

Abstract
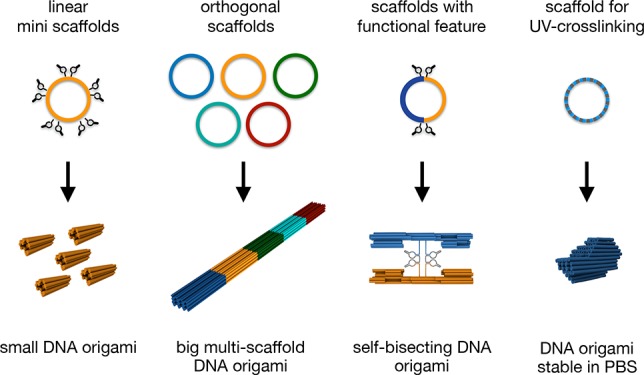
DNA origami nano-objects are usually designed around generic single-stranded “scaffolds”. Many properties of the target object are determined by details of those generic scaffold sequences. Here, we enable designers to fully specify the target structure not only in terms of desired 3D shape but also in terms of the sequences used. To this end, we built design tools to construct scaffold sequences de novo based on strand diagrams, and we developed scalable production methods for creating design-specific scaffold strands with fully user-defined sequences. We used 17 custom scaffolds having different lengths and sequence properties to study the influence of sequence redundancy and sequence composition on multilayer DNA origami assembly and to realize efficient one-pot assembly of multiscaffold DNA origami objects. Furthermore, as examples for functionalized scaffolds, we created a scaffold that enables direct, covalent cross-linking of DNA origami via UV irradiation, and we built DNAzyme-containing scaffolds that allow postfolding DNA origami domain separation.
Keywords: nanostructures, DNA origami, DNA nanotechnology, self-assembly, phagemid
DNA origami has become a valuable tool for basic science in a diversity of research fields, and applications in material sciences, medicine, and in other areas have begun to emerge.1 The DNA origami design methodology can be used to create custom 2D2 and 3D shapes3,4 with nanometer dimensions. DNA origami uses a set of short “staple” DNA oligonucleotides that are designed to fold a long, usually circular “scaffold” DNA single strand into a user-defined shape. The self-assembly can proceed rapidly and with high yields and quality, depending on design and assembly procedures used. During DNA origami design, the scaffold sequence is typically taken as a fixed input from a library of generic scaffold sequences that are available to the community. The sequence string is then routed through a strand diagram that encodes the connectivity of the target object to be made. The sequences for the staple strands are derived by considering local Watson–Crick base complementarity to the scaffold.2,3
Generic scaffold sequences, as others have noted previously,5 may limit the scope of applications that could be addressed with DNA origami. To understand the resulting restrictions, it is helpful to consider the M13 bacteriophage single-strand genome variants which are the most popular generic scaffolds currently used in the field. Many properties of the DNA origami built with M13-based scaffolds are naturally determined by the length and sequence of the M13 phage genome: First, the overall dimensions of a single DNA origami are by default limited by the length of the scaffold strand. Second, designers cannot control global sequence properties such as GC content or sequence redundancy, which could potentially affect the self-assembly behavior as others have speculated.6 Third, designers cannot avoid undesired sequence motifs such as potentially immunogenic CpG motifs7 which occur frequently within M13-based scaffolds. Fourth, phage genes may be undesirable in the context of in vivo applications. Fifth, custom sequence motifs can be introduced in DNA origami objects only as appendices or insertions in staple strands, which may negatively affect the yield of incorporation of such motifs,8 unless the scaffold strand itself is customized by the user. One example of sequence motifs that could be of use at hundreds of sites in the scaffold are AA motifs at all crossovers positions, as this could enable covalent stabilization of the target object via UV point-welding.9
We envision that rather than designing objects around generic scaffold sequences, as the current DNA origami paradigm posits, designers should be able to fully specify the target structure not only in terms of desired 3D shape but also in terms of the sequences used. Realizing design-specific scaffolds requires a design tool that can interpret design diagrams and algorithmically build a scaffold sequence, and it also requires scaffold production methods adapted for making fully sequence-customized scaffolds. In order to address real-world applications, it should also be possible to manufacture the resulting design-specific scaffolds in a scalable and cost-efficient process. Whereas M13 scaffolds can be efficiently produced biotechnologically, a substantial portion of the M13 phage genome cannot be modified because these portions are required for the phage lifecycle. Scaffolds for DNA origami may be produced in test tubes using enzymatic reactions3,6,10−15 or in bacterial cultures via phage-based biotechnological production.2,3,16−20 Enzymatic production in test tubes affords the possibility of choosing user-defined sequences but is unfortunately economical only on analytical (μg) scales. In general, biotechnological processes using bacterial cultures allow production on much larger scales such as those required for biomedical or materials applications of DNA origami.18,20,21
The objective of this work is to provide the missing tools and methods to realize design-specific scaffolds for DNA origami. To this end, we built a tool called “scaffold smith” that can construct design-specific scaffold sequences for DNA origami, and we developed a biotechnological approach to produce fully sequence-customized scaffolds. In total, we produced a library of 17 customized scaffold strands with attractive properties that should be useful to the community as generic scaffolds (plasmids available via Addgene, alongside a target plasmid for construction of other scaffolds). Our design tools and production methods for making design-specific scaffolds allow designing fully user-specified DNA origami while maintaining the possibility to produce materials at larger scales.
Results and Discussion
Sequence Design and Strand Production
To construct design-specific custom scaffold sequences, we created a design tool termed “scaffold smith” (Figure 1A and Supporting Information Note S1). The tool integrates with the conventional DNA origami design workflow at the point when the user has produced a caDNAno strand diagram.22 The scaffold smith generates the scaffold string that users will then use subsequently to generate the staple sequences. The user can define sequence motifs that will be excluded entirely, and the user can specify a list of sequence strings to be placed in the scaffold at desired locations in the target object. The tool also enables constructing a design-specific scaffold string for direct, modification-free UV cross-linking of the target object.9 To this end, the tool automatically identifies all scaffold base indices located at staple termini and at crossovers and places “A” or “AA”, respectively, at those positions in the scaffold string. Also, scaffolds may be produced that have fixed scaffold motifs at staple termini so that residual overhangs of staples that are produced biotechnologically and DNAzyme-digested as previously described21 can directly pair with the scaffold. The scaffold smith can either generate sequences de novo or operate on existing scaffold strings to create new variants of them that include desired motifs at desired locations. For de novo construction, the sequence is built base-by-base with a stochastic Monte Carlo process beginning at a user-defined site in the strand diagram. The algorithm controls the scaffold sequence composition in terms of the statistical weights of base pair steps (e.g., how often A should be followed by A, C, G, or T, respectively), which gives the user control over the thermodynamic properties of the scaffold to be built. It also enables directly reducing or avoiding entirely the occurrence of known immunogenic or UV-radiation-sensitive motifs such as CG or TT, respectively. The tool considers the degree of sequence redundancy that emerges during sequence construction and can build (pseudo-) De Bruijn sequences of user-defined order. It can generate sequences where all strings of a user-defined length (for example 8 bases) appear only as often as the user accepts it in the entire scaffold sequence (for example, not more than once). Finally, the tool computes the overall statistics of the generated scaffold string with respect to composition and redundancy. The user may then adjust parameters and repeat the sequence construction. To summarize, the scaffold sequence construction with the scaffold smith has a deterministic and a stochastic part. The user can define properties, which will be strictly realized, such as exclusion and site-directed inclusion of user-defined sequence motifs. All remaining sites (i.e., sites where the user makes no specific demands) will be filled up stochastically; however, the user has control over the overall statistics of the sequence built in terms of composition and redundancy. The underlying algorithms are described in more detail in Supporting Information Note S1. We created a stand-alone graphical user interface (GUI) for the scaffold smith, but it should be straightforward to introduce the underlying concepts into future caDNAno versions or into future variants of automated design solutions such as DAEDALUS,23 PERDIX,24 TALOS,25 or vHelix.26
Figure 1.

Design-specific scaffold sequences in minimum-constraint vectors for making fully user-defined DNA origami. (A) Schematic diagram of input for the scaffold smith used for creating custom scaffold sequences: exemplary caDNAno design diagram with scaffold strand indicated in blue and staple strands in multiple colors (I), user-specific constraints (II), and weighting factors for a stochastic base distribution (III). (B) Illustration of scaffold production with helper-plasmid system using phagemids with a split-ori approach (top) and a modified split-ori approach where the backbone sequence is flanked by self-cleaving DNAzymes (bottom). Zn2+ addition leads to excision of the backbone and linearization. Black, constant parts for each type of scaffold; gray, user-definable parts; light green, backbone present only in the double-stranded plasmid and not in the single-stranded product; red, self-cleaving DNAzymes.
We now focus on the question of how to practically make fully sequence-customized DNA single strands. A scalable solution for ssDNA production makes use of bacteriophages with fast growing Escherichia coli (E. coli) cells as host, but phage-based ssDNA scaffolds inevitably contain cassettes with sequences that cannot be altered because they are required for the phage production. User-defined insert sequences can only be added to these fixed parts. In fully customizable scaffolds, the length of the fixed part should be negligible compared to the total length of the scaffold. However, in the conventional M13 phage production method,18,27 the fixed part is approximately 6000 bases long, which is not negligible at all. Phagemids, in combination with helper phages28 or helper plasmids,29 allow producing ssDNA with fixed backbones of ∼2000 bases, which is still not negligible. Our goal was thus minimizing the fixed-sequence cassettes to maximize the freedom to design custom scaffold sequences while maintaining the possibility for efficient production in bacterial cultures. To this end, we developed and tested several methods with minimized constant-sequence cassettes (Supporting Information Note S2).
The production method used for most of our custom scaffolds relies on a split origin of replication (split-ori) that was originally developed to produce microphages containing comparably short 221 bases long ssDNA, in combination with helper phages.30 Here, we integrated our design-specific scaffold sequences as custom inserts into the split-ori system (Figure 1B) and identified a suitable helper plasmid that allows producing pure target ssDNA without contamination of helper phage DNA or other unwanted DNA species (Supporting Information Figure S5). The thus-produced ssDNA scaffold strands are circular with a minimal constant-sequence backbone of 234 bases (Figure 1B). This residual backbone can then also be removed entirely via Zn2+-dependent digestion when flanking self-excising DNAzyme cassettes21 are added during sequence preparation for gene synthesis. As a result, the user obtains linear scaffold molecules with virtually 100% custom sequence (except for two and seven base residuals at the two termini). In support of the robustness of the split-ori/helper-phage approach, we note that, concurrent to our work, Douglas and co-workers produced scaffolds for DNA origami by inserting coding genes or parts of the lambda phage genome into a split-ori backbone, although Douglas et al. used a different helper plasmid.31
Sequence Redundancy and Sequence Composition Rules
The commonly used M13-phage-based scaffolds have a comparably high degree of sequence redundancy, and others have speculated that this redundancy may negatively influence the self-assembly behavior of DNA origami.6 On the other hand, it has also been speculated that the M13-based sequences were particularly well-behaved and thus especially suited for DNA origami.32 In addition, the influence of sequence composition (e.g., AT vs GC content) on self-assembly remains in the dark. For designing synthetic scaffolds, it is important to understand the impact of sequence redundancy and sequence composition on self-assembly in order to arrive at relevant sequence construction criteria. To study these parameters, we constructed five synthetic 7560 bases long scaffolds (SC2–6) and compared them to a popular M13-based scaffold variant (SC1) of the same length (Figure 2). The designed portions of the custom scaffolds SC2–6 were low redundancy de Bruijn sequences of order 7, which means that sequence strings with length 7 occur exactly once or not at all.33 All of these scaffolds could be produced in shake flasks with yield and purity similar to that in conventional M13-based production (Supporting Information Note S2 and Figures S4–S6). Four of the scaffolds (SC2, SC4, SC5, SC6) have insert sequences that are orthogonal to each other and to the conventional M13-based scaffolds. Residual sequence overlaps between these four individual scaffolds are determined by details of the constant-sequence cassettes in the phagemids and have lengths between 180 and 426 bases, which is small compared to the total length (7560) of the scaffold variants. Scaffold variant SC3 had a longer 1387 bases long sequence fragment taken from the M13 genome; SC3 has thus a degree of sequence redundancy which fell between the low-redundant de Bruijn scaffolds and the highly redundant M13.
Figure 2.

Influence of base composition and sequence redundancy of custom scaffolds on DNA origami self-assembly. Blue indicates M13-based scaffolds; orange, magenta, red, cyan, and green indicate custom scaffolds. (A) Schematic representations of six different 42-helix bundles folded using the six different scaffolds. SC1, M13-based scaffold; SC2, reduced backbone phagemid scaffold with CpG-free de Bruijn insert sequence; SC3, conventional phagemid with high duplicity fragment and de Bruijn insert sequence; SC4, conventional phagemid with de Bruijn insert sequence; SC5 and SC6, split-ori based scaffold with de Bruijn sequence; L, length; GC, GC content of the corresponding scaffold. (B) Electrophoretic mobility analysis of self-assembly reactions of the 42-helix bundles shown in (A) at different temperatures and salt concentrations. SC, scaffold reference; C50 and C20, assembly reactions containing 50 nM (C50) or 20 nM (C20) scaffold, 200 nM staples, and 20 mM MgCl2 that were subjected to an annealing ramp from 60 to 44 °C (1 h per °C); temperature screen, assembly mixtures as in C50 but subjected to annealing ramps covering the temperature intervals indicated above each lane (1 h per °C); magnesium screen, assembly reactions containing 50 nM scaffold, 200 nM staples, and MgCl2 concentrations between 5 mM (M5) and 30 mM (M30). P, pocket; F, folded 42-helix bundle. All samples were loaded onto the gel at an approximate scaffold concentration of 20 nM. All temperature ramps contained an initial denaturation step at 65 °C for 15 min. Laser scanned fluorescent images of the electrophoretic analysis were autoleveled. (C) Statistics of sequence duplicates of different scaffold variants as a function of fragment length. Colors as in (A). (D) Experimentally observed optimal folding temperature intervals of the 42-helix bundles plotted against total NN energy of corresponding scaffold variant. Total NN energy was calculated using nearest-neighbor free energy parameters,36 ignoring edge effects. Dots in red indicate upper, and dots in blue indicate lower limit of the highest folding temperature interval where the sample appeared fully folded. Solid lines represent linear fits.
To test our custom scaffolds, we used them as templates for variants of a previously described brick-like 42-helix bundle (42hb)34 and synthesized the corresponding sets of staple oligonucleotides (Figure 2A). We analyzed the assembly behavior of the different 42hb variants at different temperatures and salt concentrations using a standardized folding screen.35 The assembly reactions yielded well-folded products for all six scaffold sequence variants of the 42hb object, as manifested by sharp leading bands in gel electrophoresis (Figure 2B). Contrary to what has been speculated previously,6 we did not observe systematic quality differences between the scaffold variants with higher or lower degree of sequence redundancy. In particular, we did not detect a beneficial effect on assembly behavior when using the low-redundancy de Bruijn sequences compared to the conventional, much more redundant M13-based scaffold variant (Figure 2C and Supporting Information Figure S7). Similarly, we could not detect any drawbacks of synthetically designed scaffold sequences that are not M13-based.
Sequence composition, however, did have noticeable effects on self-assembly behavior. For example, well-folded objects self-assembled already at lower salt concentrations for sequence variants with higher GC content (Figure 2B, right). As seen previously for other DNA origami objects,34 each sequence variant assembled successfully in narrowly defined temperature intervals. For our 42hb variants, we found that the sequence composition of the scaffold variant determined the temperature intervals in which the objects folded successfully (Figure 2A,B). In particular, the temperature intervals that yielded the highest folding quality correlated strongly with the scaffold sequence composition in terms of the total nearest-neighbor energy (Figure 2D).36 In the SC2 sequence, C is never followed by G. As the CG base pair step has a particularly strong stacking energy, the omission of this base pair step leads to a substantially reduced nearest-neighbor energy. Only looking at GC content as predictor is too coarse: SC2 has the lowest temperature interval but the second-lowest GC percentage (44%), whereas SC1 (=M13) has the lowest GC content but does not fold in the lowest temperature interval. Hence, the sequence composition should be considered during sequence construction at the level of base pair step composition. Our design tool scaffold smith was thus built accordingly.
Smaller DNA Origami
Depending on the target application, scaffolds shorter than the conventional M13 variants (∼8000 bases) may be desirable. With the scaffold smith, scaffold sequence strings of any length may now be designed. However, the scaffold production method must be adapted according to the length of the target strand. We thus tested the split-ori approach for its capacity to produce short scaffolds in the ∼1000 bases length range. To this end, we built a circular, 1317 bases long mini-scaffold (Supporting Information Figures S5 and S6D,E). We found that the ssDNA amount per culture volume for this short scaffold was substantially lower (0.38 mg/L) compared to the yields obtained for target strands with lengths between ∼3000 (3.6 mg/L) and ∼9000 bases (2.6 mg/L). We therefore developed an alternative method for the convenient biotechnological production of short linear scaffolds with completely user-definable sequences. The method builds on our recently reported strategy for the biotechnological production of staple strands.21 We integrated multiple copies of the same target scaffold sequence in one phagemid and interleaved them with Zn2+-dependent, self-excising DNAzyme “cassettes”. The resulting multi-insert circular DNA single strands have a total size comparable to that of the conventional M13 genome, which is presumably favorable for DNA packaging and phage particle production. Indeed, the multi-insert phagemids can be produced with satisfying yields. Upon incubation with Zn2+, the DNAzyme cassettes become catalytically active and the circular ssDNA is digested into excised DNAzyme snippets, residual backbone, and multiple copies of the linear single-stranded target scaffold (Figure 3A). Thus, the multi-insert excision approach effectively allows mass producing homotypic pools of DNA oligonucleotides (as opposed to heterotypic pools as in our previous work21). We used our multi-insert excision approach to produce three scaffold variants with lengths of 1024, 1512, and 2048 bases and used them to assemble 13-helix bundles of different lengths. All 13-helix bundle variants self-assembled with excellent yield into the desired shape, as corroborated by gel electrophoresis, transmission electron microscopy (TEM) imaging, and reference-free class averaging (Figure 3A,B). For making scaffolds with lengths between ∼3000 and ∼9000 bases, we found the conventional phagemid approach to be well-suited. As an example, we produced an additional series of synthetic-sequence scaffolds with lengths of 2873, 4536, 6048, and 9072 bases (Figure S6D,E). These variants expand the currently available set of generic scaffolds17,32 that is available to the community and that may be used to produce DNA origami with corresponding sizes.
Figure 3.

DNA origami objects with sizes ranging between 1024 bp (633 kDa) and 37800 bp (23.4 MDa) can be assembled using mini-scaffolds or in one-pot assembly reactions containing multiple scaffolds. Blue indicates M13-based scaffolds; orange, green, cyan, and red indicate custom scaffolds. (A) Schematic representation of a circular DNA single strand (top left) that, in the presence of Zn2+, cleaves itself to yield four copies of a short, linear scaffold (top right) that can subsequently be used to assemble a small DNA origami object (bottom). (B) Schematic representation (top) and average TEM images of 13-helix bundle (13hb) variants assembled from linear mini-scaffolds comprising 1024 (I), 1536 (II), or 2048 bases (III). Scale bar: 20 nm. (C) Electrophoretic mobility analysis of mini-scaffolds and 13-helix bundle variants described in (B). (D) Schematic representations, single TEM images, and average TEM images (from top to bottom) of a 42-helix bundle assembled with five scaffolds in one-pot reactions. Scale bar: 50 nm. (E) Schematic representations, single TEM images, and average TEM images (from top to bottom) of an improved 42-helix bundle design with five interlocked scaffolds. Scale bar: 50 nm. (F) Electrophoretic mobility analysis of the two 42-helix bundle versions shown in (D,E). (G) Schematic representation (top), average TEM images with corresponding model views (left), and gel electrophoretic analysis (right) of a 126-helix bundle (126hb) assembled with two interlocked scaffolds. Scale bar: 50 nm. (H) Overlay of a cryo-EM density map fragment and the corresponding scaffold routing diagram. Blue and orange paths indicate the two orthogonal scaffolds. Laser scanned fluorescent images of the electrophoretic analyses were autoleveled. P, pocket; sta, staples.
Larger DNA Origami
Many applications of DNA origami require objects whose sizes exceed the dimensions of conventional M13 scaffolds.16,37−41 Researchers have thus invested effort into building larger DNA origami to achieve greater overall dimensions and to integrate more features.41,42 One possibility to build larger DNA origami with sizes beyond 10000 base pairs is to use increasingly long scaffold chains. Consequently, other researchers have reported up to 50000 bases long scaffold strands that were constructed from biological sequences, including E. coli genomic sequences and lambda phage sequences.16,43 However, for scaffold lengths beyond 10kb assembly, cloning and plasmid handling become challenging. Moreover, when we compared the yield of production of scaffolds of different lengths, a trend emerged indicating that the yield drops for lengths approaching 10000 (Supporting Information Figure S6F), although the data are not entirely conclusive. A second possibility for making larger objects is to form higher-order assemblies from separately folded DNA origami subunits.40,41,44,45 Oligomerization of individually assembled DNA origami objects can be achieved using sticky-end interactions37,40 or via shape-complementary surface features and stacking interactions.38,39,41 When following these routes, the individual building blocks must be produced separately and usually require some type of purification, which in addition to manual labor can negatively affect the overall yield.
Here, we thus pursued a third, complementary strategy to make larger DNA origami which considers the usage of multiple scaffold chains in one-pot assembly reactions, which has been used already exemplarily in our own previous work41 and in those of others.31,46 For one-pot assembly of multiscaffold DNA origami, we anticipate that the scaffold sequences must be sufficiently distinct (“orthogonal”) to achieve productive folding of the target object. We tested these requirements experimentally and found that successful one-pot coassembly does indeed require orthogonal scaffold sequences (Supporting Information Figures S8 and S9). To enable one-pot coassembly with multiple scaffolds, we thus designed four 7560 bases long scaffolds (SC2, 4, 5, 6, compare Figure 2) that are orthogonal to each other and to the conventional M13-based scaffold (SC1). As a proof-of-concept, we designed a long pentameric 42-helix bundle object (Figure 3D) that self-assembled in a one-pot folding reaction mixture containing the five scaffold chains with distinct sequences and the several hundred staple oligonucleotides. Direct imaging with negative-staining TEM revealed the expected 42-helix bundle pentamers without visible seams between the subunits containing the individual scaffolds (Figure 3D). Reference-free class averages indicated a global twist deformation along the helical axis, which is consistent with recent findings concerning the occurrence of residual twist in honeycomb DNA origami.41 TEM imaging further revealed higher-order branched networks in which well-folded 42hb pentamers were connected with other 42hb pentamers (Supporting Information Figure S10). We attributed these connected pentamers to design flaws: For this initial demonstration, we simply designed staple strands that connect the individual single-scaffold 42hb blocks across the helical interface. Some of these connecting staple strands featured long binding segments that presumably cause the undesired branched connections. We thus made a second, distinct 42hb pentamer design in which we changed the routing of the five scaffold chains to better interlock the individual chains. We also corrected right-handed twist using base pair deletions in the design, and we included an asymmetric feature. The thus-revised object self-assembled in the expected shape as seen by TEM (Figure 3E), now with reduced twist, and it appears as a single discrete species as seen in gel electrophoresis (Figure 3F). The extent of aggregates was substantially reduced compared to the variant without interlocked scaffolds. Importantly, the folding reaction mixtures for both 42hb pentamer design variants yielded only the pentameric target object in addition to a design-dependent extent of aggregates of intact pentamers, as seen in gel electrophoresis (Figure 3F). Incomplete pentamers were absent in both design versions. To achieve complete pentamers as a single folding product, the scaffold concentrations must be adjusted such that they appear in exactly equivalent amounts in the folding reaction mixture.
To illustrate the excellent potential of using multiple orthogonal scaffold chains for efficiently constructing larger DNA origami with high yield and high quality, we designed a barrel-like 126-helix bundle (126hb) comprising 15120 base pairs distributed over two orthogonal scaffolds that are interlocked in the helical direction (Figure 3G). When the relative scaffold concentrations were properly adjusted, the object formed successfully with close to 100% yield and virtually no side products, as seen in gel electrophoretic mobility analysis and TEM imaging (Figure 3G,H and Figure S11). Reference-free class averages from single-particle micrographs were in very good agreement with the designed shape. Due to the high quality of the object, we were able to solve a structure of this object using cryo-electron microscopy, in which nearly all of the 126 constituent helices were resolved in such detail that the grooves of double helices and all connecting crossovers could be discerned. We analyzed the map with respect to systematic differences at scaffold–scaffold seams and could not find any differences between seams containing one or both scaffolds (Figure 3G). Therefore, given a suitable scaffold routing and properly calibrated strand concentrations, multiscaffold DNA origami objects can be assembled with the high yield and the high quality known from well-behaved single-scaffold DNA origami designs. One-pot assembly of multiscaffold objects represents thus a powerful route for building larger DNA origami.
Functional Scaffolds: Catalytic Motifs and Covalent Cross-Linking
The design of fully synthetic scaffolds enables exclusion of undesired motifs and the inclusion of specific sequence motifs that serve user-defined purposes. As a demonstration for motif exclusion, we built a synthetic de Bruijn scaffold on the order of 7 that lacks CG base pair steps (SC2 from Figure 2). The absence of these CpG motifs could potentially circumvent Toll-like receptor-9-mediated immunogenic reactions in organisms.7 This CG-free scaffold could be particularly advantageous when exploring in vivo applications of DNA origami. As a demonstration for the site-directed functionalization of synthetic scaffolds with functional sequences, we built two scaffolds that contain catalytic sequence motifs. We included one or two self-excising DNAzyme cassettes during sequence construction. Upon incubation with Zn2+, the DNAzymes become catalytically active, causing excision of the DNAzyme cassettes and thus linearization or bisection of the scaffold. Including these 132 bases long DNAzyme cassettes into the scaffold sequence ensures incorporation into every assembled DNA origami.
To illustrate the functionality, we used the self-bisecting scaffold to assemble a variant of a previously published DNA origami switch object (Figure 4A).38,47 The switch object consists of two rigid beams that are flexibly linked by a single scaffold crossover at the center. The switch features double-helical shape-complementary protrusions and recessions that can dock into each other, stabilizing a closed state of the switch via base stacking interactions. Due to the electrostatic repulsion of the negatively charged DNA arms, the switch will predominantly occupy its open state at low salt concentrations. At higher salt concentrations, the electrostatic repulsion is shielded, and the stacking interactions are sufficient to stabilize the closed state. In our bisectable switch variant, we placed the self-excising DNAzyme cassettes directly at the pivot point, where the scaffold chain crosses from one switch arm to the other (Figure 4A). The thus-designed objects self-assembled with high yield and predominantly populated an open state at <10 mM MgCl2 and a closed state at >10 mM MgCl2, as expected. When incubated with Zn2+, the switch objects are cut at the pivot point due to the excision of the DNA enzyme cassettes (Figure 4B). Gel electrophoretic mobility analysis (Figure 4C,D) reveals that the bisection reaction goes to completion, and that the kinetics of bisection strongly depends on the state of the switch: at high salt (closed state), the reaction is substantially slower, which we attribute to activity-reducing conformational constraints on the DNAzyme cassettes. A simple Mg2+ dependence of the reaction kinetics can be ruled out because the reaction speed is the same in the presence of 1.4 or 5 mM MgCl2. The cleavage reaction was also faster when residual staple oligonucleotides were removed by PEG precipitation48 prior to incubation with Zn2+ (Figure 4D).
Figure 4.

Self-cleaving DNA origami. (A) Schematic representations of circular scaffolds containing two self-excising DNAzyme cassettes (top left) that can be cleaved into two linear scaffolds (bottom left) or assembled into a switch object (top right). Individual switch arms (bottom right) can be obtained by cleavage of assembled switch objects or assembly using cleaved linear scaffolds. (B) Electrophoretic analysis of reaction kinetics of scaffold cleavage. Controls: cleaved scaffold (lane 1), undigested sample (lane 2), and switch arms assembled separately (lane 7) using cleaved scaffold. (C) Field-of-view TEM images of uncleaved (left) and cleaved (right) switch objects. (D) Electrophoretic analysis of cleavage reactions containing unpurified (lanes 1 and 5) and PEG-purified (lanes 2–4, 6–8) switch objects at 1.4, 4, 10, or 20 mM MgCl2. Laser scanned fluorescent images of the electrophoretic analysis were autoleveled, and the highlighted region was autoleveled individually. P, pocket; U, undigested species; D, digested species. Scale bar: 100 nm.
Synthetic scaffold design also allows integrating hundreds of user-defined motifs site-specifically into a DNA origami, which can be exploited, for example, for sequence-programmable, chemical-modification-free covalent cross-linking of DNA origami objects,9 termed UV point-welding. UV point-welded DNA origami objects are substantially more durable compared to nontreated objects and can remain stable at temperatures up to 90 °C and in pure double-distilled water with no additional cations present. In our previous work, covalent cross-linking was achieved by placing additional thymidine bases in the staple strand sequences at all termini and at all double-crossover positions.9 Irradiation of such objects with 310 nm light induces the formation of covalent cyclobutane pyrimidine dimer (CPD) bonds between colocalized thymidine bases. As a result, double-helical domains become topologically trapped, and the constituent strands of thus-treated DNA origami can no longer dissociate, unless covalent bonds are broken. The possibility of making fully customized scaffolds offers an elegant way to realize the formation of UV-induced CPD bonds at desired sites while suppressing the formation of CPD bonds at undesired sites. Using the scaffold smith tool, a scaffold sequence can be designed that does not exhibit any TT motifs and that features AA only at desired crossover sites and strand termini as specified in the strand diagram.
As a demonstration, we constructed a semigeneric scaffold that can be used to create UV-cross-linkable single- or multilayer DNA origami objects in square lattice packing. In this scaffold, AA sites simply appear in regular intervals of eight bases. Given appropriate scaffold routing, all staple crossover sites feature AA motifs on the scaffold, which therefore leads to thymidines in staple strands that can be cross-linked (Figure 5A). We produced the corresponding 7560 bases long welding scaffold using the backbone excision split-ori method described in Figure 1B and used it to assemble a variant of a previously reported multilayer DNA origami object known as the pointer.49 The UV-welding-ready pointer object self-assembled with satisfyingly high yield, as judged by electrophoretic mobility analysis (Figure 5C, lanes 2 and 12) and TEM imaging (Figure 5BI). We then irradiated the pointer object at 310 nm in the presence of 30 mM magnesium chloride. TEM images of the pointer acquired directly after exposure to UV light compared very well to those acquired prior to irradiation (Figure 5BII), indicating that the object retained its structure. We then incubated the irradiated sample for 48 h in physiological (low) ionic strength conditions (PBS buffer) at 40 °C (Figure 5BIII). Under such low ionic strength conditions, nonirradiated control pointer objects immediately dissociated into staple strands and scaffold strand as seen in gel electrophoresis (Figure 5B, left). By contrast, the irradiated samples remained fully intact, as indicated by the fact that the electrophoretic mobility did not change and by the absence of dissociated staple strand bands (Figure 5C, right). TEM imaging of the 48 h long PBS-incubated UV-welded pointer reveals well-folded objects consistent with the designed shape (Figure 5B, right). We thus conclude that the UV point-welding via scaffold-templated CPD bonds of the pointer was successful.
Figure 5.

UV point-welding of DNA origami with a custom scaffold. (A) Section of a multilayer DNA origami strand diagram with a customized scaffold featuring AA motifs every 8 base pairs, which results in adjacent Thymidines in separate staple strands that may be UV-cross-linked. Blue lines, scaffold strand; gray lines, staple strands. (B) Schematic representation (left) and average TEM images of the pointer object assembled with the welding scaffold. Average images of the pointer as obtained in the presence of 30 mM MgCl2 before irradiation (I), after irradiation for 2 h at 310 nm (II) in the presence of 30 mM MgCl2, and after irradiation for 2 h at 310 nm and 48 h long incubation in low ionic strength phosphate-buffered saline (PBS) at 40 °C (III). (C) Electrophoretic analysis of nonirradiated and irradiated pointer objects incubated over time in PBS at 40 °C. L, 1kB Ladder; NI, not irradiated; RT, room temperature; P, pocket; F, folded species; sta, staples. Scale bar: 50 nm.
Conclusion
With the tools and methods presented herein, researchers can now fully specify a target structure not only in terms of desired 3D shape and dimensions but also in terms of the sequences used. There is no longer a need to design objects around generic scaffold sequences as in the original DNA origami procedures. We demonstrated the potential of these tools and methods with a set of synthetic-sequence scaffolds which we used to explore the effects of sequence redundancy and sequence composition on the self-assembly of DNA origami, which is important input for guiding the construction of design-specific scaffolds. We built mini scaffolds as short as 1024 bases and a set of fully orthogonal scaffolds that enable efficient one-pot multiscaffold assembly of DNA origami comprising up to ∼38000 base pairs. We also made scaffolds containing functional motifs that enable DNAzyme-driven linearization and bisection of scaffolds or folded structures, which can enable constructing for example interlocked machine-like objects. Interlocked parts of these objects could be released by DNAzyme cleavage triggered by Zn2+-addition. We demonstrate that functional sequence motifs like DNAzymes, which are too long for staple strand synthesis, can be integrated in the scaffold sequence. We also constructed a CpG-free scaffold with presumably lower immunogenicity for future in vivo applications. Finally, we produced a customized scaffold with AA motifs spaced in intervals of 8 base pairs, which enables constructing square-lattice like single- or multilayer DNA origami that can be covalently cross-linked via UV point-welding right after folding. This scaffold can be considered as a demonstration of a fully design-specific scaffold, but the design was done such that the resulting scaffold can be used modularly in many other DNA origami designs.
With the currently available commercial gene synthesis services, our method allows constructing an entirely custom scaffold for less than 1000 € synthesis cost and requiring about 2 weeks of manual labor. We deposited precursor plasmids for all of our scaffolds at Addgene to make them available for the use by other researchers, along with the helper plasmids needed to produce the actual scaffold ssDNA. We also deposited a designated target plasmid containing the split-ori cassette, allowing other researchers to easily create their own custom scaffolds. Synthetic genes or gene fragments can be introduced into our target plasmid using a convenient and robust one-step Golden Gate cloning protocol.50
With custom-sequence scaffolds, DNA origami designers may rationally exploit sequence composition as a design parameter. Here, we produced mostly scaffold variants having a total nearest-neighbor energy higher than the conventional M13 variants, which led to assembly at temperatures higher than those of the M13-scaffolded object. It may be beneficial to explore whether the sequence composition may be tuned to push productive assembly temperature intervals down to physiological temperatures and without requiring a prior denaturation step. Furthermore, with full control over sequence design, sets of orthogonal scaffolds may now be produced that enable the direct and efficient assembly of oligomeric superstructures in one pot. For optimized designs such as the 126hb, we observed virtually perfect assembly yield in a one-pot reaction containing multiple scaffolds, which underlines the great potential of the multiscaffold strategy.
DNA origami applications often rely on the positioning of functionalities that typically consist of or are attached to specific ssDNA sequences. When conventional M13 scaffolds or natural sequences are used, these functional sequences must be introduced as extensions of staple strands. The incorporation yield of these extended staple strands may vary and can be unsatisfyingly low (e.g., 48%).8 If, on the other hand, the desired functional sequences are included in the scaffold strand, the incorporation yield into a folded DNA origami is 100%. As we demonstrated, custom scaffolds can be designed and produced to include functional sequences at user-defined positions. An extreme example is the welding scaffold that contained hundreds of custom AA sites while excluding undesired TT sites. As an example, we integrated self-excising DNAzyme cassettes as functional motifs into our scaffolds. Assembly of mechanically interlocked DNA origami mechanisms39,51 should become much easier with such bisectable scaffolds because detachment and component release can be achieved through Zn2+-induced excision of the DNAzyme cassettes. Self-linearizing scaffolds should be useful for designing multilayer DNA origami with odd-numbered helices and for making objects with applications such as nanopore translocation52,53 or for tethered fluorophore motion assays54 that require a linear scaffold. Future custom scaffolds might be designed to include other functional sequence motifs, such as aptamers, recognition sites for DNA-binding proteins, and indicator sites for complementary DNA strands as needed, for example, for DNA paint super-resolution microscopy.55
Another attractive aspect of creating design-specific scaffolds is that they lower the barrier to making DNA origami at larger scales. Previously, we reported how to biotechnologically mass produce pools of staple strands.21 The synthesis of the necessary plasmids with many interleaved self-cleaving DNAzyme cassettes poses an initial obstacle, which may render this method somewhat unattractive at intermediate scales and in situations where design variants will need to be iterated. However, precursor plasmids for custom-sequence scaffolds are easily synthesized as they do not, by default, contain repetitive sequences. Hence, the DNA origami concept can now be inverted: one fixed pool of staple strands could be mass-produced biotechnologically in a lab-scale (or even industrial scale) bioreactor. Then, different custom-sequence scaffolds can be made in shake flasks that fold the set of fixed-sequence staple strands into different structures, thereby allowing to iterate through design versions at scales inaccessible with DNA reagents produced via chemical synthesis. A variant of this idea has been tested presented previously with the goal to reuse chemically produced DNA oligonucleotides.19
Methods
Design and Construction of Scaffold Plasmids
For our custom scaffolds, we designed insert sequences for the variable part using either a python-based de Bruijn sequence generator or by using the scaffold smith GUI (see Supporting Information Note S1). Resulting insert sequences were split into shorter fragments to facilitate gene synthesis either manually or using a gene splitter GUI. Gene fragments were ordered either as linear gene strands or as genes in plasmids from Eurofins genomics (Ebersberg, Germany) or Twist bioscience (San Francisco, CA, USA). Full-length precursor plasmids were assembled using either Gibson assembly56 or Golden Gate cloning.50 Correct assembly was verified using restriction digest and DNA sequencing (Eurofins genomics, Ebersberg Germany). Sequences of all plasmids used in this work can be found in an Excel table in the Supporting Information.
Production of ssDNA in Shaker flasks
A detailed overview of the four different production methods is given in Supporting Information Note S2. M13 phage scaffolds were produced as previously described.18 For custom scaffolds (methods II, III, and IV), chemically competent cells (E. coli DH5α) were cotransformed with the corresponding precursor plasmid and a helper plasmid. The actual ssDNA production for methods II and IV was carried out as previously described.21 For convenience, the protocol is reprinted below:
“A single clone was picked and grown to saturation in a 5 mL pre-culture in 2xYT medium containing 5 mM MgCl2, 30 μg/mL kanamycin, and either 50 μg/mL carbenicillin or 30 μg/mL chloramphenicol, depending on the phagemid backbone used; 750 mL of the same medium was inoculated with the preculture and grown overnight at 37 °C in 2.5 L Ultra Yield flasks (Thomson). Bacteria were removed by centrifugation for 30 min at 4000 rcf. Phagemid particles were precipitated by adding 3% polyethylene glycol 8000 (PEG-8000) and 0.5 M NaCl and centrifugation for 30 min at 4000 rcf. The pellet was resuspended in 5 mL of 1× TE buffer (10 mM Tris, 1 mM EDTA, pH 8) and centrifuged again for 15 min at 16000 rcf to remove residual bacterial cell fragments.”
For method III, ssDNA production was carried out analogously but using a growth medium containing 16 g/L tryptone, 10 g/L yeast extract, 60 g/L sucrose, and 30 μg/mL kanamycin.57 For all methods, ssDNA was subsequently isolated from the phagemid particles using alkaline lysis according to standard protocols for M13 ssDNA purification.58 Purity of ssDNA was analyzed using gel electrophoresis, and sequences of custom scaffolds were verified using DNA sequencing. ssDNA concentrations were determined via the absorbance at 260 nm using extinction coefficients of 9828 M–1 cm–1 per base. Extinction coefficients for all scaffolds used in this work can be found in an Excel table in the Supporting Information.
Design, Assembly, and Purification of 3D DNA Origami
DNA origami objects were designed using caDNAno22 and the designs were evaluated using CanDo.59,60 Design diagrams can be found in Supporting Information Figures S11–S17 or in the corresponding references.34,41,49,61 Reaction mixtures contained concentrations of scaffold strands and staples that were optimized for each object (42hb monomers: 50 nM scaffold + 200 nM staples; one-pot reaction mixture of 42hb and 10hb: 10 nM scaffold + 100 nM staples; 94hb: 20 nM scaffold + 100 nM staples; 126hb: 20 nM scaffold + 220 nM staples; 42hb pentamer version I assembly setup: 10 nM scaffold + 100 nM staples; 42hb pentamer version II assembly setup: 10 nM scaffold + 200 nM staples; switch object and pointer object: 20 nM scaffold + 200 nM staples nM; 13hb variants: 40 nM scaffold + 200 nM staples). All reaction mixtures contained 5 mM Tris, 1 mM EDTA, 20 mM MgCl2, and 5 mM NaCl (pH 8). To compare different scaffolds, 42hb monomers were assembled using a standardized folding screen (for details see the caption of Figure 1), which was also used to identify optimized temperature intervals for each DNA origami object. Hence, each reaction mixture was subjected to an individually optimized thermal annealing ramp using a TETRAD (MJ Research, now Biorad), which included a preincubation step at 65 °C for 15 min. Subsequent temperature ramps were 60–40 °C at 1 h/°C for the 94hb, the switch-object, the pointer object, and for all one-pot reaction mixtures with multiple objects; 60–40 °C at 3 h/°C for the 126 and 42hb oligomers and 60–40 °C at 15 min/°C for the 13hb variants. DNA origami objects were purified via PEG precipitation.35,48 For the 13hb objects, a final PEG concentration of 10.7% (w/v), a final NaCl concentration of 535 mM, and a final MgCl2 concentration of 14.7 mM were used for precipitation. After precipitation, all pellets were resuspended in a buffer containing 5 mM Tris, 1 mM EDTA, 5 mM MgCl2, and 5 mM NaCl (pH 8).
Agarose Gel Electrophoresis
In general, scaffolds and assembled DNA origami objects were analyzed using 1–2% agarose gels in 0.5× TBE buffer (45 mM Tris base, 45 mM boric acid, 1 mM EDTA) including 5.5 mM MgCl2 and 0.5 μg/mL ethidium bromide at 90 V for 1.5 h in a water bath. Agarose concentrations were 1.5% for the 94hb in Figure S9 and the 42hb oligomers in Figure 3 and 2% for all other gels. Gels were scanned using a Typhoon FLA 9500 laser scanner (GE) at a resolution of either 50 or 25 μm/px (EtBr-channel: excitation at 535 nm, emission >575 nm). Images were inverted and autoleveled (Adobe Photoshop CS6).
Transmission Electron Microscopy Imaging and Image Processing
Samples were diluted to final concentrations between 1 and 5 nM in a buffer containing 20 mM MgCl2 and adsorbed for 30 s to 1 min on glow-discharged Formvar-supported carbon-coated Cu400 TEM grids (Science Services, Munich) and stained using a 2% aqueous uranyl formate solution containing 25 mM NaOH. Imaging was performed using a Philips CM100 EM operated at 100 kV and an AMT 4 megapixel CCD camera (magnification: 28500×) or using a Tecnai Spirit operated at 120 kV and a TVIPS F416 detector (Tietz Camera Systems) (magnification: 30000×). For reference-free class averaging, image libraries were created by individual particle picking and analyzed using Xmipp 3.0.62 TEM micrographs shown were subjected to high-pass filtering and autoleveling (Adobe Photoshop CS6).
Sample Preparation of 126hb for the Cryo-EM Study
The 126hb sample was purified and enriched via ultrafiltration with centrifugation steps at 10000 rcf for 5 min at 20 °C (Amicon Ultra 0.5 mL 50 kDa cutoff filters, Millipore). The sample was diluted 4-fold with folding buffer (1× FOB = 1 mM Tris, 1 mM EDTA, and 5 mM NaCl) prior to the first run. The filters were rinsed with 1× FOB including 5 mM MgCl2 (1× FOB5), filled with 500 μL sample each and subjected to a centrifugation step. After six washing steps consisting of removing of the flow-through, refilling of the filters to 500 μL with 1× FOB5, and a centrifugation step, the filters were placed upside down in fresh tubes and subjected to another centrifugation step. The recovered sample was pooled from the filter tubes and added to one fresh filter in steps of 500 μL each followed by a centrifugation step. The filter was placed upside down in a fresh tube and subjected to two final centrifugation steps. The final concentration of the recovered sample was measured to be 1.2 μM.
Cryo-EM: Acquisition and Processing of Data
The purified and enriched sample was applied to C-Flat 2/1 4C (Protochips) grids and plunge-frozen using a Vitrobot Mark V (FEI, now Thermo Scientific) at the following settings: temperature of 22 °C, humidity of 100%, 0 s wait time, 2 s blot time, −1 blot force, 0 s drain time. The data were acquired on a Titan Krios G2 electron microscope operated at 300 kV equipped with a Falcon 3 direct detector using the EPU software for automated data collection (FEI, now Thermo Scientific). Micrograph movies comprising 7 frames each were recorded at a calibrated magnification of 47000 and magnified pixel size of 1.39 Å, a total dose of ∼50 e–/Å2 and defocus values from −1 to −2 μm. The image processing was performed in RELION 2.163 and 3.0,64 using MotionCor265 and CTFFIND4.166 for motion correction and contrast transfer function estimation, respectively. A total of 2118 particles were manually picked and subjected to reference-free 2D classification. Four of the best classes (as judged by visual inspection) were selected as templates for automated particle picking. The particles were subjected to multiple rounds of 2D and 3D classification using a 3D de novo initial model created inside RELION as a 3D reference. A total of 123433 particles assigned to classes showing the most features were selected for a 3D refinement and further motion-corrected using the Bayesian Polishing tool. A focused refinement of a subregion was performed using the Multibody Refinement tool. The refined maps were sharpened by automatically estimated B-factors and locally filtered using the post processing and local resolution tool, respectively.
Zn-Induced Excision of DNAzyme Cassettes and Postpurification Using EtOH
Cleavage reactions were performed by incubating 20 nM phagemid ssDNA or 20 nM switch object in a buffer containing 50 mM HEPES, 100 mM NaCl, and 2 mM ZnCl2, pH 7.0 at 37 °C overnight. ssDNA cleavage products were purified by adding 0.3 vol of 3 M KOAc (pH5), 0.033 vol of 1 M MgCl2, and 2 vol of isopropyl alcohol. After 5 h incubation at −20 °C, the sample was pelleted by centrifugation for 45 min at 16000 rcf. The pellet was washed with 75% ethanol to remove residual salt, centrifuged again, and dissolved in 1× TE buffer. Switch object cleavage products were purified via PEG purification and resuspended in 1× FOB5.
UV Irradiation and Buffer Exchange to PBS and Incubation at 40 °C
The pointer object was irradiated for 120 min in folding buffer (5 mM Tris, 1 mM EDTA, 30 mM MgCl2, and 5 mM NaCl) with a 300 W xenon light source (MAX-303 from Asahi Spectra) with a high transmission band-pass filter centered around 310 nm (XAQA310 from Asahi Spectra). A light guide (Asahi Spectra) was placed directly on top of a 0.65 mL reaction tube to couple the light into the sample. For all samples (cross-linked and un-cross-linked control) buffer exchange to PBS was achieved via ultrafiltration (Amicon Ultra 0.5 mL 50 kDa cutoff filters, Millipore) with three centrifugation steps at 7000 rcf for 5 min at 20 °C (Eppendorf 5424R). Samples were then incubated at 40 °C, and aliquots were shock-frozen in liquid nitrogen after different time intervals.
Acknowledgments
The authors thank Max Honemann for technical assistance, and Klaus Wagenbauer for auxiliary experiments. We thank Dirk Weuster-Botz for providing auxiliary resources. This project was supported by European Research Council consolidator Grant No. 724261, the Deutsche Forschungsgemeinschaft through grants provided within the Gottfried-Wilhelm-Leibniz Program, and the SFB863 TPA9.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b01025.
Detailed notes on generating a design-specific scaffold sequence with the scaffold smith, overview of methods for phage-based production of ssDNA scaffolds, workflow for the design, cloning, and production of a custom scaffold, supplementary experimental data and design schematics (PDF)
Sequence information for scaffolds and plasmids used in this work (XLSX)
Tools used in this work to design and analyze scaffold sequences (ZIP)
Author Contributions
F.A.S.E. and F.P. contributed equally to this work. F.A.S.E., F.P., and C.H.W. performed the research, and H.D. designed the research. G.B., B.K., K.L.K., P.N.P., and K.L.B. performed auxiliary experiments. F.K. performed cryo-EM. T.G. designed the bisectable switch and performed auxiliary experiments. F.A.S.E., F.P., and H.D. prepared figures and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hong F.; Zhang F.; Liu Y.; Yan H. DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 2017, 117, 12584. 10.1021/acs.chemrev.6b00825. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Hogberg B.; Graf F.; Shih W. M. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature 2009, 459, 414–418. 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H.; Douglas S. M.; Shih W. M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science 2009, 325, 725–730. 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B.; Dai M.; Yin P. Complex Shapes Self-Assembled from Single-Stranded DNA Tiles. Nature 2012, 485, 623–626. 10.1038/nature11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozyra J.; Ceccarelli A.; Torelli E.; Lopiccolo A.; Gu J.-Y.; Fellermann H.; Stimming U.; Krasnogor N. Designing Uniquely Addressable Bio-Orthogonal Synthetic Scaffolds for DNA and Rna Origami. ACS Synth. Biol. 2017, 6, 1140–1149. 10.1021/acssynbio.6b00271. [DOI] [PubMed] [Google Scholar]
- Krieg A. M. Cpg Motifs in Bacterial DNA and Their Immune Effects. Annu. Rev. Immunol. 2002, 20, 709–760. 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Strauss M. T.; Schueder F.; Haas D.; Nickels P. C.; Jungmann R. Quantifying Absolute Addressability in DNA Origami with Molecular Resolution. Nat. Commun. 2018, 9, 1600. 10.1038/s41467-018-04031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerling T.; Kube M.; Kick B.; Dietz H. Sequence-Programmable Covalent Bonding of Designed DNA Assemblies. Sci. Adv. 2018, 4, eaau1157 10.1126/sciadv.aau1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducani C.; Kaul C.; Moche M.; Shih W. M.; Hogberg B. Enzymatic Production of ’Monoclonal Stoichiometric’ Single-Stranded DNA Oligonucleotides. Nat. Methods 2013, 10, 647–652. 10.1038/nmeth.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducani C.; Bernardinelli G.; Hogberg B. Rolling Circle Replication Requires Single-Stranded DNA Binding Protein to Avoid Termination and Production of Double-Stranded DNA. Nucleic Acids Res. 2014, 42, 10596–10604. 10.1093/nar/gku737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T. L.; Beliveau B. J.; Uca Y. O.; Theilmann M.; Da Cruz F.; Wu C. T.; Shih W. M. Scalable Amplification of Strand Subsets from Chip-Synthesized Oligonucleotide Libraries. Nat. Commun. 2015, 6, 8634. 10.1038/ncomms9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz J.; Yin P.; Voigt C. A. Genetic Encoding of DNA Nanostructures and Their Self-Assembly in Living Bacteria. Nat. Commun. 2016, 7, 11179. 10.1038/ncomms11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg E.; Shih W. M. Selective Nascent Polymer Catch-and-Release Enables Scalable Isolation of Multi-Kilobase Single-Stranded DNA. Angew. Chem., Int. Ed. 2018, 57, 714–718. 10.1002/anie.201710469. [DOI] [PubMed] [Google Scholar]
- Veneziano R.; Shepherd T. R.; Ratanalert S.; Bellou L.; Tao C.; Bathe M. In Vitro Synthesis of Gene-Length Single-Stranded DNA. Sci. Rep. 2018, 8, 24677. 10.1038/s41598-018-24677-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi A. N.; Saaem I.; Vogen B. N.; Brown S.; LaBean T. H. Toward Larger DNA Origami. Nano Lett. 2014, 14, 5740–5747. 10.1021/nl502626s. [DOI] [PubMed] [Google Scholar]
- Brown S.; Majikes J.; Martínez A.; Girón T.; Fennell H.; Samano E.; LaBean T. An Easy-to-Prepare Mini-Scaffold for DNA Origami. Nanoscale 2015, 7, 16621–16624. 10.1039/C5NR04921K. [DOI] [PubMed] [Google Scholar]
- Kick B.; Praetorius F.; Dietz H.; Weuster-Botz D. Efficient Production of Single-Stranded Phage DNA as Scaffolds for DNA Origami. Nano Lett. 2015, 15, 4672. 10.1021/acs.nanolett.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niekamp S.; Blumer K.; Nafisi P. M.; Tsui K.; Garbutt J.; Douglas S. M. Folding Complex DNA Nanostructures from Limited Sets of Reusable Sequences. Nucleic Acids Res. 2016, 44, e102 10.1093/nar/gkw208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kick B.; Hensler S.; Praetorius F.; Dietz H.; Weuster-Botz D. Specific Growth Rate and Multiplicity of Infection Affect High-Cell-Density Fermentation with Bacteriophage M13 for Ssdna Production. Biotechnol. Bioeng. 2017, 114, 777–784. 10.1002/bit.26200. [DOI] [PubMed] [Google Scholar]
- Praetorius F.; Kick B.; Behler K. L.; Honemann M. N.; Weuster-Botz D.; Dietz H. Biotechnological Mass Production of DNA Origami. Nature 2017, 552, 84–87. 10.1038/nature24650. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3d DNA-Origami Shapes with Cadnano. Nucleic Acids Res. 2009, 37, 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veneziano R.; Ratanalert S.; Zhang K.; Zhang F.; Yan H.; Chiu W.; Bathe M. Designer Nanoscale DNA Assemblies Programmed from the Top Down. Science 2016, 352, 1534. 10.1126/science.aaf4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun H.; Zhang F.; Shepherd T.; Ratanalert S.; Qi X.; Yan H.; Bathe M. Autonomously Designed Free-Form 2d DNA Origami. Sci. Adv. 2019, 5, eaav0655 10.1126/sciadv.aav0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun H.; Shepherd T. R.; Zhang K.; Bricker W. P.; Li S.; Chiu W.; Bathe M. Automated Sequence Design of 3d Polyhedral Wireframe DNA Origami with Honeycomb Edges. ACS Nano 2019, 10.1021/acsnano.8b08671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson E.; Mohammed A.; Gardell J.; Masich S.; Czeizler E.; Orponen P.; Hogberg B. DNA Rendering of Polyhedral Meshes at the Nanoscale. Nature 2015, 523, 441–444. 10.1038/nature14586. [DOI] [PubMed] [Google Scholar]
- Bellot G.; McClintock M. A.; Chou J. J.; Shih W. M. DNA Nanotubes for Nmr Structure Determination of Membrane Proteins. Nat. Protoc. 2013, 8, 755–770. 10.1038/nprot.2013.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J.; Messing J. Production of Single-Stranded Plasmid DNA. Methods Enzymol. 1987, 153, 3–11. 10.1016/0076-6879(87)53044-0. [DOI] [PubMed] [Google Scholar]
- Chasteen L.; Ayriss J.; Pavlik P.; Bradbury A. R. Eliminating Helper Phage from Phage Display. Nucleic Acids Res. 2006, 34, e145 10.1093/nar/gkl772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specthrie L.; Bullitt E.; Horiuchi K.; Model P.; Russel M.; Makowski L. Construction of a Microphage Variant of Filamentous Bacteriophage. J. Mol. Biol. 1992, 228, 720–724. 10.1016/0022-2836(92)90858-H. [DOI] [PubMed] [Google Scholar]
- Nafisi P. M.; Aksel T.; Douglas S. M. Construction of a Novel Phagemid to Produce Custom DNA Origami Scaffolds. Synth. Biol. 2018, 10.1093/synbio/ysy015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said H.; Schuller V. J.; Eber F. J.; Wege C.; Liedl T.; Richert C. M1.3--a Small Scaffold for DNA Origami. Nanoscale 2013, 5, 284–290. 10.1039/C2NR32393A. [DOI] [PubMed] [Google Scholar]
- De Bruijn N. G. A Combinatorial Problem. Koninklijke Nederlandse Akademie v. Wetenschappen 1946, 49, 758–764. [Google Scholar]
- Sobczak J. P.; Martin T. G.; Gerling T.; Dietz H. Rapid Folding of DNA into Nanoscale Shapes at Constant Temperature. Science 2012, 338, 1458–1461. 10.1126/science.1229919. [DOI] [PubMed] [Google Scholar]
- Wagenbauer K. F.; Engelhardt F. A.; Stahl E. K.; Hechtl V. K.; Stömmer P.; Seebacher F.; Meregalli L.; Ketterer P.; Gerling T.; Dietz H. How We Make DNA Origami. ChemBioChem 2017, 18, 1873. 10.1002/cbic.201700377. [DOI] [PubMed] [Google Scholar]
- SantaLucia J. Jr.; Hicks D. The Thermodynamics of DNA Structural Motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- Berardi M. J.; Shih W. M.; Harrison S. C.; Chou J. J. Mitochondrial Uncoupling Protein 2 Structure Determined by Nmr Molecular Fragment Searching. Nature 2011, 476, 109–113. 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerling T.; Wagenbauer K. F.; Neuner A. M.; Dietz H. Dynamic DNA Devices and Assemblies Formed by Shape-Complementary, Non-Base Pairing 3d Components. Science 2015, 347, 1446–1452. 10.1126/science.aaa5372. [DOI] [PubMed] [Google Scholar]
- Ketterer P.; Willner E. M.; Dietz H. Nanoscale Rotary Apparatus Formed from Tight-Fitting 3d DNA Components. Sci. Adv. 2016, 2, e1501209 10.1126/sciadv.1501209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhomirov G.; Petersen P.; Qian L. Fractal Assembly of Micrometre-Scale DNA Origami Arrays with Arbitrary Patterns. Nature 2017, 552, 67–71. 10.1038/nature24655. [DOI] [PubMed] [Google Scholar]
- Wagenbauer K. F.; Sigl C.; Dietz H. Gigadalton-Scale Shape-Programmable DNA Assemblies. Nature 2017, 552, 78–83. 10.1038/nature24651. [DOI] [PubMed] [Google Scholar]
- Ong L. L.; Hanikel N.; Yaghi O. K.; Grun C.; Strauss M. T.; Bron P.; Lai-Kee-Him J.; Schueder F.; Wang B.; Wang P.; Kishi J. Y.; Myhrvold C.; Zhu A.; Jungmann R.; Bellot G.; Ke Y.; Yin P. Programmable Self-Assembly of Three-Dimensional Nanostructures from 10,000 Unique Components. Nature 2017, 552, 72–77. 10.1038/nature24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Wang Q.; Peng J.; Long Q.; Yu H.; Li Z. Self-Assembly of Large DNA Origami with Custom-Designed Scaffolds. ACS Appl. Mater. Interfaces 2018, 10, 24344–24348. 10.1021/acsami.8b09222. [DOI] [PubMed] [Google Scholar]
- Iinuma R.; Ke Y.; Jungmann R.; Schlichthaerle T.; Woehrstein J. B.; Yin P. Polyhedra Self-Assembled from DNA Tripods and Characterized with 3d DNA-Paint. Science 2014, 344, 65–69. 10.1126/science.1250944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Ye T.; Su M.; Zhang C.; Ribbe A. E.; Jiang W.; Mao C. Hierarchical Self-Assembly of DNA into Symmetric Supramolecular Polyhedra. Nature 2008, 452, 198–201. 10.1038/nature06597. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Jiang S.; Wu S.; Li Y.; Mao C.; Liu Y.; Yan H. Complex Wireframe DNA Origami Nanostructures with Multi-Arm Junction Vertices. Nat. Nanotechnol. 2015, 10, 779–784. 10.1038/nnano.2015.162. [DOI] [PubMed] [Google Scholar]
- Bruetzel L. K.; Walker P. U.; Gerling T.; Dietz H.; Lipfert J. Time-Resolved Small-Angle X-Ray Scattering Reveals Millisecond Transitions of a DNA Origami Switch. Nano Lett. 2018, 18, 2672–2676. 10.1021/acs.nanolett.8b00592. [DOI] [PubMed] [Google Scholar]
- Stahl E.; Martin T. G.; Praetorius F.; Dietz H. Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions. Angew. Chem., Int. Ed. 2014, 53, 12735–12740. 10.1002/anie.201405991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X. C.; Martin T. G.; Scheres S. H.; Dietz H. Cryo-Em Structure of a 3d DNA-Origami Object. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 20012–20017. 10.1073/pnas.1215713109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C.; Kandzia R.; Marillonnet S. A One Pot, One Step, Precision Cloning Method with High Throughput Capability. PLoS One 2008, 3, e3647 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List J.; Falgenhauer E.; Kopperger E.; Pardatscher G.; Simmel F. C. Long-Range Movement of Large Mechanically Interlocked DNA Nanostructures. Nat. Commun. 2016, 7, 12414. 10.1038/ncomms12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesa C.; van Loo N.; Ketterer P.; Dietz H.; Dekker C. Velocity of DNA During Translocation through a Solid-State Nanopore. Nano Lett. 2015, 15, 732–737. 10.1021/nl504375c. [DOI] [PubMed] [Google Scholar]
- Bell N. A.; Keyser U. F. Digitally Encoded DNA Nanostructures for Multiplexed, Single-Molecule Protein Sensing with Nanopores. Nat. Nanotechnol. 2016, 11, 645–651. 10.1038/nnano.2016.50. [DOI] [PubMed] [Google Scholar]
- Schickinger M.; Zacharias M.; Dietz H. Tethered Multifluorophore Motion Reveals Equilibrium Transition Kinetics of Single DNA Double Helices. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7512. 10.1073/pnas.1800585115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann R.; Avendaño M. S.; Woehrstein J. B.; Dai M.; Shih W. M.; Yin P. Multiplexed 3d Cellular Super-Resolution Imaging with DNA-Paint and Exchange-Paint. Nat. Methods 2014, 11, 313. 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R.-Y.; Venter J. C.; Hutchison C. A. III; Smith H. O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6, 343. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Luke J.; Carnes A. E.; Hodgson C. P.; Williams J. A. Improved Antibiotic-Free DNA Vaccine Vectors Utilizing a Novel Rna Based Plasmid Selection System. Vaccine 2009, 27, 6454–6459. 10.1016/j.vaccine.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Chou J. J.; Shih W. M. DNA-Nanotube-Induced Alignment of Membrane Proteins for Nmr Structure Determination. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6644–6648. 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C. E.; Kilchherr F.; Kim D. N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods 2011, 8, 221–229. 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- Kim D. N.; Kilchherr F.; Dietz H.; Bathe M. Quantitative Prediction of 3d Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2012, 40, 2862–2868. 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfitzner E.; Wachauf C.; Kilchherr F.; Pelz B.; Shih W. M.; Rief M.; Dietz H. Rigid DNA Beams for High-Resolution Single-Molecule Mechanics. Angew. Chem., Int. Ed. 2013, 52, 7766–7771. 10.1002/anie.201302727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S. H.; Nunez-Ramirez R.; Sorzano C. O.; Carazo J. M.; Marabini R. Image Processing for Electron Microscopy Single-Particle Analysis Using Xmipp. Nat. Protoc. 2008, 3, 977–990. 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D.; Forsberg B. O.; Scheres S. H.; Lindahl E. Accelerated Cryo-Em Structure Determination with Parallelisation Using Gpus in Relion-2. eLife 2016, 5, 18722. 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J.; Nakane T.; Forsberg B. O.; Kimanius D.; Hagen W. J.; Lindahl E.; Scheres S. H. New Tools for Automated High-Resolution Cryo-Em Structure Determination in Relion-3. eLife 2018, 7, 42166. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S. Q.; Palovcak E.; Armache J. P.; Verba K. A.; Cheng Y.; Agard D. A. Motioncor2: Anisotropic Correction of Beam-Induced Motion for Improved Cryo-Electron Microscopy. Nat. Methods 2017, 14, 331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell J. A.; Grigorieff N. Accurate Determination of Local Defocus and Specimen Tilt in Electron Microscopy. J. Struct. Biol. 2003, 142, 334–347. 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.