

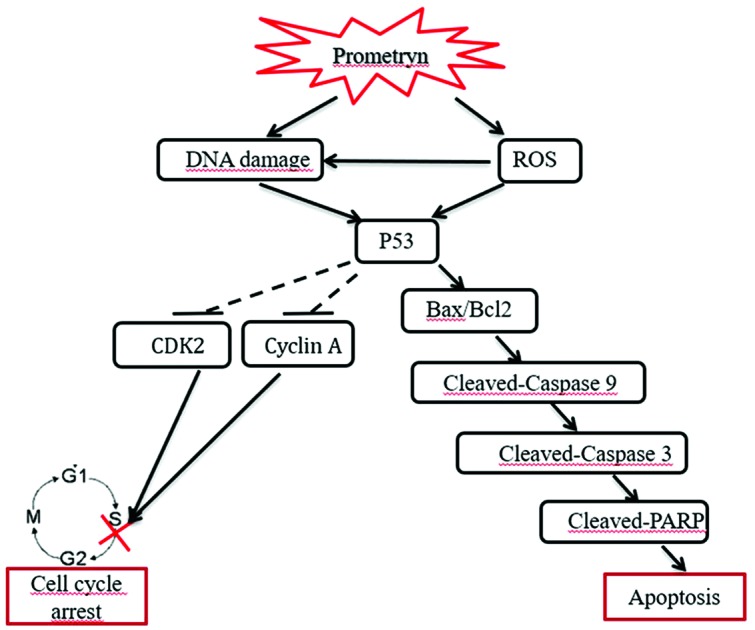
 Prometryn induces oxidative stress-mediated DNA damage; Prometryn induces S phase of cell cycle arrest; Prometryn induces Bax-dependent apoptosis in BEAS-2B cells.
Prometryn induces oxidative stress-mediated DNA damage; Prometryn induces S phase of cell cycle arrest; Prometryn induces Bax-dependent apoptosis in BEAS-2B cells.
Abstract
Prometryn is a slightly to moderately toxic herbicide belonging to the triazine family of herbicides, which are widely used in agriculture to control the growth of various weeds. Although many studies have shown that triazine herbicides have carcinogenic potential in humans, the cytotoxic effects of prometryn on human cells, and the mechanisms underlying these effects, are not yet fully understood. The lung is one of the most important organs where there is accumulation of environmental pollutants. The aim of this study was to determine the cytotoxic effects of prometryn on normal lung cells using the human bronchial epithelial cell line BEAS-2B. We found that treatment with high concentrations of prometryn arrested BEAS-2B cell growth in the S phase, while at low concentrations the cell cycle was not affected. Furthermore, we observed changes in the expression levels of cyclin-dependent kinase 2 (CDK2) and cyclin A that were consistent with the induction of cell cycle arrest in BEAS-2B cells exposed to prometryn. We also observed the increased formation of intracellular reactive oxygen species (ROS) in BEAS-2B cells, suggesting that this cell line is sensitive to prometryn. Finally, prometryn induced DNA double-strand breaks in BEAS-2B cells. In conclusion, prometryn affected key molecules involved in cell cycle regulation, induced oxidative stress, and induced DNA damage in BEAS-2B cells, which may shed light on the mechanism by which prometryn promotes lung cancer development.
1. Introduction
2,4-Bis (isopropylamino)-6-methylthio-1,3,5-triazine (prometryn) (Fig. 1a) is a widely used selective triazine herbicide, which is currently used as an alternative to atrazine-based products, and is widely applied to agriculture as it has lower acute toxicity than atrazine herbicides, stable chemical properties, and a long period of effectiveness.1
Fig. 1. Time- and concentration-dependent cytotoxicity of prometryn in BEAS-2B cells. General structure of triazine herbicides (a). BEAS-2B cells were treated with prometryn (0–200 μM) for 24 or 48 h, and cell viability was measured by MTT assay (b). BEAS-2B cells were treated with 0–200 μM of prometryn for 48 h, and cell morphology was examined under a light microscope (c).

However, prometryn can be also toxic to animals.2 In addition, triazine herbicides and their degradation products have long persistence in the environment, especially in air and water, and can damage the human immune system and endanger the health of humans, animals and plants.3,4 Last year, the United States Environmental Protection Agency (EPA) established tolerances for prometryn residues in several commodities and pointed out that prometryn increased pre- and post-natal quantitative susceptibility.5
However, the cytotoxic effects of prometryn on human cells, and the mechanisms underlying these effects, are not yet fully understood. The lung is one of the most affected organs due to the accumulation of environmental pollutants. Thus, the aim of this study was to determine the cytotoxic effects of prometryn on normal lung cells using the human bronchial epithelial cell line BEAS-2B. Specifically, cell proliferation analysis, single cell gel electrophoresis (comet assay), detection of reactive oxygen species levels, flow cytometry, and western blotting were used to evaluate the effects of prometryn on cell viability, DNA damage, intracellular ROS levels, cell cycle distribution, apoptosis and protein expression, respectively.
2. Materials and methods
2.1. Cell culture and treatment conditions
The human immortalized bronchial epithelial cell line BEAS-2B was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). BEAS-2B cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 100 U mL–1 penicillin G, 100 μg mL–1 streptomycin at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Then the cells were treated with different concentrations of prometryn (25, 50, 100 and 200 μM) or with the solvent alone (DMSO at a final concentration of 0.1%) for 48 h.
2.2. Chemicals and reagents
Analytical standard grade prometryn, 2′,7′-dichlorofluorescein diacetate (DCFH-DA), 3(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) dye, N-acetylcysteine (NAC), Triton X-100, 4′,6-diamidino-2-phenylindole (DAPI), and propidium iodide (PI) powder were purchased from Sigma Chemical Co. Ltd (St Louis, MO, USA). RPMI-1640, fetal bovine serum (FBS), trypsin, and antibiotic and antimycotic solutions (10 000 U mL–1 penicillin, 10 mg mL–1 streptomycin, 25 μg mL–1 amphotericin B) were purchased from Gibco BRL Co. Ltd (Rockville, MD, USA). Cell culture plasticware and phosphate-buffered saline (PBS) were obtained from Hangzhou Xin Yue Biotechnology Co., Ltd (Hangzhou, China). The Comet Assay Kit was purchased from Trevigen (Gaithersburg, MD, USA). The annexin V-fluorescence isothiocyanate (FITC)/PI apoptosis detection kit and cell cycle staining kit were purchased from MultiSciences (Lianke) Biotech Co., Ltd (Hangzhou, China). All other chemicals were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China) and were of analytical reagent grade.
2.3. Antibodies
Bcl-2 antibodies were purchased from Abcam (Cambridge, MA, USA). γ-H2AX antibodies were purchased from Millipore (Billerica, City, MA, USA). Cyclin A and CDK2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All the other primary antibodies were purchased from Cell Signaling Technology (Boston, MA, USA). Horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG were purchased from Abcam.
2.4. Assessment of cell viability
Cell viability was assessed by MTT assay. Briefly, BEAS-2B cells were seeded into two 96-well plates at 1 × 105 cells per well in a volume of 100 μL RPMI-1640 and incubated overnight at 37 °C. Cells were treated with varying concentrations of prometryn (0, 25, 50, 100 and 200 μM; four wells per concentration) for 48 h. MTT (10 μL) was added into each well at a concentration of 5 mg mL–1. Following 4 h incubation, 150 μL dimethylsulfoxide (DMSO) was added, and the solution was mixed thoroughly for 5 min. After the formazan generated by cell-mediated conversion of MTT was dissolved, the optical density at 490 nm was measured using a microplate reader (TECAN Infinite M200, Bio-Rad).
2.5. Cell cycle analysis
BEAS-2B cells were seeded into 12-well plates and treated with prometryn for 48 h. After treatment, cells were trypsinized and centrifuged, and the supernatant was discarded. The cells were then washed with PBS, re-suspended in 3 mL of precooled absolute ethyl alcohol, and stored at –20 °C for at least 12 h. The immobilized cells were then centrifuged again, washed with 3 mL PBS and incubated for 15 min. After the third centrifugation, 500 μL of DNA staining solution from the cell cycle staining kit was added to the cells. After gentle mixing, the cells were incubated in the dark at 37 °C for 30 min. Finally, the cells were analyzed with a FC500 MCL flow cytometer (Beckman Coulter).
2.6. Assessment of apoptosis
Approximately 1 × 105 cells were seeded into 6 cm dishes and incubated for 24 h, after which the cells were treated with various concentrations of prometryn or DMSO (as a control) for 48 h. After treatment, the cells were harvested and washed twice with PBS, then resuspended in 500 μL FACS binding buffer. The annexin V-fluorescence isothiocyanate (FITC)/PI apoptosis detection kit was used to analyze BEAS-2B cell apoptosis according to the manufacturer's protocol. Briefly, the cells were stained with 10 μL PI and 5 μL annexin V-FITC in the dark for 5 min at room temperature. The apoptotic cells were then immediately analyzed using an FC500 MCL flow cytometer and Cell-Quest software (Beckman Coulter).
2.7. Comet assay
DNA damage was measured by single-cell gel electrophoresis (comet assay). The BEAS-2B cells were treated with prometryn or DMSO alone as described above. The cells were harvested, and the rate of DNA damage was measured using a Comet Assay Kit (Trevigen, Gaithersburg, MD, USA) as described previously (Lu et al., 2005).6 All samples were observed under a fluorescence microscope (Leica, Bensheim, Germany) and analyzed using Comet Assay Software (CaspLab). Data are expressed as mean ± SD.
2.8. Measurement of Intracellular ROS
Intracellular ROS levels were fluorometrically monitored using the fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA). Briefly, cells grown in 6-well plates or 24-well plates were exposed to prometryn for 48 h, after which positive control cells were incubated with 2 mL hydrogen peroxide (30 ppm) for 15 min. The prometryn-treated and positive control cells were then centrifuged and resuspended in a 10 μM solution of DCFH-DA dye at 37 °C for 20 min in the dark. Then, a serum-free cell culture medium was used to wash the cells three times to remove any extracellular DCFH-DA. The cells grown in the 6-well plates were viewed under a fluorescence microscope (Leica, Bensheim, Germany), and the cells grown in the 24-well plates were analyzed using a FACS LSR II flow cytometer (BD Biosciences, San Jose, CA) at excitation and emission wavelengths of 488 and 525 nm, respectively. The DCF (2′,7′-dichlorofluorescein) fluorescence results are presented as the x-mean value.
2.9. Western blotting analyses
Protein expression levels were measured by western blotting with specific primary antibodies. Cells (5 × 105 mL–1) were seeded into 12-well plates. About 24 h after seeding, the cells were treated with different concentrations of prometryn for 24 h or 48 h. Then, the cells were gently rinsed three times with PBS on ice and directly lysed in an appropriate volume of cell lysis solution buffer containing 50 mM Tris–HCl (pH 7.4), 50 mM NaF, 250 mM NaCl, 5 mM EDTA, 1 mM PMS F, 0.5 mM Na3VO4, 2% SDS and 1% NP40. After the cells were completely lysed, the proteins were collected by centrifugation. The protein samples were re-suspended in SDS-PAGE loading buffer, heated at 100 °C for 5 min, and separated on a 4–20% sodium dodecyl sulfate polyacrylamide electrophoresis gradient gel at 80 V for 20 min and 120 V for 1 h. The separated proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Schleicher & Schuell; GER) at 300 mA for 80 min. The PVDF membrane was blocked with 5% dried skimmed milk in Tris-buffered saline containing 0.1% (v/v) Tween-20 (TBST) for one hour at room temperature. Then, the membranes were incubated overnight with the primary antibody at 4 °C, followed by incubation with horseradish peroxidase-conjugated (HRP) secondary antibody for 2 h at room temperature. After washing three times with TBST, the proteins on the PVDF membrane were detected by chemiluminescence (ECL-plus; Pierce, USA), and images were acquired using an Odyssey infrared imaging system.
2.10. Immunofluorescence staining of γ-H2AX
The subcellular localization of γ-H2AX, the DNA double-strand break marker, was examined in cells (1 × 106 mL–1) grown on coverslips pre-placed in 6-well plates. Immunofluorescence staining and γ-H2AX nuclear focus quantification were performed as previously described.7 Coverslips were examined under a fluorescent microscope using a green light filter at 450–490 nm for FITC and a blue light filter at 510–560 nm for DAPI. The results were then imported into Image J analysis software to merge the DAPI (blue) and γ-H2AX (green) images. γ-H2AX foci were counted in at least 100 cells for each treatment condition. Cells were considered to be positive (containing prometryn-induced γ-H2AX foci) when more than five green foci were detected.
2.11. Statistical analysis
All experiments were performed at least three times. Statistical analysis was performed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Normally distributed data are expressed as the mean ± standard deviation (SD), and counted data are shown as frequencies and percentages. Significant differences were determined by Student's t-test using SPSS software. GraphPad Prism 5 data processing software was used to perform statistical analyses and graph the data. P-Values less than 0.05 were considered significant.
3. Results
3.1. Effect of prometryn on cell viability
To investigate the cytotoxic effects induced by prometryn in BEAS-2B cells, cells were treated with different concentrations of prometryn for 24 h or 48 h, and cell viability was determined by MTT assay. Exposure to 100–200 μM prometryn for 48 h, but not 24 h, decreased BEAS-2B cell viability (Fig. 1b).
After treatment with different concentrations of prometryn for 48 h, a variety of morphological changes were observed in BEAS-2B cells. When the cells were treated with increasing concentrations of prometryn, cell volume decreased, cells became rounded, intercellular connections disappeared, adjacent cells gradually separated from each other, and the overall number of cells decreased. At concentrations of 50–200 μM, some of the cells showed obvious shrinkage, and the cell growth medium contained some floating cells and debris (Fig. 1c). In contrast, the DMSO-treated cells appeared to be healthy: they were adherent to the wall of the plates, and there were no apparent floating cells.
Taken together, these results suggest that prometryn decreased the viability of BEAS-2B cells starting at 50 μM.
3.2. Effect of prometryn on cell cycle progression
To evaluate the effects of prometryn on the cell cycle, BEAS-2B cells were stained with PI, and the cell cycle distribution was analyzed by flow cytometry. Treatment with prometryn for 24 h had no effect on the cell cycle. However, after the treatment with high concentrations (100 and 200 μM) of prometryn for 48 h, the proportion of cells in the G1 phase decreased and that of cells in the S phase increased, indicating that the cell cycle was arrested at the S phase (Fig. 2a and b).
Fig. 2. Cell cycle analysis of BEAS-2B cells treated with prometryn (0, 25, 50, 100, 200 μM) for 48 h. The treated cells were harvested and labeled with PI, then analyzed by flow cytometry (a). Quantitative data of cell cycle distribution. Results are representative of at least three independent experiments. *, P < 0.05, **, P < 0.01, ***, P < 0.001, versus vehicle-only control (Student's t-test) (b). Western blot detection of P53 and cell cycle-related proteins in BEAS-2B cells, with GAPDH used as the internal reference (c).

Cyclin-dependent kinases (CDKs), cyclins, and a few other proteins are key regulators of the cell cycle.8 Cyclin D and CDK2 play important roles in regulating the cell cycle in the G1 phase. However, after entering the S phase, the cyclin A-CDK2 complex contains the main CDK kinase.8 p53 also plays an important role in cell cycle regulation.9 Thus, we next evaluated whether cyclin A, CDK2 and p53 expression levels were modulated by prometryn. Western blotting showed that treatment with prometryn reduced cyclin A and CDK2 expression levels and increased p53 expression levels compared with the control, in a dose-dependent manner (Fig. 2c). These results suggest that prometryn induces S phase arrest in BEAS-2B cells.
3.3. Prometryn-induced apoptosis in BEAS-2B cells
Next, we used FCM and WB to determine whether prometryn induces BEAS-2B cell apoptosis. After treatment with prometryn at different concentrations for 48 h, the cells were double stained with annexin V and PI and subjected to FCM analysis. Prometryn induced apoptosis of BEAS-2B cells at several concentrations (100–200 μM) (Fig. 3a and b).
Fig. 3. Prometryn-induced apoptosis in BEAS-2B cells. Representative flow cytometric analysis of cells stained with annexin V-PI after being treated with 0, 25, 50, 100, or 200 μM of prometryn for 48 h. The lower left quadrant (Annexin V-FITC–/PI–) shows live cells, the lower right quadrant (Annexin V-FITC+/PI–) shows early apoptotic cells, the upper right quadrant (Annexin V-FITC+/PI+) shows late apoptotic cells or cells undergoing necrosis, and the upper left quadrant (Annexin V-FITC–/PI+) shows necrotic or mechanically damaged cells (a). Histograms showing the percentage of apoptotic cells. Results are presented as mean ± SD from three independent experiments (b). Effect of prometryn on Bax, Bcl2, Caspase 3, Caspase 9, and PARP expression in BEAS-2B cells. Whole-cell extracts of cells treated with 0, 25, 50, 100, or 200 μM of prometryn for 48 h were prepared. Bax, Bcl2, Caspase-3, caspase-9, and PARP expression were analyzed by western blot. GAPDH was used as a loading control (c).

To further verify that apoptosis was induced by prometryn, western blot analysis was performed. The results showed that Bcl2 expression noticeably decreased after the treatment with prometryn, while Bax expression noticeably increased. The expression of the apoptosis-related proteins Caspase 9, Caspase 3 and PARP was clearly down-regulated, and the expression of the corresponding cleaved forms of these proteins was clearly up-regulated (Fig. 3c). These results confirmed that prometryn induced apoptosis in BEAS-2B cells.
3.4. Prometryn-induced generation of intracellular ROS
To study the effect of prometryn on oxidative stress in BEAS-2B cells, ROS generation was assessed. After treatment with different concentrations of prometryn, the cells were stained with the fluorescent probe DCFH-DA. The changes in intracellular ROS levels were analyzed quantitatively by flow cytometry. The results showed that the percentage of cells producing ROS increased in a dose-dependent manner, and the average fluorescence intensity (x-mean value) reached a peak value at a concentration of 200 μM (Fig. 4a and b). Green fluorescence was clearly increased in the H2O2 positive control group, and there was a significant increase in the amount of intracellular green fluorescence in BEAS-2B cells treated with 200 μM prometryn compared with the control (Fig. 4c). Pre-treating the cells with the antioxidant NAC partially decreased ROS production. These data suggest that prometryn induced ROS generation in BEAS-2B cells.
Fig. 4. Intracellular ROS generation induced by prometryn in BEAS-2B cells. Flow cytometry was used to determine intracellular ROS levels in BEAS-2B cells after 48 h of treatment with different concentrations of prometryn (0, 25, 50, 100, and 200 μM). Untreated cells were used as a negative control, and cells treated with 1 mM H2O2 were used as a positive control (a). The mean fluorescence intensity (MFI) was measured by flow cytometry. Results are presented as MFI ± SEM (n ≥ 3). The data were analyzed for statistical significance using Student's t test (*P < 0.05, ** P < 0.01) (b). Cells were treated with prometryn at the indicated concentrations in the presence or absence of 5 mM NAC. The solvent alone (0.1% DMSO) with or without NAC was used as negative controls, and H2O2 was used as a positive control. After 48 h, cells were stained with 10 μM DCFH-DA for 20 min and then viewed with an inverted fluorescence microscope (original magnification, ×400) (c).

3.5. DNA damage
Finally, we studied the toxic effects of prometryn on BEAS-2B cell DNA. Cells were treated with different concentrations of prometryn for 48 h, and a comet assay was performed. The number of cells with a comet-like appearance and the fluorescence intensity of the comet tails (TDNA%) were quantified. The DNA of the BEAS-2B cells was clearly damaged, as the cells formed obvious comet-like tails, and the percentage of comet-like cells increased in a dose-dependent manner. In BEAS-2B cells pretreated with the antioxidant NAC and then treated with 200 μM prometryn, fewer comet-like cells were observed, and the comet tails were shortened (Fig. 5a and b).
Fig. 5. Prometryn-induced DNA damage in BEAS-2B cells. Images of comet-like BEAS-2B cells after treatment with 0–200 μM prometryn or pretreatment with 5 mM NAC followed by 0 μM or 200 μM prometryn for 48 h (a); CASP software was used to analyze the TDNA%, and a Student's t-test was used to analyze the differences, *P < 0.05, **P < 0.01, and ***P < 0.001. # Means P < 0.05 between 200 μM and 200 μM + NAC group. (b). After 48 h of treatment with the indicated concentrations of prometryn without or with 5 mM NAC pretreatment, the cells were stained with DAPI and γH2AX antibodies (fluorescein isothiocyanate, FITC) (original magnification, ×400) (c). Western blot of γH2AX expression in BEAS-2b cells treated with prometryn for 48 h (d).

Studies have shown that the formation of γ-H2AX foci is proportional to the number and severity of DSBs.10 Therefore, we assessed changes in γ-H2AX protein expression by immunofluorescence and immunoblotting. Immunofluorescence analysis showed clear γ-H2AX foci in BEAS-2B cells treated with prometryn, and as the prometryn concentration increased, the number of foci and the fluorescence intensity also increased. At concentrations of 100 and 200 μM, in addition to uniform distribution of fluorescence focal points in the nucleus, fluorescent clumps also appeared in parts of the cells (Fig. 5c). Western blot analysis showed that treatment with prometryn resulted in up-regulation of γ-H2AX protein expression in all treatment groups (Fig. 5d). These data suggest that prometryn induced DNA damage in BEAS-2B cells starting at 25 μM, and with an increased rate at 200 μM.
4. Discussion
The results from this study showed that treatment of normal lung epithelial cells with prometryn caused a decrease in cell viability, and cell cycle arrest in the S phase. In addition, prometryn affected the expression of key molecules involved in cell cycle regulation. Our study showed that normal lung epithelial cells sustained DNA damage and increase in intracellular ROS with prometryn treatment.
Treatment of BEAS-2B cells with prometryn arrested cells in the S phase, which might explain the concentration-dependent decrease in cell viability, as cell viability is closely related to cell cycle progression.
p53 was expressed at high levels in prometryn treated BEAS-2B cells, suggesting the induction of DNA damage in these cells. Earlier studies have reported abnormal p53 expression in BEAS-2B cells before their transformation into cancer cells.11,12 Therefore, increased p53 levels could be used as a sensitive indicator of prometryn cytotoxicity in vitro, as well as an early warning sign of lung injury. The increased p53 and decreased Cyclin A and CDK2 expression levels support the cell cycle results.
The apoptosis rates in BEAS-2B cells increased significantly with increasing concentrations of prometryn. Consistent with this, the expression of the anti-apoptosis protein bcl-2 significantly decreased, while the expression of the pro-apoptosis protein Bax significantly increased. These results further confirmed that prometryn induced apoptosis in BEAS-2B cells. We also showed that Caspase 9, Caspase 3 and PARP expression levels decreased with increasing prometryn concentrations, and this decrease was accompanied by a corresponding increase in the levels of the active cleaved forms of these proteins. Đikic et al.2 found a certain degree of apoptosis or necrosis in the thymus, spleen, and lymph nodes of mice that received three 185–555 mg kg–1 doses of prometryn every day for 28 days. Intermediate doses induced early apoptosis, while advanced apoptosis and early necrosis occurred only in the high-dose group. This is essentially consistent with the trends observed in our study. After treatment with different concentrations of prometryn, the BEAS-2B cells primarily exhibited early apoptosis and late apoptosis, with no obvious necrosis. In 2015, Abarikwu and Farombi13 found that the triazine herbicide atrazine induced apoptosis in human neuroblastoma cells by altering the Bax/bcl-2 ratio and stimulating the Caspase 3 signaling pathway. Therefore, we speculate that prometryn may induce BEAS-2B cell apoptosis by modifying the regulation of apoptosis-related proteins such as Bax/bcl-2, Caspase 9, Caspase 3 and PARP.
Boulahia et al.1 showed that exposing seeds to prometryn could trigger a significant oxidative stress response. When the soil was treated with prometryn at concentrations of 100 μM or more, the cells began to produce ROS, which had a harmful effect on seed growth. This is consistent with a clear overproduction of ROS in our BEAS-2B cells as a result of 200 μM prometryn exposure.
The mechanism by which prometryn induced oxidative stress in human and animal cells is not well-documented, however, some studies have reported that prometryn alters antioxidant enzyme activity in fish.14–16 Prometryn binds to the PS II D1 protein,17,18 which inhibits the plant cell photosynthetic electron chain.19 These actions eventually result in the accumulation of ROS in body cells and induce oxidative stress.1,20 Photosynthetic electron transport chains have many similarities to other electron transport chains. Therefore, we speculate that prometryn induces oxidative stress in human and animal cells in a similar manner. We hypothesize that, after entering the cells, prometryn primarily acts on the respiratory electron transport chain in the mitochondria, and damages the function of its important components or antioxidant enzymes, thus leading to ROS accumulation in the cell.
ROS production and DNA damage are closely related phenomena that interact with each other. On the one hand, excessive ROS levels can induce DNA damage.21,22 On the other hand, continued DNA damage can in turn increase oxidative stress and ROS levels.23
We found that intracellular ROS levels increased continuously in BEAS-2B cells after treatment with prometryn, and this increase was accompanied by severe DNA damage. When the BEAS-2B cells were pretreated with the antioxidant NAC, prometryn-induced intracellular ROS production was blocked, and the DNA damage was partially rescued. This suggests that the DNA damage induced by prometryn primarily occurs due to excessive ROS production, although other mechanisms and pathways may also be involved. DNA damage and ROS production could form a positive feedback loop that is adverse to cell survival. However, the precise mechanism is not clarified totally because the receptors of prometryn are still unclear.24 A previous study indicated that prometryn does not activate the aryl hydrocarbon receptor, the estrogen receptor or the thyroid receptor by in vitro assays,24 thus further work is required for elucidating the mechanisms of how prometryn produces oxidative stress.
The distribution of γ-H2AX fluorescence signals also reflects the degree of DNA damage and the repair process.10,25,26 The BEAS-2B cells exhibited scattered γ-H2AX fluorescence, indicating somewhat severe DNA damage, which was consistent with the comet assays.
Although prometryn is considered to be only a slightly to moderately toxic herbicide, the cytotoxic effects of prometryn on human cells, and the mechanism underlying these effects, have not been well studied. We observed significant changes on the molecular level and obvious DNA damage caused by prometryn in normal lung cells. However, the cytotoxic effects of prometryn on other potential target organs such as the liver and stomach still need to be explored through further research. These results suggest that measures should be taken to reduce contact with prometryn in order to avoid the potential health hazards caused by prometryn ingestion.
Author contributions
Dajing Xia and Yihua Wu had the right to grant on behalf of all authors. Qiaoyun Liu and Longsheng Wang performed all the experiments. Bo Huang, Jiawei Xu, Paul Héroux and Xinqiang Zhu were involved in designing the experiments. Qiaoyun Liu, Hanwen Chen and Ying Li contributed to statistical analyses. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81773016 and 31471297), Zhejiang Provincial Natural Science Foundation of China (No. LY18C060001), and the Fundamental Research Funds for the Central Universities.
References
- Boulahia K., Carol P., Planchais S., Abrous-Belbachir O. J. Agric. Food Chem. 2016;64:3150–3160. doi: 10.1021/acs.jafc.6b00328. [DOI] [PubMed] [Google Scholar]
- Đikić D., žIdoveclepej S., Remenar A., Bendelja K., Benković V., HorvatkneŽEvić A., Brozović G., Oršolić N. Environ. Toxicol. Pharmacol. 2009;27:182–186. doi: 10.1016/j.etap.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Santos T. G., Martinez C. B. R. Chemosphere. 2012;89:1118–1125. doi: 10.1016/j.chemosphere.2012.05.096. [DOI] [PubMed] [Google Scholar]
- Xing H., Wang Z., Gao X., Chen D., Wang L., Li S., Xu S. Ecotoxicol. Environ. Saf. 2015;113:52–58. doi: 10.1016/j.ecoenv.2014.11.027. [DOI] [PubMed] [Google Scholar]
- Environmental Protection Agency, Prometryn; Pesticide Tolerances: Final Rule., https://www.federalregister.gov/documents/2017/12/04/2017-26083/prometryn-pesticide-tolerances.
- Lu H. R., Meng L. H., Huang M., Zhu H., Miao Z. H., Ding J. Cancer Chemother. Pharmacol. 2005;55:286–294. doi: 10.1007/s00280-004-0877-z. [DOI] [PubMed] [Google Scholar]
- Gowan S. M., Hardcastle A., Hallsworth A. E., Valenti M. R., Hunter L. J., Ak D. H. B., Garrett M. D., Raynaud F., Workman P., Aherne W. Assay Drug Dev. Technol. 2007;5:391. doi: 10.1089/adt.2006.044. [DOI] [PubMed] [Google Scholar]
- Malumbres M. Genome Biol. 2014;15:122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giono L. E., Manfredi J. J. J. Cell. Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. DNA Repair. 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Hussain S. P., Amstad P., Raja K., Sawyer M., Hofseth L., Shields P. G., Hewer A., Phillips D. H., Ryberg D., Haugen A., Harris C. C. Cancer Res. 2001;61:6350–6355. [PubMed] [Google Scholar]
- Park Y. H., Kim D., Dai J., Zhang Z. Toxicol. Appl. Pharmacol. 2015;287:240–245. doi: 10.1016/j.taap.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abarikwu S. O., Farombi E. O. Pestic. Biochem. Physiol. 2015;118:90–98. doi: 10.1016/j.pestbp.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Stara A., Kouba A., Velisek J. BioMed Res. Int. 2014;2014:680131. doi: 10.1155/2014/680131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stara A., Kristan J., Zuskova E., Velisek J. Pestic. Biochem. Physiol. 2013;105:18–23. doi: 10.1016/j.pestbp.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Stara A., Machova J., Velisek J. Neuroendocrinol. Lett. 2012;33(Suppl 3):130–135. [PubMed] [Google Scholar]
- Pallett K. E., Dodge A. D. J. Exp. Bot. 1980;31:1051–1066. [Google Scholar]
- Pfister K., Radosevich S. R., Arntzen C. J. Plant Physiol. 1979;64:995–999. doi: 10.1104/pp.64.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M. M., Turnburke A. C., Lagace E. A., Steinback K. E. Plant Physiol. 1983;71:388–392. doi: 10.1104/pp.71.2.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Yang H. Ecotoxicol. Environ. Saf. 2009;72:1687–1693. doi: 10.1016/j.ecoenv.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Bonassi S., El-Zein R., Bolognesi C., Fenech M. Mutagenesis. 2011;26:93–100. doi: 10.1093/mutage/geq075. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T., Kashino G., Ono K., Watanabe M. J. Radiat. Res. 2009;50:151–160. doi: 10.1269/jrr.08109. [DOI] [PubMed] [Google Scholar]
- Zhao H., Shi P., Deng M., Jiang Z., Li Y., Kannappan V., Wang W., Li P., Xu B. Oncotarget. 2016;7:85515–85528. doi: 10.18632/oncotarget.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Casa-Resino I., Navas J. M., Fernandez-Cruz M. L. ATLA, Altern. Lab. Anim. 2014;42:25–30. doi: 10.1177/026119291404200105. [DOI] [PubMed] [Google Scholar]
- Brand M., Sommer M., Ellmann S., Wuest W., May M. S., Eller A., Vogt S., Lell M. M., Kuefner M. A., Uder M. PLoS One. 2015;10:e0127142. doi: 10.1371/journal.pone.0127142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon C., Pilch D., Rogakou E., Sedelnikova O., Newrock K., Bonner W. Curr. Opin. Genet. Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]