503–9513. [DOI] [PubMed] [Google Scholar]
- 44. Reis L.M., Tyler R.C., Abdul-Rahman O., Trapane P., Wallerstein R., Broome D., Hoffman J., Khan A., Paradiso C., Ron N. et al. (2008) Mutation analysis of B3GALTL in Peters plus syndrome. Am. J. Med. Gen., 146A, 2603–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson E.M., Winter R.M. and Baraitser M. (1993) Kivlin syndrome and Peters'-plus syndrome: are they the same disorder? Clin Dysmorphol., 2, 301–316. [PubMed] [Google Scholar]
- 46. Barbieri O., Astigiano S., Morini M., Tavella S., Schito A., Corsi A., Di Martino D., Bianco P., Cancedda R. and Garofalo S. (2003) Depletion of cartilage collagen fibrils in mice carrying a dominant negative Col2a1 transgene affects chondrocyte differentiation. Am. J. Physiol. Cell Physiol., 285, C1504–C1512. [DOI] [PubMed] [Google Scholar]
- 47. Tsang K.Y., Chan D., Cheslett D., Chan W.C., So C.L., Melhado I.G., Chan T.W., Kwan K.M., Hunziker E.B., Yamada Y. et al. (2007) Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol., e44, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chan W.C.W., Tsang K.Y., Cheng Y.W., Ng V.C.W., Chik H., Tan Z.J., Boot-Handford R., Boyde A., Cheung K.M.C., Cheah K.S.E. et al. (2017) Activating the unfolded protein response in osteocytes causes hyperostosis consistent with craniodiaphyseal dysplasia. Hum. Mol. Genet., 26, 4572–4587. [DOI] [PubMed] [Google Scholar]
- 49. Kokame K., Matsumoto M., Soejima K., Yagi H., Ishizashi H., Funato M., Tamai H., Konno M., Kamide K., Kawano Y. et al. (2002) Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc. Natl. Acad. Sci. U. S. A., 99, 11902–11907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng X.L. (2015) ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu. Rev. Med., 66, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brouillard P., Dupont L., Helaers R., Coulie R., Tiller G.E., Peeden J., Colige A. and Vikkula M. (2017) Loss of ADAMTS3 activity causes Hennekam lymphangiectasia-lymphedema syndrome 3. Hum. Mol. Genet., 26, 4095–4104. [DOI] [PubMed] [Google Scholar]
- 52. Van Damme T., Colige A., Syx D., Giunta C., Lindert U., Rohrbach M., Aryani O., Alanay Y., Simsek-Kiper P.O., Kroes H.Y. et al. (2016) Expanding the clinical and mutational spectrum of the Ehlers-Danlos syndrome dermatosparaxis type. Genet. Med., 18, 882–891. [DOI] [PubMed] [Google Scholar]
- 53. Zhou H., Hickford J.G. and Fang Q. (2012) A premature stop codon in the ADAMTS2 gene is likely to be responsible for dermatosparaxis in Dorper sheep. Anim. Genet., 43, 471–473. [DOI] [PubMed] [Google Scholar]
- 54. Dubail J., Aramaki-Hattori N., Bader H.L., Nelson C.M., Katebi N., Matuska B., Olsen B.R. and Apte S.S. (2014) A new Adamts9 conditional mouse allele identifies its non-redundant role in interdigital web regression. Genesis, 52, 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rigueur D. and Lyons K.M. (2014) Whole-mount skeletal staining. Methods Mol. Biol., 1130, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hsieh J.C., Lee L., Zhang L., Wefer S., Brown K., DeRossi C., Wines M.E., Rosenquist T. and Holdener B.C. (2003) Mesd encodes an LRP5/6 chaperone essential for specification of mouse embryonic polarity. Cell, 112, 355–367. [DOI] [PubMed] [Google Scholar]
- 57. Koo B.H., Coe D.M., Dixon L.J., Somerville R.P., Nelson C.M., Wang L.W., Young M.E., Lindner D.J. and Apte S.S. (2010) ADAMTS9 is a cell-autonomously acting, anti-angiogenic metalloprotease expressed by microvascular endothelial cells. Am. J. Pathol., 176, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kakuda S. and Haltiwanger R.S. (2014) Analyzing the posttranslational modification status of notch using mass spectrometry. Methods Mol. Biol., 1187, 209–221. [DOI] [PubMed] [Google Scholar]
- 59. Choi Y.J., Halbritter J., Braun D.A., Schueler M., Schapiro D., Rim J.H., Nandadasa S., Choi W.I., Widmeier E., Shril S. et al. (2019) Mutations of ADAMTS9 cause Nephronophthisis-related Ciliopathy. Am J. Hum. Genet., 104, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dagoneau N., Benoist-Lasselin C., Huber C., Faivre L., Megarbane A., Alswaid A., Dollfus H., Alembik Y., Munnich A., Legeai-Mallet L. et al. (2004) ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am. J. Hum. Genet., 75, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morales J., Al-Sharif L., Khalil D.S., Shinwari J.M., Bavi P., Al-Mahrouqi R.A., Al-Rajhi A., Alkuraya F.S., Meyer B.F. and Al Tassan N. (2009) Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet., 85, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aldahmesh M.A., Alshammari M.J., Khan A.O., Mohamed J.Y., Alhabib F.A. and Alkuraya F.S. (2013) The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum. Mutat., 34, 1195–1199. [DOI] [PubMed] [Google Scholar]
- 63. Peluso I., Conte I., Testa F., Dharmalingam G., Pizzo M., Collin R.W., Meola N., Barbato S., Mutarelli M., Ziviello C. et al. (2013) The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet. J. Rare Dis., 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hendee K., Wang L.W., Reis L.M., Rice G.M., Apte S.S. and Semina E.V. (2017) Identification and functional analysis of an ADAMTSL1 variant associated with a complex phenotype including congenital glaucoma, craniofacial, and other systemic features in a three-generation human pedigree. Hum. Mutat., 38, 1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Christensen A.E., Fiskerstrand T., Knappskog P.M., Boman H. and Rodahl E. (2010) A novel ADAMTSL4 mutation in autosomal recessive ectopia lentis et pupillae. Invest. Ophth. Vis. Sci., 51, 6369–6373. [DOI] [PubMed] [Google Scholar]
- 66. Sharifi Y., Tjon-Fo-Sang M.J., Cruysberg J.R. and Maat-Kievit A.J. (2013) Ectopia lentis et pupillae in four generations caused by novel mutations in the ADAMTSL4 gene. Br. J. Ophthalmol., 97, 583–587. [DOI] [PubMed] [Google Scholar]
- 67. Mularczyk E.J., Singh M., Godwin A.R.F., Galli F., Humphreys N., Adamson A.D., Mironov A., Cain S.A., Sengle G., Boot-Handford R.P. et al. (2018) ADAMTS10-mediated tissue disruption in Weill-Marchesani syndrome. Hum. Mol. Genet., 27, 3675–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang L.W., Kutz W.E., Mead T.J., Beene L.C., Singh S., Jenkins M.W., Reinhardt D.P. and Apte S.S. (2018) Adamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage. Matrix biol., 77, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jeanes E.C., Oliver J.A.C., Ricketts S.L., Gould D.J. and Mellersh C.S. (2019) Glaucoma-causing ADAMTS17 mutations are also reproducibly associated with height in two domestic dog breeds: selection for short stature may have contributed to increased prevalence of glaucoma. Canine Genet. Epidemiol., 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oichi T., Taniguchi Y., Soma K., Oshima Y., Yano F., Mori Y., Chijimatsu R., Kim-Kaneyama J.R., Tanaka S. and Saito T. (2019) Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell. Mol. Life Sci., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ataca D., Caikovski M., Piersigilli A., Moulin A., Benarafa C., Earp S.E., Guri Y., Kostic C., Arsenijevic Y., Soininen R. et al. (2016) Adamts18 deletion results in distinct developmental defects and provides a model for congenital disorders of lens, lung, and female reproductive tract development. Biol. Open, 5, 1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Collin G.B., Hubmacher D., Charette J.R., Hicks W.L., Stone L., Yu M., Naggert J.K., Krebs M.P., Peachey N.S., Apte S.S. et al. (2015) Disruption of murine Adamtsl4 results in zonular fiber detachment from the lens and in retinal pigment epithelium dedifferentiation. Hum. Mol. Genet., 24, 6958–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Le Goff C., Morice-Picard F., Dagoneau N., Wang L.W., Perrot C., Crow Y.J., Bauer F., Flori E., Prost-Squarcioni C., Krakow D. et al. (2008) ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet., 40, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ivkovic S., Yoon B.S., Popoff S.N., Safadi F.F., Libuda D.E., Stephenson R.C., Daluiski A. and Lyons K.M. (2003) Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development, 130, 2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tarr J.T., Visser T.G., Moon J.E., Hendesi H., Barbe M.F., Bradley J.P. and Popoff S.N. (2017) The pivotal role of CCN2 in mammalian palatogenesis. J. Cell Commun. Signal., 11, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bader H.L., Ruhe A.L., Wang L.W., Wong A.K., Walsh K.F., Packer R.A., Mitelman J., Robertson K.R., O'Brien D.P., Broman K.W. et al. (2010) An ADAMTSL2 founder mutation causes Musladin-Lueke syndrome, a heritable disorder of beagle dogs, featuring stiff skin and joint contractures. PLoS One, 5, 12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Delhon L., Mahaut C., Goudin N., Gaudas E., Piquand K., Le Goff W., Cormier-Daire V. and Le Goff C. (2019) Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB J., 33, 2707–2718. [DOI] [PubMed] [Google Scholar]
- 78. Mo F.E. and Lau L.F. (2006) The matricellular protein CCN1 is essential for cardiac development. Circ. Res., 99, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mo F.E., Muntean A.G., Chen C.C., Stolz D.B., Watkins S.C. and Lau L.F. (2002) CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell. Biol., 22, 8709–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Amend S.R., Uluckan O., Hurchla M., Leib D., Novack D.V., Silva M., Frazier W. and Weilbaecher K.N. (2015) Thrombospondin-1 regulates bone homeostasis through effects on bone matrix integrity and nitric oxide signaling in osteoclasts. J. Bone Miner. Res., 30, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mittaz L., Russell D.L., Wilson T., Brasted M., Tkalcevic J., Salamonsen L.A., Hertzog P.J. and Pritchard M.A. (2004) Adamts-1 is essential for the development and function of the urogenital system. Biol. Reprod., 70, 1096–1105. [DOI] [PubMed] [Google Scholar]
- 82. Boerboom D., Lafond J.F., Zheng X., Lapointe E., Mittaz L., Boyer A., Pritchard M.A., DeMayo F.J., Mort J.S., Drolet R. et al. (2011) Partially redundant functions of Adamts1 and Adamts4 in the perinatal development of the renal medulla. Dev. Dyn., 240, 1806–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Abdelrahman H.A., Al-Shamsi A., John A., Hertecant J., Lootah A., Ali B.R. and Al-Gazali L. (2018) A recessive truncating variant in thrombospondin-1 domain containing protein 1 gene THSD1 is the underlying cause of nonimmune hydrops fetalis, congenital cardiac defects, and haemangiomas in four patients from a consanguineous family. Am. J. Med. Gen., 176, 1996–2003. [DOI] [PubMed] [Google Scholar]
- 84. Stankunas K., Hang C.T., Tsun Z.Y., Chen H., Lee N.V., Wu J.I., Shang C., Bayle J.H., Shou W., Iruela-Arispe M.L. et al. (2008) Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev. Cell, 14, 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dupuis L.E., McCulloch D.R., McGarity J.D., Bahan A., Wessels A., Weber D., Diminich A.M., Nelson C.M., Apte S.S. and Kern C.B. (2011) Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev. Biol., 357, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li Y., Klena N.T., Gabriel G.C., Liu X., Kim A.J., Lemke K., Chen Y., Chatterjee B., Devine W., Damerla R.R. et al. (2015) Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature, 521, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kern C.B., Wessels A., McGarity J., Dixon L.J., Alston E., Argraves W.S., Geeting D., Nelson C.M., Menick D.R. and Apte S.S. (2010) Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix biol., 29, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.