

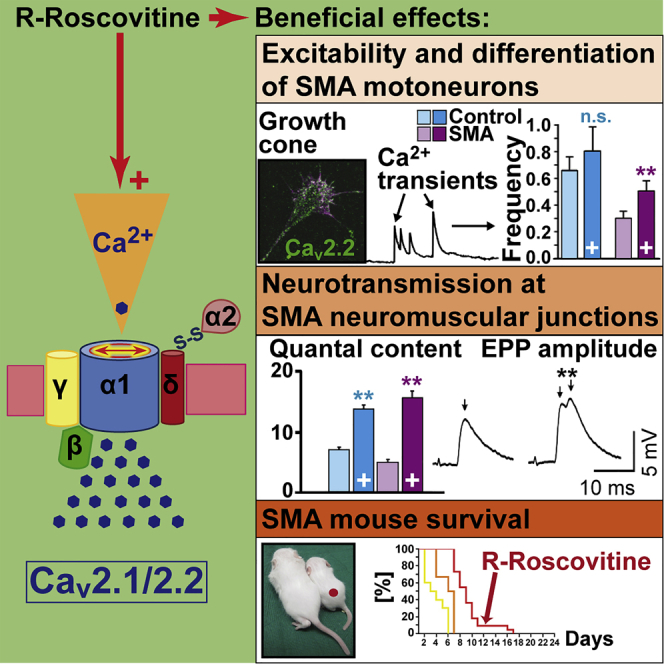
Summary
Neurotransmission defects and motoneuron degeneration are hallmarks of spinal muscular atrophy, a monogenetic disease caused by the deficiency of the SMN protein. In the present study, we show that systemic application of R-Roscovitine, a Cav2.1/Cav2.2 channel modifier and a cyclin-dependent kinase 5 (Cdk-5) inhibitor, significantly improved survival of SMA mice. In addition, R-Roscovitine increased Cav2.1 channel density and sizes of the motor endplates. In vitro, R-Roscovitine restored axon lengths and growth cone sizes of Smn-deficient motoneurons corresponding to enhanced spontaneous Ca2+ influx and elevated Cav2.2 channel cluster formations independent of its capability to inhibit Cdk-5. Acute application of R-Roscovitine at the neuromuscular junction significantly increased evoked neurotransmitter release, increased the frequency of spontaneous miniature potentials, and lowered the activation threshold of silent terminals. These data indicate that R-Roscovitine improves Ca2+ signaling and Ca2+ homeostasis in Smn-deficient motoneurons, which is generally crucial for motoneuron differentiation, maturation, and function.
Subject Areas: Neuroscience, Clinical Neuroscience, Cellular Neuroscience
Graphical Abstract
Highlights
-
•
R-Roscovitine prolongs survival of SMA mice
-
•
R-Roscovitine increases Ca2+ influx and growth cone size of SMA motoneurons
-
•
R-Roscovitine beneficially affects neurotransmission in SMA motor nerve terminals
-
•
R-Roscovitine wakes up dormant synapses of SMA motoneurons
Neuroscience; Clinical Neuroscience; Cellular Neuroscience
Introduction
Proximal spinal muscular atrophy (SMA) is the most common form of fatal motoneuron diseases in childhood. SMA is caused by loss or compound heterozygous mutations within the survival motoneuron gene (SMN1) that results in deficiency of the SMN protein (Lefebvre et al., 1997). Although genetically issued therapeutic strategies (Hua et al., 2011, Naryshkin et al., 2014, Ottesen, 2017, Palacino et al., 2015, Van Alstyne and Pellizzoni, 2016) are under consideration, it is unknown how SMN deficiency dysregulates cellular mechanisms in motoneurons. A defective Ca2+ homeostasis was discussed in glial and neuronal cells in SMA animal models (Biondi et al., 2008, Biondi et al., 2010, Jablonka et al., 2007, Lyon et al., 2014, McGivern et al., 2013, Riessland et al., 2017, Ruiz et al., 2010, Subramanian et al., 2012). Embryonic motoneurons in primary cultures from SMA mouse models show reduced N-type (Cav2.2) voltage-gated calcium channel (VGCC) cumuli in their protrusions (Jablonka et al., 2007) that corresponds to altered axonal extension on a synapse-specific laminin isoform (laminin-221) and on a Schwann cell-specific isoform (laminin-111) (Jablonka et al., 2007, Rossoll et al., 2003). At the neuromuscular junction (NMJ), motor nerve terminals from SMA mice also display a reduction in P/Q-type (Cav2.1) VGCCs (Tejero et al., 2016), which correlates with the neurotransmission deficit (Kong et al., 2009, Ruiz et al., 2010).
To test whether the re-balancing of the dysregulated Ca2+ homeostasis compensates for motoneuron defects, we used the small molecule Roscovitine. R- and S-Roscovitine are stereoisomers originally described as equally effective inhibitors of cyclin-dependent kinases (Cdks), in particular Cdk-5 (Meijer et al., 1997, Meijer et al., 2016). In addition, R-Roscovitine slows the deactivation (closing) kinetics of Cav2.1 and Cav2.2 channels, which prolongs the open state of the channels and increases calcium influx (Yan et al., 2002). In contrast, S-Roscovitine has no effect on the deactivation of the channels and can even inhibit Ca2+ influx (Buraei and Elmslie, 2008). In vitro, R-Roscovitine increases Ca2+ influx and transmitter release at presynaptic terminals of cultured hippocampal neurons independent of its Cdk-5 inhibitory effect (Buraei et al., 2005, Yan et al., 2002). In frog neuromuscular preparations, R-Roscovitine, but not S-Roscovitine, increases the number of acetylcholine quanta released during electrical stimulation (Cho and Meriney, 2006).
Our data indicate that R-Roscovitine increases SMA mouse survival and beneficially supports motoneuron differentiation, as well as synapse maintenance in the spinal cord, and counteracts morphological defects of the NMJ in an SMA mouse model (Smn−/−;SMN2;SMNΔ7). Finally, acute R-Roscovitine application induces Ca2+ transients in vitro and increases quantal content (QC) ex vivo. We conclude that these effects are primarily mediated through the capability of R-Roscovitine to enhance Ca2+ currents.
Results
Increased Mean Lifespan of SMA Mouse Models upon Systemic R-Roscovitine Treatment
We used different severe SMNΔ7 mouse models (groups 1 and 2, Figure 1A) to investigate the effect of R-Roscovitine on their survival. To discern whether the impact of R-Roscovitine is due to its inhibitory effect on Cdk-5, we used S-Roscovitine as a control, which inhibits Cdk-5 as well as R-Roscovitine but does not increase Ca2+ influx. Pregnant heterozygous SMA mice were daily injected with R- or S-Roscovitine (2 mg/kg; see also Supplemental Information, Transparent Methods). In group 1, the mean survival of the homozygous SMA mice (Smn−/−;SMN2tg/x;SMNΔ7tg/x) treated with R-Roscovitine significantly increased (13.4 ± 6.2 days) when compared with untreated SMA mice (4.9 ± 2.0 days) (Figures 1B and 1C). Additional postnatal application of R-Roscovitine did not prolong survival of SMA mice (13.9 ± 5.5 days) (Figures 1B and 1C). To verify whether a consistent double transgenic (SMN2tgtg;SMNΔ7tgtg) background increases the mean survival after R-Roscovitine application, pregnant Smn+/-;SMN2tgtg;SMNΔ7tgtg mice were injected. The SMN2tgtg;SMNΔ7tgtg background (group 2) increased the mean survival rate up to approximately 14 days (14.0 ± 1.1 days) (Figures 1B blue curve; 1C blue bar) comparable to previous reports (Le et al., 2005). R-Roscovitine application again elevated the mean survival significantly up to 19 days (18.9 ± 1.7 days) (Figure 1B, pink curve; Figure 1C pink bar). However, the maximum value of 23 days upon R-Roscovitine application to group 1 mice (Figure 1B, green curve) was not exceeded by the double transgenic SMN2/Δ7 background (22 days) (Figure 1B, pink curve). No significant weight gain (until P9), but a non-significant delayed weight loss up to P12, had been observed following prenatal R-Roscovitine application in SMA mice from group 2 (Figure S1A). However, a slight but non-significant effect on the righting reflex between P3 and P6 was detectable (Figure S1B).
Figure 1.

Prolonged Lifespan of SMA Mice under Systemic R-Roscovitine Treatment
(A) Genotype of Smn-deficient mice used in the study.
(B and C) (B) Survival curve and (C) mean survival rate of SMA mice in group 1: untreated (black), prenatally treated with R- or S-Roscovitine (green and gray, respectively), and pre- and postnatally treated with R-Roscovitine (purple); and in group 2: untreated (blue) and treated with R-Roscovitine (pink) (**p < 0.01; ***p < 0.001, ANOVA and U Mann-Whitney).
(D and E) (D) Survival curve and (E) mean survival rate of group 1 postnatally treated SMA mice with PBS (yellow), DMSO (orange) and R-Roscovitine (red) (***p < 0.001, ANOVA).
(F–I) Representative images for each example from postnatal application depicted at days (F) 2, (G) 5, (H) 10, and (I) 12.
Bars represent mean ± SD; n defines the number of mice; n.s., no significance.
Prenatal injection of S-Roscovitine had no beneficial effect on the survival of SMA mice (5.7 ± 2.3 days) (Figure 1B gray curve, Figure 1C gray bar). To test if R-Roscovitine compensates for reduced survival of SMA mice when first clinical symptoms already appeared, Smn-deficient mouse pups from group 1 received a postnatal injection at P2 with R-Roscovitine (1.5 mg/kg) reaching a blood serum concentration of about 50–100 μM (see also Supplemental Information, Transparent Methods). Application of R-Roscovitine significantly extended the mean lifespan to 9.3 ± 2.7 days, compared with Smn-deficient pups treated with PBS (3.8 ± 1.8 days) or DMSO (5.8 ± 1.5 days) (Figures 1D and 1E). Representative examples of postnatal treated and non-treated mice from group 1 are depicted in Figures 1F–1I.
Prenatal Treatment with R-Roscovitine Decreases Loss of Spinal Motoneurons, Increases the Number of Excitatory Somatodendritic Inputs on Motoneurons, and Beneficially Affects Cav2.1 Channel Cluster Formation in SMNΔ7 Mice
To analyze the cellular effects of R-Roscovitine on motoneuron loss, the number of excitatory somatodendritic inputs, the area of NMJs, Cav2.1 cluster formation, as well as muscle fiber caliber were compared between control and SMA mice (group 2). To check whether prenatal treatment with R-Roscovitine changed the number of spinal motoneurons in SMA mice, acetyltransferase (ChAT)-positive cells were labeled and their number estimated at the upper lumbar regions (L1-L2), which are particularly vulnerable in SMA (Mentis et al., 2011). Figure 2A shows an example of the distribution of labeled neurons and the mean number of motoneurons estimated in non-treated and prenatally treated control and SMNΔ7 mice. In the absence of the drug, mutants showed a significant reduction by 40% with respect to their control littermates. However, in treated mutants the number of motoneurons was not significantly different from their controls (Figure 2A). We next checked whether the number of excitatory somatodendritic inputs on motoneurons was modified by the drug treatment. We found no significant differences in the density or the number of VGlut2-positive inputs per μm2 at the soma between mutants and their littermate controls in the group treated with R-Roscovitine, contrarily to that found in the non-treated group (Figure 2B). To test whether R-Roscovitine had an effect on the pre- and postsynaptic sides of the NMJ, we quantified the postsynaptic area (Figure 2C) and determined the Cav2.1 cluster formation in the presynaptic compartment (Figure 2D) of the transversus abdominis (TVA), one of the most affected muscles in the disease model (Tejero et al., 2016, Torres-Benito et al., 2011). In non-treated mice the endplate surface area was reduced significantly in mutants when compared with littermate controls, whereas no differences were found between the two genotypes when mice were treated with R-Roscovitine (Figure 2C). Additional effects were observed after antibody stainings against the P/Q-type VGCC (Cav2.1). R-Roscovitine beneficially affected Cav2.1 cluster formations in mutant NMJs indicated by the ratio between P/Q area and BTX area (Figure 2D upper and lower panels). Finally, we investigated muscle fiber area and perimeter of the TVA. Contrarily, no improvement in myofiber surface area or perimeter was found following R-Roscovitine treatment (Figure 2E). To figure out whether muscle innervation is influenced by R-Roscovitine treatment we used the flexor digitorum brevis (FDB3) muscle, which has been described to be one of the most denervated in Smn-deficient mice (Ling et al., 2012). Surprisingly, we found a low number of unoccupied endplates in both genotypes, and no significant changes in R-Roscovitine-treated mice compared with untreated mice (Figure S1C).
Figure 2.

Prenatal Treatment with R-Roscovitine Reduces Spinal Motoneuron Loss, Ameliorates the Decrease of Excitatory Synaptic Inputs, and Increases Endplate Surface Area and Cav2.1 Accumulation Area, but Not Muscle Fiber Size In SMNΔ7 Mice
(A) Quantification (right panel) of ChAT-positive L1-L2 spinal neurons (left panel) in untreated (left bars) and R-Roscovitine-treated (right bars) mice (**p < 0.01, ANOVA).
(B) Representative examples of excitatory synaptic inputs onto L1-L2 motoneurons in control and SMA mice; z stack projections of confocal images, 2 μm (5 optical sections) for the indicated genotypes and conditions (left panel). Densities of VGlut2-positive inputs per soma (right panel) (**p < 0.01, ANOVA) (scale bar, 10 μm).
(C) Representative images of endplates labeled with bungarotoxin (BTX) conjugated to rhodamine in control and SMA TVA muscles (left panel). Mean values of BTX surface area in all four conditions (right panel) (**p < 0.01; ***p < 0.001; ANOVA) (scale bar, 10 μm).
(D) Cav2.1 channel distribution in control and Smn-deficient neuromuscular junctions (upper panel) indicated by the ratio of the P/Q-BTX area (lower panel) (*p < 0.05; ***p < 0.001, ANOVA) (scale bar, 10 μm).
(E) The skeletal muscle membrane was evidenced in transversal slices (20 μm) stained against lectins with rhodamine-labeled WGA; representative images per condition (left panel); mean values for area and perimeter (right panel) (**p < 0.01, ANOVA) (scale bar, 50 μm).
Bars represent mean ± SEM. The second number inside bars defines the number of mice; the first number is the number of motoneurons in (A and B), the number of neuromuscular junctions in (C and D), and the number of myofibers in (E); n.s., no significance. See also Figures S1A–S1C.
R-Roscovitine Increases Calcium Signaling of Primary Cultured SMA Motoneurons
The in vivo results raised the possibility that the delay in motoneuron loss and the larger P/Q-VGCC surface area at motor nerve terminals were, in part, due to the capability of R-Roscovitine to slow down the deactivation kinetics of Cav2.2/2.1 channels and, therefore, to increase cytosolic Ca2+ levels. Thus, we analyzed acute and long-term effects of the drug on axons and axonal nerve endings of isolated embryonic motoneurons from Smn-deficient mouse embryos. As SMA motoneurons cultured on the β2 chain-containing laminin isoform (laminin-221) display a reduction in spontaneous spike-like Ca2+ transients and reduced Cav2.2 cluster formations at the growth cone (Jablonka et al., 2007), we examined whether R-Roscovitine modulates Ca2+ transients in motoneurons. We started our approach with an acute application of 5 μM R- as well as S-Roscovitine, given that primary cultured motoneurons are very vulnerable and that in cultured hippocampal neurons acute application of 10 μM R-Roscovitine effectively increases synaptic potentials (Tomizawa et al., 2002). In control motoneurons at 5 days in vitro (DIV 5) the acute application of 5 μM R-Roscovitine, but not S-Roscovitine, led to an immediate and continuous induction of Ca2+ transients both in the soma and growth cone (Figure 3A), whereas the acute application of 0.5 μM R-Roscovitine had no effect (Figure S2A). Next, we wanted to know whether the Ca2+ transients evoked by acute R-Roscovitine application might also cause a continuous/general increase of spontaneous Ca2+ transients in Smn-deficient motoneurons during chronic exposure to the drug over 5 days. Thus, we investigated the long-term effects of both drugs on motoneurons in vitro. As R- or S-Roscovitine at concentrations higher than 1 μM diminished survival in dissociated motoneurons over 5 days in culture (Figure S3A), all further long-term in vitro experiments were performed with an end concentration of 0.5 μM R- or S-Roscovitine. Chronic R-Roscovitine treatment for 5 days compensated the reduced frequency of spontaneous spike-like Ca2+ transients at Smn-deficient distal axons (Figure 3B, right and middle panel). Single values per neuron are depicted in Figure 3B, left panel. This effect was blocked when 0.3 μM ω-conotoxin (ω-CTX-MVIIC), a Cav2.2/2.1 inhibitor, was present during the cell culture period (Figure S2B). Nevertheless, permanent application of 0.5 μM S-Roscovitine for 5 days to Smn-deficient motoneurons resulted in a moderate, but not significant, increase in the frequency of spontaneous spike-like transients (Figure 3B, left, right, and middle panel). To rule out VGCC-affecting attitudes of Cdk-5, motoneurons from Cdk-5 knockout mouse embryos were cultured with and without 0.5 μM R-Roscovitine over 5 days and processed for calcium imaging studies at DIV 5. Cdk-5 deficiency had no effect on spontaneous Ca2+ transients (Figure S2C). Taking together, these observations are compatible with the calcium channel-modulating properties of R-Roscovitine through its ability to slow the closing time of Cav2.1 and Cav2.2 channels. Furthermore, the potential inhibition of Cdk-5 by 0.5 μM R-Roscovitine did not impact its capability to rescue altered excitability in axon terminals of primary cultured Smn-deficient motoneurons.
Figure 3.

Application of R-Roscovitine Leads to Enhanced Spontaneous Ca2+ Transients in Smn−/−;SMN2 Motoneurons
(A) Representative responses of control motoneurons to 40-s application of 5 μM S-Roscovitine followed by R-Roscovitine in the soma and the growth cone.
(B) Permanent application of R- or S-Roscovitine over 5 days in culture. The left panel shows the frequency of Ca2+ transients per neuron at the growth cone of control and SMA motoneurons untreated and under permanent R- or S-Roscovitine exposure (**p < 0.01, ANOVA). The red line defines the mean values. The middle panel depicts the associated bar diagram (mean ± SEM) (**p < 0.01, ANOVA). The right panels show spontaneous Ca2+ spikes indicated by red dots in their corresponding growth cones. N defines number of cells, n indicates number of experiments (N/n); n.s., no significance.
See also Figures S2A–S2C and S3A.
Chronic Application of R-Roscovitine Improves Differentiation of SMA Motoneurons in Culture
To figure out whether the increase of spontaneous Ca2+ transients might cause long-term support of cellular differentiation of Smn-deficient motoneurons, we performed a morphological analysis. Given that Smn-deficient motoneurons in culture display smaller growth cone sizes (Jablonka et al., 2007), we checked the ability of R-Roscovitine (0.01–0.5 μM, for 5 days) to compensate it. We found that the effect of R-Roscovitine acts in a dose-dependent manner, with 0.5 μM being required for full compensation (Figures 4A–4D). Strikingly, mean signal intensity of the calcium channel “clusters” measured by immunofluorescence was also restored to control level upon permanent R-Roscovitine treatment over 5 days (Figures 4A–4C and 4E). These effects of R-Roscovitine were independent of Smn protein levels as the total transcript and protein amount was unaffected (Figures S3B and S3C).
Figure 4.

Permanent Exposure to R-Roscovitine Supports Cellular Differentiation of Smn−/−;SMN2 Motoneurons
(A–C) Representative immunofluorescent images of growth cones on day 5 stained with antibodies against Cav2.2 channels (green) and phalloidin (magenta) of control (A) and SMA motoneurons, untreated (B) or permanently treated (C) with 0.5 μM R-Roscovitine (R-Rosc.) (scale bar, 5 μm).
(D) The growth cone area sizes were quantified on different concentrations (0, 0.01, 0.05, 0.1, 0.5 μM) of R-Roscovitine in SMA motoneurons (**p < 0.01; ***p < 0.001; ANOVA).
(E) Quantification of signal intensities representing Cav2.2 channel cluster formations at growth cones (***p < 0.001; ANOVA).
(F) Axon lengths of R-Roscovitine (0.5 μM) and CTX-treated (ω-CTX-MVIIC, 0.3 μM) and untreated Smn-deficient and control motoneurons cultured on laminin-221/211 or on laminin-111 on day 7 (**p < 0.01; ***p < 0.001; ANOVA).
(G) Axon lengths of S-Roscovitine (S-Rosc.)-treated Smn-deficient and control motoneurons on laminin-221/211 or laminin-111 on day 7 (*p < 0.05; ***p < 0.001, ANOVA).
(H) Comparison of axon lengths between GV-58 versus S- or R-Roscovitine-treated SMA motoneurons on laminin-221/211 after 7 days in culture (*p < 0.05, ***p < 0.001; ANOVA).
Bars represent mean ± SEM. Numbers inside bars define number of cells and number of experiments, respectively; n.s., no significance. See also Figures S3B, S3C, and S4A–S4C.
In addition, reduced spontaneous Ca2+ transients in Smn-deficient motoneurons and ω-CTX-MVIIC-treated control cells are associated with altered axon elongation on different laminin isoforms (Jablonka et al., 2007). In the presence of 0.5 μM R-Roscovitine, however, axon lengths of Smn-deficient motoneurons, both on Schwann cell-specific laminin (laminin-111) and in the synapse-specific laminin (laminin-221/211), reached control values on day 7 (DIV 7) (Figure 4F). The presence of 0.3 μM ω-CTX-MVIIC over 7 days increased the length of control axons to mutant level and prevented the R-Roscovitine-induced rescue in mutant cells on both laminin isoforms (Figure 4F). S-Roscovitine did not affect axon elongation on laminin-111 or laminin-221/211 significantly (Figure 4G), which supports the notion that R-Roscovitine modulates motoneuron differentiation via Cav2.2 channels. To further test this hypothesis, we compared axon lengths of Smn-deficient motoneurons following treatment with R- and S-Roscovitine and GV-58. GV-58, an N- and P/Q-type VGCC agonist with a 20-fold lower cyclin-dependent kinase antagonist activity (Liang et al., 2012), has already been investigated in a mouse model for Lambert-Eaton syndrome (Tarr et al., 2013). GV-58 application for 7 days adjusted axon lengths of Smn-deficient motoneurons to wild-type level on laminin-221/211, like R-Roscovitine, but not S-Roscovitine (Figure 4H). Finally, we also checked whether 0.5 μM R-Roscovitine has any effect on the Cdk-5 substrate Pak1 or the p35/25 activators and found no changes in western blot measurements (Figures S4A–S4C).
Gene Expression of Cav2.2/2.1 Channels Is Not Up- or Downregulated in Cultured SMA Motoneurons following R-Roscovitine Treatment
Previous reports have shown altered gene expression under Smn deficiency (Doktor et al., 2017, Fletcher et al., 2017, Jablonka and Sendtner, 2017, Saal et al., 2014). This poses the question of whether the beneficial effect(s) on VGCCs via R-Roscovitine is(are) due to upregulated gene expression of Cav2.2/2.1 channels. To explore this, we collected the RNA for high-throughput sequencing (RNA sequencing [RNA-Seq]) from primary Smn-deficient and control motoneurons cultured on laminin-111 and treated with and without R-Roscovitine, respectively (Figure 5, Tables S1–S7). Differential expression analysis revealed 2,404 transcripts that were significantly (p < 0.05) altered in untreated Smn−/−;SMN2 compared with untreated Smn+/+;SMN2 motoneurons (1,011 up- and 1,393 downregulated transcripts) (Figure 5A and Table S1). The transcript level of SMN2 is not affected by R-Roscovitine (Figure S3B). Addition of R-Roscovitine induced significant alterations of 612 transcripts (464 up- and 148 downregulated transcripts) in Smn−/−;SMN2 motoneurons and of 898 transcripts (582 up- and 316 downregulated transcripts) in Smn+/+;SMN2 motoneurons (Figure 5A and Tables S2 and S3). Strikingly, of the 612 transcripts whose levels were changed by R-Roscovitine in Smn-deficient motoneurons 357 were also significantly altered between Smn+/+;SMN2 and Smn−/−;SMN2 motoneurons (Figure 5B and Table S4). The magnitude of change for these 357 transcripts was strongly negatively correlated with a Pearson correlation coefficient of −0.87 (Figure 5C). As a result, we detected 305 transcripts that were downregulated in Smn−/−;SMN2 compared with Smn+/+;SMN2 motoneurons and upregulated by R-Roscovitine treatment in Smn−/−;SMN2 motoneurons (Figures 5C and Table S5). Subunits of Cav2.2, as well as Cav2.1, were not among this set of transcripts (Table S5). Gene Ontology (GO) term analysis of these 305 transcripts revealed enrichment of transcripts encoding proteins with functions in neuronal differentiation, neurotransmitter transport, and location at the synapse (Figure 5D and Table S6). We also detected 40 transcripts that were upregulated in Smn−/−;SMN2 compared with Smn+/+;SMN2 motoneurons and downregulated by R-Roscovitine treatment in Smn−/−;SMN2 motoneurons (Figure 5C and Table S7). Taken together, R-Roscovitine treatment partially rescued the transcriptome perturbations induced by reduced SMN expression, although no effect on the expression of Cav2.2, as well as Cav2.1 channels was detectable.
Figure 5.

R-Roscovitine Affects the RNA Profile of Smn-Deficient Motoneurons
(A) Volcano plots showing the significance of change (-log10(p value)) and the magnitude of change (log2(fold change), log2FC) for each transcript for the indicated differential expression analyses. Significantly altered transcripts with p < 0.05 are marked in red. For easier visualization data points for transcripts with log2FC < −10 or >10 (all of which were not significantly altered) were omitted.
(B) Overlap of transcripts significantly changed (p < 0.05) in the indicated differential expression analyses.
(C) Scatterplot showing the magnitude of change (log2(fold change), log2FC) of the transcripts indicated in (B) that are significantly altered in untreated Smn−/−;SMN2 compared with untreated Smn+/+;SMN2 motoneurons and also significantly altered in R-Roscovitine-treated Smn−/−;SMN2 compared with untreated Smn−/−;SMN2 motoneurons. Note that in the scatterplot only transcripts that were detectable under all conditions are shown (351 of the 357 transcripts indicated in (B)).
(D) Gene ontology (GO) term analysis of the 305 transcripts indicated by the red box in (C).
See also Figure S3B and Tables S1–S7.
Acute Exposition to R-Roscovitine Increases Neurotransmitter Release in Motor Nerve Terminals from SMA and Control Mice
Our data suggest that the cell-autonomous and excitability-associated defects of cellular differentiation in primary Smn-deficient motoneurons (Figures 3 and 4) and of the neuromuscular system in SMA mice (Figures 1 and 2) can be improved by the modulatory effect of R-Roscovitine on VGCCs. Thus, we decided to transfer our studies on ex vivo neuromuscular preparations from SMNΔ7 (group 2) mice. We investigated if R-Roscovitine was able to increase neurotransmitter release at the NMJ by performing intracellular recordings of postsynaptic potentials in the presence and absence of the drug. The experiments were performed in the TVA muscle of Smn-deficient and littermate control mice at P9-P11, a stage at which neurotransmission is highly affected (Kong et al., 2009, Ruiz et al., 2010, Tejero et al., 2016, Torres-Benito et al., 2011). Following the calculated concentrations achieved by subcutaneous application of R-Roscovitine in pregnant mice and postnatal mouse pups (see Supplemental Information, Transparent Methods), in each experiment, a number of fibers were first recorded in the absence of the drug (control solution) and then in the presence of R-Roscovitine (10, 50, 100 μM) after 20–30 min of incubation. 100 μM R-Roscovitine did not modify the amplitude of spontaneous miniature endplate potentials (mEPPs), neither in control nor in SMA terminals (Figure 6A), but significantly increased the mEPP frequency in both genotypes (Figure 6B). The EPP amplitudes greatly increased in control (∼280%) and SMA (∼295%) terminals in the presence of 100 μM R-Roscovitine (Figures 6C and 6E). QC, the ratio between mean EPP and mean mEPP amplitudes, was similarly elevated in the presence of the drug both in control and SMA mice (Figures 6D and 6F). In contrast, 10 μM R-Roscovitine had no effect on mEPP and EPP amplitudes or QC in control or SMA nerve terminals (Figures S2D–S2F). Although 50 μM showed no significant effect, it tended to increase EPP amplitude and QC. This is in line with the effective dose to increase neurotransmitter release at the adult frog NMJ (Cho and Meriney, 2006). S-Roscovitine and olomoucine, another structurally related cyclin-dependent kinase inhibitors, were not able to increase neurotransmitter release, similar to what has been described before at the frog NMJ (Cho and Meriney, 2006). In fact, S-Roscovitine had a significant inhibitory effect on neurotransmission, which was partially reversible (Figures 6G and 6H), whereas olomoucine (100 μM) had no effect at all (QC: 6.02 ± 0.36 in the absence, and 5.72 ± 0.21 in the presence of the drug, six fibers in each condition).
Figure 6.

R-Roscovitine Increases Spontaneous and Evoked Neurotransmitter Release at Control and SMNΔ7 Mouse Motor Nerve Terminals
(A and B) (A) Amplitude and (B) frequency of spontaneous events (mEPPs) in both genotypes with and without R-Roscovitine (100 μM) (***p < 0.001, ANOVA).
(C and D) Average EPP size (C) and quantum content (D) during 100 stimuli at 0.5 Hz in control and in SMA terminals with and without R-Roscovitine.
(E and F) R-Roscovitine increases (E) mean EPP amplitude and (F) quantal content in both genotypes at low frequency of stimulation (0.5 Hz) (*p < 0.05, **p < 0.01, ***p < 0.001; ANOVA).
(G) Representative EPP traces in control fibers before and after application of S-Roscovitine at the indicated concentrations.
(H) Quantal content declines with S-Roscovitine concentrations higher than 10 μM (***p < 0.001, ANOVA). Bars represent mean ± SEM. N = number of muscle fibers, n = number of mice; n.s., no significance.
See also Figures S2D–S2F.
Acute Application of R-Roscovitine Increases the Probability of Neurotransmitter Release and Unmasks Silenced Terminals in SMA and Control Nerve Terminals
To explore whether R-Roscovitine changes the probability of vesicle release, we measured the synaptic responses to two consecutive stimuli (pair pulse protocol, interstimulus interval 50 ms). In the absence of R-Roscovitine, the mean amplitude of the second EPP was larger than that of the first, indicating an increase in release probability between stimuli (short-term facilitation) in control and SMA terminals (Figure 7A, upper traces). In the presence of 100 μM R-Roscovitine, the synaptic facilitation significantly disappeared in both control and SMA mouse (Figures 7A lower panels, and 7B), suggesting a general increase in release probability.
Figure 7.

R-Roscovitine Reduces Facilitation and Increases the Number of Active Motor Nerve Terminals in Control and SMNΔ7 Mice
(A) Representative EPP traces from control and mutant muscle fibers before and after the application of 100 μM R-Roscovitine.
(B) Quantification of paired-pulse ratio of response amplitudes in control and mutant muscle fibers with and without R-Roscovitine (*p < 0.05, ANOVA).
(C) Representation of single (left) and double (right) EPPs in a control mouse. The arrows indicate the peak of each EPP.
(D) Number of active terminals in the same control fiber before and after application of R-Roscovitine.
(E) Proportion of recorded fibers with double EPPs with respect to the total number of fibers recorded, in control and mutant mice (**p < 0.01, ANOVA). Bars represent mean ± SEM. N = muscle fibers, n = number of mice (N/n); n.s., no significance.
Remarkably, the acute application of R-Roscovitine also significantly increased the number of muscle fibers that responded with double EPPs to a single nerve stimulus (Figures 7C and 7D). Double EPPs are a typical sign of multi-innervation, and the different timings of the peaks (Figure 7C) result from the distinct conduction velocities of each active axon terminal. Double EPPs are often recorded perinatally but become less frequent by two weeks of age as most muscle fibers become mono-innervated (Sanes and Lichtman, 1999). At P9-11, only a small percentage of the entire recorded fibers in the control solution presented double EPPs (Figure 7E, control ∼7%; mutant: ∼22%). By R-Roscovitine exposure, the fraction of recorded fibers with double EPPs increased in control and SMA mutants (Figure 7E, control: ∼ 48%; mutant: ∼67%).
R-Roscovitine Effect on Neurotransmitter Release Requires the Activation of P/Q-type Voltage-Gated Calcium Channels
We next explored whether the R-Roscovitine effect on the NMJ required the activation of P/Q-type channels (Cav2.1), the main VGCC at the age at which the recordings were made (Rosato Siri and Uchitel, 1999, Santafe et al., 2001). For this purpose, we used 200 nM ω-Agatoxin IVA (ω-Aga.) as a specific blocker of P/Q-type channels (Rosato Siri and Uchitel, 1999) and found that the mean amplitude of EPPs was reduced by ∼ 99.2% (Figure 8A, left bar graph) and the QC by ∼ 99.1% (Figure 8A, right bar graph). Adding 100 μM R-Roscovitine to terminals with ω- Agatoxin IVA still being present did not increase the EPP amplitude or the QC (Figure 8A). Accordingly, in control mouse terminals where exposition to R-Roscovitine resulted in ∼84% mean increase in neurotransmitter release, subsequent application of ω- Agatoxin IVA completely blocked the evoked response (Figure 8B). In Smn-deficient mice, similar effects occurred; ω- Agatoxin IVA completely blocked neurotransmitter release after R-Roscovitine had increased EPP amplitude and QC (Figure 8B). These results indicate that R-Roscovitine was not able to affect motor terminal release when P/Q-type VGCCs were blocked with ω-Agatoxin IVA, suggesting that R-Roscovitine does not increase release through the activation of a different calcium source.
Figure 8.

R-Roscovitine Effect on Evoked Neurotransmitter Release Requires the Activation of P/Q-type Calcium Channels in Control and SMNΔ7 Mice
(A) Representative EPP traces recorded in a control fiber in physiological solution (untreated), in the presence of ω-Agatoxin IVA (ω-Aga.), and with R-Roscovitine (R-Rosc.) (100 μM) plus ω-Agatoxin IVA (ω-Aga.) (upper panel). ω-Agatoxin IVA reduced the mean amplitude of EPPs (left graph) and the quantal content (right graph), and later application of R-Roscovitine did not increase the amplitude of the response (***p < 0.001, ANOVA).
(B) Example of EPPs recorded in control and SMA mutants in control solution, with R-Roscovitine and with ω-Agatoxin IVA plus R-Roscovitine. Terminals treated with ω-Agatoxin IVA show a complete block of the evoked response elicited in the presence of R-Roscovitine as illustrated in EPP amplitude (left graph) and quantal content (right graph), both in control (p***<0.0005, ANOVA) and in SMA mice (p***<0.0005, ANOVA). Numbers inside bars represent the number of muscle fibers and the number of mice, respectively; n.s., no significance.
Discussion
Here, we demonstrated that in vivo treatment of SMNΔ7 mice with R-Roscovitine increases survival, beneficially affects formation of Cav2.1 channel clusters at the motor nerve terminal, increases the size of the NMJ, and tends to counteract loss of motoneuron and excitatory somatodendritic inputs on spinal motoneurons. Improved preservation of the neuromuscular system corresponds to increased frequency of spontaneous calcium transients, ameliorated axonal length defects, increased growth cone sizes, and modifications of some of the transcriptome perturbations in Smn-deficient motoneurons. Acute application of R-Roscovitine increases neurotransmitter release in control and SMA motor nerve terminals and favors the recruitment of silenced terminals. Together, these data reveal that R-Roscovitine is a substance that in general supports motoneuron differentiation, function, and maintenance.
R-Roscovitine Treatment Prolongs the Survival of SMA Mice
R-Roscovitine improves cellular differentiation and maturation of SMA motoneurons in vitro and increases neurotransmission at the NMJ in ex vivo neuromuscular preparations. Based on our data, the possibility that R-Roscovitine mediates its beneficial effects on the neuromuscular system via direct modulation of the Cav2.1 and Cav2.2 channels are comprehensible and convincing. However, we cannot discard additional mechanisms, especially regarding the improvement of SMA mouse survival. R-Roscovitine binds with high affinity to Cdk-1, 2, 5, 7, and 9 and a few kinases such as CaM kinase 2 (CaMK2A), extracellular signal-regulated kinases 1 and 2 (ERK1/2), and PDXK (pyridoxal kinase) (Dhorajiya et al., 2012). Inhibitors of cyclin-dependent kinases impact cell cycle mechanisms and are used for anti-neoplastic therapies (Bruyere and Meijer, 2013). Furthermore, targeting CaMK2, Erk1, and Erk2 by R-Roscovitine or its analogs might influence motoneuron differentiation/maturation. In addition, inhibition of Cdk-5/p35 by Roscovitine could partially contribute to enhanced activity of the Cav channels by increasing their interaction with some SNARE proteins (Tomizawa et al., 2002). Finally, it should be mentioned that COOH-R-Roscovitine, the main metabolite of R-Roscovitine, although has less Cdk and kinase inhibition activity (Nutley et al., 2005), may have adverse or beneficial side effects that need to be illustrated separately in future long-term approaches on SMA animal models. In principle, the complete survival phenotype is not necessarily explained by the herein studied cellular/molecular mechanisms of R-Roscovitine action, and additional work would be needed to understand this phenotype.
Long-term Exposition to R-Roscovitine Improves Differentiation of Primary Cultured Smn-Deficient Motoneurons
Our systematic R- and S-Roscovitine concentration gradient study on the survival of motoneurons (Figure S3A) discovered that the optimal dose for in vitro chronic treatment with R-Roscovitine is 0.5 μM, which is not toxic but still compensates morphological and functional abnormalities. The parameters used to assess cellular differentiation were growth cone sizes (Figures 4A–4D) and axon lengths (Figures 4F–4H) on one side, and calcium channel density at SMA growth cones (Figure 4E) on the other. The observed improvements correlate with the marked enhanced spontaneous Ca2+ influx in embryonic SMA motoneurons (Smn−/−;SMN2) chronically treated with R-Roscovitine (Figure 3B). We found no effects on wild-type motoneurons, probably because their cellular differentiation already proceeded correctly and thus is not further improvable by calcium increment. Upon S-Roscovitine treatment (0.5 μM), which inhibits Cdk-1, Cdk-2, and Cdk-5 enzymatic activity with a similar IC50 as R-Roscovitine, we observed a slight but not significant increase of spontaneous Ca2+ transients after long-term application (Figure 3B) without compensation of morphological and functional abnormalities in Smn-deficient motoneurons (Figure 4G). These results, together with the lack of effect of S-Roscovitine on mouse survival (Figure 1) and neurotransmitter release (Figure 6) imply that the inhibition of cyclin-dependent kinases by Roscovitine, or by its metabolites, is not enough to improve the SMA phenotype and argues for the importance of R-Roscovitine on Ca2+ influx, probably by its direct action on calcium channels. This idea is further supported by two of our observations. First, we discovered, that GV-58, another Cav2.1 and Cav2.2 agonist with a 20-fold lower Cdk-1, Cdk-2, and Cdk-5 activity than R-Roscovitine (Tarr et al., 2013), leads to a significantly reduced axon elongation in Smn-deficient motoneurons on laminin-221/211, comparable to R-Roscovitine (Figure 4H). Second, Cdk-5 depletion in primary cultured motoneurons does not impact spontaneous Ca2+ influx in long-term studies (Figure S2C), arguing for an R-Roscovitine mechanism being primarily focused on cellular excitability mediated through Cav2.1 as well as Cav2.2. However, Roscovitine has been shown to affect other ion conductances; for example, it can inhibit L-type VGCCs and several types of potassium channels (Buraei et al., 2007, Yarotskyy et al., 2010). Indeed, some L-type antagonists, such as nifedipine, increase neurotransmitter release at the NMJ (Balezina et al., 2007, Rosato Siri and Uchitel, 1999, Sugiura and Ko, 1997). Nevertheless, the fact that both R- and S-Roscovitine increase the inactivation of L-type channels (Yarotskyy et al., 2010) makes less likely the hypothesis that the effects on EPP amplitude and QC in our system correspond to its effect on L-type VGCCs given the lack of positive modulation by S-Roscovitine in our experiments. On the other side, potassium channel blockers enhance secretion by broadening the action potential, which results in a larger Ca2+ influx (Cho and Meriney, 2006). Interestingly, 4-aminopyridine, an efficient Kv channel blocker, can directly target presynaptic voltage-gated Ca2+ channels independent of its effect on Kv channels (Wu et al., 2009), which would lead to the potentiation of neurotransmitter release by a double mechanism. All these possibilities need to be verified in the future by additional work.
R-Roscovitine Does Not Affect Cav2.2/2.1 Gene Expression in Smn-Deficient Motoneurons
Smn deficiency affects transcript levels in various cellular systems and in vivo (Doktor et al., 2017, Fischer et al., 1997, Fletcher et al., 2017, Imlach et al., 2012, Li et al., 2014, Liu et al., 1997, Lotti et al., 2012, Pellizzoni et al., 1998, Saal et al., 2014, See et al., 2014, Wishart et al., 2014, Zhang et al., 2008). Thus, to investigate the eventuality that R-Roscovitine somehow alters cellular transcription levels of VGCCs, an RNA-Seq approach was performed (Figure 5). Interestingly, 305 transcripts with functions in neuronal differentiation and neurotransmission are downregulated by Smn deficiency and upregulated by R-Roscovitine in Smn-deficient motoneurons, not including those of N-type or P/Q-type calcium channels (Tables S5 and S6).
The auxiliary subunit α2δ2 (Cacna2d2) of Cav2 channels is one candidate (Table S5 and categorized in the “plasma membrane” Table S6), which might support proper Cav2.2/2.1 cluster formations. Cacna2d2 is one of four subunits α2δ1-4 (Cacna2d1-4) that are loosely associated with VGCC complexes and modulate their trafficking (Dolphin, 2012, Hoppa et al., 2012). There is growing evidence that these auxiliary subunits may also have roles in the nervous system not directly linked to calcium channel function. In D. melanogaster embryos, the α2δ3 subunit plays a role in synaptogenesis independent of its association with calcium channels (Kurshan et al., 2009). Other studies describe α2δ2 as a developmental switch that regulates axon growth and regeneration, as α2δ2 gene deletion or silencing promotes axon growth in vitro (Tedeschi et al., 2016). In vivo, pharmacological blockade of α2δ2 through pregabalin administration enhances axon regeneration in adult mice after spinal cord injury (Tedeschi et al., 2016). Therefore, it remains an open issue whether altered gene expression of these auxiliary VGCC subunits modifies Cav2.1/2.2 cluster formations and in turn affects neurotransmission in SMA mice. SV2A and SV2C (Table S5) are two other candidates related to the regulation of the synaptic vesicles' readily releasable pool size and the SNARE complex assembly. Tejero et al. (2016) could already show that the signal intensity of all three SV2 paralogs (A, B, and C) is significantly reduced in motor nerve terminals of skeletal muscles of SMA mice. Apart from Smn deficiency, impaired BDNF/trkB signaling causes a calcium-related phenotype in growth cones (Dombert et al., 2017) similar to that observed in SMA motoneurons (Jablonka et al., 2007), which opens the view on more complex pathways probably affected by Smn deficiency leading to dysregulated motoneuron differentiation. Based on these data, we conclude that long-term application of R-Roscovitine might have effects on gene expression affected by Smn deficiency. For a final conclusion that cellular targets up- or downregulated by R-Roscovitine have an impact on the SMA phenotype, additional experiments with relevant knockout or transgenic animal models are required.
Acute Application of R-Roscovitine Improves Synaptic Transmission at the NMJ
In our in vitro assays, acute application of 5 μM R-Roscovitine increased Ca2+ transients frequency (Figure 3), a concentration in line with experiments on cultured hippocampal neurons (Tomizawa et al., 2002). However, postnatal motor nerve terminals required 100 μM R-Roscovitine to increase neurotransmitter release (Figures 6, 7, and 8), in line with experiments in frog NMJs (Cho and Meriney, 2006). The difference in the effective concentrations among preparations could be partially explained by their respective state of development—EC50 for Cav2.2 channels, predominant in cultured motoneurons, is about 4-fold lower than for Cav2.1 channels, the most abundant at the NMJ (Buraei et al., 2007, Tarr et al., 2013)—and by the complexity of the postnatal motor nerve terminal architecture, although additional factors cannot be ruled out.
At postnatal stages, when SMNΔ7 mice are highly affected, synaptic transmission is positively modulated by R-Roscovitine. This effect seems to be presynaptic as there was a reduction of facilitation (Figures 7A and 7B), indicative of an increase in vesicle release probability. Previous works also demonstrated that R-Roscovitine increases neurotransmitter release at the NMJ (Cho and Meriney, 2006) and central synapses (Kim and Ryan, 2010, Kim and Ryan, 2013, Monaco and Vallano, 2005, Tomizawa et al., 2002, Yan et al., 2002). This effect has been shown to be mediated by modulation of VGCCs as R-Roscovitine slows the deactivation kinetics of N- and P/Q-type calcium channels (Buraei et al., 2005, Buraei and Elmslie, 2008, Buraei et al., 2007, Yan et al., 2002). The agonistic effect of R-Roscovitine is Cdk independent because intracellularly applied R-Roscovitine failed to affect Cav2.2 channels (i.e., to slow deactivation), in contraposition to its extracellular application (Buraei and Elmslie, 2008). At mouse motor nerve terminals, P/Q-type (Cav2.1) channels are the main channel involved in exocytosis (Katz et al., 1997, Rosato Siri and Uchitel, 1999, Santafe et al., 2001, Tejero et al., 2016). By using a specific toxin, we were able to show that R-Roscovitine effects require the activation of P/Q-type calcium channels as ω-Agatoxin IVA totally abolished neurotransmission. R-Roscovitine may directly interact on the calcium channel (Yan et al., 2002, Cho and Meriney, 2006, Tarr et al., 2013) or through the inhibition of Cdk-5. The activation of Cdk-5 phosphorylates Cav2.1 channels, which decreases its interaction with SNARE proteins and depresses neurotransmission (Tomizawa et al., 2002). However, we found that S-Roscovitine decreased secretion and olomoucine had no effect at all, suggesting that the second mechanism is less efficient in our conditions. We found that R-Roscovitine was also able to wake up motor nerve terminals, which are silenced under control conditions (Figure 7D). At the age our recordings were made (P 9–11), muscle fibers were in the process of changing from multi-to mono-innervation (Murthy et al., 2009, Ruiz et al., 2010). It is not well known, however, how this process is regulated. One possibility we favor is by shutting down the calcium channels implicated in neurotransmitter release (Miyazaki et al., 2004). The fast agonistic effect via reduced Cav2.1 channel closing time favors R-Roscovitine as a drug able to support neurotransmission rapidly in SMA.
In summary, our results show that R-Roscovitine in SMA regulates/modulates calcium influx, motoneuron differentiation, enhances neurotransmitter release, and impacts gene expression. Although the data indicate that R-Roscovitine may exert many of the observed in vitro and ex vivo effects by modulating Cav2.1 and Cav2.2 channels, we cannot exclude the participation of other targets as well, and thus additional experiments such as genetic manipulation of the calcium channels are needed. Moreover, the observed benefit on survival is not necessarily explained by the described calcium enhancement and will require additional work directed to identify other potential mechanisms. Everything considered, R-Roscovitine might support the current SMA therapies with antisense oligonucleotides (Ottesen, 2017) or SMN gene transfer via an adeno-associated virus (Mendell et al., 2017), in particular at disease onset, over a defined period, to support neurotransmission in a fast manner before SMN upregulation gives rise to its beneficial effects. Beyond this, R-Roscovitine could be useful in all motoneuron diseases with defects in Ca2+ homeostasis.
Limitations of the Study
The beneficial effects of R-Roscovitine on the survival of SMA mice is not entirely explained by its cellular/molecular mechanisms, and further work is needed to understand this phenotype. Especially in the case of the voltage-gated calcium channels, knockout or knockdown approaches will narrow down the specific effects of R-Roscovitine on Cav2.2 and Cav2.1. Also, the transcriptome-affecting effects of R-Roscovitine should be taken into account in further therapeutic applications.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (JA1823/3-1 to S.J. and BL567/3-2 to R.B.), by Cure SMA (JAB1920) to S.J., by Families of SMA and Initiative Forschung und Therapie für SMA (JAB0709) to S.J., and by the Spanish Ministry of Science and Innovation/FEDER (BFU2013-43763-P & BFU2016-78934-P) to L.T.. We are grateful to Nicole Rachor, Regine Sendtner, and Mara Guerra for skillful technical support. We thank Michael Sendtner for helpful discussions concerning the manuscript. S.B. was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg.
Author Contributions
Conceptualization and Project Administration, S.J. and L.T.; Methodology, S.J., L.T., R.B., S.A., S.B., and R.T.; Investigation, R.T., S.B., J.O., J.F.-E., L.H., B.D., H.D., J.-D.C., L.T.-B., L.S.-B., and R.B.; Resources, S.J., L.T., R.B., S.A., and M.B.; Formal Analysis, R.T., S.B., J.O., J.F.-E., L.T.-B., S.A., and M.B.; Writing – Original Draft, S.J., L.T., S.B., and R.T.; Funding Acquisition, S.J. and L.T.; Supervision, S.J. and L.T.
Declaration of Interests
The authors declare no competing interests.
Published: February 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100826.
Contributor Information
Lucia Tabares, Email: ltabares@us.es.
Sibylle Jablonka, Email: jablonka_s@ukw.de.
Supplemental Information
References
- Balezina O.P., Bogacheva P.O., Orlova T.Y. Effect of L-type calcium channel blockers on activity of newly formed synapses in mice. Bull. Exp. Biol. Med. 2007;143:171–174. doi: 10.1007/s10517-007-0041-y. [DOI] [PubMed] [Google Scholar]
- Biondi O., Grondard C., Lecolle S., Deforges S., Pariset C., Lopes P., Cifuentes-Diaz C., Li H., della G.B., Chanoine C., Charbonnier F. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 2008;28:953–962. doi: 10.1523/JNEUROSCI.3237-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi O., Branchu J., Sanchez G., Lancelin C., Deforges S., Lopes P., Pariset C., Lecolle S., Cote J., Chanoine C., Charbonnier F. In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J. Neurosci. 2010;30:11288–11299. doi: 10.1523/JNEUROSCI.1764-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruyere C., Meijer L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr. Opin. Cell Biol. 2013;25:772–779. doi: 10.1016/j.ceb.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Buraei Z., Elmslie K.S. The separation of antagonist from agonist effects of trisubstituted purines on CaV2.2 (N-type) channels. J. Neurochem. 2008;105:1450–1461. doi: 10.1111/j.1471-4159.2008.05248.x. [DOI] [PubMed] [Google Scholar]
- Buraei Z., Anghelescu M., Elmslie K.S. Slowed N-type calcium channel (CaV2.2) deactivation by the cyclin-dependent kinase inhibitor roscovitine. Biophys. J. 2005;89:1681–1691. doi: 10.1529/biophysj.104.052837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z., Schofield G., Elmslie K.S. Roscovitine differentially affects CaV2 and Kv channels by binding to the open state. Neuropharmacology. 2007;52:883–894. doi: 10.1016/j.neuropharm.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Cho S., Meriney S.D. The effects of presynaptic calcium channel modulation by roscovitine on transmitter release at the adult frog neuromuscular junction. Eur. J. Neurosci. 2006;23:3200–3208. doi: 10.1111/j.1460-9568.2006.04849.x. [DOI] [PubMed] [Google Scholar]
- Doktor T.K., Hua Y., Andersen H.S., Broner S., Liu Y.H., Wieckowska A., Dembic M., Bruun G.H., Krainer A.R., Andresen B.S. RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 2017;45:395–416. doi: 10.1093/nar/gkw731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhorajiya B.D., Patel J.R., Malani M.H., Dholakiya B.Z. Plant product (R) - roscovitine valuable inhibitor of CDKs as an anti-cancer agent. Der Pharm. Sin. 2012;3(1):131–143. [Google Scholar]
- Dolphin A.C. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 2012;13:542–555. doi: 10.1038/nrn3311. [DOI] [PubMed] [Google Scholar]
- Dombert B., Balk S., Luningschror P., Moradi M., Sivadasan R., Saal-Bauernschubert L., Jablonka S. BDNF/trkB induction of calcium transients through Cav2.2 calcium channels in motoneurons corresponds to F-actin assembly and growth cone formation on beta2-chain laminin (221) Front. Mol. Neurosci. 2017;10:346. doi: 10.3389/fnmol.2017.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U., Liu Q., Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- Fletcher E.V., Simon C.M., Pagiazitis J.G., Chalif J.I., Vukojicic A., Drobac E., Wang X., Mentis G.Z. Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat. Neurosci. 2017;20:905–916. doi: 10.1038/nn.4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa M.B., Lana B., Margas W., Dolphin A.C., Ryan T.A. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F., Krainer A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach W.L., Beck E.S., Choi B.J., Lotti F., Pellizzoni L., McCabe B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–439. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka S., Sendtner M. Developmental regulation of SMN expression: pathophysiological implications and perspectives for therapy development in spinal muscular atrophy. Gene Ther. 2017;24:506–513. doi: 10.1038/gt.2017.46. [DOI] [PubMed] [Google Scholar]
- Jablonka S., Beck M., Lechner B.D., Mayer C., Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 2007;179:139–149. doi: 10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E., Protti D.A., Ferro P.A., Rosato Siri M.D., Uchitel O.D. Effects of Ca2+ channel blocker neurotoxins on transmitter release and presynaptic currents at the mouse neuromuscular junction. Br. J. Pharmacol. 1997;121:1531–1540. doi: 10.1038/sj.bjp.0701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Ryan T.A. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Ryan T.A. Balance of calcineurin Aalpha and CDK5 activities sets release probability at nerve terminals. J. Neurosci. 2013;33:8937–8950. doi: 10.1523/JNEUROSCI.4288-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Wang X., Choe D.W., Polley M., Burnett B.G., Bosch-Marce M., Griffin J.W., Rich M.M., Sumner C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan P.T., Oztan A., Schwarz T.L. Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat. Neurosci. 2009;12:1415–1423. doi: 10.1038/nn.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- Li D.K., Tisdale S., Lotti F., Pellizzoni L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev. Biol. 2014;32:22–29. doi: 10.1016/j.semcdb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M., Tarr T.B., Bravo-Altamirano K., Valdomir G., Rensch G., Swanson L., DeStefino N.R., Mazzarisi C.M., Olszewski R.A., Wilson G.M. Synthesis and biological evaluation of a selective N- and p/q-type calcium channel agonist. ACS Med. Chem. Lett. 2012;3:985–990. doi: 10.1021/ml3002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K.K., Gibbs R.M., Feng Z., Ko C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Fischer U., Wang F., Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D., Pellizzoni L. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon A.N., Pineda R.H., Hao L.T., Kudryashova E., Kudryashov D.S., Beattie C.E. Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Hum. Mol. Genet. 2014;23:1990–2004. doi: 10.1093/hmg/ddt595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGivern J.V., Patitucci T.N., Nord J.A., Barabas M.E., Stucky C.L., Ebert A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia. 2013;61:1418–1428. doi: 10.1002/glia.22522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L., Borgne A., Mulner O., Chong J.P., Blow J.J., Inagaki N., Inagaki M., Delcros J.G., Moulinoux J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Meijer L., Nelson D.J., Riazanski V., Gabdoulkhakova A.G., Hery-Arnaud G., Le Berre R., Loaëc N., Oumata N., Galons H., Nowak E. Modulating innate and adaptive immunity by (R)-Roscovitine: potential therapeutic opportunity in cystic fibrosis. J. Innate Immun. 2016;8:330–349. doi: 10.1159/000444256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- Mentis G.Z., Blivis D., Liu W., Drobac E., Crowder M.E., Kong L., Alvarez F.J., Sumner C.J., O'Donovan M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T., Hashimoto K., Shin H.S., Kano M., Watanabe M. P/Q-type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J. Neurosci. 2004;24:1734–1743. doi: 10.1523/JNEUROSCI.4208-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco E.A., III, Vallano M.L. Roscovitine triggers excitotoxicity in cultured granule neurons by enhancing glutamate release. Mol. Pharmacol. 2005;68:1331–1342. doi: 10.1124/mol.105.012732. [DOI] [PubMed] [Google Scholar]
- Murthy V., Taranda J., Elgoyhen A.B., Vetter D.E. Activity of nAChRs containing alpha9 subunits modulates synapse stabilization via bidirectional signaling programs. Dev. Neurobiol. 2009;69:931–949. doi: 10.1002/dneu.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naryshkin N.A., Weetall M., Dakka A., Narasimhan J., Zhao X., Feng Z., Ling K.K., Karp G.M., Qi H., Woll M.G. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- Nutley B.P., Raynaud F.I., Wilson S.C., Fischer P.M., Hayes A., Goddard P.M., McClue S.J., Jarman M., Lane D.P., Workman P. Metabolism and pharmacokinetics of the cyclin-dependent kinase inhibitor R-roscovitine in the mouse. Mol. Cancer Ther. 2005;4:125–139. [PubMed] [Google Scholar]
- Ottesen E.W. ISS-N1 makes the first FDA-approved drug for spinal muscular atrophy. Transl. Neurosci. 2017;8:1–6. doi: 10.1515/tnsci-2017-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino J., Swalley S.E., Song C., Cheung A.K., Shu L., Zhang X., Van H.M., Shin Y., Chin D.N., Keller C.G. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015;11:511–517. doi: 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L., Kataoka N., Charroux B., Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. doi: 10.1016/s0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- Riessland M., Kaczmarek A., Schneider S., Swoboda K.J., Lohr H., Bradler C., Grysko V., Dimitriadi M., Hosseinibarkooie S., Torres-Benito L. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am. J. Hum. Genet. 2017;100:297–315. doi: 10.1016/j.ajhg.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato Siri M.D., Uchitel O.D. Calcium channels coupled to neurotransmitter release at neonatal rat neuromuscular junctions. J. Physiol. 1999;514(Pt 2):533–540. doi: 10.1111/j.1469-7793.1999.533ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W., Jablonka S., Andreassi C., Kroning A.K., Karle K., Monani U.R., Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R., Casanas J.J., Torres-Benito L., Cano R., Tabares L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010;30:849–857. doi: 10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal L., Briese M., Kneitz S., Glinka M., Sendtner M. Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA. 2014;20:1789–1802. doi: 10.1261/rna.047373.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes J.R., Lichtman J.W. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Santafe M.M., Garcia N., Lanuza M.A., Uchitel O.D., Tomas J. Calcium channels coupled to neurotransmitter release at dually innervated neuromuscular junctions in the newborn rat. Neuroscience. 2001;102:697–708. doi: 10.1016/s0306-4522(00)00507-8. [DOI] [PubMed] [Google Scholar]
- See K., Yadav P., Giegerich M., Cheong P.S., Graf M., Vyas H., Lee S.G., Mathavan S., Fischer U., Sendtner M., Winkler C. SMN deficiency alters Nrxn2 expression and splicing in zebrafish and mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2014;23:1754–1770. doi: 10.1093/hmg/ddt567. [DOI] [PubMed] [Google Scholar]
- Subramanian N., Wetzel A., Dombert B., Yadav P., Havlicek S., Jablonka S., Nassar M.A., Blum R., Sendtner M. Role of Nav1.9 in activity-dependent axon growth in motoneurons. Hum. Mol. Genet. 2012;21:3655–3667. doi: 10.1093/hmg/dds195. [DOI] [PubMed] [Google Scholar]
- Sugiura Y., Ko C.P. Novel modulatory effect of L-type calcium channels at newly formed neuromuscular junctions. J. Neurosci. 1997;17:1101–1111. doi: 10.1523/JNEUROSCI.17-03-01101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr T.B., Malick W., Liang M., Valdomir G., Frasso M., Lacomis D., Reddel S.W., Garcia-Ocano A., Wipf P., Meriney S.D. Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J. Neurosci. 2013;33:10559–10567. doi: 10.1523/JNEUROSCI.4629-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A., Dupraz S., Laskowski C.J., Xue J., Ulas T., Beyer M., Schultze J.L., Bradke F. The calcium channel subunit Alpha2delta2 suppresses axon regeneration in the adult CNS. Neuron. 2016;92:419–434. doi: 10.1016/j.neuron.2016.09.026. [DOI] [PubMed] [Google Scholar]
- Tejero R., Lopez-Manzaneda M., Arumugam S., Tabares L. Synaptotagmin-2, and -1, linked to neurotransmission impairment and vulnerability in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016;25:4703–4716. doi: 10.1093/hmg/ddw297. [DOI] [PubMed] [Google Scholar]
- Tomizawa K., Ohta J., Matsushita M., Moriwaki A., Li S.T., Takei K., Matsui H. Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J. Neurosci. 2002;22:2590–2597. doi: 10.1523/JNEUROSCI.22-07-02590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Benito L., Neher M.F., Cano R., Ruiz R., Tabares L. SMN requirement for synaptic vesicle, active zone and microtubule postnatal organization in motor nerve terminals. PLoSOne. 2011;6:e26164. doi: 10.1371/journal.pone.0026164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Alstyne M., Pellizzoni L. Advances in modeling and treating spinal muscular atrophy. Curr. Opin. Neurol. 2016;29:549–556. doi: 10.1097/WCO.0000000000000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart T.M., Mutsaers C.A., Riessland M., Reimer M.M., Hunter G., Hannam M.L., Eaton S.L., Fuller H.R., Roche S.L., Somers E. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J. Clin. Invest. 2014;124:1821–1834. doi: 10.1172/JCI71318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.Z., Li D.P., Chen S.R., Pan H.L. Aminopyridines potentiate synaptic and neuromuscular transmission by targeting the voltage-activated calcium channel beta subunit. J. Biol. Chem. 2009;284:36453–36461. doi: 10.1074/jbc.M109.075523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Chi P., Bibb J.A., Ryan T.A., Greengard P. Roscovitine: a novel regulator of P/Q-type calcium channels and transmitter release in central neurons. J. Physiol. 2002;540:761–770. doi: 10.1113/jphysiol.2001.013376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarotskyy V., Gao G., Du L., Ganapathi S.B., Peterson B.Z., Elmslie K.S. Roscovitine binds to novel L-channel (CaV1.2) sites that separately affect activation and inactivation. J. Biol. Chem. 2010;285:43–53. doi: 10.1074/jbc.M109.076448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.