

Abstract
Mitochondrial fission and fusion are important for mitochondrial function, and dynamin 1‐like protein (DNM1L) is a key regulator of mitochondrial fission. We investigated the effect of mitochondrial fission on mitochondrial function and inflammation in fibroblast‐like synoviocytes (FLSs) during rheumatoid arthritis (RA). DNM1L expression was determined in synovial tissues (STs) from RA and non‐RA patients. FLSs were isolated from STs and treated with a DNM1L inhibitor (mdivi‐1, mitochondrial division inhibitor 1) or transfected with DNM1L‐specific siRNA. Mitochondrial morphology, DNM1L expression, cell viability, mitochondrial membrane potential, reactive oxygen species (ROS), apoptosis, inflammatory cytokine expression and autophagy were examined. The impact of mdivi‐1 treatment on development and severity of collagen‐induced arthritis (CIA) was determined in mice. Up‐regulated DNM1L expression was associated with reduced mitochondrial length in STs from patients with RA and increased RA severity. Inhibition of DNM1L in FLSs triggered mitochondrial depolarization, mitochondrial elongation, decreased cell viability, production of ROS, IL‐8 and COX‐2, and increased apoptosis. DNM1L deficiency inhibited IL‐1β–mediated AKT/IKK activation, NF‐κBp65 nuclear translocation and LC3B‐related autophagy, but enhanced NFKBIA expression. Treatment of CIA mice with mdivi‐1 decreased disease severity by modulating inflammatory cytokine and ROS production. Our major results are that up‐regulated DNM1L and mitochondrial fission promoted survival, LC3B‐related autophagy and ROS production in FLSs, factors that lead to inflammation by regulating AKT/IKK/NFKBIA/NF‐κB signalling. Thus, inhibition of DNM1L may be a new strategy for treatment of RA.
Keywords: dynamin 1‐like protein, fibroblast‐like synoviocyte, inflammation, mitochondrial fission, rheumatoid arthritis
1. INTRODUCTION
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease and is characterized by progressive destruction of multiple joints and synovial.1 Despite significant therapeutic advances, RA remains a huge threat for human health due to the high rate of patient disability. During the pathogenic process of RA, fibroblast‐like synoviocytes (FLSs) in the intimal lining can produce high levels of pro‐inflammatory cytokines and contribute to cartilage destruction.2 FLSs have been considered therapeutic targets for RA interventions. However, how FLSs contribute to the pathogenesis of RA has not been clarified.
Hypoxia can alter mitochondrial morphology and lead to mitochondrial dysfunctions, with reduced levels of adenosine triphosphate (ATP) production, excessive reactive oxygen species (ROS) production, increasing mitochondrial DNA mutations and oxidative damages, which in turn enhance inflammation in the joint, contributing to the pathogenesis of RA.3, 4, 5 Mitochondria are the main inducer and target of ROS.6 A previous study has shown that excessive ROS induces mitophagy,7 and feedback can remove damaged mitochondria to reduce ROS accumulation.8 ROS can activate the NF‐κB signalling to produce pro‐inflammatory cytokines, promoting synovial inflammation.9 It is notable that a defect in the mitochondrial respiratory chain (MRC) complex IV, and increased autophagy (mediated by LC3 and Beclin 1) are associated with the development of RA.10, 11 Inhibition of mitochondrial function can aggravate inflammatory responses in FLSs.12 Inhibition of mitochondrial functions exacerbates inflammatory responses in the joint in vivo.13 In addition, treatment with anti‐tumour necrosis factor (TNF) α can suppress mitochondrial mutagenesis in inflammatory arthritis.14 Mitochondrial membrane depolarization in peripheral blood lymphocytes has been reported in patients with RA.15 Accordingly, protecting mitochondrial function may be an effective intervention for RA.
Mitochondrial dynamics (fusion and fission) are crucial for multiple mitochondrial functions, including respiratory capacity, apoptosis, mitochondrial DNA stability, response to cellular stress and autophagy.16 Notably, dynamin 1‐like protein (DNM1L, a GTPase) and mitochondrial fission protein 1 (FIS1) regulate mitochondrial fission, whereas mitofusin 1 (MFN1), MFN2 and OPA1 regulate mitochondrial fusion.17 Previous studies have shown that increased mitochondrial fission can promote ROS production, leading to oxidative stress.18, 19, 20, 21, 22, 23 Hence, targeting mitochondrial fission may represent a therapeutic strategy to inhibit oxidative stress‐related diseases. DNM1L is conservatively expressed in the cytoplasm of many types of cells and is crucial for mitochondrial fission.24 Following its activation, DNM1L can be translocated into the mitochondrial outer membrane and bind to mitochondrial adaptor protein FIS1 to drive the process of mitochondrial fission.24 Accumulated evidence has shown that excessive expression of DNM1L is associated with development of neurodegenerative diseases, diabetes, tumours, kidney diseases, sepsis and cardiac diseases.17, 25, 26, 27, 28, 29, 30 However, it is still unclear whether excessive expression of DNM1L‐mediated mitochondrial fission is associated with the progression of RA.
In this study, DNM1L expression was examined in synovial tissues (STs) of RA and non‐RA patients. The effects of DNM1L deficiency on RA FLSs and in mice with collagen‐induced arthritis (CIA) were studied. The results indicated that up‐regulated DNM1L expression in STs was associated with RA severity, and DNM1L deficiency altered mitochondrial morphology and reduced ROS production, inflammatory responses, and autophagy in FLSs by down‐regulating the AKT/IKK/NFKBIA/NF‐κB signalling. Therefore, DNM1L may be a new therapeutic target, and inhibition of mitochondrial fission may be an effective intervention for RA.
2. MATERIALS AND METHODS
2.1. Reagents
The specific reagents used in this study included antibodies against DNM1L (A2586), GAPDH (AC002), AKT (A3145), IKK (A2087), NF‐kBp65 (A2547), IL‐8 (A2541) from ABclonal (Wuhan, China); antibodies against p‐AKT (4060), p‐IKK (2697T), LC3B (3868T), COX‐2 (12282T) from Cell Signaling Technology; antibodies against NFKBIA (10268‐1‐AP) from Proteintech; the DNM1L inhibitor Mdivi‐1, a mitochondrial division inhibitor 1 (M0199) from Sigma‐Aldrich; IL‐1β (200‐01B) from Peprotech; MitoTracker Green, BCA protein assay kit and Annexin V‐FITC (C1048, P0010S, C1063) from Beyotime; DNM1L‐specific small interfering RNA (siRNA) and control scramble siRNA from Guangzhou RiboBio; the Genomic DNA Removal Kit, SYBR Green PCR Kit from ABM; the CCK8 kit from MEC; ROS Fluorescent Probe‐DHE from Vigorous; primers from Sangon Biotech; and SDS‐PAGE gels from Epizyme.
2.2. Tissue collection and cell culture
Synovial tissues (STs) were obtained from patients with RA, who underwent a joint replacement (hit or knee) surgery in the Department of Orthopaedics of Tangdu Hospital, the Second Affiliated Hospital of Air Force Medical University from January 2018 to June 2018. Two RA markers (anti‐cyclic peptide containing citrulline [anti‐CCP] antibody, rheumatoid factor [RF]) and 2 inflammation markers (erythrocyte sedimentation rate [ESR], and high‐sensitivity C‐reactive protein [hs‐CRP]) were measured in each patient. Disease activity was calculated using the Disease Activity Score 28 joint count (DAS 28).15, 31, 32, 33 Control STs were obtained from patients with meniscus injuries without a history of acute or chronic arthritis, who underwent an arthroscopic surgery during the same period. There were four male and six female RA patients with ages ranging from 42 to 72 years (53.1 ± 11.53). There were three male control patients with ages ranging from 21 to 30 years (26.3 ± 4.75).
The partial fresh STs were washed with PBS and cut into small pieces. Subsequently, the tissue pieces were digested with 4 mg/mL of collagenase I (Invitrogen) at 37°C for 2 hours and filtered through a cell strainer (70 µm), followed by centrifugation. The obtained cells were cultured in DMEM/F12 supplemented with 10% fetal bovine serum (FBS), 100 U/mL of penicillin and 100 µg/mL of streptomycin. The cells at passages of 3‐6 were used as FLSs for experiments. All patients signed the written informed consent agreements before participation. The experimental protocols were approved by the Ethics Committee of Tangdu Hospital, the Second Affiliated Hospital of Air Force Medical University.
2.3. Immunohistochemistry (IHC)
Another part of STs was fixed in 10% formalin overnight and paraffin‐embedded. The ST sections were deparaffinized, rehydrated and treated with 3% H2O2 in methanol for 10 minutes, followed by antigen retrieval in a microwave. After being blocked with 5% BSA (AR0004; Boster), the sections were probed with anti‐DNM1L (1:300) at 4°C overnight, and the bound antibodies were detected with horseradish peroxidase (HRP)‐conjugated goat anti‐rabbit antibodies (1:1000, AS014; ABclonal), followed by visualization using diaminobenzidine. Images were obtained under a light microscope (Olympus).
2.4. Transfection of siRNA
Fibroblast‐like synoviocytes (1 × 105 cells/mL) were cultured in 6‐well plates overnight and then transfected with 100 nmol/L DNM1L‐specific or control siRNA using lipofectamine 3000 (Invitrogen), according to the manufacturer's instruction. Three days later, DNM1L expression was determined by qRT‐PCR and Western blot assays.
2.5. Quantitative real‐time polymerase chain reaction (qRT‐PCR)
Fibroblast‐like synoviocytes (1 × 105 cells/mL) were treated with vehicle control, 50 µmol/L mdivi‐1 or siRNA for 72 hours. Total RNA was extracted from the different groups of cells or fresh STs using TRIzol Reagent (Invitrogen). After removing genomic DNA using the Genomic DNA Removal Kit (ABM), each RNA sample (2 µg) was reversely transcribed into cDNA. The relative levels of target mRNA transcripts were determined by qRT‐PCR using the SYBR Green PCR Kit (ABM). Specific primers (Table 1) were used with the Rotor‐Gene Q RT‐PCR instrument (Qiagen GmbH, Hilden, Germany). All mRNA levels are expressed relative to the control (GAPDH) using the 2−ΔΔCt method.34
Table 1.
The sequences of primers
| Human | |
| GAPDH | forward: 5′‐GGAGCGAGATCCCTCCAAAAT‐3′ |
| reverse: 5′‐GGCTGTTGTCATACTTCTCATGG‐3′ | |
| MFN1 | forward: 5′‐TGGCTAAGAAGGCGATTACTGC‐3′ |
| reverse: 5′‐ TCTCCGAGATAGCACCTCACC‐3′ | |
| DNM1L | forward: 5′‐TAGCAGCAGTGACAGCGAGGA‐3′ |
| reverse: 5′‐GGGAGTAAGCCCTGAACCAAT‐3′ | |
| IL‐6 | forward: 5′‐ ACTCACCTCTTCAGAACGAATTG‐3′ |
| reverse: 5′‐CCATCTTTGGAAGGTTCAGGTTG‐3′ | |
| IL‐8 | forward: 5′‐ACTGAGAGTGATTGAGAGTGGAC‐3′ |
| reverse: 5′‐AACCCTCTGCACCCAGTTTTC‐3′ | |
| COX‐2 | forward: 5′‐TTCAAATGAGATTGTGGGAAAATTGCT‐3′ |
| reverse: 5′‐AGTTCA TCTCTGCCTGAGTATCTT‐3′ | |
| MMP13 | forward: 5′‐GACCCTGGAGCACTCATGT‐3′ |
| reverse: 5′‐ CATTTGTCTGGCGTTTTTGGAT‐3′ | |
| Mouse | |
| GAPDH | forward: 5′‐AGGCCGGTGCTGAGTATGTC‐3′, |
| reverse: 5′‐TGCCTGCTTCACCACCTTCT; | |
| NLRP3 | forward: 5′‐CTGGCTGCGGATGGAATTTG‐3′ |
| reverse: 5′‐CTGGTCCTTTCCTCACGGTC‐3′ | |
| TNF‐α | forward: 5′‐ATGTCTCAGCCTCTTCTCATTC‐3′ |
| reverse: 5′‐GCTTGTCACTCGAATTTTGAGA‐3′ | |
| MMP13 | forward: 5′‐CTTCCTGATGATGACGTTCAAG |
| reverse: GTCACACTTCTCTGGTGTTTTG. | |
| IL‐6 | forward: 5′‐CTCCCAACAGACCTGTCTATAC‐3′ |
| reverse: 5′‐CCATTGCACAACTCTTTTCTCA‐3′ | |
| IL‐10 | forward: 5′‐TTCTTTCAAACAAAGGACCAGC‐3 |
| reverse: 5′‐GCAACCCAAGTAACCCTTAAAG −3 | |
| COX‐2 | forward: 5′‐GGTCATTGGTGGAGAGGTGTA‐3′ |
| reverse: 5′‐TGAGTCTGCTGGTTTGGAATAG‐3′ | |
2.6. Transmission electron microscopy (TEM) and confocal microscopy
The different groups of FLSs and STs were fixed in glutaraldehyde at 4°C for 2 hours and post‐fixed in 1% citric acid‐0.1 mol/L phosphate buffer (PB) for 2 hours, followed by embedded. The ultra‐thin sections (70 nm) were stained with uranyl acetate and lead citrate. Images were collected using TEM (Hitachi, HT7700). In addition, the mitochondrial morphology of different groups of FLSs was examined by confocal microscopy (Zeiss 800) after staining with fluorescent MitoTracker Green, according to the manufacturer's instructions. In particular, cells (1 × 104 cells/ well) were cultured in a chamber for 24 hours, stained with MitoTracker Green at 37°C in the dark for 40 minutes, and then examined under a confocal microscope. The different groups of cells were also stained with JC‐1 using the JC‐1 assay kit (Beyotime, C2006), according to the manufacturer's instruction. The fluorescent signals from this stain were also examined under a confocal microscope to determine mitochondrial membrane potential.
2.7. Western blot analysis
ST samples (100 mg wet‐weight/sample) were homogenized and centrifuged. FLSs (2.5 × 105 cells/well) were transfected with DNM1L‐specific siRNA and control siRNA or treated with 50 µmol/L mdivi‐1 for 72 hours, followed by treatment with/without, IL‐1β (10 ng/mL) for 10 minutes. The different groups of cells were lysed with RIPA buffer containing proteinase and phosphatase inhibitors and centrifuged. The tissue homogenates or cell lysates (30 µg/lane) were separated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) on 10% or 12.5% gels and electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% fat‐free milk powder in TBST and probed with primary antibodies at 4°C overnight. After being washed, the bound antibodies were detected with HRP‐goat anti‐rabbit (1:2000) or antimouse (1:5000) for 1 hour at room temperature and visualized using the enhanced chemiluminescent reagents. The levels of each target protein relative to the loading control (GAPDH) were determined by densitometric analysis using ImageJ software. The primary antibodies were diluted by 1:1000 for DNM1L, IKK and LC3B; 1:10 000 for GAPDH, 1:500 for p‐AKT, p‐IKK, AKT, NFKBIA, COX‐2 and IL‐8.
2.8. Cell viability and apoptosis assays
The cell viability was measured using the CCK‐8 assay kit. Briefly, FLSs (1 × 104 cells/well) were cultured in 96‐well plates overnight and treated in triplicate with vehicle or different concentrations (10, 20, 30, 40 and 50 µmol/L) of mdivi‐1 for 24 hours. During the last 2‐hours culture, 10 µL of CCK8 solution was added to each well. The absorbance at 450 nm was measured using a microplate reader.
The apoptosis of different groups of cells was determined by flow cytometry using the Annexin V‐FITC apoptosis Detection Kit. Briefly, FLSs (5 × 105 cells/tube) were stained with 5 µL Annexin V‐FITC and 10 µL PI in the dark for 10 minutes at room temperature. The percentages of apoptotic cells were determined by flow cytometry (Epics XL‐MCL, Beckman). In addition, following transfection with DNM1L‐specific miRNA or control scramble siRNA for 72 hours, cells viability and apoptosis were determined.
2.9. Reactive oxygen species (ROS) detection
The contents of cellular ROS were determined using ROS fluorescent probe‐dihydroethidium (DHE), according to the manufacturer's protocols. Briefly, FLSs were reacted with DHE (20 µmol/L) at 37°C in the dark for 30 minutes and then stained with DAPI. The fluorescent signals were observed in an inverted fluorescent microscope (Olympus, IX‐53), with excitation at 480 nm and emission at 590 nm.
2.10. Immunofluorescence staining
The NF‐κBp65 nuclear accumulation was examined by immunofluorescence. Briefly, FLSs (0.5 × 104 cells/well) were cultured in 24‐well culture plates and treated with vehicle or the DNM1L inhibitor for 24 hours. The cells were fixed with 4% paraformaldehyde and blocked with confining liquid (containing Triton X‐100 and 5% BSA). After being washed, the cells were probed with anti–NF‐κBp65 (1:50) at 4°C overnight, stained with Cy3‐conjugated secondary antibody (1:500), and then, the nuclei were stained using DAPI. The fluorescent signals were observed in an inverted fluorescence microscope.
2.11. GTPase activity assay
Different concentrations of mdivi‐1 (0, 10, 25 and 50 µmol/L) were incubated with DNM1L (human) recombinant protein (Abnova, P01) for 30 minutes at 4°C. A GTPase Assay Kit (Abnova, KA1610) was then used to determine the GTPase activity of DNM1L according to the manufacturer's protocol.
2.12. Induction of collagen‐induced arthritis (CIA) and mdivi‐1 treatment
Male DBA/1 mice (7‐week‐old) were obtained from Beijing Weitong Lihua Experimental Animal Technology Co and were housed in a specific pathogen‐free facility with free access to food and water. The mice were immunized with 200 µg bovine type II collagen (Chondrex, 20022) in 50% Freund's complete adjuvant (FCA) (Sigma, F5881) at the tail base. On day 21, the mice were injected intradermally with bovine type II collagen in 50% incomplete Freund's adjuvant (IFA) (Chondrex, 7002). On day 28, the mice were randomized and treated intraperitoneally with DMSO (vehicle, n = 7) or mdivi‐1 (0.2 mg/mouse, n = 7) every other day for two weeks. The animal study was approved by the Ethics Committee of Tangdu Hospital, The Second Affiliated Hospital of Air Force Medical University (Xi'an, China).
2.13. Clinical and histological assessment of arthritis
The mice were monitored for arthritis signs every other day post‐treatment. The severity of arthritis was scored from 0 to 4.35 The paw thickness of individual mice was measured using a Vernier caliper. On day 50, the mice were killed, and their knee joints, liver and renal tissues were collected and fixed in 10% formalin. The tissue sections (4 µm) were stained with haematoxylin and eosin (H&E) or toluidine blue. In addition, their STs were used for analysing the expression of cytokine mRNAs. The ROS contents in crystal synovial membrane sections were determined by DHE staining.13
2.14. Statistical analysis
Data are presented as the means ± SD of three or more separate experiments. The difference between two groups was analysed by Student's t test. Correlation was determined from Pearson's correlation coefficient. All statistical analyses were performed using the SPSS software 19.0 (SPSS). A P‐value below .05 was considered statistically significant.
3. RESULTS
3.1. Enhanced mitochondrial fission in STs of RA patients is correlated with disease severity
To investigate the mitochondrial dynamics in STs during the pathogenic process of RA, the morphologic changes of mitochondria were investigated in STs from RA and non‐RA patients and examined their FLSs by TEM. As shown in Figure 1A, mitochondrial length in the RA group was shorter than non‐RA group. Similarly, the length of mitochondria in FLSs from patients with RA was also shorter than that of FLSs from non‐RA patients. Furthermore, qRT‐PCR analysis indicated more DNM1L mRNA transcripts in the STs from the RA patients than non‐RA patients (Figure 1B). IHC and Western blot analysis exhibited that the levels of DNM1L expression in STs from patients with RA were remarkably up‐regulated, compared with that in the non‐RA patients (Figure 1C,D). Interestingly, the ratio of DNM1L‐to‐MFN1 (determined by qRT‐PCR) had significantly positive correlations with the serum anti‐CCP level (r = .733, P = .016), DAS28 score (r = .089, P = .004), and ESR (r = .693, P = .026, Figure 1E). However, the DNM1L‐to‐MFN1 ratio had no significant correlations with RF level, hs‐CRP level or disease duration (data not shown). In addition, there were no significant differences in the expression of MFN2, FIS1 and OPA1 mRNAs among RA and non‐RA individuals (data not shown). Hence, some markers of enhanced mitochondrial fission in the STs of RA patients correlated with disease severity.
Figure 1.
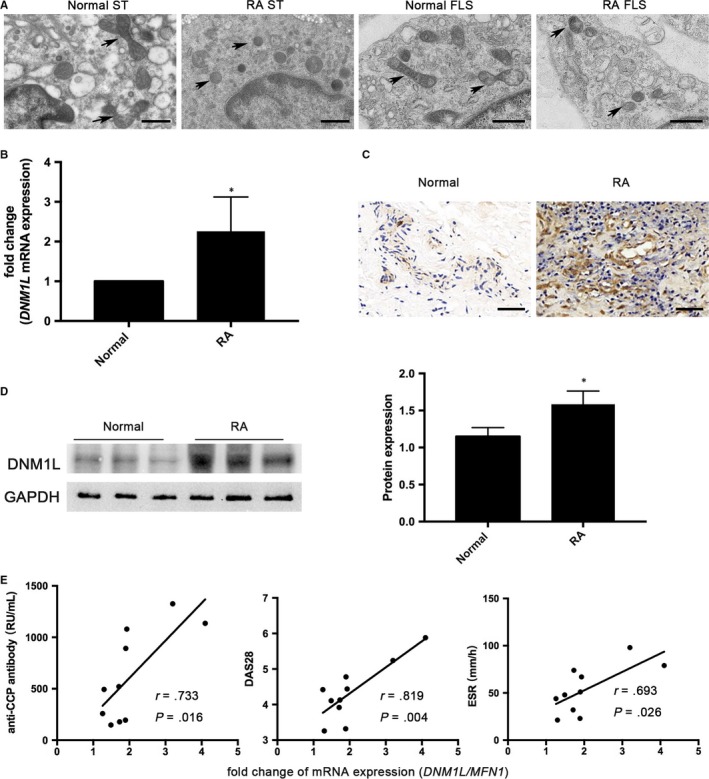
Enhanced mitochondrial fission in STs of RA patients correlates with disease severity. A, Representative TEM images of mitochondrial morphology in STs and FLS. Scale bars: 1 µm. B, qRT‐PCR analysis of DNM1L in STs. C, IHC analysis of DNM1L expression in STs. Scale bars: 50 µm. D, Western blot analysis of DNM1L in STs. E, Correlation of the ratio of DNM1L‐to‐MFN1 with the level of serum anti‐CCP, DAS28 and ESR in RA patients. Data are the mean ± SD of each group. N = 10 RA patients and n = 3 non‐RA patients. *P < .05 vs the control
3.2. DNM1L deficiency alters mitochondria morphology and mitochondrial membrane potential in FLSs
DNM1L is crucial for mitochondrial fission. Next, we examined how DNM1L deficiency could affect mitochondrial morphology and membrane potential in FLSs. In comparison with the control cells transfected with scramble siRNA, transfection with DNM1L‐specific siRNA significantly decreased DNM1L protein expression by about 30% and DNM1L mRNA transcripts by 55% in FLSs (Figure 2A,B). We also measured GTPase activity of DNM1L after mdivi‐1 treatment. The results indicate that mdivi‐1 inhibited the GTPase activity of DNM1L in a dose‐dependent manner (Figure 2C). Following staining with MitoTracker Green, treatment of mdivi‐1 (50 µmol/L) or DNM1L silencing by transfection with DNM1L‐specific siRNA significantly increased the length of mitochondrial in FLSs (Figure 2D). Following JC‐1 staining, treatment of mdivi‐1 or DNM1L silencing reduced the ratios of red to green fluorescent signals, a hallmark of depolarization in FLSs (Figure 2E). Collectively, such data indicated that DNM1L deficiency altered mitochondrial morphology and induced mitochondrial membrane depolarization in FLSs.
Figure 2.
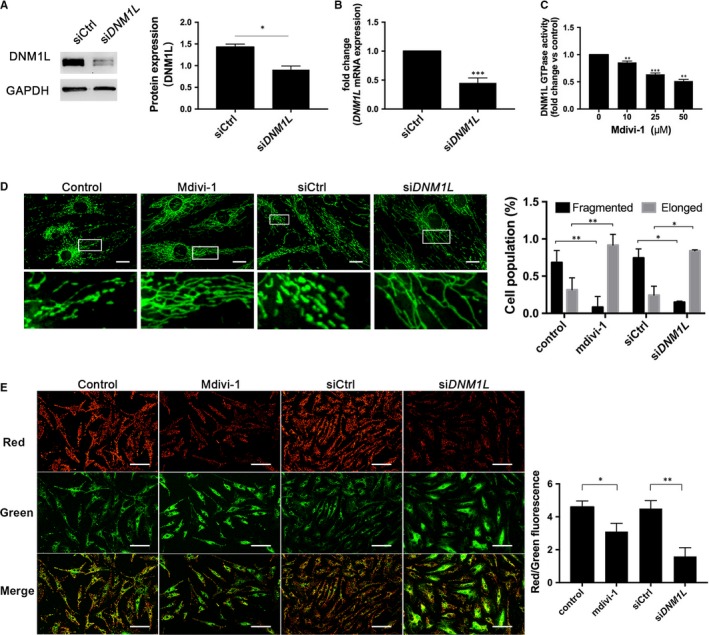
DNM1L deficiency alters mitochondria morphology and mitochondrial membrane potential in FLSs. (A, B) FLSs from RA patients were transfected with control (siCtrl) or DNM1L‐specific siRNA (siDNM1L) for 72 h, and the relative levels of DNM1L expression were determined by Western blot and qRT‐PCR. C, Effect of mdivi‐1 concentration on DNM1L GTPase activity. D, Representative confocal microscopy images of mitochondrial morphology in different groups of FLSs, with staining by MitoTracker Green and quantitation of these data. Scale bars: 20 µm. E, Representative confocal microscopy images of the effect of DNM1L deficiency on mitochondrial membrane depolarization (JC‐1 fluorescence) and quantitation of these data. The ratio of red to green signal was determined for quantitative analysis. Scale bars: 100 µm. Data are representative images or expressed as the mean ± SD of each group from three separate experiments. *P < .05; **P < .01; ***P < .001
3.3. DNM1L deficiency in FLSs reduces their viability and production of pro‐inflammatory cytokines and increases apoptosis
Treatment with 10 µmol/L mdivi‐1 did not affect the viability of FLSs while treatment with 20‐50 µmol/L mdivi‐1 significantly reduced the viability of FLSs (Figure 3A). Similarly, DNM1L silencing also significantly reduced the viability of FLSs by nearly 45%. Furthermore, treatment with mdivi‐1 (50 µmol/L) or DNM1L silencing significantly decreased the relative levels of COX‐2 and IL‐8 expression in FLSs (Figure 3B,C). In addition, treatment with 50 µmol/L mdivi‐1 or DNM1L silencing significantly increased the percentages of apoptotic FLSs (Figure 3D). Thus, DNM1L deficiency reduced the viability of FLSs and their production of pro‐inflammatory cytokines by triggering apoptosis.
Figure 3.
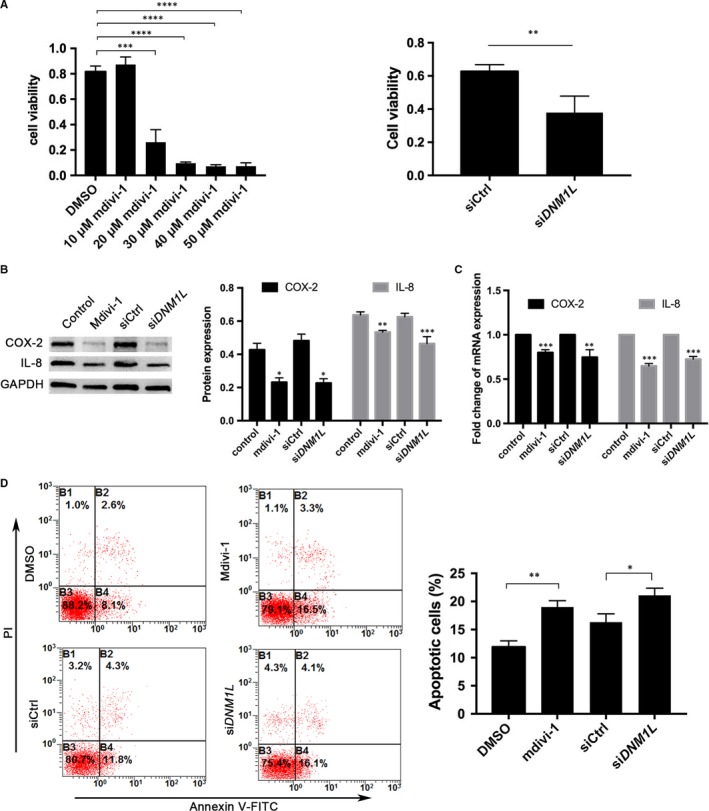
DNM1L deficiency in FLSs reduces their viability and production of pro‐inflammatory cytokines, and increases apoptosis. A, Cell viability was determined using the CCK‐8 assay. (B, C) Western blot and qRT‐PCR analyses of COX‐2 and IL‐8 expression in FLSs (mdivi‐1 concentration = 50 µmol/L). D, Representative flow cytometry data of apoptotic FLSs after staining with FITC‐Annexin V and PI, and quantitation of these data. Data are representative flow cytometry charts, images or expressed as mean ± SD of each group from three separate experiments. *P < .05; **P < .01; ***P < .001; ***P < .0001
3.4. DNM1L deficiency reduces ROS production and LC3B‐related autophagy in FLSs
To investigate the impact of DNM1L deficiency on ROS production, FLSs were treated with mdivi‐1 (50 µmol/L) or transfected with DNM1L‐specific siRNA for 72 hours. The levels of intracellular ROS were measured by DHE staining. As expected, treatment with mdivi‐1 or DNM1L silencing significantly reduced the levels of ROS in FLSs (Figure 4A). Furthermore, while treatment with IL‐1β and H2O2 significantly induced AKT activation, treatment with mdivi‐1 abrogated the IL‐1β– and IL‐1β/H2O2–induced AKT expression and phosphorylation, although the inhibitory effect of mdivi‐1 on the IL‐1β/H2O2–induced AKT expression was less than that of IL‐1β–induced AKT activation in FLSs (Figure 4B). Moreover, treatment with mdivi‐1 or DNM1L silencing significantly decreased the ratio of LC3B‐II to LC3B‐I and the IL‐1β–increased ratios of LC3B‐II to LC3B‐I in FLSs (Figure 4C). The results suggest that ROS and LC3B‐related autophagy may play a key role in DNM1L‐mediated proliferation, inflammation and apoptosis in RA FLS.
Figure 4.
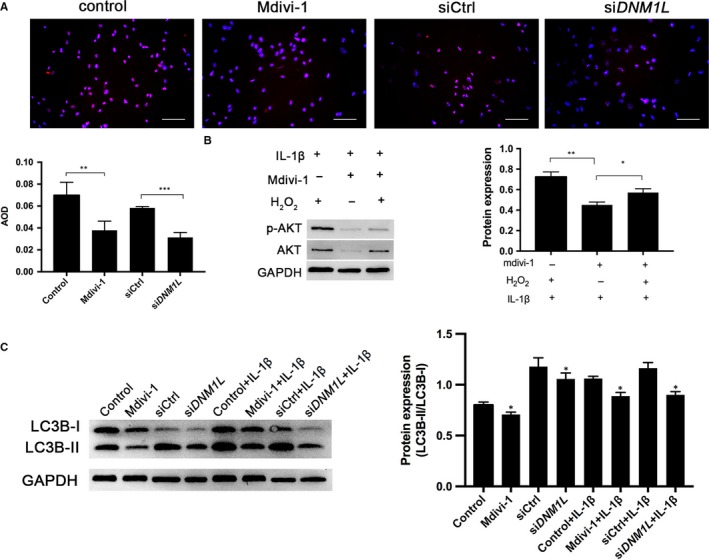
DNM1L deficiency reduces ROS production and autophagy in FLS. A, Representative DHE and DAPI staining, and quantitation of these data. AOD (average optical density) indicates the absolute absorption of DHE signal. Scale bars: 100 µm. B, Effect of mdivi‐1 on IL‐1β– and IL‐1β/H2O2–mediated AKT activation in FLSs. FLSs were treated with mdivi‐1 in the presence of IL‐1β for 10 min, and with or without, 100 µmol/L H2O2 for 12 h. C, Western blot analysis of LC3B‐I and LC3B‐II in the different groups of FLSs. Data are representative images or expressed as the mean ± SD of each group of cells from three separate experiments. *P < .05; **P < .01; ***P < .001
3.5. DNM1L deficiency attenuates the IL‐1β–induced NF‐κB activation in FLSs
To further explore the role of DNM1L in RA FLS inflammation, we examined the effect of DNM1L deficiency on the IL‐1β–induced AKT and IKK activation and NFKBIA expression in FLSs. We found that treatment with mdivi‐1 or DNM1L silencing mitigated the IL‐1β–induced AKT and IKK activation, but enhanced the IL‐1β–up‐regulated NFKBIA expression in FLSs (Figure 5A). Further immunofluorescence studies revealed that treatment with mdivi‐1 attenuated the IL‐1β–induced NF‐κBp65 nuclear translocation in FLSs (Figure 5B). Together, such data indicated that DNM1L deficiency attenuated the IL‐1β–induced NF‐κB activation in FLSs by down‐regulating AKT/IKK/NFKBIA signalling.
Figure 5.
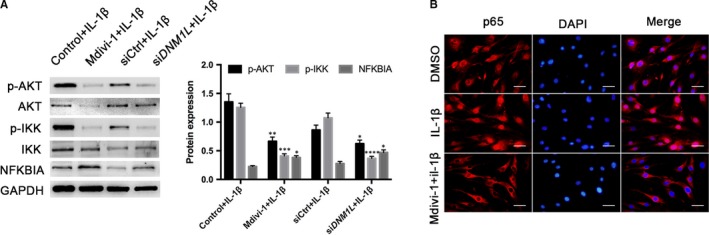
DNM1L deficiency attenuates the IL‐1β–induced NF‐κB activation in FLSs. A, The relative levels of AKT, IKK and NFKBIA expression and AKT and IKK phosphorylation were determined by Western blot. B, The nuclear translocation of NF‐κBp65 in the different groups of FLSs was determined by immunofluorescent microscopy. Scale bars: 50 µm. Data are representative images or expressed as the mean ± SD of each group of cells from three separate experiments. *P < .05; **P < .01; ***P < .001; ***P < .0001
3.6. Treatment with mdivi‐1 ameliorates the severity of CIA in mice
Finally, we examined the potential therapeutic effect of mdivi‐1 treatment on the development and severity of CIA in mice. Compared with the control (DMSO) CIA mice, treatment with mdivi‐1 (0.2 mg/mouse) significantly decreased the clinical scores, paw thickness and affected paws and ameliorated the overt symptoms (Figure 6A‐D). Furthermore, treatment with mdivi‐1 significantly mitigated the CIA‐mediated upregulation of MMP‐13, NLRP3, TNF‐α and COX‐2, but not IL‐6, and increased the expression of IL‐10 in the STs (Figure 6E). While control CIA mice displayed joint damages, treatment with mdivi‐1 preserved the intact joint architecture (Figure 6F). In addition, treatment with mdivi‐1 also decreased ROS production in the STs of mice (Figure 6H). However, histological examination revealed that treatment with mdivi‐1 did not change the morphology of the liver and kidney tissues in mice (Figure5G). Therefore, treatment with mdivi‐1 to impaired DNM1L activity and ameliorated the severity of CIA in mice by modulating the pro‐inflammatory and anti‐inflammatory responses, but had no effect on non‐target tissues (liver and kidney).
Figure 6.
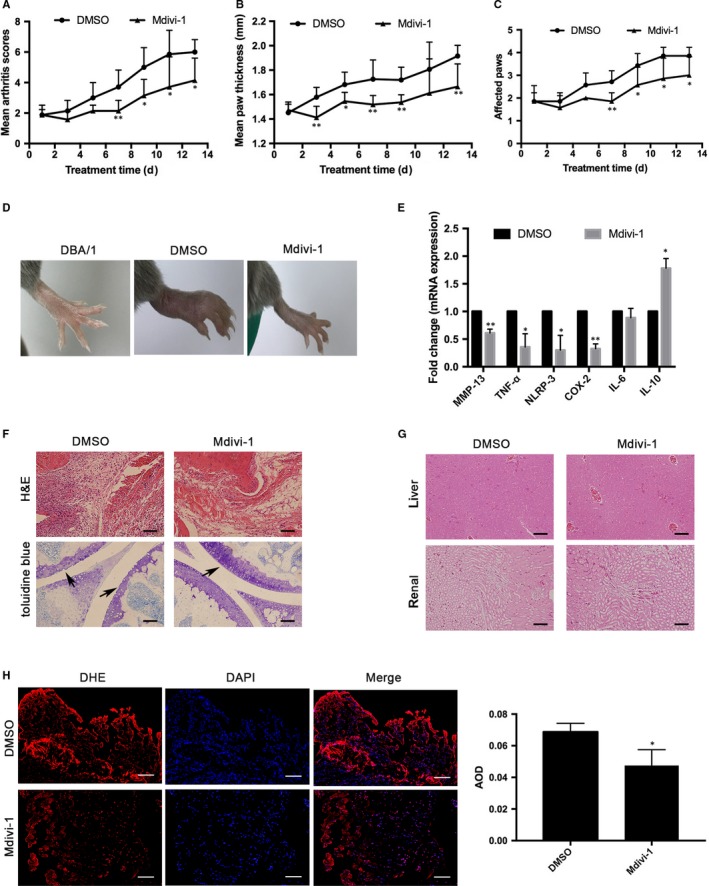
Treatment with mdivi‐1 ameliorates the severity of CIA in mice. After established CIA, the mice were randomized and treated with DMSO or 0.2 mg mdivi‐1 every other day for 2 wk. (A‐C) Their CIA clinical scores, paw thickness and affected paws were measured. D, Representative images of paws in the different groups of mice. E, The relative levels of inflammatory molecules in the joints of different groups of mice were determined by qRT‐PCR. F, Histological examination of the hind paws after H&E or toluidine blue staining. Scale bar: 100 µm. G, Histological examination of the liver and kidney tissues after H&E staining. Scale bar: 100 µm. H, DHE staining of ROS contents in the crystal synovial membranes of mice. Scale bar: 200 µm. Data are representative images or expressed as the mean ± SD of each group of mice (n = 7). *P < .05; **P < .01
4. DISCUSSION
In the present study, we found that up‐regulated DNM1L expression in STs from patients with RA was associated with the severity of RA. Such findings support the notion that up‐regulated mitochondrial fission is associated with the development of different types of diseases, including inflammatory diseases.4, 17, 25, 26, 27, 28, 29, 36, 37, 38, 39, 40 To the best of our knowledge, this was the first report on up‐regulated DNM1L expression in STs from patients with RA and the correlations suggest that DNM1L expression may be a new biomarker to evaluate the severity of RA.
DNM1L is crucial for mitochondrial fission. MFN1 knockout mice display fragmented mitochondria.41 In this study, we found that up‐regulated DNM1L expression was associated with decreased length of mitochondria in STs, consistent with previous observations.4, 17, 42 Similarly, DNM1L silencing and treatment with mdivi‐1 also increased the length of mitochondria in FSLs. Hence, up‐regulated DNM1L expression reflected enhanced mitochondrial fission, which should promote mitochondrial membrane depolarization and in tune enhance ROS production. Actually, a previous study has shown that mitochondrial membrane depolarization in peripheral lymphocytes is positively correlated with serum CRP levels and DAS28 values in patients with RA.15 DNM1L deficiency promoted mitochondrial membrane depolarization and decreased the levels of ROS and pro‐inflammatory COX‐2 and IL‐8 in RA FLSs, similar to that in synoviocytes.12 We also found that the ratio of DNM1L‐to‐MFN1 transcripts correlated with the level of serum anti‐CCP antibodies, DAS28 and ESR values in patients with RA. Such data suggest that DNM1L‐related mitochondrial fission may contribute to the pathogenesis of RA.
Mdivi‐1 inhibits DNM1L GTPase activity and is a potent inhibitor of mitochondrial fission. Previous studies demonstrated that mdivi‐1 can inhibit myocardial infarction, stroke, neurodegenerative diseases, and cancers.22, 25, 26, 36, 43, 44, 45, 46 In the present study, we found that mdivi‐1 inhibited DNM1L GTPase activity in a dose‐dependent manner, consistent with previous studies.43, 47 Furthermore, treatment with mdivi‐1 significantly reduced the severity of CIA in mice by decreasing pro‐inflammatory cytokine production, but increasing anti‐inflammatory IL‐10 production. These, together with its inhibition on ROS and pro‐inflammatory COX‐2 and IL‐8 production in FLSs, suggest that inhibition of mitochondrial fission may modulate the balance of pro‐inflammatory and anti‐inflammatory responses in joint lesions. Given that mitochondria are crucial for nutritional metabolisms, it is possible that inhibition of mitochondrial fission may differentially modulate glucose and lipid metabolisms to affect pathogenic and regulatory T‐cell responses during the pathogenic process of RA.
It is well known that mitochondrial dysfunction can induce excessive ROS production, which promotes autophagy17 and exacerbates synovial inflammation by activating the NF‐κB signalling to promote the expression of pro‐inflammatory cytokines.12, 48 Furthermore, mitochondrial dysfunction can activate PI3K/AKT signalling, which is crucial for inflammation in RA FLSs.17, 49 In addition, the activated AKT can induce IKK phosphorylation and NF‐κB, which is down‐regulated by NFKBIA.50, 51 In this study, we found that treatment with mdivi‐1 or DNM1L silencing effectively decreased ROS production and LC3B‐related autophagy in FLSs, extending the previous observation in osteoblasts.42 Moreover, DNM1L deficiency also mitigated the IL‐1β–induced AKT and IKK phosphorylation and NF‐κBp65 nuclear translocation, but increased NFKBIA expression in RA FLS. Therefore, anti‐oxidants and inhibition of mitochondrial fission to attenuate the AKT/IKK/NF‐κB signalling may be promising strategies for treatment of RA.
This study had limitations, including a small sample size and the lack of studies on how inhibition of mitochondrial fission alters mitochondrial DNA and glucose and lipid metabolisms that affect pathogenic and regulatory T‐cell responses. Hence, future investigations in a bigger population are warranted to determine molecular mechanisms by which the mitochondrial dynamic factors regulate the pathogenesis of RA.
In conclusion, our results indicated that up‐regulated DNM1L expression in STs was associated with the severity of RA. Treatment with mdivi‐1 or DNM1L silencing altered mitochondrial morphology, promoted mitochondrial membrane depolarization and apoptosis, and decreased cell viability, ROS, LC3B‐related autophagy and pro‐inflammatory cytokine production in FLSs. The same treatment also mitigated the IL‐1β–mediated LC3B‐related autophagy and activation of AKT, IKK and NF‐κB in FLSs by up‐regulating NFKBIA expression. Treatment with mdivi‐1 significantly reduced the severity of CIA in mice by decreased ROS and modulating the balance of pro‐inflammatory and anti‐inflammatory responses. Therefore, mitochondrial fission may be new target for treatment of RA.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Wang Xiaoyan and Chen Zhufeng performed the research and wrote the paper. Fan Xuemei, Qu Jiaqi and Li Wei analysed the data. Dong Chuan, Wang Zhixue and Ji Zhenwei provided samples in the study. Li Yang and Wang Xiaoyan designed the research study.
ACKNOWLEDGEMENTS
The authors thank for supports from the Department of Rheumatology and Immunology, the Second Affiliated Hospital of Harbin Medical University (Harbin, China) and Department of Orthopaedics, Tangdu Hospital, the Second Affiliated Hospital of Air Force Medical University (Xi'an, China). This work was supported by a grant from the National Natural Science Foundation of China (grant 81373202).
Wang X, Chen Z, Fan X, et al. Inhibition of DNM1L and mitochondrial fission attenuates inflammatory response in fibroblast‐like synoviocytes of rheumatoid arthritis. J Cell Mol Med. 2020;24:1516–1528. 10.1111/jcmm.14837
DATA AVAILABILITY STATEMENT
All data generated or analysed during this study are included in this article.
REFERENCES
- 1. McInnes IB, Schett G. The pathogenesis of Rheumatoid Arthritis. N Engl J Med. 2011;365:2205‐2219. [DOI] [PubMed] [Google Scholar]
- 2. Bartok B, Firestein GS. Fibroblast‐like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ng CT, Biniecka M, Kennedy A, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheumat Dis. 2010;69:1389‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo F, Herrup K, Qi X, Yang Y. Inhibition of Drp1 hyper‐activation is protective in animal models of experimental multiple sclerosis. Exp Neurol. 2017;292:21‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stuart JA, Brown MF. Mitochondrial DNA maintenance and bioenergetics. Biochim Biophys Acta. 2006;1757:79‐89. [DOI] [PubMed] [Google Scholar]
- 6. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483‐495. [DOI] [PubMed] [Google Scholar]
- 7. Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu W, Fu YC, Wang W. Cellular and molecular effects of resveratrol in health and disease. J Cell Biochem. 2012;113:752‐759. [DOI] [PubMed] [Google Scholar]
- 9. Zhang J, Song X, Cao W, et al. Autophagy and mitochondrial dysfunction in adjuvant‐arthritis rats treatment with resveratrol. Sci Rep. 2016;6:32928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biniecka M, Fox E, Gao W, et al. Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheumat. 2011;63:2172‐2182. [DOI] [PubMed] [Google Scholar]
- 11. Xu K, Xu P, Yao JF, Zhang YG, Hou WK, Lu SM. Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm Res. 2013;62:229‐237. [DOI] [PubMed] [Google Scholar]
- 12. Valcarcel‐Ares MN, Riveiro‐Naveira RR, Vaamonde‐Garcia C, et al. Mitochondrial dysfunction promotes and aggravates the inflammatory response in normal human synoviocytes. Rheumatology. 2014;53:1332‐1343. [DOI] [PubMed] [Google Scholar]
- 13. Vaamonde‐Garcia C, Loureiro J, Valcarcel‐Ares MN, et al. The mitochondrial inhibitor oligomycin induces an inflammatory response in the rat knee joint. BMC Musculoskelet Disord. 2017;18:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biniecka M, Kennedy A, Ng CT, et al. Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia‐induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res Ther. 2011;13:R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moodley D, Mody G, Patel N, Chuturgoon AA. Mitochondrial depolarisation and oxidative stress in rheumatoid arthritis patients. Clin Biochem. 2008;41:1396‐1401. [DOI] [PubMed] [Google Scholar]
- 16. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241‐1252. [DOI] [PubMed] [Google Scholar]
- 17. Huang Q, Zhan L, Cao H, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS‐modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12:999‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vega RB, Horton JL, Kelly DP. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ Res. 2015;116:1820‐1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fan X, Hussien R, Brooks GA. H2O2‐induced mitochondrial fragmentation in C2C12 myocytes. Free Radic Biol Med. 2010;49:1646‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jendrach M, Mai S, Pohl S, Voth M, Bereiter‐Hahn J. Short‐ and long‐term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion. 2008;8:293‐304. [DOI] [PubMed] [Google Scholar]
- 21. Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission‐fusion proteins. FEBS J. 2011;278:941‐954. [DOI] [PubMed] [Google Scholar]
- 22. Gan X, Huang S, Wu L, et al. Inhibition of ERK‐DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell. Biochim Biophys Acta. 2014;1842:220‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gan X, Wu L, Huang S, et al. Oxidative stress‐mediated activation of extracellular signal‐regulated kinase contributes to mild cognitive impairment‐related mitochondrial dysfunction. Free Radic Biol Med. 2014;75:230‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872‐884. [DOI] [PubMed] [Google Scholar]
- 25. Rehman J, Zhang HJ, Toth PT, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao J, Zhang J, Yu M, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814‐4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pal AD, Basak NP, Banerjee AS, Banerjee S. Epstein‐Barr virus latent membrane protein‐2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis. 2014;35:1592‐1601. [DOI] [PubMed] [Google Scholar]
- 28. Wan YY, Zhang JF, Yang ZJ, et al. Involvement of Drp1 in hypoxia‐induced migration of human glioblastoma U251 cells. Oncol Rep. 2014;32:619‐626. [DOI] [PubMed] [Google Scholar]
- 29. Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256‐1268. [DOI] [PubMed] [Google Scholar]
- 30. Huang S, Wang Y, Gan X, et al. Drp1‐mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes. 2015;64:1728‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Veale DJ, Orr C, Fearon U. Cellular and molecular perspectives in rheumatoid arthritis. Semin Immunopathol. 2017;39:343‐354. [DOI] [PubMed] [Google Scholar]
- 32. Rantapaa‐Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741‐2749. [DOI] [PubMed] [Google Scholar]
- 33. Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380‐386. [DOI] [PubMed] [Google Scholar]
- 34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 35. Yan M, Liu X, Dang Q, Huang H, Yang F, Li Y. Intra‐articular injection of human synovial membrane‐derived mesenchymal stem cells in murine collagen‐induced arthritis: assessment of immunomodulatory capacity in vivo. Stem Cells Int. 2017;2017:9198328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rappold PM, Cui M, Grima JC, et al. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun. 2014;5:5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song W, Chen J, Petrilli A, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin‐related protein‐1 and increases its enzymatic activity. Nat Med. 2011;17:377‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Disatnik MH, Ferreira JC, Campos JC, et al. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long‐term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057‐9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zepeda R, Kuzmicic J, Parra V, et al. Drp1 loss‐of‐function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia‐reperfusion injury. J Cardiovasc Pharmacol. 2014;63:477‐487. [DOI] [PubMed] [Google Scholar]
- 41. Patrushev MV, Mazunin IO, Vinogradova EN, Kamenski PA. Mitochondrial fission and fusion. Biochem. 2015;80:1457‐1464. [DOI] [PubMed] [Google Scholar]
- 42. Gan X, Huang S, Yu Q, Yu H, Yan SS. Blockade of Drp1 rescues oxidative stress‐induced osteoblast dysfunction. Biochem Biophys Res Commun. 2015;468:719‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cassidy‐Stone A, Chipuk JE, Ingerman E, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak‐dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferrari LF, Chum A, Bogen O, Reichling DB, Levine JD. Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J Neurosci. 2011;31:11404‐11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012‐2022. [DOI] [PubMed] [Google Scholar]
- 47. Bordt EA, Clerc P, Roelofs BA, et al. The putative Drp1 inhibitor mdivi‐1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40(6):583‐594.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vaamonde‐Garcia C, Riveiro‐Naveira RR, Valcarcel‐Ares MN, Hermida‐Carballo L, Blanco FJ, Lopez‐Armada MJ. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012;64:2927‐2936. [DOI] [PubMed] [Google Scholar]
- 49. Camps M, Ruckle T, Ji H, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936‐943. [DOI] [PubMed] [Google Scholar]
- 50. An J, Wang X, Guo P, Zhong Y, Zhang X, Yu Z. Hexabromocyclododecane and polychlorinated biphenyls increase resistance of hepatocellular carcinoma cells to cisplatin through the phosphatidylinositol 3‐kinase/protein kinase B pathway. Toxicol Lett. 2014;229:265‐272. [DOI] [PubMed] [Google Scholar]
- 51. Factor V, Oliver AL, Panta GR, Thorgeirsson SS, Sonenshein GE, Arsura M. Roles of Akt/PKB and IKK complex in constitutive induction of NF‐kappaB in hepatocellular carcinomas of transforming growth factor alpha/c‐myc transgenic mice. Hepatology. 2001;34:32‐41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this article.