

Despite the sporadic detection of fluoroquinolone-resistant Shigella in Asia in the early 2000s and subsequent global spread of ciprofloxacin-resistant (cipR) Shigella sonnei from 2010, fluoroquinolones remain the recommended therapy for shigellosis1–7. The potential for cipR S. sonnei to develop resistance to alternative second-line drugs may further limit future treatment options8. Here, we aimed to understand the evolution of novel antimicrobial resistant (AMR) S. sonnei variants after introduction into Vietnam. We found that cipR S. sonnei displaced the resident ciprofloxacin-susceptible (cipS) lineage while rapidly acquiring additional resistance to multiple alternative antimicrobial classes. We identified several independent acquisitions of XDR/MDR-inducing plasmids, likely facilitated by horizontal transfer from commensals in the human gut. By characterizing commensal E. coli from Shigella-infected and healthy children, we identified an extensive array of AMR genes and plasmids, including an identical MDR plasmid isolated from both S. sonnei and E. coli in the gut of a single child. We additionally found that antimicrobial usage may impact plasmid transfer between commensal E. coli and S. sonnei. These results suggest that in a setting with high antimicrobial use and a high prevalence of AMR commensals, cipR S. sonnei may be propelled towards pan-resistance by adherence to outdated international treatment guidelines.
The fluoroquinolones were highly effective for the treatment of shigellosis until sporadic cases of cipR S. dysenteriae, S. flexneri, and S. boydii were detected in the early 2000s 1–4. Fully cipR S. sonnei emerged soon after 2,9. These S. sonnei carried the classical chromosomal point mutations in the quinolone resistance determining regions (QRDRs) at codon 83 (serine to leucine) and codon 87 (aspartic acid to glycine/asparagine) in gyrA, and at codon 80 (serine to isoleucine) in parC 4,9. Our previous work demonstrated that all cipR S. sonnei globally were clonal and emerged once from a single lineage arising in South Asia 10. Here, we describe the expansion of a single lineage of cipR S. sonnei in southern Vietnam by providing phylo-temporal insights into its extant clonal dynamics using clinical samples obtained from S. sonnei-infected children presenting to hospitals in Ho Chi Minh City (HCMC), Vietnam. We observe the replacement of the resident cipS clone by the novel cipR lineage, as well as the concurrent and independent acquisition of a diverse range of resistance plasmids, leading to MDR (multi-drug resistant) and XDR (extensively-drug resistant) phenotypes. Through a detailed analysis of the plasmid content from commensal E. coli and S. sonnei isolated from a single patient and a series of conjugation experiments, we provide compelling evidence that MDR and XDR plasmids are transferred from commensal E. coli into S. sonnei in the guts of infected children, which may be enhanced by antimicrobial exposure.
Between January 2014 and July 2016, we isolated 79 S. sonnei from children hospitalized with dysenteric diarrhea; 75.9% (60/79) were cipR. Phylogenetic reconstruction demonstrated that all except one cipR S. sonnei comprised a distinct clade, which was distantly related to the cipS isolates (Fig. 1). The most recent common ancestor (MRCA) of the cipR clade in Vietnam was estimated to date back to late 2008 (95% highest posterior density (HPD): 2007.1–2010.3), prior to the first detected cases of cipR S. sonnei in Vietnam in October 2013. The phylogeny depicted a clonal expansion from a single cipR organism that originated in South Asia and disseminated internationally 10. All organisms within the cipR clade were classical triple mutants (gyrA-S83L, gyrA-D87G, and parC-S80I) associated with high-level ciprofloxacin resistance (MIC ≥8 μg/ml). Conversely, isolates belonging to the resident cipS S. sonnei clade harbored a single mutation in gyrA (either S83L (16 isolates) or D87Y (4 isolates)).
Figure 1. The phylogenetic structure of Shigella sonnei in Vietnam, 2014-2016.

Maximum clade credibility phyloeny showing two distinct clades corresponding to the resident and cipR S. sonnei populations. The black asterisks indicate posterior probability support ≥70% on internal nodes. The red circles at terminal leaves highlight the ESBL-positive isolates. Triangles indicate plasmid gain/loss events. Sixteen plasmid acquisitions (PAs) were reconstructed across the tree and designated as PA1-PA16. The columns on the right correspond to: the resistance phenotype to key antimicrobials (turquoise), the presence of AMR genes (red), and the presence of different plasmid groups (multiple colors), respectively.
We estimated the median substitution rate of the S. sonnei population to be 8.2×10-7 substitutions base-1 year-1 (95% HPD; 5.9×10-7 to 10.8×10-7), comparable to previous estimates of the mutation rate within the resident cipS S. sonnei population in Vietnam 11. The cipR isolates exhibited a substantially lower median pairwise SNP distance (15 SNPs, IQR: 10-20 SNPs) than the resident cipS S. sonnei isolates (96 SNPs, IQR: 69-111 SNPs). During the sampling period, the proportion of cipR S. sonnei significantly increased from 60.7% (17/28) in 2014 to 93.8% (15/16) in 2016 (p=0.01; Chi-squared test). These results provide evidence that cipR S. sonnei underwent a rapid clonal expansion and successfully persisted following introduction to southern Vietnam, displacing the resident cipS S. sonnei as the dominant S. sonnei lineage.
Almost all isolates carried AMR genes on a chromosomally integrated class II integron (dfrA1, sat-2A) and a small spA plasmid (strAB, sulII, tetAR) encoding resistance to tetracycline, streptomycin, and co-trimoxazole. During their circulation in southern Vietnam, the cipR S. sonnei acquired resistance to additional antimicrobial classes, including third-generation cephalosporins, macrolides, and aminoglycosides, consequently creating XDR variants. In 2014, the proportions of co-resistance against ceftriaxone, ceftriaxone-azithromycin, and ceftriaxone-azithromycin-gentamicin were 59% (10/17), 11.8% (2/17), and 5.9% (1/17), respectively. By 2016, these respective proportions were 87% (13/15), 47% (7/15), and 40% (6/15). Our analyses show that co-resistance in cipR S. sonnei was generated by sustained and independent acquisitions of ESBL-encoding plasmids (plasmid acquisition events (PAs); Fig. 1). This phenomenon was not characterized by selection of the same plasmid/clone combination; we identified at least thirteen independent acquisitions of ESBL-encoding plasmids across the phylogenetic tree (Fig. 1), with plasmids of incompatibility groups IncB/O and IncI the most common vehicles associated with blaCTX-M.
IncB/O plasmids were independently acquired on at least seven occasions; these plasmids carried an array of ESBL genes, including blaCTX-M-55 (PA3, 5, 16), blaCTX-M-14 (PA10, 12), blaCTX-M-15 (PA1), and blaCTX-M-24 (PA13). Critically, in 6/7 IncB/O PAs, the blaCTX-M gene was associated with mphA and acc6-IIa genes, leading to an XDR phenotype incorporating resistance to third-generation cephalosporins, macrolides, and aminoglycosides. IncI plasmids were acquired on four independent occasions and carried only a blaCTX-M gene (blaCTX-M-15 (PA9) and blaCTX-M-55 (PA6, 11, 15)). Furthermore, blaCTX-M genes were acquired on IncFI (blaCTX-M14 (PA7)) and IncAnco3 (blaCTX-M-15 (PA14)) plasmid backbones. Three cipR non-ESBL isolates also acquired an mphA gene associated with IncFII (PA4, 8) and IncK (PA2) plasmids.
The acquisition of a resistance plasmid was sporadically followed by continued circulation of the resistant sub-clone, as observed for the IncB/O/blaCTX-M-55 (PA3), IncB/O/blaCTX-M-55-mphA-aac6-IIa (PA16), IncI/blaCTX-M-55 (PA6), and IncI/blaCTX-M-15 plasmids (PA9) (Fig. 1). The inferred time from the most recent common ancestor (TMRCA) to the most recent isolate in each resistant sub-clone was estimated to be at least three years.
When a blaCTX-M/mphA plasmid was acquired, it rarely became established in the population, suggesting that these structures could have a fitness disadvantage in the absence of antimicrobial pressure. Conversely, the successful maintenance of four independent XDR S. sonnei sub-clones warrants further investigation into their potential fitness, plasmid stability, and future evolutionary trajectories. Additionally, the selected IncB/O and IncI ESBL-encoding conjugative plasmids acquired and maintained by cipR S. sonnei suggest possible plasmid tropisms in this species. IncI and IncB/O plasmids belong to the IncI-complex (IncI, IncB/O, IncK, IncZ), which have comparable antisense RNA plasmid replication control mechanisms 12. Our results concur with previous reports that proposed a commonality of IncI-complex plasmids associated with the blaCTX-M element in S. sonnei from countries at various stages of economic development 8,11,13–17. The sampling of these plasmids by cipR S. sonnei could be attributed to several factors, including the close genetic relatedness between Shigella and E. coli, the propensity of S. sonnei to acquire AMR plasmids, and the circulation of highly transmissible AMR plasmids in commensal E. coli.
We assessed the plasmid contents of all S. sonnei isolates by comparing undigested crude plasmid extracts; all cipR isolates exhibited a distinct plasmid profile from that of the resident Vietnamese S. sonnei (Extended Data Fig. 1). Additionally, all cipR S. sonnei isolates carrying blaCTX-M and/or mphA consistently harbored a 90-110 kb plasmid. An EcoRI digestion of these ESBL-encoding plasmids showed two major independent clusters, consistent with IncI and IncB/O plasmid backbones (Fig. 2a). The genetic structure within each plasmid group was highly conserved, with the IncI and IncB/O plasmids sharing ~70% and ~60% similarity within their respective restriction patterns.
Figure 2. The diversity of ESBL-encoding plasmids in ciprofloxacin-resistant Shigella sonnei.

a) Dendrogram showing the similarities in restriction digestion patterns of ESBL-encoding plasmids associated with independent plasmid acquisitions. Black lines separate lanes that were not contiguous in a gel. Data were obtained from a single experiment. b) BLAST comparisons of IncI plasmids associated with four independent acquisitions (PA6, 9, 11, 15). The central circle indicates the full reference sequence of the IncI plasmid associated with PA9, with similarity between the reference sequence and other IncI plasmids shown as concentric rings. c) BLASTN comparison between IncI and IncB/O plasmid structures, in which the central circle indicates the IncI plasmid (PA9). d) BLAST comparisons of IncB/O plasmids associated with seven independent acquisitions (PA1, 3, 5, 10, 12, 13, 16). The central circle indicates the full reference sequence of the IncB/O plasmid associated with PA3, with similarity between the reference sequence and other IncB/O plasmids shown as concentric rings.
Additional plasmid sequencing and comparative analyses found that the IncI plasmids, acquired on four occasions (PA6, 9, 11, 15), shared a conserved backbone of ~84 kb (coverage: 80-100%, nucleotide identity: 99-100%). This conserved region contained typical structures associated with self-transmissible IncI plasmids, including a type IV pil operon (pilI-PilV), traABC regulatory genes, the tra/trb type IV secretion system genes, the origin of transfer (oriT including nikA and nikB), and conjugal leading region (ssb, psiA-psiB, parB homolog, ardA). The IncI/blaCTX-M-15 plasmid belonged to sequence type 16 (ST16) and was nearly identical to the previously described S. sonnei IncI plasmid pKHSB1 (NC_020991), which has been maintained in the resident Vietnamese S. sonnei population since 2006 11. Alternatively, the IncI/blaCTX-M-55 plasmids belonged to ST167 (one allele from ST16) and did not harbor the Tn3 transposon-mediated ISecp1-blaCTX-M-15, but had an insertion of ISecp1-blaCTX-M-55 (Fig. 2b). The emergence and expansion of cipR XDR S. sonnei clones associated with ISecp1-blaCTX-M-55 raise a major concern regarding the epidemic potential of this novel CTX-M ESBL variant in S. sonnei 18–20.
The IncB/O plasmids, acquired on seven occasions (PA1, 3, 5, 10, 12, 13, 16), also shared a conserved genetic structure of ~90 kb (coverage: 75-100%, nucleotide identity 99-100%) (Fig. 2d). In comparison to plasmid sequences in GenBank, our IncB/O plasmid backbone shared the highest similarity with IncB/O plasmids from an E. coli (pECAZ161: CP19011), a S. flexneri (pSF150802: CP030917.1) and a S. sonnei (p866: CO022673.1); overall synteny ranged from 72% to 94%, with 99% sequence identity. The IncB/O plasmid backbone contained comparable conjugative IncI plasmid modules; however, the pil operon (~12 kb) exhibited extremely low sequence identity to that of the IncI plasmid (coverage 1%, identity 78%) (Fig. 2c). The sizes of IncB/O plasmids varied from 95-110kb, depending on the resistance gene cassettes. These plasmids carried a wide range of blaCTX-M mobile elements, including ISecp1-IS5-blaCTX-M-55, IS66-blaCTX-M-55-orf-tnpA, ISecp1-blaCTX-M-15-orf-tnpA, IS5-blaCTX-M-14-ISecp1, and ISecp1b-blaCTX-M-24. Additionally, these IncB/O plasmids also contained other transposable elements associated with mphA (IS6-mphA-mrx-mphr) and aac6-IIa (IS4-aac6-IIa-tmrB) adjacent to blaCTX-M-carrying elements. One IncB/O plasmid additionally carried the ermB gene associated with the ISCR3 family (ISCR3-groEL-ermB-ermC).
Given the diversity of the AMR plasmids observed in cipR S. sonnei, their similarity to E. coli plasmids, and the fact that humans are the only natural reservoir for S. sonnei, we speculated that these plasmids had been transferred from commensal E. coli into S. sonnei during infection. Therefore, we performed additional characterization of AMR genes and plasmid diversity in commensal Enterobacteriaceae isolated from the same fecal samples that contained S. sonnei and from rectal swabs taken from healthy children. Metagenomic sequencing of these mixed bacterial populations (lacking Shigella) from MC plates indicated that E. coli was the most commonly isolated commensal Enterobacteriaceae (47/48 pooled colonies). Sequence data identified a substantial quantity of AMR genes and plasmid backbones with a particularly high prevalence of cipR in the commensal Enterobacteriaceae (Fig. 3a and Extended Data Fig. 2). Furthermore, a number of different AMR determinants were found to be present in both the commensal bacteria and the cipR S. sonnei. For example, blaCTX-M, mphA, aac3-IIa, and ermB were found to be present in cipR S. sonnei and 92% (44/48), 75% (36/48), 52% (25/48), and 38% (18/48) of pooled commensal Enterobacteriaceae, respectively (Fig. 3a). IncF (IncFII, IncFIA, IncFIB, and IncFIC) plasmids were found to be the most prevalent replicon types in the commensal Enterobacteriaceae. However, we additionally identified IncI and IncB/O plasmids in E. coli from the fecal samples of three children infected with S. sonnei and three healthy children, respectively.
Figure 3. Antimicrobial resistance genes and plasmids in human commensal bacteria and Shigella sonnei.

a) The first column highlights the four different sample types. Fecal/rectal swab cultures with ciprofloxacin-resistant and ESBL-producing isolates are highlighted in turquoise and red (second and third columns, respectively). The remaining columns show the presence of key antimicrobial resistance genes and plasmid groups (blue) in commensal bacteria and S. sonnei. b) The central circle indicates the full sequence of plasmid p01-0123 assembled from Nanopore sequences of commensal E. coli. The next ring shows the depth of coverage from raw Illumina reads of ciprofloxacin-resistant Shigella sonnei strain 01-0123 mapped onto the central reference sequence. Graph height is proportional to the number of reads mapping at each nucleotide position in the reference genome from 0 to 30x coverage. Regions with plasmid coverage greater than 30x are shown as solid blue bands. The turquoise ring shows the BLASTN comparison between assembled sequences of Shigella sonnei strain 01-0123 and the central reference sequence. The red ring indicates the gene annotations of the central reference sequence.
We aimed to identify comparable plasmid structures between E. coli and S. sonnei. The IncB/O plasmids found in E. coli from three healthy children (22889, 22959, and 22274) exhibited high levels of sequence similarity to the IncB/O plasmid backbone acquired by cipR S. sonnei (coverage/identity: 76/99%, 94/99%, and 99/97%; respectively). Similarly, among the three E. coli samples carrying IncI plasmids, we identified an IncI plasmid in an E. coli without a Tn3 transposon-mediated blaCTX-M-15, with high sequence similarity (coverage 81%, identity 98%) to an IncI plasmid from cipR S. sonnei. Most notably, an E. coli originating from a patient infected with a cipR S. sonnei (01-0123) carried an analogous IncI/blaCTX-M-15 plasmid. We isolated a single ESBL-producing E. coli from this MC plate and subjected the plasmid to long-read Nanopore sequencing. The sequencing resulted in a 90,786 bp circularized plasmid sequence, harboring blaCTX-M-15 and blaTEM1 on a Tn3 transposon. The raw IncI plasmid sequence from the corresponding cipR S. sonnei 01-0123 was mapped against the E. coli plasmid sequence and produced a plasmid with 100% coverage. The assembled plasmid contigs from cipR S. sonnei 01-0123 shared 100% sequence identity (Fig. 3b). These data suggest that this resistance plasmid may have been transferred between E. coli and cipR S. sonnei 01-0123 within the gut of this child. Plasmid transfer between commensal Gram-negative bacteria and Shigella spp. in the human gut has been suggested previously 21,22. We could not resolve the directionality of plasmid movement or discount the role of other components of the human microbiome as the original donor of this plasmid. However, the large biomass of Enterobacteriaceae in the human gastrointestinal tract and the apparently common circulation of IncI and IncB/O plasmids in commensal bacteria in the gastrointestinal tracts of Vietnamese children suggests that the direction of plasmid transfer is more likely to be from commensal bacteria to S. sonnei. The routine acquisition of a wide variety of ESBL-encoding plasmids by cipR S. sonnei reflects extensive interspecies gene flow from a substantial local gene pool, possibly as a result of bacterial response to selective pressures exerted by widespread and uncontrolled antimicrobial use. These plasmids likely originate from bacterial hosts that share the same ecological niche as S. sonnei.
Our data illustrate that commensal E. coli are an important reservoir of AMR genes that may be transferred to S. sonnei in vivo. Furthermore, the high diversity of AMR plasmids observed in this single S. sonnei lineage is atypical and has not been observed previously in a geographically restricted clonal expansion. The reason for this observation is unclear, but we suspect it is associated with the combination of a permissive circulating clone, widespread fluoroquinolone use, and a wide variety of AMR plasmids in the resident commensal population. To assess the potential for plasmid transfer between commensal E. coli and S. sonnei, we identified cipS/ESBL+ commensal E. coli donors and attempted to mobilize plasmids into a cipR/ESBL- S. sonnei recipient. We identified 13 commensal cipS (MIC ≤1mg/L)/ESBL+ E. coli isolates, nine of which were derived from children infected with S. sonnei and four from healthy children. ESBL plasmids from 9/13 of the commensal E. coli could be transferred into the cipR/ESBL- S. sonnei at frequencies ranging from 4x10-7 to 1.6x10-3/recipient cells. When conjugation media was supplemented with 0.5x MIC ciprofloxacin (cipS E. coli donor), 4/9 commensal E. coli (22784, 01-0123, 02-1936, and 22959) demonstrated respective increases in conjugation frequencies of ESBL plasmids to cipR S. sonnei of 3, 6, 11, and 36-fold, in comparison to media without ciprofloxacin (Fig. 4). These respective frequencies increased to 4, 10, 25 and 42-fold with 0.75x MIC ciprofloxacin. These preliminary observations suggest that exposure to sub-inhibitory concentrations of ciprofloxacin may impact the transfer of AMR plasmids between commensal E. coli and cipR S. sonnei. Transconjugant plasmid sequencing demonstrated that E. coli 01-0123 (ciprofloxacin MIC: 0.25 mg/L) carried an IncI/blaCTX-M-15 plasmid. E. coli 02-1936 (ciprofloxacin MIC: 0.016 mg/L) and 22784 (ciprofloxacin MIC: 0.38 mg/L) carried IncF/blaCTX-M-27 plasmids with high similarity to the IncF plasmid pC15 in GenBank (AY458016, ~ 92 kb) (coverage: 75% and 85%, identity: 99% and 98%, respectively). E. coli 22959 (ciprofloxacin MIC: 0.38 mg/L) harbored an IncB/O/blaCTX-M-27 plasmid exhibiting high genetic similarity to the IncB/O plasmid acquired by cipR S. sonnei (coverage 94%, identity 99%). E. coli 22978 (ciprofloxacin MIC: 0.5 mg/L) carried an IncF/blaCTX-M-27 similar to the IncF plasmid pDA33135 in GenBank (CP029577.1, ~ 139 kb) (coverage 94%, identity 99%). In the clinical study in which the S. sonnei were isolated, the majority of S. sonnei-infected children (85%, 67/79) were treated empirically with ciprofloxacin. The durations of illness between children infected with cipR (51 cases) versus cipS S. sonnei (16 cases) were comparable (median 4 days (IQR: 3-6.5 days) versus 3 days (IQR: 2-4)) 23. These data raise questions regarding the effect of ciprofloxacin on the composition of commensal bacterial and the transfer dynamics of AMR determinants in the human gut under antimicrobial pressure.
Figure 4. The conjugation efficiency of ESBL-encoding plasmids from human commensal E. coli to ciprofloxacin-resistant Shigella sonnei with and without supplementation with ciprofloxacin.

Boxplots showing the conjugation frequencies (log10) between five commensal E. coli isolates and cipR S. sonnei strain 03-0520 with and without supplementing conjugation media with 0.25x, 0.5x, and 0.75x MIC of the donor cipS commensal E. coli. Each conjugation experiment was performed in triplicate in each condition. Boxes represent the interquartile range, with the internal line indicating the data median. Maxima and minima shown extend from the box +/- 1.5*IQR, with data falling outside of that range shown as points (n = 3 biologically independent experiments, 3 technical replicates of each).
Here, by decoding the genomic sequences of S. sonnei isolated in Vietnam between 2014 and 2016, we provide unparalleled and highly concerning insights into the local clonal establishment and AMR dynamics of cipR S. sonnei as it entered a new human population. Our work outlines the progression and co-circulation of multiple XDR S. sonnei clones in Vietnam, some of which have gained resistance to all antimicrobial therapies currently recommended by WHO for the treatment of Shigella. To combat the emergence and circulation of AMR Gram-negative bacteria, a better understanding of plasmid-host interactions, plasmid stability, and the role of plasmids in the fitness of cipR S. sonnei are now critical. A significant burden of shigellosis, high prevalence of AMR among Gram-negative commensal bacteria, and unregulated purchasing of antimicrobials in the community appear to be the key factors contributing to the emergence and maintenance of XDR S. sonnei in Vietnam.
In conclusion, multiple XDR sub-clones of S. sonnei have emerged and are co-circulating in Vietnam. Commensal E. coli in the gastrointestinal tracts of Vietnamese children display an exceptionally high degree of diversity in AMR genes and plasmid composition, and evidence suggests that these are the most likely reservoir for the maintenance and transfer of MDR/XDR plasmids to cipR S. sonnei. Our data further suggest that in vivo plasmid transfer between commensal E. coli and cipR S. sonnei may occur during infection, and that this may be facilitated by antimicrobial usage. We advocate for the continued surveillance of XDR S. sonnei in Vietnam and suggest an urgent international re-evaluation of empirical antimicrobial use for gastrointestinal infections.
Methods
Study Design
The S. sonnei used in this study were isolated during a 2-year observational study conducted at three tertiary hospitals (Children’s Hospital 1, Children’s Hospital 2, and the Hospital for Tropical Diseases) in Ho Chi Minh City (HCMC), Vietnam, between May 2014 and April 2016, as previously described (Supplementary Table 1) 23. In brief, children aged <16 years admitted to one of the three study hospitals with diarrhea (defined as ≥3 passages of loose stools within 24 hours) and >1 loose stool containing blood and/or mucus were recruited. A fecal sample was collected and processed within 24 hours after enrolment. All hospitalized patients received standard of care treatment at each of the study sites. Treatment and clinical outcomes (e.g. patient recovery status (three days after enrolment) and duration of hospital stay) were recorded by clinical staff at the study sites. For the phylogenetic analyses, we additionally included two cipR S. sonnei isolated from children attending the Hospital for Tropical Diseases in HCMC in October 2013.
Ethical approval was provided by the ethics committees of all three participating hospitals in HCMC and the University of Oxford Tropical Research Ethics Committee (OxTREC No.1045-13). Written consent from parents or legal guardians of all participants was obtained prior to enrolment.
Microbiological methods
Fecal samples were inoculated onto MacConkey agar (MC agar; Oxoid) and xylose-lysine-deoxycholate agar (XLD agar, Oxoid) and incubated at 37°C. Non-lactose fermenting colonies grown on MC agar and/or XLD agar were sub-cultured on nutrient agar and identified biochemically (API20E, Biomerieux). Serological identification was performed by slide agglutination with somatic (O) antigen grouping sera following the manufacturer’s recommendations (Denka Seiken). Additionally, colony sweeps from MC agar were collected and suspended in 20% glycerol and stored at -80°C for further characterization.
Antimicrobial susceptibility testing was initially performed by the Kirby-Bauer disc diffusion method against ampicillin, chloramphenicol, trimethoprim-sulfamethoxazole, tetracycline, nalidixic acid, ciprofloxacin, azithromycin, gentamicin, amikacin, imipenem, and ceftriaxone. Subsequently, minimal inhibitory concentrations (MICs) against ciprofloxacin, azithromycin, gentamicin, and ceftriaxone were measured using the E-test (AB Biodisk), according to the manufacturer’s instructions. All antimicrobial testing was performed on Mueller-Hinton agar and susceptibility criteria were interpreted following the CLSI 2016 guidelines 15. MDR was defined as acquired non-susceptibility to at least one agent in three or more antimicrobial categories; XDR was defined as non-susceptibility to at least one agent in all but two or fewer antimicrobial categories (i.e. bacterial isolates remain susceptible to only one or two categories) 24. Detection of Extended Spectrum Beta Lactamase (ESBL) activity was performed for all isolates that were resistant to ceftriaxone using the combination disc method (cefotaxime, 30μg; ceftazidime, 30μg; with and without clavulanic acid, 10μg). ESBL-producing organisms were defined as those exhibiting a >5mm increase in the size of the zone of inhibition for the beta-lactamase inhibitor combinations in comparison to a third-generation cephalosporin without the beta-lactamase inhibitor.
Isolation of commensal bacteria
For the purposes of this study, we defined commensal organisms as organisms isolated from the stool samples of children thought not to be associated with the observed episode of diarrheal disease. In children with and without Shigella infections (i.e. symptomatic and asymptomatic children), non-Shigella organisms grown on MC plates were considered to be commensals. Colony sweeps from MC agar from Shigella-infected children were serially diluted and plated onto the MC agar without antimicrobial selection. All single colonies with a different color and morphology from that of S. sonnei were harvested, identified, and homogenized in 20% glycerol. Subsequently, DNA was extracted from these commensal bacteria by boiling and was then subjected to qualitative real-time PCR with primers and probes specific for Shigella to detect if these samples were contaminated with Shigella 25. To determine the AMR gene and plasmid diversity in human commensal bacteria, we also included a subset of commensal bacteria recovered from the rectal swabs of 18 healthy children enrolled in a longitudinal cohort study with active surveillance for diarrheal disease in HCMC between 2014 and 2016 26.
Whole genome sequencing (WGS)
Genomic DNA from S. sonnei isolates and commensal bacteria was extracted using the Wizard Genomic DNA Extraction Kit (Promega, Wisconsin, USA) following the manufacturer’s recommendations. 50ng of genomic DNA from each sample was subjected to library construction using a Nextera kit, followed by whole genome sequencing on an Illumina MiSeq platform (Illumina, CA, USA) to generate 150 bp paired-end reads. Raw sequence data are available in the European Nucleotide Archive (project: PRJEB30967).
SNP calling for S. sonnei was performed as previously described 27. In brief, raw Illumina reads were mapped against S. sonnei reference genome Ss046 chromosome (accession number NC_007382) and virulence plasmid pSs046 (NC_007385.1) using SMALT (v0.7.4, http://www.sanger.ac.uk/resources/software/smalt/). SNPs were called against the reference sequence and filtered using SAMtools (v1.3) 28. The allele at each locus in each isolate was determined by reference to the consensus base in that genome using SAMtools mpileup and removal of low confidence alleles with consensus base quality ≤20, read depth ≤5, or a heterozygous base call. SNPs occurring in non-conserved regions including prophages or repetitive sequences were removed. Subsequently, Gubbins (v2.3.2) 29 was used to identify recombinant regions from the whole genome alignment produced by SNP-calling isolates, and SNPs detected within these regions were also removed, resulting in a final set of 1,219 chromosomal SNPs.
Phylogenetic analyses
The best-fit evolutionary model for the SNP alignment of all S. sonnei isolates was determined based on the Bayesian Information Criterion in jModelTest implemented in IQ-TREE (v1.4.4) 30. A maximum likelihood phylogeny was subsequently reconstructed using IQ-TREE under the best-fit model (TVM). Support for the maximum likelihood tree was assessed via 1,000 pseudo-replicates. To explore the temporal signal in the data, the relationship between genetic divergence and date of sampling was estimated by using TempEst (v1.5) 31 to perform a linear regression analysis of root-to-tip distances, taken from the maximum likelihood tree, against the year of isolation (Extended Data Fig. 3). Temporal phylogenetic inference was then performed using Bayesian Markov Chain Monte Carlo (MCMC) implemented in BEAST software (v1.8.4) 32. For BEAST analysis, we first identified best-fit model combinations by performing multiple BEAST runs using the TVM nucleotide substitution model with constant, exponential growth or Bayesian skyline demographic models 33, in combination with either a strict or a relaxed molecular clock (uncorrelated lognormal distribution) 34. For each BEAST run, path sampling and stepping-stone sampling approaches 35,36 were used to obtain the marginal likelihood estimates for model comparison. Bayes factor (the ratio of marginal likelihoods of two models) comparisons indicated that the TVM substitution model with a relaxed lognormal molecular clock and Bayesian skyline demographic model showed the best fit to the data (Bayes factor >200). The standard deviation (SD) of inferred substitution rates across branches was 0.3 (95% highest posterior probability (HPD) = 0.14-0.47), providing additional support for a relaxed molecular clock. For the final analyses, we performed three independent runs with the best-fit model using a continuous 150 million generation MCMC chain with samples taken every 15,000 generations, and parameter convergence (indicated by effective sample size values >500) was assessed in Tracer (v1.7). LogCombiner (v1.8.4) was used to combine triplicate runs, with removal of 10 % burn-in.
Resistome analysis of S. sonnei and commensal enterobacteria
From raw Illumina reads of S. sonnei and commensal enterobacteria Short Read Sequence Typer-SRST2 (v0.2.0) 37 was used to identify the acquired resistance genes and their precise alleles using the ARG-Annot database 38, as well as the plasmid replicons using the PlasmidFinder database 39. Multilocus sequence typing (MLST) of IncI plasmids 40 was also determined using SRST2. Raw Illumina reads were de novo assembled using Velvet (v1.2), with the parameters optimized by Velvet Optimizer (v2.2.5) 41. Contigs <300 bp in size were discarded from further analyses and assembled contigs were annotated with Prokka (v1.5) 42. For the assembled sequences of S. sonnei, ABACAS (v1.3.1) 43 was used to map all the contigs against a concatenated reference sequence containing S. sonnei Ss046 chromosome (NC_007382), virulence plasmid pSs046 (NC_007385.1) and three small plasmids commonly found in S. sonnei belonging to global lineage III: spA (NC_009345.1), spB (NC_009346.1), and spC (NC_009347.1). The unmapped assembled sequences presumably containing the blaCTX-M/mphA plasmid were subjected to manual investigation using BLASTN searching with the plasmid sequences available in GenBank, and comparative analysis was performed and visualized using ACT 44. For the assembled sequences of commensal bacteria carrying IncI and IncB/O plasmids, ABACAS was used to map contigs against full-length sequences of IncI and IncB/O plasmids identified in cipR S. sonnei and sequence comparisons were visualized using ACT. Nucleotide sequence homology between the mapped contigs and reference plasmid was subsequently identified using BLASTN. Taxonomic labels of each pooled sample of commensal bacteria were assigned using Kraken, a k-mer based classification tool 45.
Plasmid profiling
Crude plasmid extractions from all S. sonnei isolates were performed using a modified Kado and Liu method 46. The resulting plasmid DNA was subjected to electrophoresis in 0.7 % agarose gel at 90 V for 3 hours, stained with ethidium bromide and photographed. E. coli strain 39R861 containing four plasmids with known sizes (7 kb, 36 kb, 63 kb, and 147 kb) was used as a marker. Plasmid profiles were compared using Bionumerics v5.1 software (Applied Maths, Austin, TX).
ESBL plasmid digestion and sequencing
E. coli transconjugants resulting from conjugation between an ESBL-positive S. sonnei isolate and E. coli J53 (sodium azide resistance) were subjected to plasmid extraction using a plasmid Midi kit (Qiagen). For plasmid digestion, 500ng of each extracted plasmid DNA was digested with EcoRI enzyme (10 U/μl) (Fermentas), followed by electrophoresis on 0.8% agarose gel at 100V for 4 hours with 1 kb plus DNA ladder (Invitrogen). Plasmid restriction patterns were compared, and cluster analysis was performed using the UPGMA method and Jukes-Cantor correction using Bionumerics v5.1 software. For plasmid sequencing, 50ng of each plasmid DNA was subjected to library construction with a Nextera kit and sequenced using the MiSeq Illumina platform to generate 2x250 bp paired-end reads. De novo assembly was subsequently performed using SPADES v3.11 47 and assembled contigs were annotated using Prokka v1.11 42.
Nanopore sequencing
Plasmid DNA extracted from the commensal E. coli carrying the IncI/blaCTX-M-15 plasmid was initially sequenced using Illumina MiSeq to generate 2x250bp paired-end reads (accession number: ERS3050916). However, the de novo assembly failed to produce a complete plasmid sequence. To improve the plasmid assembly, we then performed a single run on a MinION to generate longer reads. For MinION library preparation and sequencing, we used the rapid 1D sequencing kit SQK-RAD001 (Oxford Nanopore Technologies, Oxford, UK), following the manufacturer’s recommendations. We used the MinION Mk1 sequencer, FLO-MIN106 flow cell, and MinKNOW software v1.1.20 for sequencing, and protocol script NC_48Hr_Sequencing_Run_FLO-MIN106_SQK-RAD001_plus_Basecaller.py for local base-calling. MinION reads were converted from fast5 to fastq format using the script fast52fastq.py. SPADES version 3.11 was subsequently used to produce a hybrid assembly of MinION data and Illumina data. Raw MinION reads were deposited in ENA (accession number: ERS3050922).
Bacterial conjugation
Bacterial conjugation was first performed between each of the 40 blaCTX-M/mphA-carrying S. sonnei isolates associated with all the plasmid acquisitions and E. coli J53 (sodium azide resistant) by combining equal volumes (5mL) of overnight Luria-Bertani (LB) cultures. Bacteria were conjugated for 12 hours in LB broth at 37°C and E. coli transconjugants were selected on medium containing sodium azide (100 mg/l) plus ceftriaxone (6 mg/l) or sodium azide (100 mg/l) plus azithromycin (24 mg/l). To measure plasmid transfer from commensal E. coli to cipR S. sonnei and investigate the effect of ciprofloxacin on the conjugation efficiency, we first screened commensal E. coli isolates for ESBL activity and ciprofloxacin susceptibility from the pooled colony sweeps on MC agar. Subsequently, bacterial conjugation was performed between each of the 13 cipS ESBL-positive commensal E. coli isolates (donor) and the cipR ESBL-negative S. sonnei 03-0520 (recipient) in LB broth with and without supplementation of ciprofloxacin (0.25, 0.5, 0.75 x MIC of the donor organism). Successful transconjugants were selected on MC agar containing ciprofloxacin (4 mg/l) and ceftriaxone (6 mg/l). For all conjugation experiments, the conjugation frequency was calculated as the number of transconjugants per recipient cell.
Extended Data
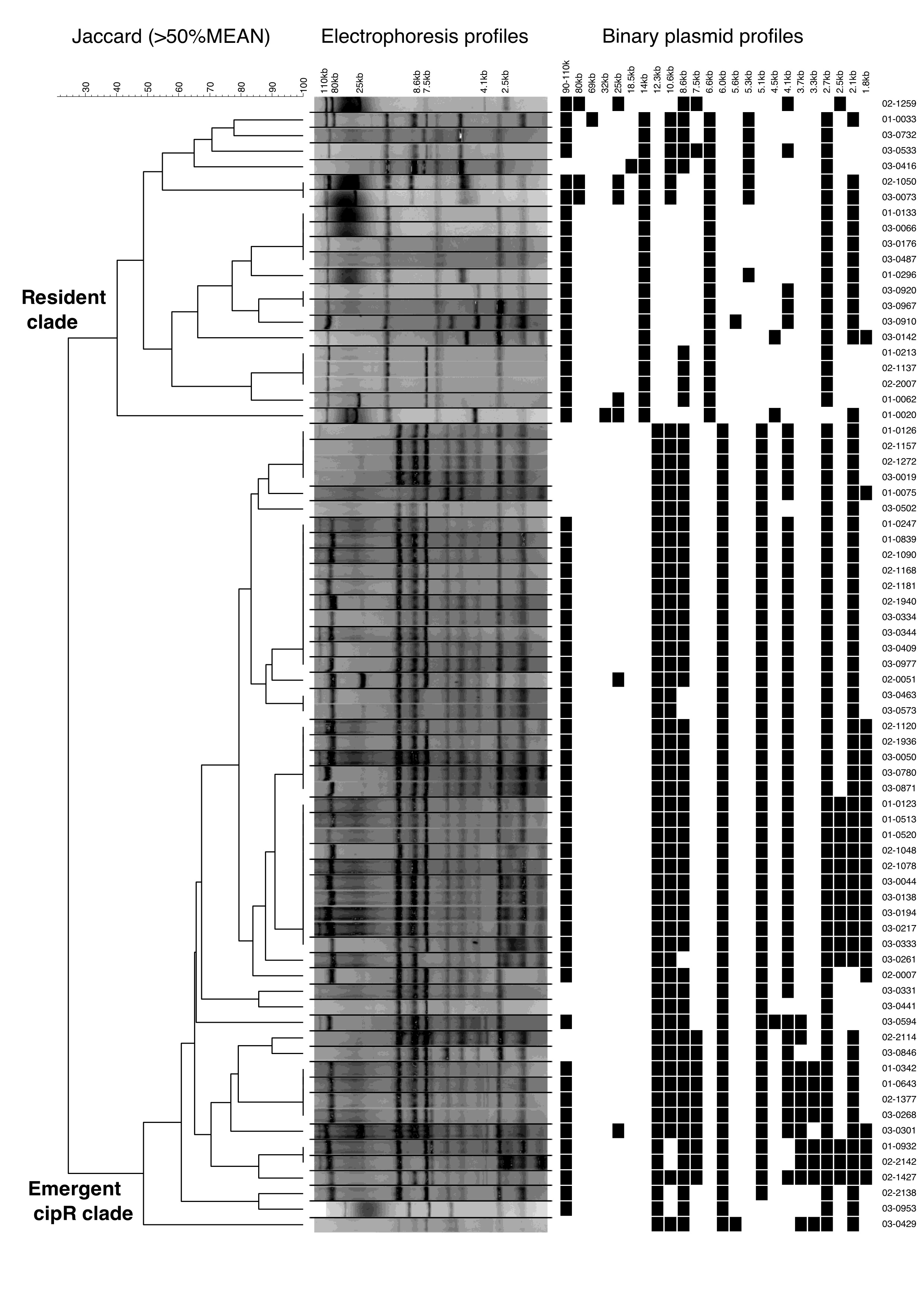
Extended Data Figure 1. Plasmid profiling of Shigella sonnei in Vietnam, 2014-2016.

Dendrogram shows the difference in plasmid electrophoresis patterns between the resident S. sonnei clade and the cipR S. sonnei clade in Vietnam. Black lines separate lanes that were not contiguous in a gel. Data were obtained from a single experiment. Cluster analysis was performed with Bionumerics by using the Jaccard coefficient and the unweighted pair group mathematical average (UPGMA) clustering algorithm.
Extended Data Figure 2. Distribution of antimicrobial resistance genes and plasmid groups in Shigella sonnei and human commensal bacteria.

The first column highlights the four different isolate types in different colors. Fecal/rectal swab cultures with ciprofloxacin-resistant and ESBL-producing isolates are highlighted in turquoise and red (second and third columns, respectively). The remaining columns show the distribution of all antimicrobial resistance genes and plasmid groups identified in commensal bacteria and S. sonnei. AMR genes are grouped together based on the class of antimicrobial agents to which they are resistant, with different variants of an AMR gene shown in different shades of blue.
Extended Data Figure 3. Root-to-tip regression for the maximum likelihood tree of Shigella sonnei in Vietnam, 2014-2016.

Each point on the plot corresponds to a measurement of genetic distance from the inferred root to each tip in the tree (n = 81 biologically independent samples). The solid line is the regression line fitted using the ordinary least squares method. The slope of the line is a crude estimate of the evolutionary rate, the x-intercept corresponds to the inferred date of the most recent common ancestor, and the R2 value measures the degree of clock-like behavior.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was funded by a Senior Research Fellowship funded by the Wellcome Trust to SB (100087/Z/12/Z) and an Oak Leader Fellowship to DPT.
Footnotes
Data Availability
All raw genomic data that support the findings of this study have been deposited in the European Nucleotide Archive (project: PRJEB30967) and can be accessed via this link (https://www.ebi.ac.uk/ena/data/search?query=PRJEB30967). Details about accession numbers of S. sonnei isolates can be found in Supplementary Table 1. The S. sonnei reference genome Ss046 chromosome (accession number: NC_007382), virulence plasmid pSs046 (accession number: NC_007385.1) and three small plasmids commonly found in global lineage III S. sonnei: spA (accession number: NC_009345.1), spB (accession number: NC_009346.1), and spC (accession number: NC_009347.1) were downloaded from GenBank. Raw MinION reads for plasmid p01_0123 are deposited in ENA (accession number: ERS3050922). Source data for main figures [Figs. 2a, 3a, 4] and Extended Data [ED Figs. 1, 2] are provided with the paper. Additional data that support the findings of this study are available from the corresponding author upon request.
Author contributions
PTD, MAR, and SB designed the study. PTD performed data analysis and interpreted the results under the scientific guidance of MAR and SB. PTD drafted the paper, with MAR and SB revising and structuring the paper. PTD and HNDT performed whole genome sequencing. PTD and TNTN performed plasmid isolation, digestion and sequencing. PTD, FA, and TNTN performed the plasmid conjugation experiments. HTT performed basic microbiology work. HCT and CB assisted in the design of laboratory experiments. DVT recruited patients and performed the clinical work required for the study. HCT, CB, and GET contributed to the editing of the paper. All authors read and approved the final draft.
Additional information
None
Competing interests
I declare that the authors have no competing interests as defined by Nature Research, or other interests that might be perceived to influence the results and/or discussion reported in this paper
References
- 1.Pazhani GP, Ramamurthy T, Mitra U, Bhattacharya SK, Niyogi SK. Species diversity and antimicrobial resistance of Shigella spp. isolated between 2001 and 2004 from hospitalized children with diarrhoea in Kolkata (Calcutta), India. Epidemiol Infect. 2005;133:1089–95. doi: 10.1017/S0950268805004498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shakya G, Acharya J, Adhikari S, Rijal N. Shigellosis in Nepal: 13 years review of nationwide surveillance. J Health Popul Nutr. 2016;35:36. doi: 10.1186/s41043-016-0073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Von Seidlein L, Deok RK, Ali M, et al. A multicentre study of Shigella diarrhoea in six Asian countries: Disease burden, clinical manifestations, and microbiology. PLoS Med. 2006;3:1556–69. doi: 10.1371/journal.pmed.0030353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pazhani GP, Niyogi SK, Singh AK, et al. Molecular characterization of multidrug-resistant Shigella species isolated from epidemic and endemic cases of shigellosis in India. J Med Microbiol. 2008;57:856–63. doi: 10.1099/jmm.0.2008/000521-0. [DOI] [PubMed] [Google Scholar]
- 5.De Lappe N, O’Connor J, Garvey P, McKeown P, Cormican M. Ciprofloxacin-resistant Shigella sonnei associated with travel to India. Emerg Infect Dis. 2015;21:894–6. doi: 10.3201/eid2105.141184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowen A, Hurd J, Hoover C, et al. Importation and Domestic Transmission of Shigella sonnei Resistant to Ciprofloxacin - United States, May 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64:318–20. [PMC free article] [PubMed] [Google Scholar]
- 7.Nüesch-Inderbinen M, Heini N, Zurfluh K, Althaus D, Hächler H, Stephan R. Shigella antimicrobial drug resistance mechanisms, 2004–2014. Emerg Infect Dis. 2016;22:1083–5. doi: 10.3201/eid2206.152088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung The H, Rabaa MA, Thanh DP, et al. Introduction and establishment of fluoroquinolone-resistant Shigella sonnei into Bhutan. Microb Genomics. 2015;1 doi: 10.1099/mgen.0.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajpara N, Nair M, Chowdhury G, et al. Molecular analysis of multidrug resistance in clinical isolates of Shigella spp. from 2001-2010 in Kolkata, India: role of integrons, plasmids, and topoisomerase mutations. Infect Drug Resist. 2018;11:87–102. doi: 10.2147/IDR.S148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung The H, Rabaa MA, Pham Thanh D, et al. South Asia as a Reservoir for the Global Spread of Ciprofloxacin-Resistant Shigella sonnei: A Cross-Sectional Study. PLoS Med. 2016;13:1–12. doi: 10.1371/journal.pmed.1002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holt KE, Thieu Nga TV, Thanh DP, et al. Tracking the establishment of local endemic populations of an emergent enteric pathogen. Proc Natl Acad Sci U S A. 2013;110:17522–7. doi: 10.1073/pnas.1308632110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Praszkier J, Pittard AJ. Control of replication in I-complex plasmids. Plasmid. 2005;53:97–112. doi: 10.1016/j.plasmid.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Seral C, Rojo-Bezares B, Garrido A, Gude MJ, Sáenz Y, Castillo FJ. [Characterisation of a CTX-M-15-producing Shigella sonnei in a Spanish patient who had not travelled abroad.] Enferm Infecc Microbiol Clin. 2012:2011–3. doi: 10.1016/j.eimc.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 14.Ma Q, Xu X, Luo M, et al. A waterborne outbreak of shigella sonnei with resistance to azithromycin and third-generation cephalosporins in China in 2015. Antimicrob Agents Chemother. 2017;61 doi: 10.1128/AAC.00308-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JS, Kim J, Jeon SE, et al. Complete nucleotide sequence of the IncI1 plasmid pSH4469 encoding CTX-M-15 extended-spectrum β-lactamase in a clinical isolate of Shigella sonnei from an outbreak in the Republic of Korea. Int J Antimicrob Agents. 2014;44:533–7. doi: 10.1016/j.ijantimicag.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Folster JP, Pecic G, Krueger A, et al. Identification and characterization of CTX-M-producing Shigella isolates in the United States. Antimicrob Agents Chemother. 2010;54:2269–70. doi: 10.1128/AAC.00039-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allué-Guardia A, Koenig SSK, Quirós P, Muniesa M, Bono JL, Eppinger M. Closed genome and comparative phylogenetic analysis of the clinical multidrug resistant Shigella sonnei strain 866. Genome Biol Evol. 2018 doi: 10.1093/gbe/evy168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zurita J, Ortega-Paredes D, Barba P. First description of Shigella sonnei harboring blaCTX-M-55 outside Asia. J Microbiol Biotechnol. 2016;26:2224–7. doi: 10.4014/jmb.1605.05069. [DOI] [PubMed] [Google Scholar]
- 19.Lee W, Chung HS, Lee H, et al. CTX-M-55-type extended-spectrum β-lactamase-producing Shigella sonnei isolated from a Korean patient who had travelled to China. Ann Lab Med. 2013;33:141–4. doi: 10.3343/alm.2013.33.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu F, Ying Z, Zhang C, et al. Plasmid-encoding extended-spectrum β-lactamase CTX-M-55 in a clinical Shigella sonnei strain, China. Future Microbiol. 2014;9:1143–50. doi: 10.2217/fmb.14.53. [DOI] [PubMed] [Google Scholar]
- 21.Bratoeva MP, John JF. In vivo R–plasmid transfer in a patient with a mixed infection of shigella dysentery. Epidemiol Infect. 1994;112:247–52. doi: 10.1017/s0950268800057654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rashid H, Rahman M. Possible transfer of plasmid mediated third generation cephalosporin resistance between Escherichia coli and Shigella sonnei in the human gut. Infect Genet Evol. 2015;30:15–8. doi: 10.1016/j.meegid.2014.11.023. [DOI] [PubMed] [Google Scholar]
- 23.Duong VT, Tuyen HT, Van Minh P, et al. No Clinical benefit of empirical antimicrobial therapy for pediatric diarrhea in a high-usage, high-resistance setting. Clin Infect Dis. 2018;66:504–11. doi: 10.1093/cid/cix844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–81. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 25.Thiem VD, Sethabutr O, Von Seidlein L, et al. Detection of Shigella by a PCR Assay Targeting the ipaH Gene Suggests Increased Prevalence of Shigellosis in Nha Trang, Vietnam. J Clin Microbiol. 2004;42:2031–5. doi: 10.1128/JCM.42.5.2031-2035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson CN, Anders KL, Nhi LTQ, et al. A cohort study to define the age-specific incidence and risk factors of Shigella diarrhoeal infections in Vietnamese children: A study protocol. BMC Public Health. 2014;14 doi: 10.1186/1471-2458-14-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt KE, Baker S, Weill F-X, et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. 2012;44 doi: 10.1038/ng.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evol. 2016;2 doi: 10.1093/ve/vew007. vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7 doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 34.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed Phylogenetics and Dating with Confidence. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie W, Lewis PO, Fan Y, Kuo L, Chen MH. Improving marginal likelihood estimation for bayesian phylogenetic model selection. Syst Biol. 2011;60:150–60. doi: 10.1093/sysbio/syq085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lartillot N, Philippe H. Computing Bayes factors using thermodynamic integration. Syst Biol. 2006;55:195–207. doi: 10.1080/10635150500433722. [DOI] [PubMed] [Google Scholar]
- 37.Inouye M, Dashnow H, Raven L-A. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6:90. doi: 10.1186/s13073-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta SK, Padmanabhan BR, Diene SM, et al. ARG-annot, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2014;58:212–20. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carattoli A, Zankari E, Garciá-Fernández A, et al. In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.García-Fernández A, Chiaretto G, Bertini A, et al. Multilocus sequence typing of IncI1 plasmids carrying extended-spectrum β-lactamases in Escherichia coli and Salmonella of human and animal origin. J Antimicrob Chemother. 2008;61:1229–33. doi: 10.1093/jac/dkn131. [DOI] [PubMed] [Google Scholar]
- 41.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. [Google Scholar]
- 43.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25:1968–9. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carver T, Berriman M, Tivey A, et al. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–6. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wood DE, Salzberg SL. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15 doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kado CI, Liu ST. Rapid Procedure for Detection and Isolation of Large and Small Plasmids. J Bacteriol. 1981;145:1365–73. doi: 10.1128/jb.145.3.1365-1373.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bankevich A, Nurk S, Antipov D, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.