

Abstract
Investigation of aeroponically grown Physalis peruviana resulted in the isolation of 11 new withanolides, including perulactones I–L (1–4), 17-deoxy-23β-hydroxywithanolide E (5), 23β-hydroxywithanolide E (6), 4-deoxyphyperunolide A (7), 7β-hydroxywithanolide F (8), 7β-hydroxy-17-epi-withanolide K (9), 24,25-dihydro-23β,28-dihydroxywithanolide G (10), and 24,25-dihydrowithanolide E (11), together with 14 known withanolides (12–25). The structures of 1–11 were elucidated by the analysis of their spectroscopic data, and 12–25 were identified by comparison of their spectroscopic data with those reported. All withanolides were evaluated for their cytotoxic activity against a panel of tumor cell lines including LNCaP (androgen-sensitive human prostate adenocarcinoma), 22Rv1 (androgen-resistant human prostate adenocarcinoma), ACHN (human renal adenocarcinoma), M14 (human melanoma), SK-MEL-28 (human melanoma), and normal human foreskin fibroblast cells. Of these, the 17β-hydroxywithanolides (17-BHWs) 6, 8, 9, 11–13, 15, and 19–22 showed selective cytotoxic activity against the two prostate cancer cell lines LNCaP and 22Rv1, whereas 13 and 20 exhibited selective toxicity for the ACHN renal carcinoma cell line. These cytotoxicity data provide additional structure–activity relationship information for the 17-BHWs.
Graphical Abstract
Prostate cancer (PC), a hormone-sensitive cancer that is influenced by androgens such as testosterone,1 is one of the most frequent solid tumors in older men and the second leading cancer killer of all men in North America.2 Androgen deprivation is still the standard therapy used to treat the metastatic disease,3 but patients invariably relapse with more aggressive androgen-insensitive PC. Despite the success of recently approved PC therapies targeting androgen receptor (AR) signaling (e.g., abiraterone)4 and second-generation antiandrogens (e.g., enzulatamide),5 durable responses appear to be limited, presumably due to acquired resistance. Moreover, androgen-insensitive PC resumes expression of multiple AR-regulated genes, such as prostate-specific antigen (PSA), suggesting that AR transcriptional activity becomes reactivated at this stage of the disease.6 Therefore, targeting both androgen-sensitive and androgen-insensitive cancer cells is important in PC drug discovery efforts. Renal carcinoma (RC) is among the 10 most common cancers in the U.S.7 Current therapies for RC include antiangiogenic and immunotherapies, which have shown only modest success for patients with advanced RC.8 Thus, the search for effective drugs to treat advanced PC and RC is considered a priority in anticancer drug discovery.
Physalis peruviana L. (family: Solanaceae) is a member of a genus with about 120 species,9 of which some are employed in traditional medicine in Asia and South America.10 Over 160 steroidal lactones belonging to the withanolide group have been reported from plants of this genus, and the ethnopharmacological activities of many Physalis species used in traditional medicines have been attributed to the presence of these withanolides.9,10 Previous phytochemical investigations of P. peruviana have resulted in the isolation of over 40 withanolides.11 Of these, 4β-hydroxywithanolide E (12), withanolide E (13), phyperunolide A (14), and withanolide C (27) have been reported to be cytotoxic to cell lines derived from lung cancer (A549),11f liver carcinomas (Hep G2 and Hep 3B),11f and breast adenocarcinomas (MCF-7 and MDA-MB-231).11f In addition, 12 was found to inhibit proliferation of MCF-7,11j MDA-MB-231,11j H1299 (lung cancer),11h Ca9–22 (oral cancer),11k and HT-29 (colorectal cancer)11n cells. We have recently reported that withanolide E (13), isolated from wild-crafted P. peruviana, was able to eliminate long-term survival of RC cells (ACHN, Caki-1, and SN-12-C) in the presence of TRAIL (tumor necrosis factor-related apoptosis-inducing ligand).12 In our continuing search for natural-product-based anticancer agents,13 we have investigated aeroponically grown P. peruviana, and herein we report the isolation of 25 withanolides, of which some exhibited selective cytotoxicity for the androgen-sensitive PC (LNCaP), androgen-insensitive PC (22Rv1), and RC (ACHN) cell lines. Eleven of the withanolides encountered are new, and these were identified as perulactones I–L (1–4), 17-deoxy-23β-hydroxywithanolide E (5), 23β-hydroxywithanolide E (6), 4-deoxyphyperunolide A (7), 7β-hydroxywithanolide F (8), 7β-hydroxy-17-epi-withanolide K (9), 24,25-dihydro-23β,28-dihydroxywithanolide G (10), and 24,25-dihydrowithanolide E (11). Comparison of spectroscopic data with those reported led to the identification of previously known withanolides as 4β-hydroxywithanolide E (12),14 withanolide E (13),14 phyperunolide A (14),11f withangulatin E (15),15 withanolide S 5-methyl ether (16),16 physalolactone (17),17 withanolide S (18),18 withaperuvin L (19),11i physapruin A (20),19 withaperuvin J (21),11i withanolide F (22),20 4-deoxyphysalolactone (23),17 perulactone B (24),14 and perulactone H (25).14
RESULTS AND DISCUSSION
Withanolides 1–4 were obtained as colorless, amorphous solids. Their 1H NMR and 13C NMR data (Tables 1 and 3) indicated the presence of a γ-lactone side chain similar to those of perulactone and perulactone B (24),11i,14 and therefore 1–4 were designated as perulactones I–L. The molecular formula of perulactone I (1) was determined to be C28H40O8 based on its HRESIMS and NMR data, suggesting nine degrees of unsaturation. Comparison of its molecular formula and NMR data with those of perulactone B (24) (C28H40O7) also encountered in this study suggested that the ring B 5,6-ene moiety in 24 was probably replaced by a 5β,6β-epoxide moiety [δH 3.19 (1H, d, J = 2.0 Hz, H-6); δC 62.1 (C-5) and 64.0 (C-6)] in 1. In addition, its HMBC data (Figures S4 and S59, Supporting Information) showed the presence of long-range correlations of H-19 (δH 1.26)/C-1 (δC 203.8), H-3 (δH 6.85)/C-1, H-19/C-5 (δC 62.1), H-4β (δH 2.96)/C-6 (δC 64.0), H-18 (δH 1.08)/C-14 (δC 83.2), H-18/C-17 (δC 88.3), H-21 (δH 1.22)/C-17, H-21/C-22 (δC 72.3), H-27 (δH 1.16)/C-26 (δC 180.8), and H-28 (δH 4.39 and 4.06)/C-25 (δC 37.7), further supporting the presence of a 5,6-epoxide moiety and a perulactone-type side chain in 1. The β-orientation of the 5,6-epoxide was confirmed by a NOESY correlation between H-6 (δH 3.19) and H-4α (δH 1.92) (Figures S5 and S60, Supporting Information). The cis-linkage of A and B rings in 1 was established by the strong positive Cotton effect at 340 nm in its electronic circular dichrosim (ECD) spectrum21 (Figure S61, Supporting Information). Epoxidation of perulactone B (24) with m-CPBA22 afforded a major product that was identical with 1. Perulactone I was thus identified as (24S,25R)-5β,6β-epoxy-14α,17β,20β,22α,28-pentahydroxy-1-oxo-ergosta-2-en-26-oic acid γ- lactone (1).
Table 1.
1H NMR Data (400 MHz, δ, Hz) for Perulactones I–L (1–4) in CDCl3a
| position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 2 | 5.99 dd (10.0, 2.8) | 5.87 dd (10.0, 2.0) | 6.01 dd (10.0, 2.4) | 2.72 dd (20.0, 4.4) |
| 3.29 brd (20.0) | ||||
| 3 | 6.85, ddd (10.0, 6.4, 2.0) | 6.77 ddd (10.0, 4.8, 2.4) | 6.84 ddd (10.0, 6.4, 2.4) | 5.61 ddd (9.6, 4.4, 2.8) |
| 4 | 1.92 m | 2.83 dd (21.2, 4.8) | 1.90 m | 6.02 dd (9.6, 2.8) |
| 2.96 brd (19.2) | 3.27 brd (21.2) | 2.98 dt (18.8, 2.8) | ||
| 6 | 3.19 d (2.0) | 5.59 brd (5.6) | 3.14 d (2.8) | 5.66 t (4.0) |
| 7 | 1.97 m | 2.03 m | 2.08 m | 2.09 m |
| 1.80 m | 1.82 m | 1.34 m | 2.03 m | |
| 8 | 1.86 m | 1.84 m | 1.78 m | 1.99 m |
| 9 | 2.33 m | 2.00 m | 1.19 m | 2.36 m |
| 11 | 2.08 m | 2.20 m | 2.13 m | 1.93 m |
| 1.62 m | 1.54 m | 1.54 m | 1.48 m | |
| 12 | 2.20 m | 1.80 m | 1.97 m | 1.73 m |
| 1.47 m | 1.55 m | 1.49 m | 1.56 m | |
| 14 | 1.32 m | |||
| 15 | 1.67 m | 1.79 m | 2.10 m | 2.08 m |
| 1.52 m | 1.52 m | 1.89 m | 1.27 m | |
| 16 | 2.62 m | 1.66 m | 5.71 dd (2.4, 1.2) | 2.67 m |
| 1.43 m | 1.85 m | 1.44 m | ||
| 17 | 2.25 m | |||
| 18 | 1.08 s | 1.03 s | 0.94 s | 1.12 s |
| 19 | 1.26 s | 1.23 s | 1.25 s | 1.38 s |
| 21 | 1.22 s | 1.20 s | 1.27 s | 1.23 s |
| 22 | 4.01 brd (10.4) | 3.46 dd (10.8, 1.6) | 3.65 dt (10.4, 2.0) | 4.11 brd (10.4) |
| 23 | 2.12 m | 1.63 m | 1.70 m | 2.38 m |
| 1.33 m | 1.24 m | 1.47 m | 1.67 m | |
| 24 | 2.71 q (7.2) | 2.70 m | 2.60–2.75 m | 2.53 m |
| 25 | 2.65 m | 2.65 m | 2.60–2.75 m | 2.52 m |
| 27 | 1.16 d (7.2) | 1.16 d (7.6) | 1.16 d (7.2) | 1.13 d (7.6) |
| 28 | 4.06 dd (8.8, 8.4) | 4.10 dd (9.2, 8.4) | 4.14 dd (9.2, 7.6) | 4.03 dd (8.8, 7.6) |
| 4.39 dd (8.8, 7.2) | 4.43 dd (9.2, 7.2) | 4.43 dd (9.2, 6.8) | 4.31 dd (8.8, 6.8) |
Assignments based on DEPT, HSQC, and HMBC data.
Table 3.
13C NMR Data (100 MHz) of Withanolides 1–11 in CDCl3a
| position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 203.8 C | 204.1 C | 203.3 C | 211.0 C | 203.5 C | 202.9 C | 202.8 C | 203.7 C | 209.9 C | 204.2 C | 203.0 C |
| 2 | 129.2 CH | 127.9 CH | 129.3 CH | 39.8 CH2 | 129.4 CH | 129.8 CH | 130.0 CH | 127.9 CH | 39.6 CH2 | 128.0 CH | 129.9 CH |
| 3 | 144.7 CH | 145.3 CH | 144.3 CH | 126.8 CH | 144.3CH | 143.6 CH | 143.5 CH | 144.9 CH | 123.9 CH | 145.3 CH | 143.7 CH |
| 4 | 33.0 CH2 | 33.4 CH2 | 33.0 CH2 | 129.2 CH | 33.0 CH2 | 32.9 CH2 | 32.8 CH2 | 32.8 CH2 | 128.6 CH | 33.5 CH2 | 32.8 CH2 |
| 5 | 62.1 C | 135.2 C | 62.1 C | 141.0 C | 61.9 C | 62.2 C | 62.2 C | 136.3 C | 141.6 C | 135.1 C | 62.1 C |
| 6 | 64.0 CH | 124.8 CH | 63.2 CH | 121.9 CH | 64.0 CH | 64.1 CH | 63.8 CH | 129.8 CH | 130.7 CH | 125.0 CH | 64.1 CH |
| 7 | 26.3 CH2 | 25.3 CH2 | 30.9 CH2 | 25.8 CH2 | 26.2 CH2 | 26.3 CH2 | 26.0 CH2 | 65.9 CH | 66.3 CH | 25.3 CH2 | 26.2 CH2 |
| 8 | 34.0 CH | 35.1 CH | 28.5 CH | 35.6 CH | 32.3 CH | 34.1 CH | 31.2 CH | 45.1 CH | 44.4 CH | 35.3 CH | 34.1 CH |
| 9 | 36.9 CH | 36.3 CH | 44.9 CH | 34.1 CH | 37.4 CH | 36.7 CH | 37.0 CH | 34.4 CH | 33.3 CH | 36.2 CH | 36.8 CH |
| 10 | 48.5, C | 50.8 C | 48.5 C | 52.4 C | 48.5 C | 48.6 C | 48.6 C | 50.2 C | 52.0 C | 50.7 C | 48.6 C |
| 11 | 23.2 CH2 | 22.1 CH2 | 23.4 CH2 | 21.7 CH2 | 22.4 CH2 | 22.7 CH2 | 21.8 CH2 | 22.8 CH2 | 21.6 CH2 | 22.2 CH2 | 22.9 CH2 |
| 12 | 30.0 CH2 | 32.5 CH2 | 36.0 CH2 | 32.3 CH2 | 32.1 CH2 | 30.1 CH2 | 28.4 CH2 | 30.2 CH2 | 30.0 CH2 | 32.3 CH2 | 30.4 CH2 |
| 13 | 54.2 C | 47.4 C | 46.9 C | 54.6 C | 47.6 C | 54.8 C | 52.3 C | 54.8 C | 54.9 C | 47.3 C | 54.7 C |
| 14 | 83.2 C | 84.6 C | 58.1 CH | 83.7 C | 84.9 C | 82.1 C | 84.5 C | 81.7 C | 81.4 C | 84.4 C | 81.9 C |
| 15 | 32.5 CH2 | 32.0 CH2 | 30.9 CH2 | 31.6 CH2 | 32.5 CH2 | 32.3 CH2 | 39.9 CH2 | 34.9 CH2 | 34.6 CH2 | 32.2 CH2 | 32.4 CH2 |
| 16 | 37.6 CH2 | 20.9 CH2 | 126.9 CH | 37.7 CH2 | 21.3 CH2 | 37.8 CH2 | 124.3 CH | 37.6 CH2 | 37.7 CH2 | 20.8 CH2 | 37.8 CH2 |
| 17 | 88.3 C | 49.4 CH | 158.9 C | 88.3 C | 49.0 CH | 88.1 C | 156.4 C | 86.7 C | 86.9 C | 49.8 CH | 87.7 C |
| 18 | 20.6 CH3 | 17.4 CH3 | 17.9 CH3 | 20.6 CH3 | 17.5 CH3 | 20.4 CH3 | 20.6 CH3 | 20.5 CH3 | 20.5 CH3 | 17.7 CH3 | 20.6 CH3 |
| 19 | 15.2 CH3 | 18.9 CH3 | 14.9 CH3 | 20.2 CH3 | 15.2 CH3 | 14.6 CH3 | 14.2 CH3 | 18.5 CH3 | 19.9 CH3 | 18.9 C | 14.5 CH3 |
| 20 | 78.5 C | 77.5 CH | 76.7 C | 79.0 C | 77.1 C | 79.5 C | 74.7 C | 78.6 C | 78.6 C | 77.2 C | 79.2 C |
| 21 | 19.3 CH3 | 20.2 CH3 | 23.1 CH3 | 19.0 CH3 | 24.1 CH3 | 19.3 CH3 | 22.41 CH3 | 19.1 CH3 | 19.2 CH3 | 20.8 CH3 | 17.2 CH3 |
| 22 | 72.3 CH | 75.2 CH | 74.6 CH | 72.6 CH | 85.6 CH | 82.4 CH | 79.4 CH | 80.7 CH | 80.5 CH | 76.7 CH | 78.9 CH |
| 23 | 31.9, CH2 | 28.8 CH2 | 27.3 CH2 | 29.9 CH2 | 67.1 CH | 66.4 CH | 29.8 CH2 | 34.3 CH2 | 34.3 CH2 | 81.4 CH | 33.2 CH2 |
| 24 | 38.1 CH | 38.1 CH | 37.9 CH | 38.1 CH | 151.7 C | 153.4 C | 149.3 C | 151.3 C | 151.2 C | 37.2 CH | 31.8, CH |
| 25 | 37.7 CH | 37.7 CH | 37.9 CH | 37.8 CH | 121.9 C | 121.1 C | 127.6 C | 121.1 C | 121.2 C | 49.6 CH | 40.7 CH |
| 26 | 180.8 C | 180.4 C | 180.3 C | 180.8 C | 164.1 C | 164.2 C | 165.2 C | 167.2 C | 167.0 C | 177.8 C | 175.4 C |
| 27 | 10.5 CH3 | 10.5 CH3 | 10.5 CH3 | 10.6 CH3 | 12.9 CH3 | 12.8 CH3 | 12.4 CH3 | 12.1 CH3 | 12.2 CH3 | 14.3 CH3 | 14.4 CH3 |
| 28 | 72.5 CH2 | 72.2 CH2 | 72.4 CH2 | 73.0 CH2 | 15.7 CH3 | 15.5 CH3 | 22.4 CH3 | 20.6 CH3 | 20.6 CH3 | 63.2 CH2 | 21.4 CH3 |
Assignments based on DEPT, HSQC, and HMBC data.
Assignments are interchangeable.
Perulactone J (2) was determined to have the molecular formula C28H40O6, corresponding to nine degrees of unsaturation. The 1H NMR spectrum of 2 contained three olefinic protons [δH 6.77 (1H, ddd, J = 10.0, 4.8, 2.4 Hz, H-3), 5.87 (1H, dd, J = 10.0, 2.0 Hz, H-2), 5.59 (1H, brd, J = 5.6 Hz, H-6)], indicating the presence of ring A 2,3-en-1-one and ring B 5,6-ene moieties as in perulactone B (24).14 Comparison of the 13C NMR data of 2 and 24 revealed that the typical tetrasubstituted oxygenated carbon (C-17) signal at δC 87.0 in 2414 was replaced by a methine signal at δC 49.4 in 2. The long-range correlations observed for H3-18 (δH 1.03)/C-17 (δC 49.7) and H3-21 (δH 1.20)/C-17 in the HMBC spectrum of 2 (Figure 2) confirmed the presence of the CH-17 moiety. The NOE correlation between CH3-18 and CH3-21 in the 1D NOESY spectrum (Figure 2) suggested that the orientation of the side chain is β. Thus, the orientation of H-17 should be α. The ECD spectrum of 2 showed a negative Cotton effect at 339 nm, which is the opposite of that observed for 1. This difference may be due to the conformational changes of rings A and B of these two compounds caused by the replacement of the 5β,6β-epoxide with a 5,6-ene moiety. The structure of perulactone J was thus elucidated as (17S,24S,25R)-14α,20β,22α,28-tetrahydroxy-1-oxo-ergosta-2,5-dien-26-oic acid γ-lactone (2).
Figure 2.
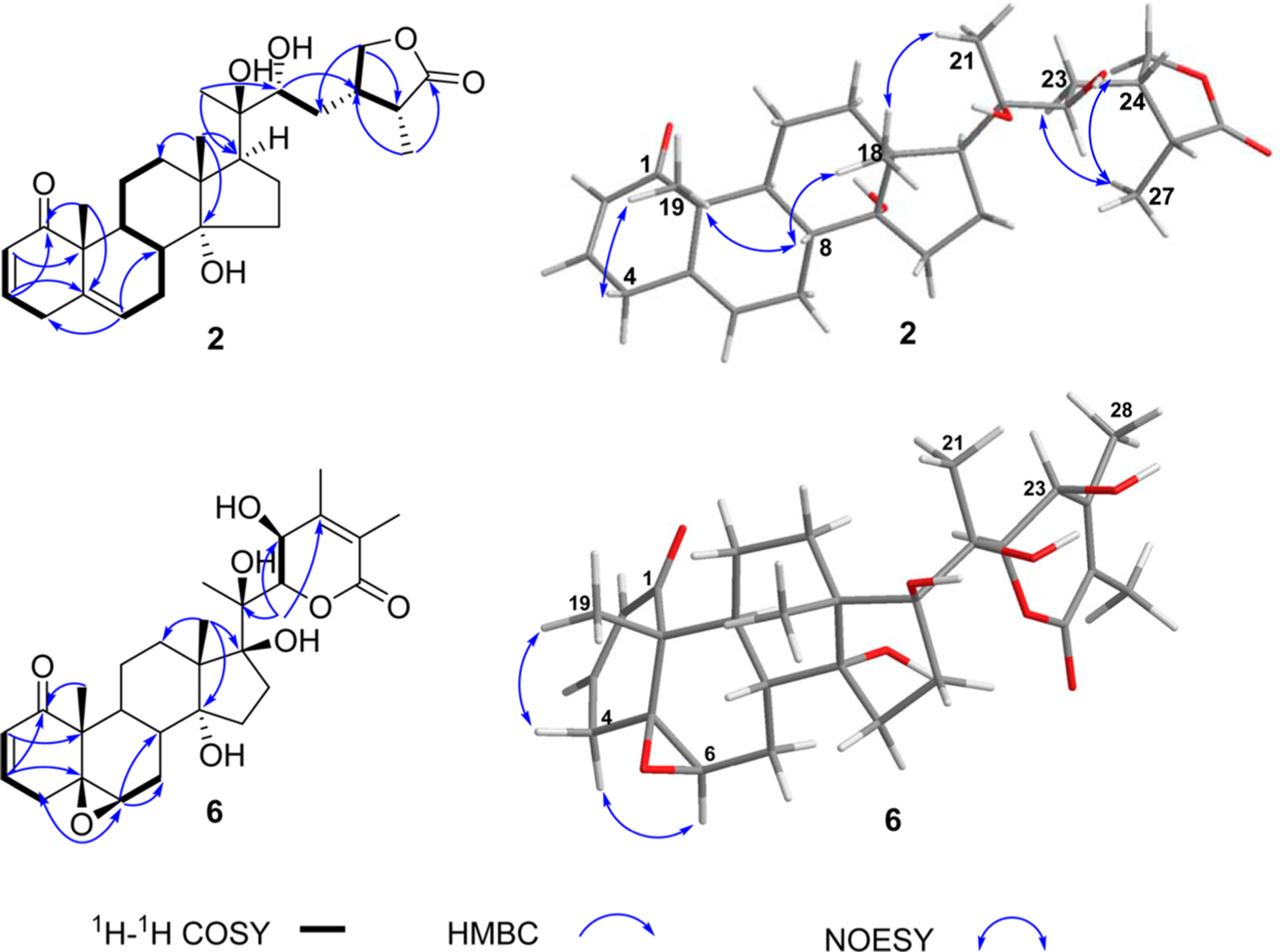
Key HMBC, 1H–1H COSY, and NOE correlations of withanolides 2 and 6.
The molecular formula of perulactone K (3) (C28H38O6) and its 1H and 13C NMR data (Tables 1 and 3) suggested that it is a dehydrated analogue of perulactone I (1) (C28H40O8) lacking an oxygen atom. Comparison of its 1H NMR data with those of 1 suggested that 3 contains a signal due to an olefinic proton [δH 5.71 (1H, dd, J = 2.4, 1.2 Hz, H-16)] besides signals due to four methyl groups, 2,3-en-1-one, 5β,6β-epoxide, and the side chain γ-lactone moieties. The 13C NMR data (Table 3) of 3 also contained signals typical of other perulactones, including 2,3-en-1-one [δC 203.3 (C-1), 144.3 (C-3), 129.3 (C-2)], 5β,6β-epoxide [δC 63.2 (C-6), 62.1 (C-5)], and the side chain γ- lactone [δC 180.3 (C-26), 72.4 (C-28)]. The 13C NMR spectrum 3 indicated the presence only two oxygenated carbons [δC 76.7 (C-20), 74.6 (C-22)]. The HMBC correlations observed for H3-18 (δH 0.94)/C-14 (δC 58.1), H3-21 (δH 1.27)/C-17 (δC 158.9), and H-16 (δH 5.71)/C-13 (δC 46.9), while confirming the absence of a hydroxy group at C-14, suggested the presence of a 16,17-ene moiety in 3 (Figures S15 and S59, Supporting Information). The ECD spectrum of 3 showed a positive Cotton effect at 342 nm, which confirmed the stereochemistry of the ring A/B linkage as being the same as that in 1.21 On the basis of the foregoing evidence, the structure of perulactone K was determined as (24S,25R)-5β,6β-epoxy-20β,22α,28-trihydroxy-1-oxo-ergosta-2,16-dien-26-oic acid γ-lactone (3).
The HREIMS and 1H and 13C NMR data of perulactone L (4) were consistent with the molecular formula C28H40O7 and indicated that it is an isomer of perulactone B (24). The 1H and 13C NMR spectra of 4 (Tables 1 and 3), assigned with the help of HSQC data (Supporting Information, Figures S19), were almost identical with those of perulactone B (24) except for the absence of signals due to the 2(3)-en-1-one moiety in ring A of 24. Instead, the 1H NMR spectrum of 4 exhibited signals due to a tetrasubstituted 3(4),5(6)-conjugated diene moiety [δ 6.02 (1H, dd, J = 9.6 and 2.8 Hz), 5.61 (1H, ddd, J = 9.6, 4.4, and 2.8 Hz), and 5.66 (1H, t, J = 4.0 Hz)]. These data suggested that 4 is isomeric with 24 and that they differ from each other by virtue of the position of the olefinic double bond in ring A. Finally, treatment of 24 with NaCN–MeOH resulted in the isomerization of its 1-oxo-2(3),5(6)-diene moiety to a 1-oxo-3(4),5(6)-diene unit, affording 4. Thus, the structure of perulactone L was determined as (24S,25R)-14α,17β,20β,22α,28-pentahydroxy-1-oxo-ergosta-3,5-dien-26-oic acid γ-lactone (4).
Withanolide 5 was determined to have the molecular formula C28H38O7, based on its HRESIMS and NMR data. The 1H NMR spectrum of 5 (Table 2) showed signals due to H-3 [δH 6.83 (1H, ddd, J = 10.0, 6.4, 2.4 Hz)], H-2 [δH 6.00 (1H, dd, J = 10.0, 2.0 Hz)], H-6 [δH 3.18 (1H, d, J = 2.0 Hz)], H-22 [δH 4.04 (1H, d, J = 8.8 Hz)], five methyl groups, and an oxygenated methine group [δH 4.38 (1H, brd, J = 8.8 Hz)]. Comparison of the 13C NMR data of 5 (Table 3) with those of withanolide E (13)14 revealed that the CH2-23 group in 13 is replaced by an oxygenated methine [δC 67.1], and the presence of a methine signal at δC 49.0 suggested that C-17 of 5 is not oxygenated. A long-range correlation observed for H3-28 and C-23 in its HMBC spectrum (Figure S25, Supporting Information) further supported the presence of OH-23 in 5, whereas long-range correlations of H3-18 (δH 1.01)/C-17 and H3-21 (δH 1.33)/C-17 suggested the presence of a methine proton at C-17, confirming that 5 is a 17-deoxy-23-hydroxy analogue of withanolide E (13). The large coupling constant (J = 8.8 Hz) observed for H-22/H-23 of 5 was consistent with the relevant dihedral angle of 45° for the favorable conformation adopted by ring E with a β-OH at C-23 as in 23β-hydroxyphysacoztolide.13 The β-orientation of its side chain at C-17 was deduced from the NOE correlation observed for H3-18/H3-21 (Figure S26, Supporting Information). The ECD spectrum of 5 exhibited positive Cotton effects at 265 and 339 nm, suggesting the 22R configuration21 and the cis-linkage of rings A/B.21 Thus, the structure of 5 was established as 17-deoxy-23β-hydroxywithanolide E [(17S,20S,22R)-5β,6β-epoxy-14α,20,23β-trihydroxy-1-oxowitha-2,24-dienolide].
Table 2.
1H NMR Data (400 MHz, δ, Hz) for Withanolides 5–11 in CDCl3a
| position | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 2 | 6.00 dd (10.0, 2.0) | 6.00 dd (10.0, 2.4) | 6.01 dd (10.0, 2.8) | 5.81,dd (10.0, 2.0) | 2.70 dd (20.2, 4.8) | 5.86 ddd (10.0, 3.0, 1.0) | 6.01 dd (10.0, 3.0) |
| 3.24 dt (20.0, 2.4) | |||||||
| 3 | 6.83 ddd (10.0, 6.4, 2.4) | 6.79 ddd (10.0, 6.4, 2.4) | 6.80 ddd (10.0, 6.4, 2.0) | 6.73 ddd (10, 4.8, 2.4) | 5.67 ddd (10.0, 4.4, 2.8) | 6.76 ddd (10.0, 5.0, 2.5) | 6.80 ddd (10.0, 6.5, 2.2) |
| 4 | 1.90 m | 1.84 m | 1.83 dd (18.4, 6.4) | 2.81 dd (21.2, 4.8) | 6.01 dd (10.0, 2.4) | 2.82 dd (21.2, 5.0) | 1.84 dd (18.4, 6.5) |
| 2.96 dt (18.8, 2.8) | 2.92 dt (18.4, 2.8) | 2.92 ddd (18.4, 2.8, 2.0) | 3.24 dt (21.2, 2.4) | 3.27 brd (21.2) | 2.93 dt (18.4, 2.6) | ||
| 6 | 3.18 d (2.0) | 3.17 brs | 3.17 brs | 5.43 brs | 5.50 d (2.8) | 5.59 brd (5.5) | 3.17 brs |
| 7 | 1.92 m | 1.95 m | 2.08 m | 4.28 d (8.8) | 4.38 brd (6.4) | 2.08 m | 1.94 m |
| 1.82 m | 1.93 m | 1.81 m | |||||
| 8 | 1.95 m | 1.91 m | 1.98 m | 1.67 dd (12.0, 8.8) | 1.79 m | 1.82 m | 1.89 m |
| 9 | 1.61 m | 1.88 m | 1.88 m | 2.33 m | 2.54 m | 2.09 m | 1.88 m |
| 11 | 1.98 m | 2.03 m | 2.02 m | 2.28 m | 1.91 m | 2.18 m | 2.06 m |
| 1.52 m | 1.57 m | 1.56 m | 1.56 m | 1.37 m | 1.56 m | 1.63 m | |
| 12 | 1.67–1.75 m | 2.19 dt (4.0, 12.0) | 2.12 m | 2.38 m | 2.41 m | 1.97 m | 2.33 m |
| 1.67 m | 1.43, m | 1.26 m | 1.24 m | 1.72 m | 1.39 m | ||
| 15 | 1.57 m | 1.67 m | 2.19 dd (14.4, 3.6) | 1.89 m | 1.93 m | 1.52 m | 1.63 m |
| 1.48 m | 1.60 m | 2.31 dd (14.4, 1.2) | 1.78 m | 1.75 m | 1.54 m | ||
| 16 | 2.05 m | 2.77 ddd (14.8, 11.6, 8.8) | 5.77 dd (3.6, 1.2) | 2.62 m | 2.66 m | 1.82 m | 2.68 m |
| 1.83 m | 1.53 m | 1.41 m | 1.43 m | 1.40 m | |||
| 17 | 2.50 t (9.2) | 2.64 t (9.4) | |||||
| 18 | 1.01 s | 1.07 s | 1.12 s | 1.05 s | 1.07 s | 1.01 s | 1.09 s |
| 19 | 1.24 s | 1.21 s | 1.23 s | 1.20 s | 1.36 s | 1.23 s | 1.24 s |
| 21 | 1.33 s | 1.45 s | 1.27 s | 1.35 s | 1.36 s | 1.33 s | 1.35 s |
| 22 | 4.04 d (8.8) | 4.76 d (10.8) | 4.39 dd (12.8, 3.6) | 4.88 dd (12.4, 4.4) | 4.87 dd (12.0, 4.8) | 3.55 d (7.4) | 4.89 dd (11.0, 3.4) |
| 23 | 4.38 brd (8.8) | 4.27 brd (10.8) | 2.78 brt (15.2) | 2.40–2.55 m | 2.40–2.55 m | 4.18 t (7.4) | 1.92 m |
| 2.14 m | 1.82 m | ||||||
| 24 | 2.32 m | 1.70 m | |||||
| 25 | 2.38 m | 2.16 m | |||||
| 27 | 1.88 s | 1.89 s | 1.85 brs | 1.82 s | 1.82 s | 1.28 d (6.8) | 1.21 d (6.8) |
| 28 | 1.95 s | 1.97 s | 1.95 s | 1.89 s | 1.89 s | 3.86 brd (11.0) | 1.12 d (6.8) |
| 3.65 dd (11.0, 7.8) |
Assignments based on DEPT, HSQC, and HMBC data.
The HREIMS and NMR data of withanolide 6 were consistent with the molecular formula C28H38O8. Its 1H NMR spectrum (Table 2) exhibited signals due to H-3 [δH 6.79 (1H, ddd, J = 10.0, 6.4, 2.4 Hz)], H-2 [δH 6.00 (1H, dd, J = 10.0, 2.4 Hz)], H-6 [δH 3.17(1H, brs)], H-22 [δH 4.76 (1H, d, J = 10.8 Hz)], five methyl groups, and an oxygenated methine group [δH 4.27 (1H, brd, J = 10.8 Hz)]. Comparison of the 13C NMR data of 6 (Table 3) with those of withanolide E (13)14 revealed that the CH2-23 in 13 is replaced by an oxygenated methine (δC 66.4) in 6. Therefore, 6 was suspected to be a 23-hydroxy analogue of withanolide E. In its HMBC spectrum, the presence of a correlation between H3-28 and C-23 supported the presence of OH-23 in 6 (Figure S31, Supporting Information and Figure 2). The large coupling constant observed for H-22/H-23 (J = 10.8 Hz) of 6 similar to those of 5 and 23β-hydroxyphysacoztolide13 suggested that the orientation of OH-23 is β. The ECD spectrum of 6 showed positive Cotton effects at 265 and 339 nm, suggesting a 22R configuration21 and cis-linkage of rings A/B.21 On the basis of the foregoing evidence, the structure of withanolide 6 was elucidated as 23β-hydroxywithanolide E [(20S,22R)-5β,6β-epoxy-14α,17β,20,23β-tetrahydroxy-1-oxowitha-2,24-dienolide].
The molecular formula of withanolide 7 was determined to be C28H36O6 using a combination of HRESIMS and 13C NMR data and accounted for 11 degrees of unsaturation. Comparison of its 1H and 13C NMR data (Tables 2 and 3) with those of withanolide E (13) and phyperunolide A (14) indicated that 7 contains 2,3-en-1-one, 5β,6β-epoxide, and side chain δ-lactone moieties similar to those in 13 and a 16,17-ene moiety as in 14. The 1H and 13C NMR data [δH 5.77 (1H, dd, J = 3.6, 1.2 Hz); δC 124.3 (CH) and 156.4 (C)] further suggested the presence of a 16,17-double bond in 7, which was confirmed by the correlations of H3-18 [δH 1.12 (3H, s)]/C-17 [δC 156.4 (C)] and H3-21 [δH 1.27 (3H, s)]/C-17 observed in its HMBC spectrum (Figure S36, Supporting Information). The absolute configuration of C-22 was determined as R from the positive Cotton effect at 257 nm in its ECD spectrum.21 Thus, withanolide 7 was identified as 4-deoxyphyperunolide A [(20S,22R)-5β,6β-epoxy-14α,20-dihydroxy-1-oxowitha-2,16,24-trienolide].
Withanolides 8 and 9 were determined to have the same molecular formula (C28H38O7) by the analysis of their HREIMS and 13C NMR data, indicating that they are isomeric with each other and both are mono-oxygenated analogues of withanolide F (22),20 a compound also encountered in the same plant extract. Comparison of the 1H NMR and 13C NMR data of 8 (Tables 2 and 3) with those of 22 suggested them to be very similar except that one of the carbocyclic ring CH2 groups of 22 underwent hydroxylation, leading to a CH-OH [δH 4.28 (1H, d, J = 8.8 Hz); δC 65.9 (CH)] moiety present in 8. The HMBC correlations of H-7 (δH 4.28)/C-5 (δC 136.3) and H3-19 (δH 1.20)/C-5 observed for 8 (Figure S41, Supporting Information) suggested that this OH is attached to C-7, and the NOE correlation of H-7/H-9α (δH 2.33) (Figure S42, Supporting Information) helped to determine the orientation of OH-7 as β. The strong positive Cotton effect at 252 nm in the ECD spectrum suggested a 22R configuration.21 The structure of withanolide 8 was thus established as 7β-hydroxywithanolide F [(20S,22R)-7β,14α,17β,20-tetrahydroxy-1-oxowitha-2,5,24-trienolide (8)]. The 1H NMR spectrum of withanolide 9 lacked resonances due to the ring A 2,3-en-1-one moiety, but exhibited signals due to a tetrasubstituted 3(4),5(6)-conjugated diene moiety [δ 6.01 (1H, dd, J = 10.0 and 2.4 Hz), 5.67 (1H, ddd, J = 10.0, 4.4, and 2.8 Hz), and 5.50 (1H, d, J = 2.8 Hz)], similar to that of perulactone L (4). In its 1H NMR spectrum, 9 showed the presence of a CHOH group [δH 4.38 (1H, brd, J = 6.4)], as in 8. The HMBC correlations of H-7 (δH 4.38)/C-5 (δC 141.6) and H3-19 (δH 1.36)/C-5 (δC 141.6) for 9 (Figure S46, Supporting Information) suggested that this CHOH group is located at C-7, and the NOE correlation of H-7/H-9α (δH 2.54) (Figure S47, Supporting Information) confirmed its orientation as β. The absolute configuration of C-22 was determined as R from the strong positive Cotton effect at 254 nm in its ECD spectrum21 (Figure S61, Supporting Information). Thus, the structure of withanolide 9 was elucidated as 7β-hydroxy-17-epi-withanolide K [(20S,22R)-7β,14α,17β,20-tetrahydroxy-1-oxowitha-3,5,24-trienolide].
The HRESIMS and 13C NMR data of withanolide 10 were consistent with the molecular formula C28H40O7. The1 H and 13C NMR spectra of 10 (Tables 2 and 3), assigned with the help of the HSQC and HMBC data (Figures S51 and S52, Supporting Information), suggested that 10 contains ring A 2,3-en-1-one, ring B 5,6-ene, 14α-OH, and 20-OH moieties similar to those of withanolide G,20 but the structure of the side chain δ-lactone moiety differed from any of the known withanolides. The presence of only four methyl signals including one doublet and three singlets in its 1H NMR spectrum suggested that ring E of 10 constituted a saturated δ-lactone and that one of the side chain methyl groups (CH3-27 or CH3-28) has undergone oxidation to a CH2OH [δH 3.86 (1H, brd, J = 11.0 Hz), 3.65 (1H, dd, J = 11.0, 7.8 Hz)]. The 13C NMR spectrum of 10 (Table 3), assigned with the help of HSQC data, while confirming the presence of ring A 2,3-en-1-one [δC 204.2 (C-1), 145.3 (CH-3), 128.0 (CH-2)], ring B 5,6-ene [δC 135.1 (C-5), 125.0 (CH-6)], and four methyls [δC 20.8 (CH3-21), 18.9 (CH3-19), 17.7 (CH3-18), 14.3 (CH3-27)], suggested that it contains a saturated δ-lactone [δC 177.8 (C-26)], five oxygenated carbons [δC 84.4 (C-14), 81.4 (CH-23), 77.2 (C-20), 76.7 (CH-22), 63.2 (CH2-28)], and a methine carbon (C-17) at 49.8 ppm. These assignments were supported by the long-range correlations of H3-19/C-1, H3-19/C-6, H-3/C-1, and H-4/C-6 for rings A/B; H3-18/C-14, H3-18/C-17, and H-17/C-21 for ring D; and H3-27/C-26, H3-27/C-24, H-23/C-25, H-23/C-28, and H-22/C-21 for ring E in the HMBC spectrum of 10 (Figure S52, Supporting Information). The orientation of the side chain at C-17 was determined to be β by the NOESY correlation between H3-18 and H3-21. Additional NOESY correlations observed for the ring E protons, H-23α with H-22α, H2-28α, and H-25α of 10 (Figure S53, Supporting Information), suggested that OH-23, H-24, and CH3-25 are β-oriented. The positive Cotton effect at 278 nm in the ECD spectrum of 10 (Figure S61, Supporting Information) suggested a 22R configuration.21 Thus, withanolide 10 was identified as 24,25-dihydro-23β,28-dihydroxywithanolide G [(20S,22R)-14α,20,23β,28-tetrahydroxy-1-oxowitha-2,5-dienolide].
The molecular formula of withanolide 11 (C28H40O7) and its 1H and 13C NMR data (Tables 2 and 3) indicated that it is an analogue of withanolide E (13) in which the double bond of ring E has undergone reduction. The 13C NMR data for 11 suggested the presence of a carbonyl of a saturated δ-lactone ring (δC 175.4), and its 1H NMR spectrum showed doublets for CH3-27 [δH 1.21 (3H, d, J = 6.8 Hz)] and CH3-28 [δH 1.12 (3H, d, J = 6.8 Hz)]. These assignments for CH3-27 and CH3-28 were confirmed by the long-range correlations of H3-27/C-26, H2-23/C-25, and H2-23/C-28 observed in its HMBC spectrum (Figure S57, Supporting Information). The relative configuration of these methyl groups was established by NOESY correlations of H3-28/H-25 [δH 2.16 (1H, m)], H3-28/H-22, and H-22/H-25 (Figure S58, Supporting Information), suggesting that the saturated δ-lactone of ring E of 11 possesses the same twist boat conformation, with equatorial orientations of CH3-27 and CH3-28, as in philadelphicalactone A.23 The absolute configuration of C-22 was determined as R from the positive Cotton effect at 278 nm in its ECD spectrum (Figure S61, Supporting Information).21 On the basis of the foregoing evidence, the structure of withanolide 11 was established as 24, 25 - dihydrowithanolide E [(20S,22R,24S,25R)-5β,6β-epoxy-14α,17β,20-trihydroxy-1-oxowitha-2-enolide].
Withanolides 1–25 were evaluated for their cytotoxic activity in a panel of selected tumor cell lines consisting of LNCaP (androgen-sensitive human prostate adenocarcinoma), 22Rv1 (androgen-resistant human prostate adenocarcinoma), ACHN (human renal adenocarcinoma), M14 (human melanoma), SK-MEL-28 (human melanoma), and normal HFF (human foreskin fibroblast) cells. Physachenolide C (26), which we have previously shown to exhibit selective and potent activity for LNCaP cells,15,22 was used for comparison purposes. Of the withanolides tested, only 17β-hydroxywithanolides (17-BHWs), 6, 8, 9, 11–13, 15, 19–22, and 26, exhibited cytotoxicity to LNCaP cells at a concentration below 2.0 μM. The IC50 data for the active withanolides are presented in Table 4. Interestingly, the order of activity of these 17-BHWs (13 > 20 > 22 > 12 > 21 > 11 > 8 > 15 > 19 > 9) was found to be almost the same for both PC cell lines, LNCaP and 22Rv1, except for 6, with a β-OH group at C-23, which was active against 22Rv1 cells with an IC50 of 0.89 ± 0.1 μM, but had no activity for LNCaP cells at 2.0 μM. The cytotoxicity data for LNCaP cells for these 17-BHWs suggested that a ring A 2,3-en-1-one and α-oriented unsaturated δ-lactone side chain and/or a β-OH at C-17 are essential for their activity and acetoxylation at C-18 leads to 3-fold enhancement of activity (26 vs 13). These data also provided additional structure–activity relationship (SAR) information for 17-BHWs, including decreases in activity due to (i) isomerization of the 2,3-ene to 3,4-ene (8 vs 9); (ii) β-hydroxylation at C-4 (12 vs 13); (iii) β-methoxylation at C-4 (15 vs 13); (iv) replacement of the 5β,6β-epoxide with 5,6-ene (22 vs 13); (v) β-hydroxylation at C-7 (19 vs 13 and 8 vs 22); (vi) reduction of the 24,25-double bond of the side chain (11 vs 13); and (vii) hydroxylation at C-28 (21 vs 22). Intriguingly, of the 26 withanolides tested, only three (13, 20, and 26) showed selectivity for renal carcinoma (ACHN) cells (Table 4). Although there appears to be no direct correlation between cytotoxic activities for ACHN and LNCaP cells, it is noteworthy that withanolides 13, 20, and 26, found to be active against ACHN cells, are those with the highest potency for LNCaP cells. Nonetheless, the prostate cancer cell lines were still much more sensitive to the effects of active 17-BHWs. In addition to supporting our previous findings that α-orientation of the side chain and/or the presence of a β-OH at C-17 of withanolides may have a significant influence on their selectivity to certain cancer cell lines, these SAR data for 17-BHWs suggest the importance of the structure of the side chain for their activity and potency.
Table 4.
Cytotoxicity Data of Withanolides from Aeroponically Cultivated Physalis peruviana against a Panel of Selected Tumor Cell Lines and Normal Cellsa
| cell lineb | ||||||
|---|---|---|---|---|---|---|
| compound | LNCaP | 22Rv1 | ACHN | M14 | SK-MEL-28 | HFF |
| 6 | >2 | 0.89 ± 0.10 | >2 | >2 | >2 | >2 |
| 8 | 0.33 ± 0.04 | 0.56 ± 0.01 | >2 | >2 | >2 | >2 |
| 9 | 0.94 ± 0.10 | 0.99 ± 0.08 | >2 | >2 | >2 | >2 |
| 11 | 0.29 ± 0.05 | 0.21 ± 0.02 | >2 | >2 | >2 | >2 |
| 12 | 0.16 ± 0.04 | 0.09 ± 0.01 | >2 | >2 | >2 | >2 |
| 13 | 0.06 ± 0.01 | 0.07 ± 0.01 | 0.46 ± 0.06 | >2 | >2 | >2 |
| 15 | 0.33 ± 0.02 | 0.24 ± 0.03 | >2 | >2 | >2 | >2 |
| 19 | 0.33 ± 0.02 | 0.26 ± 0.02 | >2 | >2 | >2 | >2 |
| 20 | 0.11 ± 0.01 | 0.07 ± 0.01 | 1.0 ± 0.08 | >2 | >2 | >2 |
| 21 | 0.24 ± 0.02 | 0.21 ± 0.02 | >2 | >2 | >2 | >2 |
| 22 | 0.13 ± 0.10 | 0.07 ± 0.02 | >2 | >2 | >2 | >2 |
| 26 | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.57 ± 0.06 | >2 | >2 | >2 |
| doxorubicin | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.09 ± 0.01 | 0.25 ± 0.01 | 0.15 ± 0.03 |
Results are expressed as IC50 values in μM. Doxorubicin and DMSO were used as positive and negative controls. Withanolides 1–5, 7, 10, 14, 16–18, and 23–25 had no activity up to 5.0 μM.
Key: LNCaP = androgen-sensitive human prostate adenocarcinoma; 22Rv1 = androgen-resistant human prostate adenocarcinoma; ACHN = human renal adenocarcinoma; M14 = human melanoma; SK-MEL-28= human melanoma; HFF = human foreskin fibroblast.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured with a JASCO Dip-370 polarimeter using MeOH as the solvent. UV spectra were recorded with a Shimadzu UV 2601 spectrophotometer. ECD spectra were measured with a JASCO J-810 spectropolarimeter. 1D and 2D NMR spectra were recorded in CDCl3 using residual solvent as the internal standard on a Bruker Avance III 400 spectrometer at 400 MHz for 1H NMR and 100 MHz for 13C NMR, respectively. The chemical shift values (δ) are given in parts per million (ppm), and the coupling constants (J values) are in Hz. Low-resolution MS were recorded on a Shimadzu LCMS-QP8000α and high-resolution MS on a Agilent G6224A TOF mass spectrometer. Analytical thin-layer chromatography (TLC) was carried out on silica gel 60 F254 aluminum-backed TLC plates (Merck), and preparative TLC was performed on Analtech silica gel 500 μm glass plates. Compounds were visualized with short-wave UV and by spraying with anisaldehyde-sulfuric acid reagent and heating until the spots appeared. Silica gel flash chromatography was accomplished using 230–400 mesh silica gel. Sephadex LH-20 for gel-permeation chromatography was obtained from Amersham Biosciences. HPLC purifications were carried out using a 10 × 250 mm Phenomenex Luna 5 μm C18 (2) column for reversed-phase (RP) chromatography and a 10 × 250 mm Econosil Si (10 μ) column for normal-phase (NP) chromatography, with a Waters Delta Prep system consisting of a PDA 996 detector. When required, MM2 energy minimizations of all possible conformers were carried out using Chem3D 15.0 from PerkinElmer, Inc.
Aeroponic Cultivation and Harvesting of P. peruviana.
The seeds of P. peruviana obtained from Trade Wind Fruit (P.O. Box 1102, Windsor, CA, USA) were germinated in 1.0 in. Grodan rock-wool cubes in a Barnstead Lab-line growth chamber kept at 28 °C with 16 h of fluorescent lighting and maintaining 25–50% humidity. After ca. 4 weeks in the growth chamber, seedlings with an aerial length of ca. 5.0 cm were transplanted to aeroponic culture boxes for further growth, as described previously for Withania somnifera and Physalis crssifolia.13,24 A herbarium sample was deposited at the University of Arizona Natural Products Center (accession number NPC-DB-11/5/10). Aerial parts of areoponically grown plants were harvested when fruits were almost mature (ca. 2 months under aeroponic growth conditions). Harvested plant materials were dried in the shade, powdered, and stored at 5 °C prior to extraction.
Extraction and Isolation of Withanolides.
Dried and powdered aerial parts of aeroponically grown P. peruviana (1.2 kg) were divided into four portions, and each portion was extracted with 60% aqueous MeOH (3.0 L) in an ultrasonic bath at 25 °C for 2 h and then allowed to stand for 8 h. After filtration, the resulting filtrates were combined and passed through a column packed with HP-20SS resin (2.0 kg). The column was first eluted with 60% MeOH(aq) (4.0 L) followed by MeOH (5.0 L). The crude withanolide extract (24.5 g) obtained by concentrating the MeOH eluates was subjected to column chromatography on RP C18 (400.0 g) and eluted with 2.0 L each of 50%, 60%, and 70% MeOH(aq) and MeOH to afford five fractions, A–E: A (3.5 g) eluted with 50% MeOH(aq); B (4.2 g) with 60% MeOH(aq); C (3.1 g) with 60–70% MeOH(aq); D (4.3 g) with 70% MeOH(aq); and E (8.2 g) with MeOH. Of these, fractions B–D, which contained withanolides, were further purified as described below. Fraction B (4.2 g) on purification by column chromatography over silica gel (200 g) and elution with EtOAc gave 4β-hydroxywithanolide E (12) (1.6 g) and an impure fraction (242 mg). This impure fraction was purified by RP HPLC [60% MeOH(aq)] followed by silica gel preparative TLC [CHCl3–MeOH (95:5)] to afford 1 (6.2 mg, Rf 0.4) and additional 4β-hydroxywithanolide E (12) (181 mg). A portion of fraction C (2.0 g) was further fractionated by silica gel (250 g) column chromatography and elution with EtOAc (1.0 L), EtOAc–MeOH (98:2) (1.0 L), and MeOH (500 mL), affording seven subfractions, C1–C7: C1 (125.0 mg) eluted with EtOAc; C2 (610.0 mg) with EtOAc; C3 (95.0 mg) with EtOAc; C4 (285.0 mg) with EtOAc; C5 (195.0 mg) with EtOAc–MeOH (98:2); C6 (67 mg) with EtOAc–MeOH (98:2); and C7 (550.0 mg) with MeOH. A portion of subfraction C2 (400.0 mg) was subjected to further fractionation by silica gel (20 g) column chromatography and elution with CHCl3–MeOH (99:1) (200 mL), CHCl3–MeOH (98:2) (200 mL), and CHCl3–MeOH (95:5) (200 mL). The resulting fractions were combined based on their TLC profiles to afford nine subfractions (C2‑1–C2‑9). On further purification by RP HPLC, subfraction C2‑2 (8.5 mg) afforded 14 [4.0 mg, tR 25.0 min, 55% MeOH(aq)]; subfraction C2‑4 (37.0 mg) gave 22 [1.4 mg, tR 33.0 min, 65% MeOH(aq)], 25 [2.0 mg, tR 22.0 min, 65% MeOH(aq)], and 13 [16.0 mg, tR 17.0 min, 65% MeOH(aq)]; subfraction C2‑5 (26.0 mg) afforded 3 [8.8 mg, tR 24.0 min, 60% MeOH(aq)] and 23 [2.1 mg, tR 14.0 min, 60% MeOH(aq)]. Subfraction C2‑6 (95.0 mg) was fractionated by column chromatography on silica gel (20.0 g), and elution with EtOAc provided three subfractions, C2‑6‑1 (40.0 mg), C2‑6‑2 (17.0 mg), and C2‑6‑3 (17.0 mg). Further purification of subfraction C2‑6‑1 (40 mg) by RP HPLC afforded 17 [3.3 mg, tR 11.0 min, 65% MeOH(aq)] and 24 [20.0 mg, tR 18.0 min, 65% MeOH (aq)]. When purified by RP HPLC, subfraction C2‑6‑1 gave 5 [1.2 mg, tR 24.0 min, 55% MeOH(aq)], 6 [3.3 mg, tR 32.0 min, 55% MeOH(aq)], and 3 [1.5 mg, tR 36.0 min, 55% MeOH(aq)]. Fraction C2‑7 (170.0 mg) was subjected to column chromatography on silica gel (20.0 g) and eluted with CHCl3–MeOH (95:5) to afford a withanolide-containing fraction (152.0 mg). Further purification of this fraction (20.0 mg) by silica gel preparative TLC (EtOAc) gave 20 (11.6 mg, Rf 0.4) and 21 (1.8 mg, Rf 0.5). Subfraction C2‑8 (24.0 mg) was subjected to column chromatography on silica gel (8 g), and elution with EtOAc afforded 8 (15.0 mg) and a fraction (5.2 mg) that was further purified by RP HPLC [60% MeOH(aq)] to give 9 (2.7 mg, tR 29.0 min). A portion of subfraction C4 (150.0 mg) was purified by column chromatography over silica gel (8 g) and elution with CH2Cl2–MeOH (95:5) to obtain a fraction (52.6 mg), which, on further purification by RP HPLC using 60% MeOH(aq), afforded 19 (27.0 mg, tR 20.0 min) and 8 (11.0 mg, tR 25.0 min). Purification of subfraction C5 by column chromatography over silica gel (8 g) and elution with CH2Cl2–MeOH (9:1) afforded 18 (35.0 mg). Fraction D (4.0 g) was separated by column chromatography over silica gel (250 g), and elution with EtOAc afforded withanolide E (13, subfraction D2, 2.2 g) and two subfractions, D1 (245.0 mg) and D3 (108.0 mg). A portion (100.0 mg) of subfraction D1 was fractionated by RP HPLC using 65% MeOH(aq) to yield three subfractions, D1‑1 (35.8 mg, tR 18.0 min), D1‑2 (6.2 mg, tR 22.0 min), and D1‑3 (4.7 mg, tR 25.0 min). Subfraction D1‑1 was further purified by column chromatography on silica gel (8 g), and elution with CHCl3–MeOH (95:5) afforded 3 (1.5 mg), 7 (3.3 mg), and 24 (20 mg). Purification of subfractions D1‑2 by preparative TLC [silica gel; CHCl3–MeOH (95:5)] gave 2 (4.8 mg), and purification of D1‑3 by the same procedure afforded 4 (2.1 mg) and 25 (2.0 mg). Further purification of subfraction D3 by RP HPLC using 65% MeOH(aq) yielded three subfractions: D3‑1 (50.1 mg), D3‑2 (25.2 mg), and D3‑3 (24.4 mg), and subsequent purification of these subfractions by column chromatography over silica gel with CHCl3–MeOH (99:1 and 98:2) gave 16 (10.5 mg) and 18 (6.1 mg) from subfraction D3‑1, 10 (3.5 mg) and 11 (1.5 mg) from subfraction D3‑2, and 15 (1.4 mg) and 25 (10.2 mg) from subfraction D3‑3.
Perulactone I (1):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 223 (3.81) nm; ECD (MeOH) [θ] −4345 (242 nm), +3766 (340 nm); 1H and 13C NMR data, see Tables 1 and 3, respectively; positive HRESIMS m/z 527.2620 [M + Na]+ (calcd for C28H40O8Na, 527.2621).
Perulactone J (2):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 224 (3.76) nm; ECD (MeOH) [θ] +7461 (254 nm), −7491 (339 nm); 1H and 13C NMR data, see Tables 1 and 3, respectively; positive HRESIMS m/z 495.2720 [M + Na]+ (calcd for C28H40O6Na, 495.2723).
Perulactone K (3):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 222 (3.89) nm; ECD (MeOH) [θ] −2170 (242 nm), +1331 (264 nm), +2031 (342 nm); 1H and 13C NMR data, see Tables 1 and 3, respectively; positive HRESIMS m/z 493.2554 [M + Na]+ (calcd for C28H38O6Na, 493.2566).
Perulactone L (4):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 230 (3.84) nm; ECD (MeOH) [θ] +10 095 (245 nm), −1400 (310 nm), −710 (342 nm, sh.); 1H and 13C NMR data, see Tables 1 and 3, respectively; positive HRESIMS m/z 511.2670 [M + Na]+ (calcd for C28H40O7Na, 511.2672).
17-Deoxy-23β-hydroxywithanolide E (5):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 223 (4.02) nm; ECD (MeOH) [θ] −13 854 (231 nm), +440 (266 nm), +1858 (339 nm); 1H and 13C NMR data, see Tables 1 and 3, respectively; positive HRESIMS m/z 509.2502 [M + Na]+ (calcd for C28H38O7Na, 509.2515).
23β-Hydroxywithanolide E (6):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 221 (3.97) nm; ECD (MeOH) [θ] −8240 (239 nm), +187 (264 nm), +2728 (339 nm); 1H and 13C NMR data, see Tables 2 and 3, respectively; positive HRESIMS m/z 525.2449 [M + Na]+ (calcd for C28H38O8Na, 525.2464).
4-Deoxyphyperunolide A (7):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 228 (4.06) nm; ECD (MeOH) [θ] +11 244 (257 nm), +6149 (341 nm); 1H and 13C NMR data, see Tables 2 and 3, respectively; positive HRESIMS m/z 491.2408 [M + Na]+ (calcd for C28H36O6Na, 491.2410).
7β-Hydroxywithanolide F (8):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 225 (4.12) nm; ECD (MeOH) [θ] +27 006 (252 nm), −7184 (338 nm); 1H and 13C NMR data, see Tables 2 and 3, respectively; positive HRESIMS m/z 509.2496 [M + Na]+ (calcd for C28H38O7Na, 509.2515).
7β-Hydroxy-17-epi-withanolide K (9):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 224 (4.15) nm; ECD (MeOH) [θ] +22 189 (254 nm), −2517 (307 nm); 1H and 13C NMR data, see Tables 2 and 3, respectively; positive HRESIMS m/z 509.2509 [M + Na]+ (calcd for C28H38O7Na, 509.2515).
24,25-Dihydro-23β,28-dihydroxywithanolide G (10):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 223 (3.65) nm; ECD (MeOH) [θ] +5674 (254 nm), −8022 (307 nm); 1H and 13C NMR data, see Tables 2 and 3, respectively; positive HRESIMS m/z 511.2667 [M + Na]+ (calcd for C28H40O7Na, 511.2672).
24,25-Dihydrowithanolide E (11):
amorphous, colorless powder; ; UV (MeOH) λmax (log ε) 224 (3.76) nm; ECD (MeOH) [θ] −2660 (242 nm), +1541 (278 nm), +3916 (339 nm); 1H and 13C NMR data, see Tables 2 and 3, respectively; positive HRESIMS m/z 511.2660 [M + Na]+ (calcd for C28H40O7Na, 511.2672).
Epoxidation of Perulactone B (24).
To a stirred solution of 24 (2.0 mg) in CH2Cl2 (1.0 mL) was added m-chloroperbenzoic acid (2.0 mg), and the mixture was stirred at 25 °C. After 3 h (TLC control), the reaction mixture was partitioned between CHCl3 (5.0 mL) and water (10.0 mL). The CHCl3 layer was washed with water (10.0 mL), dried (Na2SO4), and subjected to purification by silica gel preparative TLC [CHCl3−MeOH (95:5)] to afford the major β-epoxidation product as an off-white, amorphous powder (1.0 mg). The 1H and 13C NMR and APCIMS data of this product were identical with those of 1 obtained above.
Conversion of Perulactone B (24) to Perulactone L (4).
To a stirred solution of 24 (2.0 mg) in MeOH (1.0 mL) was added NaCN (1.0 mg), and the mixture was stirred at 25 °C. After 2 h (TLC control), the reaction mixture was concentrated using a flow of N2, and the residue thus obtained was dissolved in CHCl3 (2.0 mL). The CHCl3 solution was concentrated and was separated by silica gel preparative TLC [CHCl3–MeOH (95:5)] to afford the product as an off-white, amorphous powder (1.9 mg). The APCIMS and 1H and 13C NMR data of this product were identical with those of 4 obtained above.
Cytotoxicity Assay.
The ACHN, M14, and SK-MEL-28 cells were all obtained from the Developmental Therapeutics Program (NCI, Frederick, MD, USA). LNCaP and HFF cells were purchased from ATCC (Manassas, VA, USA), and the 22Rv1 cells were kindly provided by Dr. Len Neckers (NCI, Bethesda, MD, USA). Cell lines were maintained as recommended by the source institution. Cell numbers were estimated using the MTS dye assay (Promega, Madison, WI, USA).25 The MTS dye assay was used for evaluating cytotoxicity of withanolides 1–26 against androgen-sensitive human prostate adenocarcinoma (LNCaP), androgen-resistant human prostate adenocarcinoma (22Rv1), human renal adenocarcinoma (ACHN), human melanoma (M14 and SK-MEL-28) cell lines, and human foreskin fibroblast cells. Doxorubicin and DMSO were used as positive and negative controls, respectively. For LNCaP, ACHN, 22Rv1, and HFF cells, the assay was performed in RPMI with 5% fetal calf serum, 2.0 mM l-glutamine, 1× nonessential amino acids, 1.0 mM sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin, 10 mM HEPES, and 5 × 10−5 M 2-mercaptoethanol. For M14 and SK-MEL-28 cells, DMEM replaced RPMI. Briefly, 10 000 cells (LNCaP) or 5000 cells (22Rv1, ACHN, M14, SK-MEL-28, HFF) were incubated overnight at 37 °C in 96-well microtiter plates. Serial dilutions of compounds in DMSO or vehicle control (DMSO) were added to triplicate wells, and the microtiter plates were incubated for a further 72 h. Viable cell number was determined by the addition of CellTiter 96 AQueous One Solution cell proliferation assay solution (MTS), plates were incubated for 2 h, and then absorbance (A) at 490 nM was measured. Cytotoxicity was calculated as in the following formula: % cytotoxicity = [(AMedium – ATreatment)/AMedium)] × 100. The IC50 values and standard deviations (±) were determined using Microsoft Excel software from dose–response curves obtained from at least three independent experiments.
Supplementary Material
Figure 1.
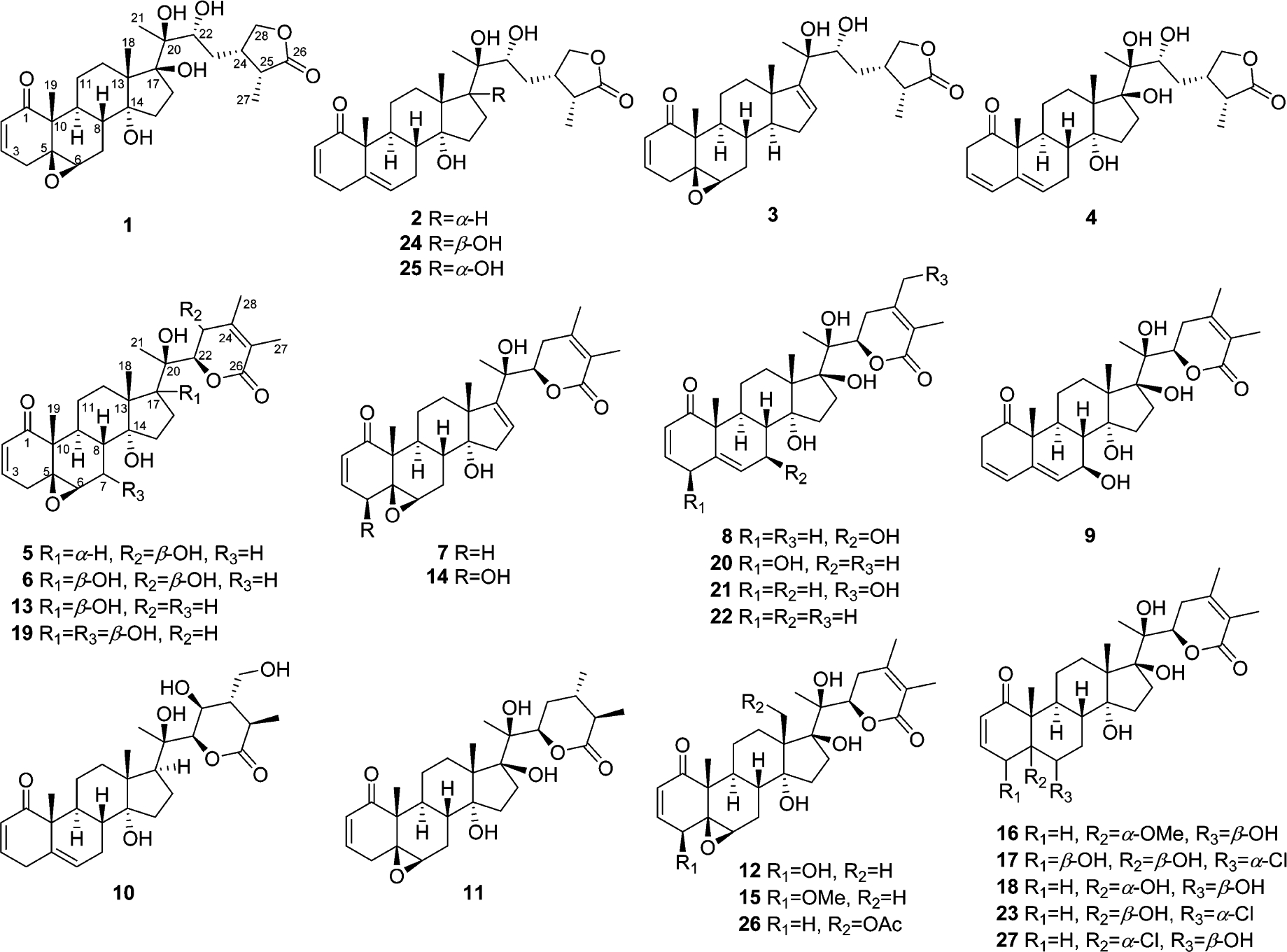
Structures of withanolides 1–25 from aeroponically grown P. peruviana, physachenolide C (26), and withanolide C (27).
ACKNOWLEDGMENTS
Financial support for this work from the College of Agriculture and Life Sciences and the School of Natural Resources and the Environment of the University of Arizona is gratefully acknowledged. This project has also been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN26120080001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. We thank Mr. D. Bunting for his help with germination and aeroponic cultivation of P. peruviana used in this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.6b01129.
1H, 13C, and 2D NMR spectra of withanolides 1–11, key HMBC and COSY correlations for 1, 3, 5, and 7–11, key NOESY correlations for 1, 3–5, and 7–11, and ECD spectra for 1–11 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Schrijvers D; Van Erps P; Cortvriend J Adv. Ther 2010, 27, 285–296. [DOI] [PubMed] [Google Scholar]
- (2).Jemal A; Bray F; Center MM; Ferlay J; Ward E; Forman D Ca-Cancer J. Clin 2011, 61, 69–90. [DOI] [PubMed] [Google Scholar]
- (3).Phillips C NCI Cancer Bull. 2012, 9, 2. [Google Scholar]
- (4).Asangani IA; Dommeti VL; Wang X; Malik R; Cieslik M; Yang R; Escara-Wilke J; Wilder-Romans K; Dhanireddy S; Engelke C; Iyer MK; Jing X; Wu Y-M; Cao X; Qin ZS; Wang S; Feng FY; Chinnaiyan AM Nature 2014, 510, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sanford M Drugs 2013, 73, 1723–1732. [DOI] [PubMed] [Google Scholar]
- (6).(a) Zegarra-Moro OL; Schmidt LJ; Huang H; Tindall DJ Cancer Res. 2002, 62, 1008–1013. [PubMed] [Google Scholar]; (b) Chen CD; Welsbie DS; Tran C; Baek SH; Chen R; Vessella R; Rosenfeld MG; Sawyers CL Nat. Med 2004, 10, 33–39. [DOI] [PubMed] [Google Scholar]; (c) Attard G; Richards J; de Bono JS Clin. Cancer Res 2011, 17, 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tan MHE; Li J; Xu HE; Melcher K; Yong E Acta Pharmacol. Sin 2015, 36, 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Key Statistics about Kidney Cancer. American Cancer Society Home Page. http://www.cancer.org/cancer/kidneycancer/detailedguide/kidney-cancer-adult-key-statistics.
- (8).(a) McDermott DI; Atkins MB I Semin. Oncol 2006, 33, 583–587. [DOI] [PubMed] [Google Scholar]; (b) Cohen RB; Oudard S Invest. New Drugs 2012, 30, 2066–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang W-N; Tong W-Y Chem. Biodiversity 2016, 13, 48–65. [DOI] [PubMed] [Google Scholar]
- (10).Chen LX; He H; Qiu F Nat. Prod. Rep 2011, 28, 705–740. [DOI] [PubMed] [Google Scholar]
- (11).(a) Baumann TW; Meier CM Phytochemistry 1993, 33, 317–321. [Google Scholar]; (b) Dinan LN; Sarker SD;Šik V Phytochemistry 1997, 44, 509–512. [Google Scholar]; (c) Ahmad S; Malik A; Muhammad P; Gul W; Yasmin R; Afza N Fitoterapia 1998, 69, 433–436. [Google Scholar]; (d) Ahmad S; Yasmin R; Malik A Chem. Pharm. Bull 1999, 47, 477–480. [Google Scholar]; (e) Fang S-T; Li B; Liu J-K Helv. Chim. Acta 2009, 92, 1304–1308. [Google Scholar]; (f) Lan Y-H; Chang F-R; Pan M-J; Wu C-C; Wu S-J; Chen S-L; Wang S-S; Wu M-J; Wu Y-C Food Chem. 2009, 116, 462–469. [Google Scholar]; (g) Fang S-T; Liu J-K; Bo LJ Asian Nat. Prod. Res 2010, 12, 618–622. [DOI] [PubMed] [Google Scholar]; (h) Yen C-Y; Chiu C-C; Chang F-R; Chen JY-F; Hwang C-C; Hseu Y-C; Yang H-L; Lee AY-L; Tsai M-T; Guo Z-L; Cheng Y-S; Liu Y-C; Lan Y-H; Chang Y-C; Ko Y-C; Chang H-W; Wu Y-C BMC Cancer 2010, 10, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fang S-T; Liu J-K; Li B Steroids 2012, 77, 36–44. [DOI] [PubMed] [Google Scholar]; (j) Wang H-C; Tsai Y-L; Wu Y-C; Chang F-R; Liu M-H; Chen W-Y; Wu C-C PLoS One 2012, 7, e37764. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Chiu C-C; Huang J-W; Chang F-R; Huang K-J; Huang H-M; Huang H-W; Chou C-K; Wu Y-C; Chang H-W PLoS One 2013, 8, e64739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) You B-J; Wu Y-C; Lee C-L; Lee H-Z Food Chem. Toxicol 2014, 65, 205–212. [DOI] [PubMed] [Google Scholar]; (m) Sang-Ngern M; Youn UJ; Park E-J; Kondratyuk TP; Simmons CJ; Wall MM; Ruf M; Lorch SE; Leong E; Pezzuto JM; Chang LC Bioorg. Med. Chem. Lett 2016, 26, 2755–2759. [DOI] [PubMed] [Google Scholar]; (n) Park E-J; Sang-Ngern M; Chang LC; Pezzuto JM Mol. Nutr. Food Res 2016, 60, 1482–1500. [DOI] [PubMed] [Google Scholar]
- (12).Henrich CJ; Brooks AD; Erickson KL; Thomas CL; Bokesch HR; Tewary P; Thompson CR; Pompei RJ; Gustafson KR; McMahon JB; Sayers TJ Cell Death Dis. 2015, 6, e1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xu YM; Bunting DP; Liu MX; Bandaranayake HA; Gunatilaka AA L. J. Nat. Prod 2016, 79, 821–830. [DOI] [PubMed] [Google Scholar]
- (14).(a) Sahai M; Gottlieb HE; Ray AB; Ali A; Glotter E; Kirson IJ Chem. Res. (S) 1982, 346–347. [Google Scholar]; (b) Gottlieb HE; Kirson I; Glotter E; Ray AB; Sahai M; Ali AJ Chem. Soc., Perkin Trans I 1986, 229–231. [Google Scholar]; (c) Ozawa M; Morita M; Hirai G; Tamura S; Kawai M; Tsuchiya A; Oonuma K; Maruoka K; Sodeoka M ACS Med. Chem. Lett 2013, 4, 730–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Damu AG; Kuo P-C; Su C-R; Kuo T-H; Chen T-H; Bastow KF; Lee K-H; Wu T-SJ Nat. Prod 2007, 70, 1146–1152. [DOI] [PubMed] [Google Scholar]
- (16).Zviely M; Goldman A; Kirson I; Glotter EJ Chem. Soc., Perkin Trans 1 1986, 229–231. [Google Scholar]
- (17).Frolow F; Ray AB; Sahai M; Glotter E; Gottlieb HE; Kirson IJ Chem. Soc., Perkin Trans 1 1981, 1029–1032. [Google Scholar]
- (18).(a) Glotter E; Abraham A; Günzberg G; Kirson IJ Chem. Soc., Perkin Trans 1 1977, 341–346. [Google Scholar]; (b) Vitali G; Conte L; Nicoletti M Planta Med. 1996, 62, 287–288. [DOI] [PubMed] [Google Scholar]
- (19).Shingu K; Miyagawa M; Yahara S; Nohara T Chem. Pharm. Bull 1993, 41, 1873–1875. [Google Scholar]
- (20).Velde VV; Lavie D; Budhiraja RD; Sudhir S; Garg KN Phytochemistry 1983, 22, 2253–2257. [Google Scholar]
- (21).Moiseeva GP; Vasina OE; Abubakirov NK Khim. Prir. Soedin 1990, 3, 371–376. [Google Scholar]
- (22).Xu YM; Liu MX; Grunow N; Wijeratne EMK; Paine-Murrieta G; Felder S; Kris RM; Gunatilaka AA L. J. Med. Chem 2015, 58, 6984–6993. [DOI] [PubMed] [Google Scholar]
- (23).Su B-N; Misico R; Park EJ; Santarsiero BD; Mesecar AD; Fong HHS; Pezzuto JM; Kinghorn AD Tetrahedron 2002, 58, 3453–3466. [Google Scholar]
- (24).Xu YM; Gao S; Bunting DP; Gunatilaka AA L. Phytochemistry 2011, 72, 518–522. [DOI] [PubMed] [Google Scholar]
- (25).Brooks AD; Jacobsen KM; Li W; Shankar A; Sayers TJ Mol. Cancer Res 2010, 8, 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.